

Abstract
Coxsackievirus B3 (CVB3), an enterovirus in the Picornavirus family, is the most common human pathogen associated with myocarditis and idiopathic dilated cardiomyopathy. We found upregulation of the cysteine-rich protein gene (cyr61) after CVB3 infection in HeLa cells with a cDNA microarray approach, which is confirmed by Northern blot analysis. It is also revealed that the extracellular amount of Cyr61 protein was increased after CVB3 infection in HeLa cells. cyr61 is an early-transcribed gene, and the Cyr61 protein is secreted into the extracellular matrix. Its function is related to cell adhesion, migration, and neuronal cell death. Here, we show that activation of the cyr61 promoter by CVB3 infection is dependent on JNK activation induced by CVB3 replication and viral protein expression in infected cells. To explore the role of Cyr61 protein in infected HeLa cells, we transiently overexpressed cyr61 and infected HeLa cells with CVB3. This increased CVB3 growth in the cells and promoted host cell death by viral infection, whereas down-expression of cyr61 with short interfering RNA reduced CVB3 growth and showed resistance to cell death by CVB3 infection. In conclusion, we have demonstrated a new role for cyr61 in HeLa cells infected with CVB3, which is associated with the cell death induced by virus infection. These data thus expand our understanding of the physiological functions of cyr61 in virus-induced cell death and provide new insights into the cellular factors involved.
Coxsackievirus B3 (CVB3) is a picornaviruses with a single-stranded, positive-sense RNA about 7,400 nucleotides in length. Its genome encodes four capsid proteins and seven nonstructural proteins (43). CVB3 induces acute and chronic viral myocarditis in children and young people (18), causing about 5% to 50% of all cases, and, its end-stage, dilated cardiomyopathy (33). However, the pathophysiological mechanism of viral myocarditis is still a matter of debate. Direct apoptosis induced by CVB3 and indirect autoimmune responses induced by cytokines in the infected heart remain attractive but unconfirmed hypotheses to explain the outcome. Infecting viruses are likely to use existing cell signals and factors for their growth. Generally, viruses in infected cells interact with host cellular proteins and interfere with cellular protein production to enable viral replication and propagation. Therefore, understanding the virus-host relationship is important if we are to understand the cellular responses against viral infections and the pathological mechanisms and processes of viral diseases.
HeLa cells infected with CVB3 induce a direct cytopathic effect and cell apoptosis (7, 20). Cytopathic effect colocalized with viral replication in cells is responsible for direct damage. The immediate host gene responses to CVB3 infection may have a role in determining the severity of myocarditis and disease progression to dilated cardiomyopathy (41, 47). Despite many molecular and serological studies, little is known about the pathogenesis of CVB3 or CVB3-host interactions because of the technical problems in assessing many cellular gene profiles simultaneously. The recent development of cDNA microarray techniques promises to overcome this challenge (6). This technique has been applied to the study of the effects of viral infection on the expression of cellular genes (13, 36) and has been applied to cardiac muscle tissues of mice infected with CVB3 (39, 41).
To elucidate the host cellular genes related to viral infection, we used a cDNA microarray approach to study HeLa cells infected with CVB3 and found that cyr61, a gene encoding cysteine-rich protein 61 (Cyr61), was gradually upregulated. This was confirmed by Northern blot analysis. cyr61 is an immediate-early gene and is one member of the CCN family, which comprises cyr61/CCN1, connective tissue growth factor/CCN2, nephroblastoma overexpressed/CCN3, and Wnt-induced secreted protein-1/CCN4, -2/CCN5, and -3/CCN6 (4). Cyr61 is a secreted growth regulating and heparin-binding protein that is associated with the cell surface and extracellular space (21, 37, 40). Moreover, Cyr61 is a ligand for integrins and a signaling molecule through integrin receptors (8, 14, 21). The cellular physiological roles of Cyr61 include the regulation of adhesion, migration, proliferation, differentiation, and chondrogenesis, as well as growth factor-induced mitogenesis and endothelial tubule formation (21, 30, 46). It is also involved in tumor growth (3, 25, 35) and is a deregulated in endometriosis (1). This protein also plays an important role during the process of neuronal cell death via the JNK signal pathway (20).
Here, to study the role of cyr61 in infected HeLa cells, we regulated its expression and showed that overexpression of this gene promoted viral growth and increased cell death in HeLa cells infected with CVB3, whereas down-expression of cyr61, performed with silencing RNA (siRNA) (11, 17), reduced the growth of CVB3 and cell death. We also found that the JNK signal pathway mediated transcriptional activation of cyr61 after CVB3 infection. Thus, cyr61 appears to play a role in the cell death induced by CVB3 infection in HeLa cells. As far as we know, this is the first paper to show the role of cyr61 in viral infection.
MATERIALS AND METHODS
Cells and viruses.
The cervical HeLa-UVM cancer cell line (5) (here simply termed HeLa) was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. CVB3, an H3 Woodrupt strain (donated by E. Jeon, Samsung Medical Center, Seoul, Korea), was grown and titered with HeLa cells. Viral stocks were prepared by infecting 90% confluent cultures of HeLa cells at a multiplicity of infectivity (MOI) of 10. After infection for 16 h at 37°C, the suspension was freeze-thawed three times. The cell lysates were centrifuged and the supernatants were harvested and stored at −75°C. The amount of virus was measured by plaque assay. The titer of CVB3 H3 was 109 PFU/ml.
Reagents.
DMEM, fetal bovine serum, and Lipofectamine reagents were purchased from Invitrogen (Rockville, Md.). The JNK inhibitor SP600125 was from Calbiochem (San Diego, Calif.). Protein G-Sepharose was from Amersham Biosciences (Piscataway, N.J.). The polyclonal anti-Cyr61 antibody was from Santa Cruz Biotechnology (Santa Cruz, Calif.). The monoclonal antibody to enterovirus VP1 was from Novocastra Laboratories (Newcastle, United Kingdom). Polyclonal phosphorylated anti-JNK1/2 and total anti-JNK1/2 antibodies were from Cell Signaling Technology (Beverly, Mass.). Brefeldin A, blocking protein transport, was from Sigma (St. Louis, Mo.).
Virus infection and inhibitor treatments.
HeLa cells were grown in complete medium upon reaching 70 to 80% confluence. The cells were infected with CVB3 and sham treated with serum-free medium for 1 h. Cells were washed with 1× phosphate-buffered saline (PBS) and cultured in serum-free DMEM. For inhibitor experiments, cells were incubated with the JNK inhibitor SP600125 for 1 h, infected with CVB3 for 1 h, washed with PBS, and placed in serum-free medium containing fresh inhibitor unless otherwise specified.
Transfection.
The HeLa cells were grown to 80% confluence in 35-mm dishes (Nunc, Roskilde, Denmark). Transfection was performed with the Lipofectamine reagent (Invitrogen). For each well in a transfection, Lipofectamine reagent (4 μl) was gently mixed with 10 μl of expression plasmid (approximately 1 μg) and 200 μl of OptiMEM medium (Invitrogen). The samples were incubated for 30 min at room temperature. The entire volume was added to HeLa cells that had been washed with OptiMEM and added to 800 μl of OptiMEM. The cultures were incubated at 37°C for 5 h. The transfection mixture was then removed and washed with PBS. Fresh complete medium was added to each dish. After 24 to 48 h from the start of transfection, the cells were infected with CVB3. Cell extracts were prepared at the indicated times, and the viral titers in harvested supernatants were determined by plaque assays.
Silencing RNA.
To silence the expression of cyr61, the following target regions were used: 5′-TAT AAC TCC AGA ATC TAC C-3′ (si-cyr61 1) and 5′-CTG TAA ACA TCA GTG CAC A-3′ (si-cyr61 2) (nucleotides in human cyr61). The pSilencer cyr61 silencing RNA (siRNA) sequences were constructed by ligation of the following oligonucleotide pairs to pSilencer 2.1-U6 RNAi vector (Ambion): 5′-GAT CCC G(19)TTC AAG AGA(19)TTT TTT GGA AA-3′ and 5′-AGC TTT TCC AAA AAA(19)TCT CTT GAA(19)CGG-3′. The annealed oligonucleotide duplex was transfected into cells with Lipofectin reagent (Invitrogen). Cells were seeded in 35-mm dishes and transfected at 80% confluency. Transfection efficiency was controlled with pGFP-N1 (Clontech, Palo Alto, Calif.). Experiments were performed 24 to 48 h after transfection of the siRNA. Cells were then infected with CVB3. The cell extracts were analyzed with Western blotting and fluorescence-activated cell sorting (FACS) analysis.
Western blot analysis.
For analysis by Western blotting, cell monolayers were washed with Tris-buffered saline (TBS) and scraped into ice-cold TBS with a rubber policeman. Cell were lysed in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) sample buffer and subjected to SDS-PAGE on 10% acrylamide gels. Samples were transferred to nitrocellulose by semidry blotting and membranes were blocked in 5% skim milk. The blots were then analyzed with incubation with the appropriate antibody directed against Cyr61 (1:1,000, vol/vol), VP1(1:2,000, vol/vol), and p-JNK1/2 and total JNK1/2 (1:1,000, vol/vol), followed by the corresponding secondary antibody, and developed with ECL reagents (Amersham Biosciences).
Metabolic labeling and immunoprecipitation.
Cells were seeded in 60-mm dishes at 1.5 × 106 cells per dish and grown overnight. They were incubated for 1 h in DMEM with 10% fetal bovine serum lacking methionine prior to labeling. The cells were labeled with 10 μCi of [35S] methionine (PerkinElmer, Tokyo, Japan, NEG009A) per ml and infected with CVB3 for the indicated times. The cell lysate was harvested and the recovered culture medium was concentrated with Centricon YM-30 tubes (30-kDa cutoff). The cell lysates and medium were immunoprecipitated with the polyclonal anti-Cyr61 serum and analyzed as described (9).
Plaque assay.
CVB3 titers in culture medium were determined from monolayers of HeLa cells by an agar overlay plaque assay in triplicate as previously described (2). Briefly, samples were serially diluted 10-fold, overlaid on 95% to 100% confluent monolayers of HeLa cells in six-well plates, and incubated for 1 h. The medium was removed, and 2 ml of complete DMEM containing 1% agar was overlaid in each well. Cells were incubated at 37°C for 72 h, fixed with 10% formalin for 1 h 30 min at room temperature, and stained with 1% crystal violet. Plaques were counted, and concentration was calculated as PFU per milliliter.
Fluorescence-activated cell sorting analysis.
Cell death was analyzed by staining with propidium iodide. At 24 h after transfection, cells were infected for 4 to 10 h. Fixed cells (106) were harvested by centrifugation and washed twice with PBS. Washed cells were suspended in 200 of 1× PBS containing 1 mM EDTA and treated with 0.1 mg of RNase A per ml for 30 min at room temperature. These cells were stained with 100 μg of propidium iodide per ml and analyzed within 3 h on a FACS Vantage flow cytometer (Becton Dickinson, San Jose, Calif.).
CAT as a reporter.
Cells were seeded at 3.5 × 105 cells per 35-mm dish and grown overnight. The cells were transfected with the cyr61/chloramphenicol acetyltransferase (CAT) promoter plasmids. At 24 h after the start of transfection, cells were infected with CVB3 for the indicated times. After the washing step with cold PBS, cell extracts were carefully prepared and immediately stored at −75°C. The CAT assay was carried out with an enzyme-linked immunosorbent CAT assay kit (Roche Molecular Biochemicals, Mannheim, Germany), according to the manufacturer's protocol.
Northern blot analysis.
For Northern blot analysis, total RNA was extracted with Trizol reagent (Invitrogen). Fifteen micrograms of RNA per sample was used for RNA gel electrophoresis, RNA was then blotted for 2 h onto a Hybond-N+ nylon membrane (Amersham) and cross-linked to the membrane. Hybridization was performed in a solution containing 1% bovine serum albumin, 1 mM EDTA, 1 M Na2HPO4, and 7% SDS at 65°C. For a hybridization probe, we used 350-bp amplified products of cyr61 fragments labeled with [32P]cytosine. The membranes were washed in 6× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) twice for 30 min at room temperature. Finally, this membrane was exposed to X-ray film overnight.
RESULTS
CVB3 infection in HeLa cells induces the cyr61 activation.
In this study, we used the cervical cancer cell line HeLa for CVB3 infection and to explore the relationship between host cellular factors and CVB3 because this cell line is a well-established model for CVB3 studies and moreover, some results obtained in HeLa cells have been reproduced in vivo (7, 12, 15, 19, 31, 32, 38). Therefore, to examine the molecular relationship between host and CVB3, we initially performed a cDNA microarray technique comparing infected and mock-infected HeLa cells and analyzed which cellular genes were up- or down-regulated in HeLa cells infected with CVB3 (S.-M. Kim, unpublished data).
We found that although most cellular genes were down-regulated at the time of major cell death after CVB3 infection, cyr61 was still about 1.6-fold up-regulated at 9 h postinfection after CVB3 infection in HeLa cells. To exclude any possible errors of cDNA microarray, we confirmed the increase of cyr61 RNA level with Northern blot analysis. As shown in Fig. 1A and B, cyr61 was slightly increased at 4 h and 10 h postinfection with CVB3. Furthermore, we studied the changes in extracellular secretion of the Cyr61 protein after CVB3 infection because Cyr61 protein is secreted in several cell types (46). HeLa cells were metabolically labeled with [35S]methionine and infected with CVB3 (multiplicity of infection [MOI] = 20), and cell lysates and culture medium were prepared by centrifugation. The cell lysates and culture medium were immunoprecipitated with the Cyr61 polyclonal antibodies. Figure 1C and D show that the cytoplasmic concentration of Cyr61 in cell lysates was not increased but decreased following infection time. However, the level of secreted Cyr61 in culture medium was increased at 4 to 8 h postinfection and maintained up to 10 h postinfection (Fig. 1C and D). Thus, CVB3 infection of HeLa cells up-regulates cyr61 mRNA in cells and Cyr61 protein in culture medium. Moreover, once induced inside the cells, most Cyr61 protein appears to be translocated gradually into the culture medium.
FIG. 1.

CVB3 infection induces cyr61RNA and protein. (A) HeLa cells were infected with CVB3 (MOI = 20) and total RNA was purified for Northern blot analysis to detect the expression of cyr61 in infected HeLa cells. For Northern blot analysis, 10 μg of total RNA was extracted from the cells and hybridized to a 350-bp 32P-labeled cyr61 cDNA fragment. The loading controls were 28S and 18S rRNAs which had been amplified by reverse transcription-PCR. (B) Each bar represents the amount of cyr61 RNA (after normalization to 28S and 18S rRNA) in HeLa cells infected with CVB3. The cyr61 RNA content at 0 h is set at 100% and compared with the percentage converted by the cyr61 RNA amount. (C) After labeling with [35S]methionine (see details in Materials and Methods), the HeLa cells were infected with CVB3 and harvested at the indicated times. Cyr61 protein in the cell lysates and culture medium (CM) were immunoprecipitated with polyclonal anti-Cyr61 immunoglobulin G, resolved by SDS-PAGE (10.0% gel), and visualized by autoradiography. Cyr61 synthesis in cell lysates and medium is shown in the individual autoradiographs. The loading control used was tubulin from cell lysates. (D) Each bar represents the amount of Cyr61 protein (after normalization to tubulin) in HeLa cells infected with CVB3. The Cyr61 protein contents in cell lysates and medium at 0 h were set at 100% and 1%, respectively, and compared with the percentage converted by the Cyr61 protein amount.
Induction of cyr61 expression in HeLa cells infected with CVB3 occurs via a c-Jun N-terminal kinase (JNK)-dependent pathway. Some toxic stimuli, such as etoposide, can induce the expression of cyr61 during neuronal cell death via the activation of JNK, a stress-activated protein kinase. This was shown from activation in response to inhibition of protein synthesis, and serum response factor (20). Kim et al. constructed several plasmids containing a cyr61 promoter fused with a CAT reporter (20). We used two of their plasmid constructs to assay the activation of cyr61 by CVB3 infection; a ΔBglII plasmid, containing a domain responsive to a JNK-dependent signaling pathway in its serum response factor-binding domain (SRE or CArG box), and the −1763 plasmid, which has no SRE domain. After both plasmids were transfected into HeLa cells and incubated for 24 h, CVB3 was infected, and we measured CAT activity to represent the transcriptional activation of the cyr61 promoter.
Interestingly, CAT activity in ΔBglII-transfected HeLa cells was dramatically increased up to 6 h postinfection (more than doubled compared with the control), whereas in the −1763-transfected HeLa cells there was no CAT activity after CVB3 infection (about 25% compared with controls: Fig. 2A). Thus, CVB3 infection transcriptionally induces the activation of the cyr61 promoter either by directly binding with some viral proteins or via indirect effects with some cellular transcription factors which are activated by CVB3 infection. Interestingly, because the ΔBglII plasmid contains the CArG box, but −1763 plasmid do not, which is a prerequisite domain for JNK activation of cyr61 (20), we anticipate that CVB3 infection might affect the cyr61 promoter via the JNK signaling pathway. Moreover, when ΔBglII-transfected HeLa cells were pretreated with the JNK inhibitor, SP60092 (SP), CAT activity was not induced after CVB3 infection (Fig. 2A; ΔBglII+SP). To validate these results, we cotransfected cells with the ΔBglII plasmid and with JNK1 and JNK2 dominant negative mutants, which are mammalian expression vectors encoding for a kinase-deficient JNK1 (pCMV5-mJNK1) and JNK2 (pSr-HA-JNK2) (kindly provided by S-J. Lee, Korea Institute of Radiological and Medical Sciences). These cotransfected HeLa cells showed dramatically reduced CAT activity compared with the ΔBglII-transfected cells (Fig. 2A; ΔBglII+JNK1 and ΔBglII+JNK2) after CVB3 infection. Furthermore, we explored the amount of induced cyr61 RNA by reverse transcription-PCR to confirm that induction of cyr61 RNA by CVB3 infection and reduction of this effect by SP. SP clearly reduced more than two- or threefold cyr61 RNA induction by CVB3 infection (CVB3 versus CVB3+SP) (Fig. 2B and C). Above all, the data suggest that CVB3 infection of HeLa cells transcriptionally activates cyr61 via a JNK-dependent signal pathway.
FIG. 2.

CVB3 infection transcriptionally induces the activation of cyr61. (A) CAT assay of cyr61/CAT promoter constructs after CVB3 infection in HeLa cells. ΔBglII plasmids, which contain serum response factor-binding domains (SRE or CArG box) as JNK-sensitive domains, and −1763 plasmids, which have no SRE domains, were transiently expressed in HeLa cells (1 μg of plasmid) for 24 h, and cells were infected with CVB3 (MOI = 20). SP indicates that SP600125, an inhibitor of the JNK signal pathway, was used to treat the cells for 30 min before transfection of the ΔBglII plasmid and infection with CVB3. JNK1/DN and JNK2/DN indicate the dominant negative plasmids of kinase-deficient JNK1/2, which were cotransfected with the ΔBglII plasmid for 24 h before infecting cells with CVB3. CAT activity was measured at the indicated times with an enzyme-linked immunosorbent CAT assay kit according to the manufacturer's protocol. The control was the CAT activity of uninfected HeLa cells 24 h after transfection with these recombinant plasmids. CAT activity is represented as 100% of the CAT activity of the ΔBglII plasmid in controls as the standard. The results shown are the averages of three independent assays, with error bars representing the standard deviations. (B) Effects of JNK inhibitor on cyr61 RNA induction by CVB3 infection. Cells were treated with SP for 30 min before CVB3 infection, and specific cyr61 gene expression was detected with the primer pairs cyrF/286 (5′-GAG.GGC.AGA.CCC.TGT.GAA.TA-3′) and cyrR/868 (5′-TGG.TCT.TGC.TGC.ATT.TCT.TG-3′). (C) Each bar represents the amount of cyr61 RNA after normalization to glyceraldehhyde-3-phosphate dehydrogenase (GAPDH) RNA in HeLa cells infected with CVB3. The cyr61 RNA content at 0 h was set at 100% and compared with the percentage converted by the cyr61 RNA amount.
CVB3 infection induces JNK in HeLa cells.
It was previously reported that there is a role of JNK1/2 in the activation of cyr61 (20), and herpes simplex virus infection has been shown to activate JNK and enhance viral replication (34). Moreover, the activation of JNK has been related to the induction of apoptosis (16, 26). We therefore hypothesized that CVB3 infection would induce the JNK1/2 signaling pathway. To explore this, we harvested HeLa cells infected with CVB3 at various times after infection. As shown in Fig. 3, JNK1 and -2 activation by phosphorylation was indicated by the altered molecular sizes of JNK1 and -2 (46.5 and 54 kDa, respectively) at 6 h postinfection and was maintained at 8 h postinfection. Viral protein VP1 was also first shown at 4 h postinfection (Fig. 3). This implies that JNK activation seems likely to occur after the start of CVB3 replication and the production of viral proteins, thus JNK activation might depend on CVB3 gene expression.
FIG. 3.

CVB3 infection activates the JNK1/2 signaling pathway in HeLa cells. CVB3 was used to infect HeLa cells (MOI = 20) for 1 h; infected cells were washed twice with PBS and incubated with complete DMEM (with 10% fetal bovine serum). Cell lysates were harvested at the indicated times postinfection (pi), separated by SDS-10% PAGE, and transferred to nitrocellulose membranes. JNK1/2 activations are represented by the analysis of JNK1/2 phosphorylation. The expression of CVB3 capsid protein VP1 indicated viral replication and growth in infected HeLa cells. Tubulin was used as an internal control.
Overexpression of cyr61 promotes viral growth in HeLa cells infected with CVB3.
To examine the role of cyr61 in HeLa cells infected with CVB3, we overexpressed cyr61 in HeLa cells by transfecting them with a pCA vector containing the cyr61 gene following a cytomegalovirus promoter. pCA is modified from pCK, a highly expressed mammalian vector (29). After transfection, HeLa cells overexpressed cyr61, as shown in Fig. 4A. The cells were then infected with CVB3, and the efficiency of infection in the cytoplasm was measured with Western blot analysis of the major capsid protein VP1 of CVB3, which reflects the amount of intracellular virus. HeLa cells overexpressing cyr61 (cyr61-HeLa) and infected with CVB3 (MOI = 10), showed an increase in viral protein production compared with control HeLa cells at 6 h postinfection (Fig. 4B and C). This demonstrated an increase in intracellular viral content. Moreover, when infected with 1 MOI of CVB3, the effect of timing was delayed in that the amount of viral protein produced in cyr61-HeLa cells was higher than controls at 24 h postinfection (Fig. 4B). In addition, the viral titer in culture medium of cyr61-HeLa cells was approximately two times higher than that of controls at 6 h after infection with 10 MOI of CVB3 (Fig. 4D). These data suggest that cyr61 expression promotes the proliferation of CVB3.
FIG. 4.

Overexpression of cyr61 promotes viral growth in HeLa cells infected with CVB3. (A) cyr61 was overexpressed in HeLa cells (cyr61-HeLa) by transfection with the cyr61 gene in pCA, and cell lysates were harvested 48 h after transfection. Controls were HeLa cells transfected with pCA as the vehicle. Tubulin was used as an internal control. (B) HeLa cells transiently overexpressing cyr61 were infected with CVB3 (MOI = 10 and 1) for 1 h and washed twice with PBS, and then cell lysates were harvested at indicated times postinfection (pi). VP1 protein production indicates CVB3 replication and growth. Tubulin was used as an internal control. (C) Each bar represents the mean VP1 content (after normalization to tubulin) at 6 h postinfection in HeLa cells infected with CVB3 (MOI = 10). The VP1 content in controls is expressed as 100% and compared with the percentage converted by the VP1 expression of cyr61-HeLa cells. (D) Viral titers in culture medium from the control and from cyr61-HeLa cells after CVB3 infection (MOI = 10) were compared. Viral titration in the extracellular space was performed by plaque assay (see details in Materials and Methods). The results shown are the averages of three independent assays, with error bars representing the standard deviations.
Down-expression of cyr61 reduces the viral growth in HeLa cells infected with CVB3.
We also down-expressed cyr61 in HeLa cells, which was performed with an siRNA technique and infected with CVB3 to confirm the results with overexpression. We used the siRNA target Finder and Design Tool provided by Ambion, at http://www.ambion.com/techlib/misc/siRNA_finder.html, to design the inserts with an inverted repeat containing two 19-nucleotide coding sequences interrupted by a 6-nucleotide loop sequence in the siRNA target sites of cyr61. We selected two target regions of cyr61 (si-cyr61-1 represented nucleotides 510 to 528, and si-cyr61-2 represented nucleotides 554 to 572) and examined the effects of siRNA. Gene silencing with si-cyr61-1 was more effective than with si-cyr61-2 (Fig. 5A, third compared with fourth lane). CVB3 was infected into HeLa cells transfected with si-cyr61-1 for 48 h, and we analyzed both intracellular and extracellular viral contents. As expected, the total viral content in HeLa cells down-expressing cyr61 (si-cyr61-HeLa) was reduced compared with controls 6 h after infection with 10 MOI of CVB3 (Fig. 5B, C, and D). These results clearly confirm the data from HeLa cells overexpressing cyr61 indicating that Cyr61 protein plays a role in CVB3 growth.
FIG. 5.

Down-expression of cyr61 reduces viral growth in HeLa cells infected with CVB3. (A) cyr61 was down-expressed in HeLa cells (si-cyr61-HeLa) performed with silencing RNA. Cells were transfected with two siRNA plasmids (1 and 2) for cyr61 genes (see details in Materials and Methods). After 48 h of transfection with siRNA plasmids, cyr61 expression was assayed. Controls were HeLa cells transfected with pCA as a vehicle. Tubulin was used as an internal control. (B) HeLa cells transiently down-expressing cyr61 were infected with CVB3 (MOI = 10) for 1 h after 48 h of transfection with siRNA-1 plasmids (indicated by si-cyr61 HeLa/1), washed twice with PBS, and then cell lysates were harvested at the indicated times postinfection (pi). VP1 protein production indicated CVB3 replication and growth. Tubulin was used as an internal control. (C) Each bar represents the mean VP1 expression (after normalization to tubulin) at 6 h postinfection in HeLa cells infected with CVB3 (MOI = 10). VP1 production in controls is expressed as 100% and compared with the percentage converted by the VP1 production from si-cyr61-HeLa/1 cells. (D) The viral titers in cell medium from control and si-cyr61-HeLa/1 cells after CVB3 infection (MOI = 10) were compared. Viral titration in the extracellular space was performed by plaque assay (see details in Materials and Methods). The results shown are the averages of three independent assays, with error bars representing the standard deviations.
cyr61 mediates cell death in HeLa cells infected with CVB3.
Because cyr61 expression seems to affect CVB3 growth, we believed that cyr61 might also be associated with the cell death caused by CVB3 infection. To examine this, CVB3 was infected into control HeLa cells and into HeLa cells over- and down-expressing cyr61 (cyr61-HeLa and si-cyr61-HeLa), and cell death was measured by FACS analysis with propidium iodide viability staining. As we expected, HeLa cells overexpressing cyr61 (cyr61-HeLa) were more liable to cell death by CVB3 infection, in contrast to those down-expressing cyr61 (si-cyr61-HeLa) at 10 h after CVB3 infection (Fig. 6). Thus, cell death caused by CVB3 infection was rescued by cyr61 down-expression. Interestingly, before CVB3 infection (0 h), HeLa cells overexpressing cyr61 showed slightly higher rates of cell death compared with controls (P < 0.05, Fig. 6B). This implies that cyr61 expression may make the cells fragile to cell death by several other stimuli.
FIG. 6.

cyr61-mediated cell death in HeLa cells occurs after infection with CVB3. (A) Representative phase contrast microscopy of HeLa cells after 10 h of CVB3 infection (MOI = 10). Controls were HeLa cells transfected with pCA as a vehicle. si-cyr61-HeLa indicates HeLa cells down-expressing the cyr61 gene with siRNA. cyr61-HeLa cells overexpress cyr61. (B) Cell death was analyzed by staining cells with propidium iodide after CVB3 infection (MOI = 10) at the indicated times.
Brefeldin A prevents Cyr61 secretion and viral protein synthesis.
Since Cyr61 is a secreted protein, we explored the effect of blocking Cyr61 secretion induced by CVB3 infection. Since brefeldin A, a metabolite synthesized by a variety of fungi, effectively blocks secretion of various proteins in cell lines and appears to disrupt protein transport from the endoplasmic reticulum to the Golgi apparatus in many cell types (22), we used brefeldin A to prevent Cyr61 secretion. Metabolically labeled HeLa cells were infected with CVB3 in the absence or presence of 5.0 μg of brefeldin A per ml and harvested at 8 h postinfection. In culture medium, brefeldin A blocked Cyr61 secretion induced by CVB3 infection (Fig. 7). Interestingly, brefeldin A also completely blocked the synthesis of viral protein VP1 (Fig. 7), indicating that brefeldin A prevents CVB3 growth in infected cells.
FIG. 7.
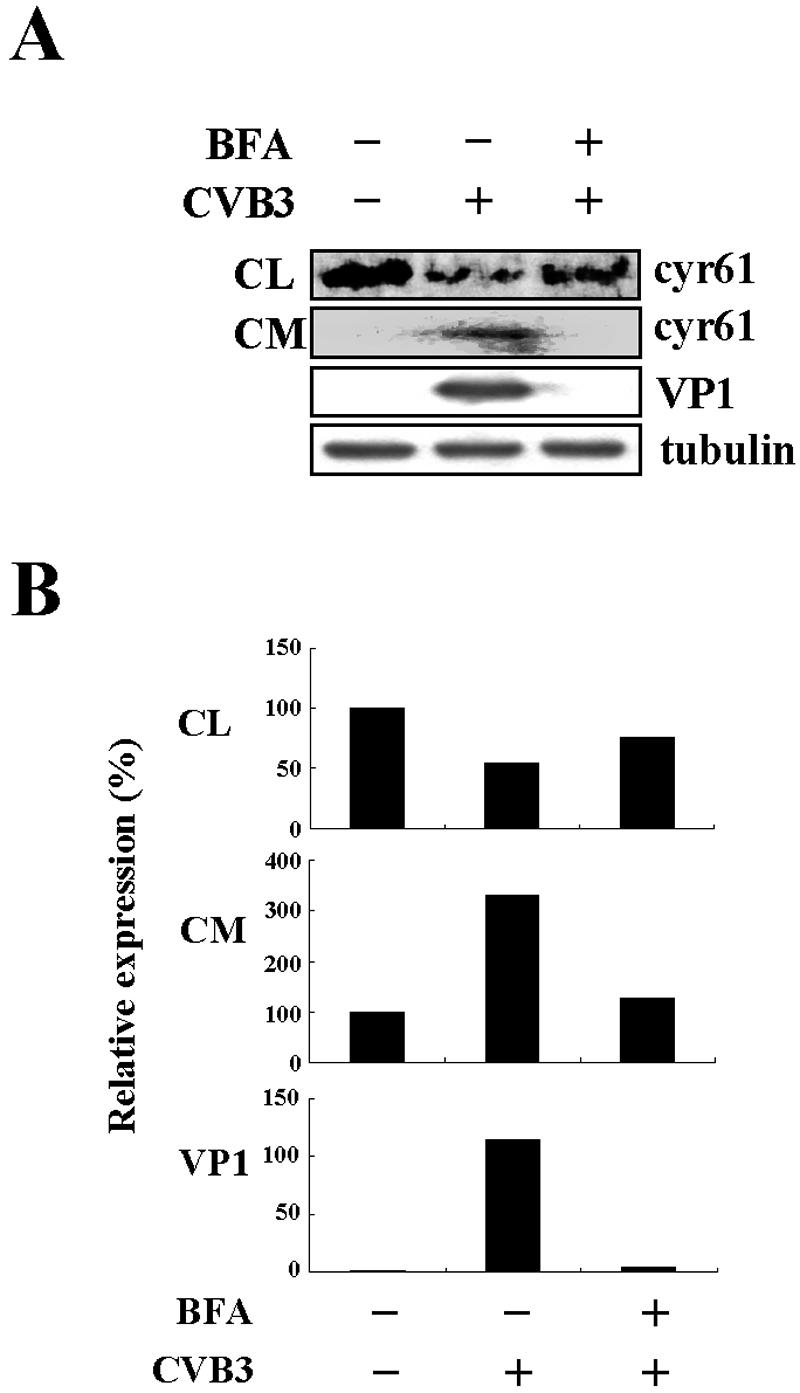
Brefeldin A inhibits Cyr61 secretion and viral protein synthesis. (A) Effects of brefeldin A (BFA) on Cyr61 secretion and viral protein synthesis. After labeling with [35S]methionine, HeLa cells were infected with CVB3 (MOI = 20) in the absence or presence of 5.0 μg of brefeldin A per ml and harvested at 8 h postinfection. Cyr61 protein in cell lysates (CL) and culture medium (CM) was immunoprecipitated with polyclonal anti-Cyr61 immunoglobulin G, resolved by SDS-PAGE (10.0% gel), and visualized by autoradiography. Cyr61 synthesis in cell lysates and culture medium is shown in individual autoradiographs. VP1 expression in infected cells was detected to determine viral protein synthesis in the absence or presence of brefeldin A. Tubulin from cell lysates was used as a loading control. (B) Each bar represents the amount of Cyr61 protein and VP1 (after normalization to tubulin) in HeLa cells infected with CVB3. Cyr61 protein content in cell lysates and culture medium at 0 h is set at 100%, and VP1 content in cells at 0 h is set at 1%.
DISCUSSION
CVB3 infection generally induces cell death both in permissive tissue culture and in tissues such as the myocardium in vivo, and this may contribute to the progress of myocarditis and dilated cardiomyopathy (23, 24, 44). Therefore, it is very important to determine how CVB3 infection induces cell death and which cellular factors are related. Several cellular factors related to CVB3 infection have been reported. CVB3 infection induces Siva, which is involved in CD27/CD70-transduced apoptosis (15). In addition, bag-1, known to be involved in the inhibition of apoptosis, is down-regulated in the CVB3-infected mouse heart, as is nip21 (39, 48). However, CVB3 infection-induced cell death is still not completely explained by these cellular factors. It is possible that other cellular factors could be involved in cell death caused by viral infection. Here, we showed that CVB3 infection in HeLa cells induced the RNA and protein levels of cyr61 (Fig. 1 and 2) and that Cyr61 protein is secreted gradually into the extracellular medium (Fig. 1C and D). Thus, we anticipate that induced cyr61 has some role in CVB3 infection.
For the analysis of transcriptional activation of cyr61, we used the cyr61 promoter fused with CAT as the reporter. The cyr61 gene has an upstream promoter, a 2-kb 5′-flanking DNA fragment that functions as a serum-inducible promoter (27). This promoter has serum response element (SRE)-like sequences (SRE or CArG Box), which mediate the induction of c-fos in response to the growth factors, cytokines, and other extracellular stimuli which activate the mitogen-activated protein kinase pathways (45). This SRE of the cyr61 promoter is especially responsive to a JNK-dependent signaling pathway; the construct containing an intact SRE (ΔBglII plasmid) showed a significant increase in cyr61 promoter activity, whereas deletion of the SRE (−1763 plasmid) resulted in a complete loss of cyr61 activity following treatment with etoposide (20). Thus, we used the ΔBglII plasmid as a positive control and the −1763 plasmid as a negative control to analyze the transcriptional activity of cyr61 in HeLa cells infected with CVB3.
As shown in Fig. 2A, CVB3 infection transcriptionally induced CAT activity in ΔBglII-transfected HeLa cells, which confirmed the transcriptional activation of cyr61 after CVB3 infection, whereas in HeLa cells transfected with −1763 did not show any cyr61 promoter activity. Because only the ΔBglII plasmid contains the SRE domain, which is dependent on JNK activation, this strongly suggests that the induction of cyr61 is dependent on JNK transcriptional activation. Moreover, when we cotransfected the ΔBglII plasmids and JNK1 or -2 dominant negative mutant plasmids into HeLa cells or treated them with a chemical inhibitor of JNK before CVB3 infection, cyr61 promoter activity was not induced compared with that in ΔBglII-transfected HeLa cells infected with CVB3 (Fig. 2A). Supporting this, we found that CVB3 infection induces JNK activation after the expression of viral proteins in HeLa cells (Fig. 3). Thus, CVB3 infection transcriptionally induces cyr61 in HeLa cells via a JNK signaling pathway related with transcriptional binding to the cyr61 upstream promoter and the induced Cyr61 protein is secreted into extracellular matrix. In addition, cyr61 RNA was induced by CVB3 infection, and this was dramatically reduced (more than two- or threefold) by SP600125, a JNK inhibitor (Fig. 2B and C). However, the reduction by SP600125 was not complete, in that cyr61 RNA induction in the presence of SP600125 was higher than the control (0 h), indicating that although JNK was the major factor, there is a possible that other signaling factors may also influence cyr61 induction by CVB3 infection.
To explore the role of cyr61 in infected HeLa cells, we over- and down-expressed cyr61 and analyzed the relation between CVB3 growth and/or cell death and the expression of cyr61. Interestingly, overexpression of cyr61 enhanced cell death after CVB3 infection, whereas down-expression of cyr61 reduced it twofold compared with controls (Fig. 6), which is a significant difference because the regulation of cyr61 was performed by transient expression (Fig. 4 and 5). These results suggest that cyr61 plays a role in CVB3 growth and cell death induced by CVB3 infection.
It has been previously reported that overexpression of Cyr61 in lung cancer cells causes G1 arrest (42). A recent report also describes that cells that have been arrested at the G1 and G1/S phase show high expression of CVB3 protein (13). Therefore, based on both previous reports, we speculate that overexpression of Cyr61 may arrest G1 phase and this arrest may increase production of viral proteins. In addition, it was recently reported that the RNA and the protein levels of cyr61are increased during etoposide-induced neuronal cell death (20). These clearly showed that cyr61 related to cell death induced by external stimuli. Our results that cyr61 is involved in CVB3-induced cell death are thus consistent with previous data. However, cyr61 expression is no longer affected by viral growth at late time (over 12h postinfection with 10 MOI of infection) (data not shown). This means that Cyr61 protein may be involved in early times of CVB3 growth events.
CVB3 infection induces a biphasic pattern of activity of extracellular signal-regulated kinases 1 and 2 (ERK1/2) in HeLa cells. This ERK1/2 activation is very important for CVB3 replication and contributes to virus-mediated cytopathogenesis (31). However, it is still unclear whether CVB3 infection induces other mitogen-activated protein kinase signaling pathways including JNK and p38 and thus modulates their role in infected cells. Here we focused on the relation between JNK and cyr61, because it previously reported that cyr61 is induced by some toxic stimuli only via JNK, not via ERK or p38 (20). Moreover, we found that ERK1/2 and p38 inhibitors did not affect the transcriptional activation of cyr61 by CVB3 infection on CAT reporter assay (data not shown). We show here that CVB3 infection induces the JNK1/2 signaling pathways (Fig. 3), and one of the downstream signals of JNK1/2 is the activation of the cyr61 promoter in HeLa cells infected with CVB3 (Fig. 2). JNK1/2 activation has various downstream functions which are associated with cell death and/or cell defense mechanisms (16, 26). Therefore, this induction of JNK by CVB3 infection might trans-activate the promoters of several cellular genes to involve virus-mediated cellular changes. Interestingly, another mitogen-activated protein kinase signal pathway, p38, is also activated at 6 h postinfection in HeLa cells infected with CVB3, like JNK (data not shown). However, the functional role of JNK and p38 in CVB3 infection is not clear. Therefore, we are currently investigating the more detailed roles and upstream and downstream mechanisms of JNK and p38 activations in CVB3 infection.
To explore the effect of Cyr61 secretion in infected HeLa cells, we used brefeldin A as an inhibitor for protein transport. Brefeldin A blocks secretion of Cyr61 and completely prevents CVB3 growth (Fig. 7), suggesting that Cyr61 may have outward effects on infected cells for CVB3 growth. However, it is well known that brefeldin A blocks secretion, vesicular assembly, antigen presentation, trans- and endocytosis, viral assembly, and budding in various cell types (22). Furthermore, brefeldin A completely inhibits replication of poliovirus (10), which, like CVB3, belongs to the genus Enterovirus. Thus, although brefeldin A blocks CVB3 growth, it is not clear that this effect originates from inhibition of Cyr61 secretion because brefeldin A has a variety of effects on protein transport and has antiviral properties.
It was previously reported that induction of Cyr61 protein level in stressed tissues likely plays a role as an early marker and a potent effector of the remodeling process in smooth muscle compartments (14, 28, 40). These data make us anticipate that the cyr61 gene may be regulated in the heart directly or indirectly damaged by CVB3 infection. However, we need to investigate the function of the Cyr61 protein in more detail.
In conclusion, we have demonstrated a new role for cyr61 in HeLa cells infected with CVB3, which is associated with the cell death induced by virus infection. Moreover, we show that activation of the cyr61 promoter is dependent on JNK activation induced by CVB3 replication and viral protein expression in infected cells. These data expand our understanding of the physiological functions of cyr61 in virus-induced cell death and provide new insights into the cellular factors involved.
Acknowledgments
This work was supported by the intramural fund of the National Institute of Health, Korea (348-6111-213-000-207).
REFERENCES
- 1.Absenger, Y., H. Hess-Stumpp, B. Kreft, J. Kratzschmar, B. Haendler, N. Schutze, P. A. Regidor, and E. Winterhager. 2004. Cyr61, a deregulated gene in endometriosis. Mol. Hum. Reprod. 10:399-407. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, D. R., J. E. Wilson, C. M. Carthy, D. Yang, R. Kandolf, and B. M. McManus. 1996. Direct interactions of coxsackievirus B3 with immune cells in the splenic compartment of mice susceptible or resistant to myocarditis. J. Virol. 70:4632-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babic, A. M., M. L. Kireeva, T. V. Kolesnikova, and L. F. Lau. 1998. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 95:6355-6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boshart, M., L. Gissmann, H. Ikenberg, A. Kleinheinz, W. Scheurlen, and H. zur Hausen. 1984. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 3:1151-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brigstock, D. R. 2003. The CCN family: a new stimulus package. J. Endocrinol. 178:169-175. [DOI] [PubMed] [Google Scholar]
- 6.Brown, P. O., and D. Botstein. 1999. Exploring the new world of the genome with DNA microarrays. Nat. Genet. 1:33-37. [DOI] [PubMed] [Google Scholar]
- 7.Carthy, C. M., D, J. Granville, K. A. Watson, D. R. Anderson, J. E. Wilson, D. Yang, D. W. Hunt, and B. M. McManus. 1998. Caspase activation and specific cleavage of substrates after coxsackievirus B3-induced cytopathic effect in HeLa cells. J. Virol. 72:7669-7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, N., C. C. Chen, and L. F. Lau. 2000. Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin alpha 6 beta 1 and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 275:24953-24961. [DOI] [PubMed] [Google Scholar]
- 9.Chung, K. C., I. Gomes, D. Wang, L. F. Lau, and M. R. Rosner. 1998. Raf and fibroblast growth factor phosphorylate Elk1 and activate the serum response element of the immediate early gene pip92 by mitogen-activated protein kinase-independent as well as -dependent signaling pathways. Mol. Cell. Biol. 18:2272-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crotty, S., M. C. Saleh, L. Gitlin, O. Beske, and R. Andino. 2004. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J. Virol. 78:3378-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498. [DOI] [PubMed] [Google Scholar]
- 12.Esfandiarei, M., H. Luo, B. Yanagawa, A. Suarez, D. Dabiri, J. Zhang, and B. M. McManus. 2004. Protein kinase B/Akt regulates coxsackievirus B3 replication through a mechanism which is not caspase dependent. J. Virol. 78:4289-4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geiss, G. K., R. E. Bumgarner, M. C. An, M. B. Agy, A. B. van't Wout, E. Hammersmark, V. S. Carter, D. Upchurch, J. I. Mullins, and M. G. Katze. 2000. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology 266:8-16. [DOI] [PubMed] [Google Scholar]
- 14.Grzeszkiewicz, T. M., D. J. Kirschling, N. Chen, and L. F. Lau. 2001. CYR61 stimulates human skin fibroblast migration through Integrin alpha vbeta 5 and enhances mitogenesis through integrin alpha vbeta 3, independent of its carboxyl-terminal domain. J. Biol. Chem. 276:21943-21950. [DOI] [PubMed] [Google Scholar]
- 15.Henke, A., H. Launhardt, K. Klement, A. Stelzner, R. Zell, and T. Munder. 2000. Apoptosis in coxsackievirus B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein siva. J. Virol. 74:4284-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichijo, H. 1999. From receptors to stress-activated MAP kinases. Oncogene 18:6087-6093. [DOI] [PubMed] [Google Scholar]
- 17.Kapadia, S. B., A. Brideau-Andersen, and F. V. Chisari. 2003. Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl. Acad. Sci. USA 100:2014-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawai, C. 1999. From myocarditis to cardiomyopathy: mechanism of inflammation and cell death. Circulation 99:1091-1100. [DOI] [PubMed] [Google Scholar]
- 19.Kerekatte, V., Keiper, C. Badorff, A. Cai, K. U. Knowlton, and R. E. Rhoads. 1999. Cleavage of poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J. Virol. 73:709-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim, K. H., Y. K. Min, J. H. Baik, L. F. Lau, B. Chaqour, and K. C. Chung. 2003. Expression of angiogenic factor cyr61 during neuronal cell death via the activation of c-Jun N-terminal kinase and serum response factor. J. Biol. Chem. 278:13847-13854. [DOI] [PubMed] [Google Scholar]
- 21.Kireeva, M. L., F. E. MO, G. P. Yang, and L. F. Lau. 1996. cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol. Cell. Biol. 16:1326-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klausner, R. D., J. G. Donaldson, and J. Lippincott-Schwartz. 1992. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116:1071-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klingel, K., P. Rieger, G. Mall, H. C. Selinka, M. Huber, and R. Kandolf. 1998. Visualization of enteroviral replication in myocardial tissue by ultrastructural in situ hybridization: identification of target cells and cytopathic effects. Lab. Investig. 78:1227-1237. [PubMed] [Google Scholar]
- 24.Knowlton, K. U., and C. Badorff. 1999. The immune system in viral myocarditis: maintaining the balance. Circ. Res. 85:559-561. [DOI] [PubMed] [Google Scholar]
- 25.Kolesnikova, T. V., and L. F. Lau. 1998. Hum. CYR61-mediated enhancement of bFGF-induced DNA synthesis in human umbilical vein endothelial cells. Oncogene 16:747-754. [DOI] [PubMed] [Google Scholar]
- 26.Kyriakis, J. M., P. Banerjee, E. Nikolakaki, T. Dai, E. A. Rubie, M. F. Ahmad, J. Avruch, and J. R. Woodgett. 1994. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 369:156-160. [DOI] [PubMed] [Google Scholar]
- 27.Latinkic, B. V., T. P. O'Brien, and L. F. Lau. 1991. Promoter function and structure of the growth factor-inducible immediate early gene cyr61. Nucleic Acids Res. 19:3261-3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lau, L. F., and S. C. Lam. 1999. The CCN family of angiogenic regulators: the integrin connection. Exp. Cell Res. 248:44-57. [DOI] [PubMed] [Google Scholar]
- 29.Lee, Y., E. J. Park, S. S. Yu, D. K. Kim, and S. Kim. 2000. Improved expression of vascular endothelial growth factor by naked DNA in mouse skeletal muscles: implication for gene therapy of ischemic diseases. Biochem. Biophys. Res. Commun. 272:230-235. [DOI] [PubMed] [Google Scholar]
- 30.Leu, S. J., S. C. Lam, and L. F. Lau. 2002. Pro-angiogenic activities of CYR61 (CCN1) mediated through integrins alphavbeta3 and alpha6beta1 in human umbilical vein endothelial cells. J. Biol. Chem. 277:46248-46255. [DOI] [PubMed] [Google Scholar]
- 31.Luo, H., B. Yanagawa, J. Zhang, Z. Luo, M. Zhang, M. Esfandiarei, C. Carthy, J. E. Wilson, D. Yang, and B. M. McManus. 2002. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase signaling pathway. J. Virol. 76:3365-3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo, H., J. Zhang, F. Dastvan, B. Yanagawa, M. A., H. Reidy, H. M. Zhang, D. Yang, J. E. Wilson, and B. M. McManus. 2003. Ubiquitin-dependent proteolysis of cyclin D1 is associated with coxsackievirus-induced cell growth arrest. J. Virol. 77:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maisch, B., A. D. Risti, G. Hufnagel, and S. Pankuweit. 2002. Pathophysiology of viral myocarditis: The role of humoral immune response. Cardiovasc. Pathol. 11:112-122. [DOI] [PubMed] [Google Scholar]
- 34.McLean, T. I., and S. L. Bachenheimer. 1999. Activation of cJUN N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 73:8415-8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menendez, J. A., I. Mehmi, D. W. Griggs, and R. Lupu. 2003. The angiogenic factor CYR61 in breast cancer: molecular pathology and therapeutic perspectives. Endocr. Relat. Cancer 10:141-152. [DOI] [PubMed] [Google Scholar]
- 36.Nam, J. H., C. H. Yu, K. A. Hwang, S. Kim, S. H. Ahn, J. Y. Shin, W. Y. Choi, Y. R. Joo, and K. Y. Park. 2002. Application of cDNA microarray technique to detection of gene expression in host cells infected with viruses. Acta Virol. 46:141-146. [PubMed] [Google Scholar]
- 37.O'Brien, T. P., G. P. Yang, L. Sanders, and L. F. Lau. 1990. Expression of cyr61, a growth factor-inducible immediate-early gene. Mol. Cell. Biol. 10:3569-3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opavsky, M. A., T. Martino, M. Rabinovitch, J. Penninger, C. Richardson, M. Petric, C. Trinidad, L. Butcher, J. Chan, and P. P. Liu. 2002. Enhanced ERK-1/2 activation in mice susceptible to coxsackievirus-induced myocarditis. J. Clin. Investig. 109:1561-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng, T., T. Sadusky, Y. Li, G. R. Coulton, H. Zhang, and L. C. Archard. 2001. Altered expression of Bag-1 in coxsackievirus B3 infected mouse heart. Cardiovasc. Res. 50:46-55. [DOI] [PubMed] [Google Scholar]
- 40.Tamura, I., J. Rosenbloom, E. Macarak, and B. Chaqour. 2001. Regulation of cyr61 gene expression by mechanical stretch through multiple signaling pathways. Am. J. Physiol. Cell Physiol. 281:1524-1532. [DOI] [PubMed] [Google Scholar]
- 41.Taylor, L. A., C. M. Carthy, D. Yang, K. Saad, D. Wong, G. Schreiner, L. W. Stanton, and B. M. McMannus. 2000. Host gene regulation during coxsackievirus B3 infection in mice: assessment by microarrays. Circ. Res. 87:328-334. [DOI] [PubMed] [Google Scholar]
- 42.Tong, X., D. Xie, J. O'Kelly, C. W. Miller, C. Muller-Tidow, and H. P. Koeffler. 2001. cyr61, a member of CCN family, is a tumor suppressor in non-small cell lung cancer. J. Biol. Chem. 276:47709-47714. [DOI] [PubMed] [Google Scholar]
- 43.Tracy, S., N. M. Chapman, J. Romero, and A. I. Ramsingh. 1996. Genetics of coxsackievirus B cardiovirulence and inflammatory heart muscle disease. Trends Microbiol. 4:175-179. [DOI] [PubMed] [Google Scholar]
- 44.Ukimura, A., H. Deguchi, Y. Kitaura, S. Fujioka, M. Hirasawa, K. Kawamura, and K. Hirai. 1997. Intracellular viral localization in murine coxsackievirus-B3 myocarditis. Ultrastructural study by electron microscopic in situ hybridization. Am. J. Pathol. 150:2061-2074. [PMC free article] [PubMed] [Google Scholar]
- 45.Whitmarsh, A. J., P. Shore, A. D. Sharrocks, and R. J. Davis. 1995. Integration of MAP kinase signal transduction pathways at the serum response element. Science 269:403-407. [DOI] [PubMed] [Google Scholar]
- 46.Wong, M., M. L. Kireeva, T. V. Kolesnikova, and L. F. Lau. 1997. cyr61, product of a growth factor-inducible immediate-early gene, regulates chondrogenesis in mouse limb bud mesenchymal cells. Dev. Biol. 192:492-508. [DOI] [PubMed] [Google Scholar]
- 47.Yang, D., J. Yu, Z. Luo, C. M. Carthy, J. E. Wilson, Z. Liu, and B. M. McMannus. 1999. Viral myocarditis: identification of five differentially expressed genes in coxsackievirus B3-infected mouse heart. Circ. Res. 84:704-712. [DOI] [PubMed] [Google Scholar]
- 48.Zhang, H. M., B. Yanagawa, P. Cheung, H. Luo, J. Yuan, D. Chau, A. Wang, L. Bohunek, J. E. Wilson, B. M. McManus, and D. Yang. 2002. Nip21 gene expression reduces coxsackievirus B3 replication by promoting apoptotic cell death via a mitochondria-dependent pathway. Circ. Res. 90:1251-1258. [DOI] [PubMed] [Google Scholar]