

Highlights
-
•
ZBD-2 significantly attenuated the symptoms of chronic SCI-pain and pain-induced depressive-like behaviors.
-
•
ZBD-2 inhibited the decreases in the expression of synaptic plasticity-related signaling proteins.
-
•
ZBD-2 reversed chronic, SCI-induced gliocyte activation at the lesion site.
Keywords: Pain, Depression, Microglia, Spinal cord injury
Abstract
In addition to debilitating sensory and motor deficits, patients with spinal cord injury (SCI) may experience chronic hyperpathic pain (SCI-pain). Recent studies have revealed that translocator protein (TSPO) is involved in repairing neural cells as well as reducing anxiety and depression. However, the role of TSPO in SCI-pain and pain-induced depression remains unknown. The present study aimed to determine the effects of a new TSPO ligand, ZBD-2, on SCI-pain and consequent pain-induced depressive-like behaviors in mice. Treatment with ZBD-2 at either dose significantly attenuated the symptoms of chronic SCI-pain and pain-induced depressive-like behaviors. ZBD-2 reversed SCI-induced elevation of serum corticosterone levels, an index of hyper-activation of the hypothalamic–pituitary–adrenal (HPA) axis. Additionally, administration of ZBD-2 inhibited decreases in the expression of synaptic plasticity-related signaling proteins, including brain-derived neurotrophic factor (BDNF) and cyclic AMP-responsive element binding protein (CREB). Moreover, ZBD-2 administration reversed chronic, SCI-induced gliocyte activation at the lesion site. Therefore, ZBD-2 may improve chronic SCI-pain and pain-induced depressive-like behaviors via suppression of gliocyte activation and restoration of the synaptic plasticity-related signaling systems.
1. Introduction
Traumatic spinal cord injury (SCI) is a common cause of paralysis in civilian and military environments. In addition to primary deficits in motor and sensory functions, other catastrophic consequences are also common in SCI, including emotional and psychological distress [1], [2]. SCI can also result in the development of either diffuse of bilateral chronic, hyperpathic pain originating below the denervated spinal segment [3], [4], [5]. Pain following SCI is categorized according to the region of hypersensitivity with respect to the level of denervation: above-level (forelimbs), at-level (girdling), or below-level (hind limbs) [6], [7]. Chronic SCI-pain occurs immediately and increases in intensity over a long period of time following the initial injury [5], [8], [9]. Chronic SCI-pain, as a persistent stressor, is correlated with biochemical, physiological, and psychological changes, and has been associated with the development of multiple neuropsychiatric disorders [10]. Patients with chronic SCI-pain are particularly susceptible to depression and anxiety. This combination of neuropathic pain and neuropsychiatric illness is the most frequently recorded reason for reduced quality of life in patients with SCI [2], [11], [12].
In the central nervous system (CNS), neurons and glial cells reciprocally maintain spinal neurofunction and neuroarchitecture, along with physiological homeostasis. Activated glial cells are associated with spinal circuit dysfunction as well as maladaptive neurophysiological and neuroanatomical construction in synaptic circuits, which eventually induces abnormal sensory transmission [13], [14]. After SCI, microglia and astrocytes are activated at the lesion site, resulting in the induction and maintenance of dorsal horn neuronal hyperexcitability, which may last from several hours to a month [15], [16]. There is a dramatic transformation in the status of microglial cells, from a normally resting (but sensing) state to an activated (reactive or alerted) state in the lumbar spinal cord in response to SCI-induced pain [17], [18]. Moreover, activation of gliocytes leads to persistent up-regulation of intracellular signaling kinases in neurons, resulting in the maintenance of neuronal hyperexcitability in the CNS [19], [20].
Following SCI, patients often experience long-term disability, as well as emotional and cognitive deficits [21], [22]. Among the proteins that may be responsible for these deficits is brain-derived neurotrophic factor (BDNF), which exerts well-established neuroprotective functions, plays an important role in neuroplasticity, and is involved in the regulation of a wide range of psychopathologies such as emotional distress, depression, and post-traumatic stress disorder, which frequently occur following SCI [23], [24], [25]. SCI results in a variety of pathophysiological alterations, such as hyperactivity of the hypothalamic–pituitary–adrenal (HPA) axis, selective reduction of cerebral biosynthesis and down-regulation of BDNF, and expression of cyclic AMP-responsive element binding protein (CREB) in the hippocampus of lesioned animals, which ultimately results in depressive-like behaviors [22], [26].
Translocator protein (18 kDa) (TSPO), which has five transmembrane domain proteins, is primarily localized in the outer mitochondrial membrane [27], [28] and mainly expressed in steroid-synthesizing tissues such as reactive microglia and astrocytes [29], [30], [31], [32], [33], [34]. TSPO is involved in the transport of cholesterol from the outer to inner mitochondrial membrane, which is the rate-limiting step in neurosteroid synthesis [35]. Additionally, TSPO also plays an important role in mitochondrial proliferation and membrane biogenesis during the proliferation and repair of neural cells after injury. Upregulated expression of TSPO has been observed in many neurological and neurodegenerative disorders, such as Alzheimer’s disease, multiple sclerosis, Huntington’s disease, amyotrophic lateral sclerosis, and Parkinson’s disease [36]. ZBD-2 (N-benzyl-Nethyl-2-(7,8-dihydro-7-benzyl-8-oxo-2-phenyl-9H-purin-9-yl) acetamide) is a novel TSPO ligand synthesized by our Pharmacologic Lab (Chinese patent number 201210047188.6). The structure of ZBD-2 is analogous to XBD173, which has been reported to exert anxiolytic and antidepressant effects in animal models. Compared to the affinity of 3H-PK11195 for TSPO, the classic TSPO ligand [37], (Ki = 0.657 nM), ZBD-2 displays high affinity for TSPO in the nanomolar range (Ki = 0.463 nM) in rat brain mitochondria [38].
Given the aforementioned evidence of the role of translocator proteins in repairing neural cells and reducing anxiety and depression, we examined the effects of the novel translocator protein (TSPO) ligand ZBD-2 on SCI-pain and pain-induced depressive-like behaviors. Specifically, we aimed to investigate the underlying mechanisms of ZBD-2 in chronic SCI-pain and depressive-like behaviors by further clarifying the pathobiology of SCI-pain, identifying molecular mechanisms and signaling pathways underlying the associated gliopathy, and examining the impact of SCI on the expression of BDNF and CREB in the hippocampus. Investigation of the effects of ZBD-2 may aid in the development of novel therapeutic targets for the reduction of SCI-pain and consequent depressive-like behaviors.
2. Materials and methods
2.1. Materials
All chemicals and reagents utilized in the present study were obtained from Sigma (St. Louis, MO, USA), unless otherwise noted. ZBD-2 was synthesized at our Pharmacologic Lab with a purity of 99.9% and dissolved in saline (0.9% NaCl). ZBD-2 was intragastrically administrated (2 or 4 mg/kg) after SCI. Goat anti-IB-1 antibody was purchased from Santa Cruz Biotech (Santa Cruz, CA, USA). Rabbit anti-BDNF antibody was purchased from Abcam (Cambridge, UK). All chemicals utilized were commercially available and characterized by standard biochemical quality.
2.2. Animals
Adult male C57BL/6 mice (20–25 g) were purchased from the Laboratory Animal Center of the Fourth Military Medical University (FMMU). The animals were randomly divided into four groups: (1) Sham/Vehicle group (n = 8); (2) SCI/Vehicle (n = 8); (3) SCI + 2 mg/kg ZBD-2 group (n = 8); (4) SCI + 4 mg/kg ZBD-2 group (n = 8). Mice were housed with a light-dark cycle (lights on at 7:00 AM) at room temperature (24 ± 2 °C), with humidity between 50% and 60%, and allowed free access to food and water. Animals were allowed to accommodate to laboratory conditions for a week before the experimental procedure began. The Animal Ethics Committee of the Fourth Military Medical University approved all experiments. To induce SCI-pain, a laminectomy were performed at T8–T10 to expose a circle of dura, following which the spinal cord was moderately compressed for 15 s under anesthesia [39], [40]. A sham surgery was also executed using the same procedures but without the spinal contusion injury. After SCI, the animals received either the vehicle or ZBD-2 (2 or 4 mg/kg) via oral administration once a day for 2 weeks. Behavioral tests were conducted according to the following procedures.
2.3. Behavioral assessment
2.3.1. Basso mouse scale (BMS)
To confirm lesion severity, all animals were assessed for hind limb locomotion function in an open field (OF) preoperatively and weekly after SCI using the BMS. Hind limb function scores ranged from 0 (no ankle movement) to 9 (complete functional recovery). The scores for left and right hind paws were averaged to obtain a single value per mouse, which represents the overall mobility for that mouse. All behavioral assessments were conducted by a blinded experimenter [41], [42].
2.3.2. Inclined plane test
Neurological deficits were also estimated according to maintenance of position on an inclined plane, both preoperatively and at least weekly after injury, as previously described [43], [44]. The mice were placed horizontally on a smooth tilted board (60 cm × 80 cm) with an adjustable angle of 0–90°. The angle was then gradually elevated to the vertical position from a preliminary angle of 30°, at a speed of 2.5°/s, until the mouse could no longer remain and hold its position for 5 s without sliding. The last angle at which the mouse was able to maintain its position for 5 s was recorded.
2.3.3. Mechanical allodynia
Mice were placed in individual plastic boxes for at least 7 days prior to the daily threshold experiments, in order to habituate them to the environment. To assess the degree of tactile sensory changes after operation, the plantar surface of the hind paw was stimulated with a set of von Frey filaments using the up-down paradigm [45]. To ensure that all mice could withdraw their hind paw from unpleasant stimuli, postoperative assessment was not initiated until hind limb weight support recovered from SCI, according to BMS scores. Each von Frey hair (VFH) was applied six times (approximately 30 s apart) in ascending order [39].
2.3.4. Thermal hyperalgesia
Thermal hyperalgesia was measured with a plantar analgesia instrument (BME410A, Institute of Biological Medicine, Academy of Medical Science, China). Postoperative assessment was not initiated until hind limb weight support recovered from SCI, according to BMS scores. Thermal hyperalgesia was assessed by measuring paw withdrawal latency (PWL), defined as the time from onset of radiant heat application to withdrawal of the mouse’s hind paw, as previously described [45].
Three of the most widely used models to examine SCI-induced depressive-like behaviors in mice are the forced swim test (FST), tail suspension test (TST), and open field test (OF). These tests were performed during week 5, as previously described [46], [47], [48].
2.3.5. Forced swimming test (FST)
Mice were individually placed into five separate glass beakers (30 cm height × 18 cm diameter) filled with 40 cm of water at 23 ± 2 °C for 15 min. Twenty-four hours later, mice were subjected to a 5-min forced swim to measure the duration of immobility. A mouse was judged to be immobile when it ceased struggling (i.e., no upward-directed movements of the forepaws) and motionless when it would passively float, with only slight movements to keep the head above the water.
2.3.6. Tail suspension test (TST)
Each mouse was individually suspended at a height of 50 cm with adhesive tape placed approximately 2 cm from the tip of its tail. The duration of immobility was measured throughout a 5-min test. Immobility was defined as a state with passive hanging and complete lack of motion.
2.3.7. Open field test (OF)
The OF test was conducted in a square arena (30 cm × 30 cm × 30 cm) with clear Plexiglass walls and a white floor (JL Behv-LAM-Shanghai jiliang software, China). Mice were placed in the center of the arena for 15 min, and their exploration track was recorded using a camera fixed above the floor. The data was analyzed using a video-tracking system.
2.4. Serum corticosterone level
Blood samples were collected immediately following the sacrifice of each mouse and centrifuged at 12,000 rpm at 4 °C for 15 min. The supernatant was then collected for enzyme-linked immunosorbent assay (ELISA). Serum was stored at −80 °C until assayed for corticosterone. The serum corticosterone level was measured using an Assay Max Corticosterone ELISA kit (R&D Systems Inc., Minneapolis, MN, USA) according to the manufacturer's instructions and as previously described [48].
2.5. Hemotoxylin and eosin (HE) staining and immunohistochemistry
HE staining was performed as previously described [49]. The spinal cord tissues (T5–L1 vertebrae) and tissue samples from the amygdala dissected from brain slices under an anatomical microscope 2 weeks after SCI were infused with 4% paraformaldehyde solution with 0.1 M PBS (pH = 7.4) for 24 h at room temperature. Transverse tissue sections (5-μm thickness) were obtained and subsequently stained with HE. The extent of lesioning was determined by the volume of cavitation at the epicenter of the injury recorded by a microscope (Olympus BX61, Japan). The volume of unstained areas, defined as a cavitation, was calculated using image analysis software (Adobe Photoshop, version CS3 for Windows, San Jose, CA, USA).
Immunohistochemistry was performed on spinal cord sections transverse to the injury epicenter. Standard frozen immunocytochemistry sections (15-μm thick) were created, as previously described [50]. Rabbit anti-Iba-1 (1:1000; Wako Chemicals, Richmond, VA, USA) was used as the primary antibody. To eliminate endogenous peroxidase activity, each section was incubated in 3% hydrogen peroxide (H2O2), followed by incubation in 0.15% Triton X-100 at room temperature, and blocked with 1% goat serum albumin in Phosphate-buffered Saline with Tween® 20 (PBST) for 1 h. The sections were incubated at 4 °C for 24 h with rabbit anti-Iba-1 (1:1000; Wako Chemicals, Richmond, VA, USA). The sections were infused with horseradish peroxidase-conjugated secondary antibodies for 2 h at 37 °C and halted with 3,3-diaminobenzidine (DAB). The results were photographed by a microscope (Olympus BX61, Japan). The intensity of IB-1 immunoreactivity was determined using image analysis software (Image J, National Institute of Health, Bethesda, MD, USA).
2.6. Western blot analysis
Western blot analysis was performed as previously described [49]. The tissue samples from the T5–L1 vertebrae and the hippocampus were collected on day 35 after SCI under an anatomical microscope and then sonicated. Aliquots of 25 μg of protein were prepared for western blot analysis using the following primary antibodies: TSPO (dilution ratio 1:2000), Ib-1 (dilution ratio 1:1000), glial fibrillary acidic protein (GFAP; dilution ratio 1:1000), BDNF (dilution ratio 1:2000), Phospho-CREB (dilution ratio 1:1000), and CREB (dilution ratio 1:1000), with β-actin (dilution ratio 1:10000) as the loading control. Immune complexes were detected with appropriate secondary antibodies and chemiluminescence reagents (Pierce, Rockford, IL, USA). The densitometric analysis of the western blot was performed by ChemiDoc XRS (Bio-Rad, Hercules, CA, USA) and determined using Quantity One version 4.1.0 (Bio-Rad, Hercules, CA, USA), according to the instructions provided. For data quantification, the band intensity of each blot was calculated as a ratio relative to β-actin. The band intensity ratio of the control group was set at 100%, and the intensity of the other treatment groups was expressed as a percentage relative to that of the control group.
2.7. Data analysis
Results are presented as mean ± standard error of the mean (SEM). Statistical comparisons between two groups were performed using unpaired Student’s t-tests (SPSS version 13.0, SPSS Inc., Chicago, IL, USA). One-way analysis of variance (ANOVA) was used to compare multiple groups. Data that passed the homogeneity test were analyzed using the one-way ANOVA least significant difference (LSD) test. Data that did not pass the homogeneity test were analyzed using the one-way ANOVA Dunnett’s T3 test. In all cases, p < 0.05 was regarded as statistically significant.
3. Results
3.1. ZBD-2 improves recovery of function after SCI
We used the BMS score, as well as the angle on the inclined plane test, to evaluate the effect of ZBD-2 on functional motor recovery (Fig. 1A and B). During the early post-operative period, both the BMS score and the angle on the inclined plane test were significantly decreased, indicating that hind limb locomotion was dramatically impaired. Administration of ZBD-2 (2 and 4 mg/kg) for two weeks significantly improved both BMS score and inclined-plane angle in a dose-dependent and time-dependent manner.
Fig. 1.

ZBD-2 improves recovery of motor function after SCI. The BMS and inclined plane experiments were performed weekly following SCI (Fig. 1A and B). Both the BMS score and angle were significantly decreased, and no differences were noted among the groups during the first week following SCI. Compared to the SCI/Vehicle group, administration of ZBD-2 (either 2 or 4 mg/kg) significantly improved both BMS scores and angles in a dose-dependent and time-dependent manner two weeks post-operation. n = 8 in each group, *p < 0.05, **p < 0.01 vs. Sham/Vehicle group; #p < 0.05, ##p < 0.01 vs. SCI/Vehicle group.
3.2. Antidepressant effect of ZBD-2 in mice with SCI-induced chronic pain
SCI-pain induced depressive-like behaviors were detected using the FST, TST, and OF test on day 35 when there was a high level of motor recovery. The time in the central area and the total distance traveled in the OF tests were significantly decreased after SCI (Fig. 2A–C). Additionally, SCI resulted in significant increases in immobility times in both the FST and the TST (Fig. 2D and E). Administration of ZBD-2 (2 and 4 mg/kg) for two weeks reversed the time in the central area and the total distance in the OF tests, as well as the immobility times in the FST and TST in a dose-dependent manner when compared to the SCI/Vehicle group.
Fig. 2.

ZBD-2 reduces depressive-like behaviors. The FST, TST, and OF test were performed on day 35 post-operation. Representative types of travel strategies in the OF test (Fig. 2A). SCI mice traveled shorter distances and spent less time in the center area compared to Sham/Vehicle mice in the OF test. ZBD-2 (either 2 or 4 mg/kg) significantly reversed the reduction in distance and time compared to the SCI/Vehicle group (Fig. 2B and C). In the FST and TST, Sham/Vehicle mice exhibited reduced immobility time when compared with SCI mice. Administration of ZBD-2 (either 2 or 4 mg/kg) significantly decreased the immobility time in a dose-dependent manner compared to the SCI/Vehicle group (Fig. 2D and E). n = 8 in each group, *p < 0.05, **p < 0.01 vs. Sham/Vehicle group; #p < 0.05, ##p < 0.01 vs. SCI/Vehicle group.
3.3. ZBD-2 reduces serum corticosterone level in SCI-induced depressant mice
We measured the serum level of corticosterone to determine whether the feedback mechanism of the HPA axis was impaired following SCI. Compared to the Sham/Vehicle group, the SCI/Vehicle group had a significantly increased level of serum corticosterone (Fig. 3). However, administration of ZBD-2 (2 and 4 mg/kg) for two weeks significantly suppressed the SCI-induced elevation of corticosterone and improved depressive-like behaviors in SCI-mice.
Fig. 3.
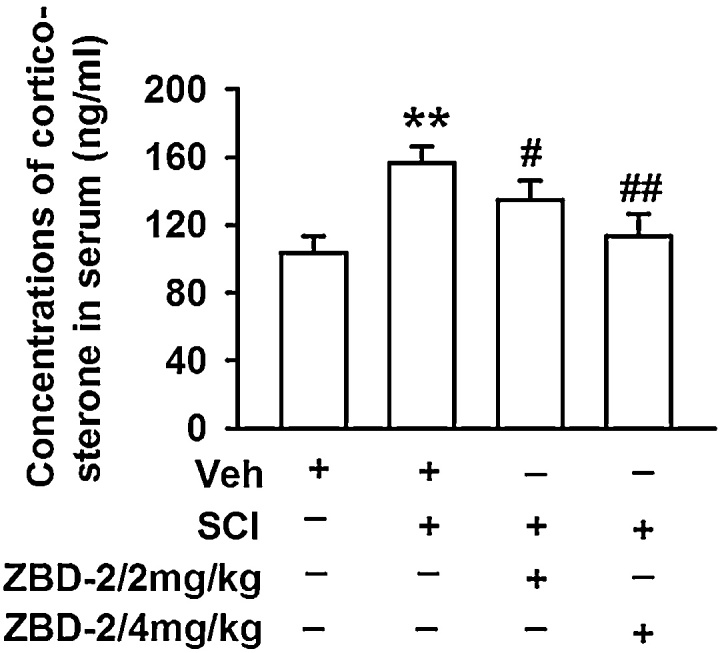
ZBD-2 reduces serum corticosterone level. The serum level of corticosterone was assessed on day 35 post-operation. SCI mice exhibited higher serum levels of corticosterone than Sham/Vehicle mice. Administration of ZBD-2 (either 2 or 4 mg/kg) significantly restored the serum level of corticosterone compared to the SCI/Vehicle group after SCI. n = 8 in each group, *p < 0.05, **p < 0.01 vs. Sham/Vehicle group; #p < 0.05, ##p < 0.01 vs. SCI/Vehicle group.
3.4. Effect of ZBD-2 on mechanical and heat hyperalgesia
We additionally considered whether the antidepressant-like effect of ZBD-2 is caused by analgesic activity in animals with chronic SCI-induced pain. Therefore, we evaluated mechanical and heat hyperalgesia in mice at least two weeks after the operation, as SCI-induced pain develops after a latent period in both humans and rodents [18], [51]. Compared to the Sham/Vehicle group, there was a significant decrease in the withdrawal threshold and latency of the right hind limb after SCI (Fig. 4A and B). However, we observed that antidepressant doses of ZBD-2 (2 and 4 mg/kg) markedly reduced mechanical allodynia (Fig. 4A) and thermal hyperalgesia (Fig. 4B) .
Fig. 4.

Effects of ZBD-2 on pain perception. Mechanical allodynia was evaluated on days 14, 21, 28, and 35, while thermal hyperalgesia was assessed on the right lower limb on day 35 after SCI. SCI mice exhibited increased mechanical allodynia and thermal hyperalgesia when compared to Sham/Vehicle mice (Fig. 4A and B). Administration of ZBD-2 (either 2 or 4 mg/kg) markedly improved the mechanical allodynia two weeks later (Fig. 4A) and hyperalgesia on day 35 after SCI (Fig. 4B). n = 8 in each group, *p < 0.05, **p < 0.01 vs. Sham/Vehicle group; #p < 0.05, ##p < 0.01 vs. SCI/Vehicle group.
3.5. ZBD-2 decreased morphological lesions on the spinal cord after SCI
We employed HE staining to assess the morphology of the injury epicenter 35 days after SCI. The volume of the spinal lesions in SCI mice was evaluated by the area of cavitation formation and analyzed using stereological techniques. The SCI/Vehicle group exhibited a larger volume of cavitation and lesion size induced by SCI (Fig. 5A and B). There was also a significant reduction in lesion size and greater preservation of the cavitation formation in SCI mice treated with ZBD-2 (2 and 4 mg/kg) than in vehicle-treated animals (Fig. 5B). This reduction occurred in both white and gray matter, suggesting an overall decrease in cavitation formation and tissue loss. This morphological result was consistent with the neurological outcome evaluated by the BMS and the angle of the inclined plane test (Fig. 1A and B).
Fig. 5.

ZBD-2 decreased morphological lesions of the lumbar dorsal horn. HE staining was employed to assess the morphology of the injury epicenter of the spinal cord on day 35 after SCI (Fig. 5A and B). SCI mice exhibited increased areas of cavitation formation when compared to Sham/Vehicle mice. Administration of ZBD-2 (either 2 or 4 mg/kg) significantly decreased the cavitation volume in a dose-dependent manner in SCI mice relative to SCI/Vehicle mice (Fig. 5B). n = 8 in each group, *p < 0.05, **p < 0.01 vs. Sham/Vehicle group; #p < 0.05, ##p < 0.01 vs. SCI/Vehicle group. Scale bars = 50 μm.
3.6. ZBD-2 suppressed gliocyte activation in the lumbar dorsal horn following SCI
Gliocyte activation plays an important role in the development and maintenance phases of chronic SCI-induced pain. To determine whether the observed attenuation of hyperexcitability in ZBD-2-treated mice is related to the suppressed activation of spinal gliocytes, we stained spinal cord lumbar sections for Iba-1 5 weeks following SCI and subsequently quantified the number of activated microglia within the dorsal horn (Fig. 6A and B). We observed that Iba-1 positive reactive microglia were hardly observed in Sham/Vehicle tissue (Fig. 6A), though Iba-1+ cells were obviously increased in the SCI/Vehicle group (Fig. 6B). Administration of ZBD-2 (2 and 4 mg/kg) for two weeks significantly reduced the number of positively-stained cells (Fig. 6A).
Fig. 6.

ZBD-2 administration alters expression of microglia immunoreactivity. Representative images of Iba-1 in the lumbar dorsal horn were obtained on day 35 after SCI (Fig. 6A and B). Quantification of immunoreactivity for Iba-1 revealed significantly increased expression of Iba-1 positive reactive microglia in SCI mice compared to the Sham/Vehicle group. This change was remarkably suppressed by administration of ZBD-2 (either 2 or 4 mg/kg) (Fig. 6B). n = 8 in each group, *p < 0.05, **p < 0.01 vs. Sham/Vehicle group; #p < 0.05, ##p < 0.01 vs. SCI/Vehicle group. Scale bars = 40 μm.
3.7. Effects of ZBD-2 on protein expression in the spinal cord and hippocampus
Upregulated expression of TSPO has been confirmed in many neurological disorders and is generally regarded as a biomarker of neural damage [34]. In the present study, the level of TSPO in the spinal cord was increased in response to SCI, and treatment with ZBD-2 (2 and 4 mg/kg) for two weeks markedly decreased the upregulation of TSPO (Fig. 7A and B). This result indicates that TSPO upregulation in the spinal cord is associated with SCI-induced pain. Chronic activation of microglia and astrocytes plays an important role in the induction and maintenance of SCI-induced pain from a few hours up to a month post-injury. There was an obvious upregulation of Iba-1 and GFAP in the spinal cord after SCI, though treatment with ZBD-2 (either 2 or 4 mg/kg) for two weeks caused an obvious decrease in the upregulation of TPSO in SCI mice (Fig. 7A, C, and D). ZBD-2-treated mice exhibited restored levels of BDNF and phospho-CREB in the hippocampus compared to the SCI/Vehicle group (Fig. 7E–G); however, no significant differences were observed in the expression of CREB (Fig. 7E and H).
Fig. 7.

Effects of ZBD-2 on protein expression in the spinal cord and hippocampus. Representative results of western blot analysis for TSPO, Iba-1, GFAP, BDNF, CREB, and phospho-CREB on day 35 after SCI (Fig. 7A). SCI mice exhibited obvious upregulation of TSPO, Iba-1, and GFAP in the spinal cord (Fig. 7B–D) and downregulation of BDNF and phospho-CREB in the hippocampus when compared to the Sham/Vehicle group (Fig. 7E–G). Administration of ZBD-2 (either 2 or 4 mg/kg) for two weeks significantly decreased the upregulation of TSPO, Iba-1, and GFAP in SCI mice and reversed the downregulation of BDNF and phospho-CREB when compared to the SCI/Vehicle group. Levels of CREB in the hippocampus remained unaltered for any group (Fig. 7H). n = 8 in each group, *p < 0.05, **p < 0.01 vs. Sham/Vehicle group; #p < 0.05, ##p < 0.01 vs. SCI/Vehicle group.
4. Discussion
In the present study, we revealed that administration of ZBD-2 (2 or 4 mg/kg, intragastric) for two weeks significantly attenuated the symptoms of chronic SCI-pain and pain-induced depressive-like behaviors in mice. First, in our SCI mouse model, ZBD-2 significantly improved recovery of motor function in a dose-dependent and time-dependent manner. Activation of TPSO with ZBD-2 had an obvious antidepressant effect in SCI mice via alteration of the nociceptive threshold, as well as the restoration of serum corticosterone levels. We further observed that ZBD-2 reduced cavitation formation and attenuated damage to the gray matter. Moreover, ZBD-2 significantly reversed chronic activation of microglia and astrocytes, as evidenced by a reduction in the number of Iba-1 positive reactive microglia and the downregulation of Iba-1 and GFAP expression in the spinal cord. Finally, ZBD-2 restored the downregulation of BDNF and phospho-CREB in the hippocampus and effectively alleviated depressive-like symptoms in SCI mice.
SCI is a devastating event that leads to motor dysfunction below the level of the lesion and to the development of chronic pain syndromes within months of the initial injury. Post-SCI-pain can produce drastic impairments in daily routines, greatly impacting quality of life and frequently leading to depression and suicidal ideation [19]. Previous research has revealed that binding to the cholesterol-binding domain of TSPO may rescue motor neurons from axotomy-induced death and improve nerve regeneration and functional recovery [52]. In the healthy nervous system, TSPO is expressed at low levels in steroid-synthesizing tissues such as dorsal root ganglia sensory neurons, microglia, and reactive astrocytes in glia [33]. TSPO also plays an important role in mitochondrial proliferation and membrane biogenesis during proliferation and repair of neural cells after injury. Therefore, TSPO is highly expressed in the injured or diseased sites in both the central and peripheral nervous systems. Activation of TSPO in order to promote neurosteroid synthesis is beneficial for many neurological and neurodegenerative disorders, such as Alzheimer’s disease, multiple sclerosis, Huntington’s disease, amyotrophic lateral sclerosis, and Parkinson’s disease [36]. Downregulation of TSPO by ZBD-2 may have therefore played a role in attenuating compensatory effects.
Gliocyte activation has been proposed as a critical mechanism for chronic and persistent SCI-pain and depressive-like behaviors [1]. Furthermore, gliocyte activation in the spinal cord following SCI has been correlated with lower nociceptive thresholds [53]. In the present study, we observed gliocyte activation in the lumbar spinal dorsal horn following SCI, although treatment with ZBD-2 markedly suppressed chronic post-traumatic gliocyte activation at lesion sites and limited the development of hyperesthesia after SCI. In addition, previous studies have indicated that the CREB-BDNF signaling system can be affected by chronic stress and is a potential target for antidepressant treatment [54]. In fact, BDNF may be a crucial factor in mediating neuronal survival, differentiation, function, and plasticity. The expression of BDNF mRNA can be modulated by signaling cascades converging into CREB [55]. Neurotrophins play a key role in plastic processes that regulate a broad range of psychopathologies, such as depression, emotional distress, and post-traumatic stress disorder, which frequently occur in patients with spinal lesions [56]. We observed that SCI selectively decreased BDNF expression in the hippocampus, resulting in the development of depressive-like symptoms, and that administration of ZBD-2 significantly ameliorated depressive-like symptoms by reversing the SCI-induced dysfunction of the BDNF-CREB signaling pathway in the hippocampus.
In conclusion, the present data provide underlyingevidence for the role of ZBD-2 in improving chronic SCI-pain and pain-induced depressive-like symptoms in SCI mice.However, from the current research, it is far from clear whether SCI-pain caused the depressive-like symptoms or they are interrelated and mutually promoted. What is more, The upregulation of TSPO by ZBD-2 is associated with SCI-induced pain, but also possibly related to variation itself in the lesion of spinal cord. Meanwhile, The issue of potential liver-related side effectsand differential dosing regimens should also be researched. The medium-term and long-term efficacy of TSPO ligands in distinct neuropsychiatric conditions are far more explicit. Since the effects of ZBD-2 on pain and other SCI consequences last longer than the treatment regimen, it would be important to follow up on long-lasting behavioral changes in these outcome measures for clinical relevance. Further more studies are required in order to confirm this finding. Nonetheless, the current study has important implications because these findings expand the clinical application of ZBD-2 in the therapy of slowing and preventing SCI-induced neuropathology and psychopathology.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No. 31500820) and the Military Medical Science and Technology Youth Training Project of China (No. 14QNP095).
Contributor Information
Yong Chen, Email: 13813889089@139.com.
Qi Yang, Email: yangqifmmu@126.com.
Jian-ning Zhao, Email: jnzhao2002@163.com.
References
- 1.Anderson C.J. Anxiety and depression in children and adolescents with spinal cord injuries. Dev. Med. Child Neurol. 2009;51(10):826–832. doi: 10.1111/j.1469-8749.2009.03268.x. [DOI] [PubMed] [Google Scholar]
- 2.Wu J. Spinal cord injury causes brain inflammation associated with cognitive and affective changes: role of cell cycle pathways. J. Neurosci. 2014;34(33):10989–11006. doi: 10.1523/JNEUROSCI.5110-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stormer S. Chronic pain/dysaesthesiae in spinal cord injury patients: results of a multicentre study. Spinal Cord. 1997;35(7):446–455. doi: 10.1038/sj.sc.3100411. [DOI] [PubMed] [Google Scholar]
- 4.Yezierski R.P. Pain following spinal cord injury: pathophysiology and central mechanisms. Prog. Brain Res. 2000;129:429–449. doi: 10.1016/S0079-6123(00)29033-X. [DOI] [PubMed] [Google Scholar]
- 5.Defrin R. Pain following spinal cord injury. Spinal Cord. 2002;40(2):96–97. doi: 10.1038/sj.sc.3101245. author reply 98–99. [DOI] [PubMed] [Google Scholar]
- 6.Sjolund B.H. Pain and rehabilitation after spinal cord injury: the case of sensory spasticity? Brain Res. Brain Res. Rev. 2002;40(1–3):250–256. doi: 10.1016/s0165-0173(02)00207-2. [DOI] [PubMed] [Google Scholar]
- 7.Vierck C.J., Jr., Light A.R. Allodynia and hyperalgesia within dermatomes caudal to a spinal cord injury in primates and rodents. Prog. Brain Res. 2000;129:411–428. doi: 10.1016/S0079-6123(00)29032-8. [DOI] [PubMed] [Google Scholar]
- 8.Wasner G. Central pain syndromes. Curr. Pain Headache Rep. 2010;14(6):489–496. doi: 10.1007/s11916-010-0140-8. [DOI] [PubMed] [Google Scholar]
- 9.Jensen M.P., Chodroff M.J., Dworkin R.H. The impact of neuropathic pain on health-related quality of life: review and implications. Neurology. 2007;68(15):1178–1182. doi: 10.1212/01.wnl.0000259085.61898.9e. [DOI] [PubMed] [Google Scholar]
- 10.Naik A. Storage study and quality evaluation of coconut protein powder. J. Food Sci. 2013;78(11):S1784–92. doi: 10.1111/1750-3841.12267. [DOI] [PubMed] [Google Scholar]
- 11.Asmundson G.J., Katz J. Understanding the co-occurrence of anxiety disorders and chronic pain: state-of-the-art. Depress. Anxiety. 2009;26(10):888–901. doi: 10.1002/da.20600. [DOI] [PubMed] [Google Scholar]
- 12.Robinson M.J. Depression and pain. Front. Biosci. (Landmark Ed) 2009;14:5031–5051. doi: 10.2741/3585. [DOI] [PubMed] [Google Scholar]
- 13.Eulenburg V., Gomeza J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res. Rev. 2010;63(1–2):103–112. doi: 10.1016/j.brainresrev.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Gwak Y.S. Spatial and temporal activation of spinal glial cells: role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp. Neurol. 2012;234(2):362–372. doi: 10.1016/j.expneurol.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang H.T. Subacute human spinal cord contusion: few lymphocytes and many macrophages. Spinal Cord. 2007;45(2):174–182. doi: 10.1038/sj.sc.3101910. [DOI] [PubMed] [Google Scholar]
- 16.Zhao P., Waxman S.G., Hains B.C. Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J. Neurosci. 2007;27(9):2357–2368. doi: 10.1523/JNEUROSCI.0138-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hains B.C. Serotonergic neural precursor cell grafts attenuate bilateral hyperexcitability of dorsal horn neurons after spinal hemisection in rat. Neuroscience. 2003;116(4):1097–1110. doi: 10.1016/s0306-4522(02)00729-7. [DOI] [PubMed] [Google Scholar]
- 18.Hains B.C., Waxman S.G. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J. Neurosci. 2006;26(16):4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hulsebosch C.E. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res. Rev. 2009;60(1):202–213. doi: 10.1016/j.brainresrev.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crown E.D. Activation of p38 MAP kinase is involved in central neuropathic pain following spinal cord injury. Exp. Neurol. 2008;213(2):257–267. doi: 10.1016/j.expneurol.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trahan E., Pepin M., Hopps S. Impaired awareness of deficits and treatment adherence among people with traumatic brain injury or spinal cord injury. J. Head Trauma Rehabil. 2006;21(3):226–235. doi: 10.1097/00001199-200605000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Migliorini C., Tonge B., Taleporos G. Spinal cord injury and mental health. Aust. N. Z. J. Psychiatry. 2008;42(4):309–314. doi: 10.1080/00048670801886080. [DOI] [PubMed] [Google Scholar]
- 23.Fumagalli F. Single exposure to erythropoietin modulates nerve growth factor expression in the spinal cord following traumatic injury: comparison with methylprednisolone. Eur. J. Pharmacol. 2008;578(1):19–27. doi: 10.1016/j.ejphar.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 24.Gomez-Pinilla F. BDNF and learning: evidence that instrumental training promotes learning within the spinal cord by up-regulating BDNF expression. Neuroscience. 2007;148(4):893–906. doi: 10.1016/j.neuroscience.2007.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ying Z. BDNF-exercise interactions in the recovery of symmetrical stepping after a cervical hemisection in rats. Neuroscience. 2008;155(4):1070–1078. doi: 10.1016/j.neuroscience.2008.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen M.S. Post-traumatic stress disorder and emotional distress in persons with spinal cord lesion. Spinal Cord. 2003;41(5):296–302. doi: 10.1038/sj.sc.3101427. [DOI] [PubMed] [Google Scholar]
- 27.Papadopoulos V. Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006;27(8):402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Papadopoulos V., Liu J., Culty M. Is there a mitochondrial signaling complex facilitating cholesterol import? Mol. Cell. Endocrinol. 2007;265-266:59–64. doi: 10.1016/j.mce.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 29.Jorda E.G. Evidence in favour of a role for peripheral-type benzodiazepine receptor ligands in amplification of neuronal apoptosis. Apoptosis. 2005;10(1):91–104. doi: 10.1007/s10495-005-6064-9. [DOI] [PubMed] [Google Scholar]
- 30.Jayakumar A.R., Panickar K.S., Norenberg M.D. Effects on free radical generation by ligands of the peripheral benzodiazepine receptor in cultured neural cells. J. Neurochem. 2002;83(5):1226–1234. doi: 10.1046/j.1471-4159.2002.01261.x. [DOI] [PubMed] [Google Scholar]
- 31.Delavoie F. In vivo and in vitro peripheral-type benzodiazepine receptor polymerization: functional significance in drug ligand and cholesterol binding. Biochemistry. 2003;42(15):4506–4519. doi: 10.1021/bi0267487. [DOI] [PubMed] [Google Scholar]
- 32.Lacapere J.J., Papadopoulos V. Peripheral-type benzodiazepine receptor: structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids. 2003;68(7–8):569–585. doi: 10.1016/s0039-128x(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 33.Chen M.K., Guilarte T.R. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol. Ther. 2008;118(1):1–17. doi: 10.1016/j.pharmthera.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rupprecht R. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010;9(12):971–988. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- 35.Papadopoulos V. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis: neuropathology and neurological disorders. Neuroscience. 2006;138(3):749–756. doi: 10.1016/j.neuroscience.2005.05.063. [DOI] [PubMed] [Google Scholar]
- 36.Girard C. Axonal regeneration and neuroinflammation: roles for the translocator protein 18 kDa. J. Neuroendocrinol. 2012;24(1):71–81. doi: 10.1111/j.1365-2826.2011.02215.x. [DOI] [PubMed] [Google Scholar]
- 37.Rupprecht R. Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science. 2009;325(5939):490–493. doi: 10.1126/science.1175055. [DOI] [PubMed] [Google Scholar]
- 38.Kita A. Antianxiety and antidepressant-like effects of AC-5216: a novel mitochondrial benzodiazepine receptor ligand. Br. J. Pharmacol. 2004;142(7):1059–1072. doi: 10.1038/sj.bjp.0705681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Detloff M.R. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp. Neurol. 2008;212(2):337–347. doi: 10.1016/j.expneurol.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nasirinezhad F. Viral vectors encoding endomorphins and serine histogranin attenuate neuropathic pain symptoms after spinal cord injury in rats. Mol. Pain. 2015;11:p2. doi: 10.1186/1744-8069-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basso D.M. Basso mouse scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J. Neurotrauma. 2006;23(5):635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- 42.Pajoohesh-Ganji A. A combined scoring method to assess behavioral recovery after mouse spinal cord injury. Neurosci. Res. 2010;67(2):117–125. doi: 10.1016/j.neures.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pearse D.D. Histopathological and behavioral characterization of a novel cervical spinal cord displacement contusion injury in the rat. J. Neurotrauma. 2005;22(6):680–702. doi: 10.1089/neu.2005.22.680. [DOI] [PubMed] [Google Scholar]
- 44.Sedy J. Methods for behavioral testing of spinal cord injured rats. Neurosci. Biobehav. Rev. 2008;32(3):550–580. doi: 10.1016/j.neubiorev.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 45.Liu S.B. Attenuation of reserpine-induced pain/depression dyad by gentiopicroside through downregulation of GluN2 B receptors in the amygdala of mice. Neuromolecular Med. 2014;16(2):350–359. doi: 10.1007/s12017-013-8280-8. [DOI] [PubMed] [Google Scholar]
- 46.Duman R.S., Monteggia L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry. 2006;59(12):1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Ito N. Antidepressant-like activity of a Kampo (Japanese herbal) medicine, Koso-san (Xiang-Su-San), and its mode of action via the hypothalamic-pituitary-adrenal axis. Phytomedicine. 2006;13(9-10):658–667. doi: 10.1016/j.phymed.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 48.Tian Z. Estrogen receptor GPR30 exerts anxiolytic effects by maintaining the balance between GABAergic and glutamatergic transmission in the basolateral amygdala of ovariectomized mice after stress. Psychoneuroendocrinology. 2013;38(10):2218–2233. doi: 10.1016/j.psyneuen.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 49.Esposito E. Glucocorticoid-induced leucine zipper (GILZ) over-expression in T lymphocytes inhibits inflammation and tissue damage in spinal cord injury. Neurotherapeutics. 2012;9(1):210–225. doi: 10.1007/s13311-011-0084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu J. Cell cycle activation contributes to increased neuronal activity in the posterior thalamic nucleus and associated chronic hyperesthesia after rat spinal cord contusion. Neurotherapeutics. 2013;10(3):520–538. doi: 10.1007/s13311-013-0198-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Falci S. Dorsal root entry zone microcoagulation for spinal cord injury-related central pain: operative intramedullary electrophysiological guidance and clinical outcome. J. Neurosurg. 2002;97(Suppl. (2)):193–200. doi: 10.3171/spi.2002.97.2.0193. [DOI] [PubMed] [Google Scholar]
- 52.Papadopoulos V., Lecanu L. Translocator protein (18 kDa) TSPO: an emerging therapeutic target in neurotrauma. Exp. Neurol. 2009;219(1):53–57. doi: 10.1016/j.expneurol.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meller S.T. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33(11):1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 54.Kuipers S.D. Chronic stress-induced changes in the rat brain: role of sex differences and effects of long-term tianeptine treatment. Neuropharmacology. 2013;75:426–436. doi: 10.1016/j.neuropharm.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 55.Tardito D. Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Pharmacol. Rev. 2006;58(1):115–134. doi: 10.1124/pr.58.1.7. [DOI] [PubMed] [Google Scholar]
- 56.Gundersen B.B. Increased hippocampal neurogenesis and accelerated response to antidepressants in mice with specific deletion of CREB in the hippocampus: role of cAMP response-element modulator tau. J. Neurosci. 2013;33(34):13673–13685. doi: 10.1523/JNEUROSCI.1669-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]