

Abstract
In the past, liver cirrhosis was considered an irreversible phenomenon. However, many experimental data have provided evidence of the reversibility of liver fibrosis. Moreover, multiple clinical studies have also shown regression of fibrosis and reversal of cirrhosis on repeated biopsy samples. As various etiologies are associated with liver fibrosis via integrated signaling pathways, a comprehensive understanding of the pathobiology of hepatic fibrogenesis is critical for improving clinical outcomes. Hepatic stellate cells play a central role in hepatic fibrogenesis upon their activation from a quiescent state. Collagen and other extracellular material components from activated hepatic stellate cells are deposited on, and damage, the liver parenchyma and vascular structures. Hence, inactivation of hepatic stellate cells can lead to enhancement of fibrolytic activity and could be a potential target of antifibrotic therapy. In this regard, continued efforts have been made to develop better treatments for underlying liver diseases and antifibrotic agents in multiple clinical and therapeutic trials; the best results may be expected with the integration of such evidence. In this article, we present the underlying mechanisms of fibrosis, current experimental and clinical evidence of the reversibility of liver fibrosis/cirrhosis, and new agents with therapeutic potential for liver fibrosis.
Keywords: Liver cirrhosis, Fibrosis, Hepatic stellate cells, Reversal, Therapy
INTRODUCTION
Many liver diseases occur as a response to injury over an extended period of time before culminating in liver cirrhosis. Although the etiologies of liver diseases may vary, fibrosis and cirrhosis develop through common signaling pathways. Cascades of reactions stimulate quiescent hepatic stellate cells (HSCs) into their activated forms, leading to the accumulation of collagen and other extracellular matrix (ECM) components. Sustained stimulation and accumulation of these materials lead to the destruction of liver structures and hepatic innervation, and decreased liver function [1]. We have recently increased our understanding of the mechanisms underlying hepatic fibrosis, which may be used as potential treatment targets for the inhibition or reversal of fibrosis. In this review, we will discuss some new aspects of the pathophysiology of fibrosis, the clinical evidence of reversibility according to etiology, and future therapeutics for fibrosis.
PATHOBIOLOGY OF HEPATIC FIBROSIS
Activation of HSCs: a key driving factor
Liver fibrogenesis is initiated by HSC activation, which is the primary effector cell orchestrating the deposition of ECM in the liver structure (Fig. 1). HSCs are located in the perisinusoidal space between the sinusoids and hepatocytes, known as the space of Disse [2]. HSCs activate the immune response through the secretion of cytokines and chemokines and through interactions with immune cells [3]. Activation of HSCs can be provoked by a range of chronic liver inflammatory factors, reactive oxygen species (ROS), and cytokines. Activated HSCs are transformed into myofibroblasts, which have profibrogenic properties; they secrete transforming growth factor β (TGF-β), α-smooth muscle actin, and type I collagen [4].
Figure 1.
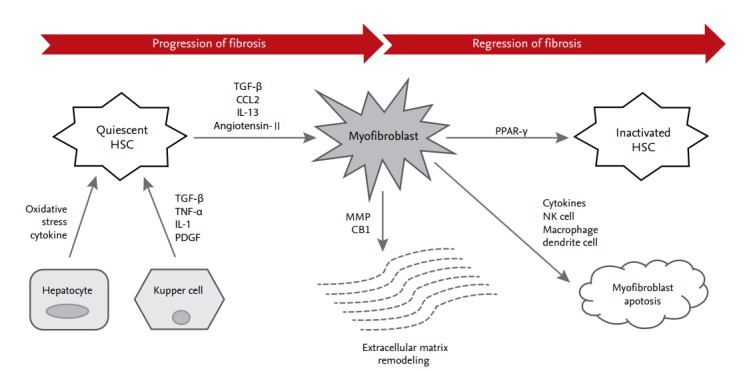
Process of hepatic stellate cell (HSC) differentiation during progression and regression of fibrosis. In the underlying pathway of liver fibrosis, HSCs undergo differentiation from quiescent cells to myofibroblasts. A neighboring environment that is characterized by multiple immune cells, cytokines, and small molecules orchestrates this process. TGF-β, transforming growth factor β; CCL2, C-C motif chemokine ligand type 2; IL, interleukin; TNF-α, tumor necrosis factor α; PDGF, platelet-derived growth factor; MMP, matrix metalloproteinase; CB1, cannabinoid receptor 1; PPAR-γ, peroxisome proliferator-activated receptor γ; NK, natural killer.
Hepatic inflammation
Chronic inflammation is the main cause of hepatic fibrogenesis and it was found to be present in the majority of chronic liver diseases such as viral hepatitis, toxic liver injury, alcoholic hepatitis, non-alcoholic steatohepatitis (NASH), and autoimmune liver diseases [5]. Hepatic inflammation results in activation of HSCs through several factors and pathways irrespective of its etiology. Initial paracrine stimulation, including exposure to apoptotic bodies of damaged hepatocytes can, in turn, activate quiescent HSCs and transform them into myofibroblasts. Lipid peroxides from Kupffer cells drive early activation and changes in the surrounding ECM [6]. Lipopolysaccharide (LPS) activates Toll-like receptor 4 signaling in Kupffer cells leading to the activation of nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB)-interferon regulatory factor 3 pathway and the subsequent transcriptional activation of proinflammatory cytokines, such as tumor necrosis factor α (TNF-α) and interferon γ (IFN-γ) [7]. This pathway leads to endothelial cell dysfunction, impaired exchange of solutes among neighboring cells, altered hepatocyte function, and subsequent non-parenchymal cell damage. In an experimental study, HSCs showed a more activated phenotype, greater proliferation rates, and increased collagen synthesis when they were co-cultured with Kupffer cells or hepatocytes, as compared to when they were cultured alone [8].
ROS production
ROS released by Kupffer cells and hepatocytes can increase oxidative stress in hepatocytes, promote their apoptosis, and further stimulate the activation of HSCs [9]. ROS are generated mainly via the mitochondrial electron transport chain or via activation of cytochrome P450, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase, or via mitochondrial damage. The production of ROS is influenced by the activity of NADPH oxidase in HSCs, macrophages, and hepatocytes and by the production of nitric oxide in Kupffer cells [10-12]. Clinically in alcoholics, there is a strong induction of cytochrome P450 2E1 leading to increased ROS and pericentral (zone 3) damage. NADPH oxidase mediates liver injury and fibrosis through the generation of oxidative stress [13].
Cytokines
Cytokines, including TGF-β, platelet-derived growth factor (PDGF), and endothelial growth factor induce the transformation of quiescent HSCs into myofibroblasts [14]. Angiotensin II, which is secreted by HSCs, enhances HSC proliferation and, in turn, contributes to the production of ECM [15]. The cannabinoid receptor cannabinoid receptor 1 (CB1) is upregulated in myofibroblasts or activated HSCs, and increases hepatic fibrosis. In contrast, the CB2 receptor on these cells demonstrates an antifibrotic effect [16,17]. Additionally, adipokines also contribute to the hepatic manifestations of obesity and fibrogenesis. Leptin, which is a circulating adipogenic hormone, promotes stellate cell fibrogenesis, enhances tissue inhibitor of metalloproteinase 1 (TIMP-1) expression, exerts its action through Janus kinase (JAK)-signal transduction, and suppresses peroxisome proliferator-activated receptor γ (PPAR-γ) [18].
Immune responses: variable roles in hepatic fibrosis
Immune interactions play an important role in driving fibrogenesis, as persistent inflammation usually precedes fibrosis.
Responses of the diverse cells of the liver
Activated HSCs secrete inflammatory cytokines, interact directly with immune cells by expressing different adhesion molecules, and modulate the adaptive immune system by functioning as antigen presenting cells [19]. Therefore, a positive feedback loop exists in which inflammatory and fibrogenic cells stimulate each other to amplify fibrosis. Other cell types that regulate the progression and resolution of fibrosis include liver sinusoidal endothelial cells (LSECs), Kupffer cells, hepatocytes, natural killer (NK) cells, T cells, monocytes, cholangiocytes, ductular cells, portal fibroblasts, and various other inflammatory cells [20].
Hepatic revascularization with LSEC activation and proliferation is highly associated with perisinusoidal fibrosis. During perisinusoidal fibrosis, activated LSECs contribute to ECM production including synthesis of basement membrane components, fibronectin, and interstitial collagen type I. They also produce cytokines that activate HSCs and secrete factors that contribute to intrahepatic vasoconstriction, which contributes to portal hypertension in cirrhosis [21].
Kupffer cell activation leads to increased NF-κB activity and subsequent secretion of proinflammatory cytokines and chemokines including TNF-α and monocyte chemoattractant protein 1 (MCP-1) [22]. In turn, HSCs respond to this stimulation by secreting macrophage colony-stimulating factor, MCP-1, interleukin 6 (IL-6), C-C motif chemokine ligand type 21 (CCL21), and C-C motif chemokine receptor type 5 (CCR5) leading to an amplified acute phase response with further activation of macrophages [23-25]. TNF-α also induces neutrophil infiltration and stimulates mitochondrial oxidant production in hepatocytes, which undergo apoptosis. Damage to hepatocytes, which occurs predominantly in liver diseases characterized by enhanced oxidative and endoplasmic reticulum stress, lysosomal activation, and mitochondrial damage, are a strong trigger for fibrogenesis [26]. Phagocytosis of damaged hepatocytes by myofibroblasts triggers their fibrogenic activation via NADPH oxidase 2 and the JAK/signal transducer and activator of transcription (STAT) and phosphoinositide 3-kinase/Akt pathways [27,28].
NK cells exert their antifibrotic activity by inhibiting and killing activated HSCs. In liver injury, NK cells induce apoptosis of HSCs through the production of IFN-γ, although this pathway cannot be activated in advanced stages of liver fibrosis [22,29]. HSCs interact directly with various immune cells by expressing adhesion molecules including intercellular adhesion molecule 1 and vascular cell adhesion molecule 1 [30,31]. The expression of both of these adhesion molecules is increased in HSCs during injury, which is mediated by TNF-α, and peaks with maximal cell infiltration. Thus, adhesion molecule-induction on HSCs facilitates the recruitment of inflammatory cells to the injured liver.
CD4+ T cells with a Th2 polarization promote fibrogenesis in the liver, lungs, and kidneys [32]. Th2 cells, in particular, produce IL-4 and IL-13, which stimulate the differentiation of potentially fibrogenic myeloid cells and activated macrophages [33]. In experimental studies, rodents with Th2-dominant T cell infiltration display rapid fibrosis progression, while CD4+ Th1 cells have an antifibrotic effect [34].
Monocytes play a key role in inflammation and fibrosis. They are also precursors of fibrocytes, macrophages, and dendritic cells (DCs), and share characteristics with myeloid suppressor cells [35]. At the interface of innate and adaptive immunity, monocytes help adaptive immune responses, and proinflammatory monocytes (CD14+ and CD16+ in humans) promote fibrogenesis [36]. Interaction between chemokines and their receptors is important in the recruitment, activation, and function of monocytes; it could be an attractive target for fibrosis modulation [26]. CCL2 and its receptor CCR2 are central to monocyte recruitment to the liver during hepatic inflammation and fibrosis [37,38]. Although their inhibition ameliorates fibrosis progression in rodent models, they also delay fibrosis reversal [35]. Monocytes are the precursors of circulating fibrocytes, which are cells that differentiate into collagen-producing fibroblasts, and are related to bone marrow (BM) mesenchymal stem cells (MSCs). Additionally, monocytes are the source of fibrolytic CD133+ cells that accumulate in the liver to induce fibrosis reversal after BM transplantation [39]. Chemokines and their receptors are important in monocyte recruitment and activation, representing attractive targets for fibrosis modulation.
Responses during systemic inflammation and intestinal dysbiosis
During systemic inflammation, the immune response is initiated when bacteria are introduced through portal flow from the intestinal lumen. Pathogen-associated molecular patterns (PAMPs) from enteric bacterial organisms and damage-associated molecular patterns (DAMPs) originating from the host tissue upon injury stimulate innate immune cells [40]. Immune recognition of bacteria and PAMPs including LPS, lipopeptides, glycopolymers, flagellin, and bacterial DNA occurs both locally in the gut-associated lymph node tissue (GALT) and in mesenteric lymph nodes (MLN) as well as systemically [41]. Furthermore, immune cells already activated in the GALT and MLN may enter the peripheral blood and spread the inflammatory response systemically. DAMPs and sterile particulates, also released from necrotic hepatocytes, might also contribute to elicit an inflammatory response and fibrosis [42].
Intracellular response during hepatic fibrogenesis
Autophagy
Autophagy participates in hepatic fibrosis by activating HSCs and may also participate by influencing other fibrogenic cells [43]. Quiescent HSCs are filled with cytoplasmic lipid droplets (LDs) that contain retinyl esters [44]. Along with the switch from LD-rich cells to myofibroblast-like cells, autophagy flux is upregulated [45]. Autophagy may supply energy for activation of HSCs by delivering triglycerides and other components in LDs from autophagosomes to lysosomes for degradation [43].
Activation of TGF-β and the Smad pathway
In the liver, the role of TGF-β is important due to its multiple effects on hepatocellular proliferation and liver regeneration, induction of parenchymal cell apoptosis, immune surveillance, and hepatic fibrogenesis. Additionally, TGF-β secreted by myofibroblasts can induce hepatocellular apoptosis after activation [46]. During fibrogenesis, tissue and blood levels of active TGF-β are elevated and overexpression of TGF-β1 can induce fibrosis. These effects, along with the ability of TGF-β to upregulate ECM expression and the presence of functional TGF-β receptors on the surface of HSCs with persistent autocrine stimulation of activated HSCs and myofibroblasts by TGF-β, are key mechanisms of liver fibrogenesis [47]. Based on the identification of downstream events of TGF-β signaling transduction over the past several years, TGF-β1 has been shown to activate Smad2 and Smad3, which are negatively regulated by Smad7, an inhibitor of TGF-β signaling, through the ubiquitin-proteasome degradation mechanism [48]. In the context of liver fibrosis, Smad3 is profibrogenic, as Smad3 knockout mice are protected against dimethylnitrosamine-induced hepatic fibrosis [49]. Although Smad7 depletion promotes hepatic fibrosis, Smad7 is protective since its overexpression protects against HSC activation and hepatic fibrosis in in vitro and in vivo studies [50].
Role of microRNAs in fibrogenesis
MicroRNAs (miRNA) represent a family of small non-coding RNAs that control the translation and transcription of many genes [51]. Dysregulation of miRNA affects a wide range of cellular processes such as cell proliferation and differentiation involved in organ remodeling processes [52]. The importance of miR-29 in hepatic collagen homeostasis is underlined by in vivo data demonstrating that experimental severe fibrosis is associated with a prominent miR-29 decrease. The loss of miR-29 is due to the response of HSCs to exposure to the profibrogenic mediators TGF-β and PDGF [53]. Several putative binding sites for the Smad proteins and the Ap1 complex are located in the miR-29 promoter, which are suggested to mediate the decrease in miR-29 in fibrosis. Other miRNAs are highly increased after profibrogenic stimulation, such as miR-21. miR-21 is transcriptionally upregulated in response to Smad3 rather than Smad2 activation after TGF-β stimulation. In addition, TGF-β promotes miR-21 expression by formation of a microprocessor complex containing Smad proteins. Elevated miR-21 may then act as a profibrogenic miRNA by repressing the TGF-β inhibitory Smad7 protein [54].
Consequences of HSC activation
The ECM is a very important component of the liver structure and undergoes highly dynamic changes during synthesis and degradation. Life-threatening pathological conditions arise when ECM remodeling becomes excessive or uncontrolled. HSCs, neutrophils, and macrophages are involved in hepatic ECM degradation. Matrix metalloproteinases (MMPs) are the main enzymes responsible for ECM degradation and TIMPs have the ability to inhibit MMPs [55]. Thus, regulation of the MMP-TIMP balance is critical for efficient ECM remodeling. Activated HSCs not only synthesize and secrete ECM proteins such as type I and type III collagen but also produce MMP1 and MMP13 [56]. Moreover, activated HSCs up-regulate the expression and synthesis of TIMP1 and TIMP2 [57]. TIMP1 not only prevents the degradation of the rapidly increasing ECM by blocking MMPs but also inhibits apoptosis of activated HSCs [58]. The net result is the deposition of mature collagen fibers within the space of Disse, resulting in scarring.
EXPERIMENTAL EVIDENCE OF THE REGRESSION OF FIBROSIS
Increase in fibrolytic activity
The induction and subsequent spontaneous resolution of fibrosis has been observed in several animal models, and constitute data that are invaluable in determining the underlying biological mechanisms of fibrosis [57,59]. In the face of ECM degradation, fibrotic ECM continues to accumulate in chronic liver injury because of inhibition of MMP activity by myofibroblast-derived TIMP-1 [60]. Several studies investigating the resolution of liver fibrosis in rats showed that levels of TIMP-1 decreased after the cessation of injury [57,61]. As the level of TIMP-1 decreased, hepatic collagenase activity increased and ECM degradation occurred. Subsequent mechanistic studies that altered TIMP to balance MMP levels in situ have confirmed the powerful influence of this ratio on the development and resolution of fibrosis in the liver [62]. In terms of restoration of macrophages, macrophages have also been shown to be pivotal in the resolution of fibrosis, which emphasizes their role as regulators of effective wound healing and organ homeostasis [37]. Located in fibrotic tissue of the liver, macrophages are ideally placed to mediate ECM degradation and are a rich source of fibrolytic MMPs, including MMP12/13 [63,64]. Macrophages also express TNF-related apoptosis-inducing ligand that promotes myofibroblast apoptosis. Furthermore, phagocytosis of apoptotic cells by macrophages induces MMP expression and augments ECM degradation in rodent models of resolving hepatocellular fibrosis [65]. Stabilization of DCs has also been investigated in the context of the resolution of liver fibrosis using Cd11c-diphtheria toxin receptor (DTR) transgenic mice to deplete hepatic DCs during the recovery phase, following CCl4-mediated injury, as well as the use of adoptive transfer protocols. DCs were shown to mediate ECM degradation, probably through enhanced MMP9 expression [66].
Apoptosis or inactivation of HSCs
Activation of HSCs in response to chronic liver injury is a key step in the pathogenesis of liver fibrosis. Recently, clinical and experimental studies have demonstrated that fibrosis resolution may occur upon eradication of the liver insult [67]. Experimental models of fibrosis recovery have consistently reported that elimination of activated HSCs by apoptosis or inactivation of fibrolytic pathways led to the regression of fibrosis. This suggests that clearance of activated HSCs is a fundamental step in the onset of fibrosis regression [68]. Myofibroblasts produce fibrous scars in hepatic fibrosis. In the CCl4 model of liver fibrosis, quiescent HSCs are activated and transformed into myofibroblasts. When the underlying etiological agent is removed, clinical and experimental fibrosis undergo a remarkable regression, with complete disappearance of these myofibroblasts. However, it was shown that a subset of the myofibroblasts escaped apoptosis during regression of liver fibrosis, down-regulated fibrogenic genes, and acquired a phenotype similar to, but distinct from, quiescent HSCs; they were able to more rapidly reactivate into myofibroblasts in response to fibrogenic stimuli and strongly contribute to liver fibrosis. Inactivation of HSCs was associated with up-regulation of antiapoptotic genes, such as Hspa1a/b, which participate in the survival of HSCs in culture and in vivo [69].
In brief, liver fibrosis usually has potential for regression. Early liver fibrosis, which lacks ECM crosslinking and marked angiogenesis, can even reverse into almost normal architecture if the underlying cause is successfully treated [70]. This is considered the best form of antifibrotic therapy and facilitates the subsequent endogenous regulation of wound healing (Fig. 1).
CLINICAL EVIDENCE OF REVERSIBILITY ACCORDING TO ETIOLOGY
Regression of fibrosis is now a reality in clinical settings. The serial assessment of biopsy samples from patients with chronic liver disease of diverse etiologies, who were successfully treated, indicates that liver fibrosis is a dynamic and bidirectional process that has an inherent capacity for recovery and remodeling [71]. Next, we will describe the evidence and research outcomes of liver fibrosis recovery in clinical practice settings according to different etiologies (Tables 1 and 2) [72-90].
Table 1.
Major studies investigating the efficacy of treatment for biopsy-proven liver fibrosis in relation to hepatitis virus infection
| Liver disease | Drugs | Study design | No. of patients | Effects | Reference |
|---|---|---|---|---|---|
| HBV | Peginterferon | Non RCT | 110 | 27% Fibrosis improvement (16-month F/U) | [72] |
| Lamivudine | Non RCT | 63 | 73% Fibrosis improvement (F4 to F2-3, 36-month F/U) | [73] | |
| Entecavir | Non RCT | 57 | 88% Fibrosis improvement (65-month F/U) | [74] | |
| Tenofovir | Non RCT | 348 | 51% Fibrosis improvement (60-month F/U) | [75] | |
| HCV | Peginterferon and ribavirin | Non RCT | 60 | 82% Fibrosis improvement (60-monthF/U) | [76] |
| Angiotensin II type 1 receptor blocker (losartan) | Non RCT | 14 | 50% Fibrosis improvement (18-month F/U) | [81] | |
| PPAR-γ agonist (Farglitazar) | RCT | 109 | No fibrosis improvement (18-month F/U) | [82] |
HBV, hepatitis B virus; RCT, randomized controlled trial; F/U, follow-up; F4, METAVIR score, fibrosis score 4; HCV, hepatitis C virus; PPAR-γ, peroxisome proliferator-activated receptor γ.
Table 2.
Major studies investigating the efficacy of treatment for biopsy-proven liver fibrosis in non-viral chronic liver diseases
| Liver disease | Drugs | Study design | No. of patients | Effects | Reference |
|---|---|---|---|---|---|
| Alcoholic hepatitis | Abstinence of alcohol | Non RCT | 100 | Survival benefit, no fibrosis F/U data | [77] |
| ACE inhibitor (candesartan) | RCT | 85 | 33% vs. 12% Fibrosis improvement | [83] | |
| NASH | PPAR-γ agonist (pioglitazone) | RCT | 74 | Fibrosis improvement | [84] |
| PPAR-γ agonist (pioglitazone) vs. vitamin E vs. placebo | RCT | 247 | Decreased fibrosis progression rate of PPAR-γ group (96-week F/U) | [78] | |
| PPAR-γ agonist (rosiglitazone) | RCT | 53 | No effect | [85] | |
| Anti-TNF (pentoxifylline) | RCT | 55 | Fibrosis improvement | [86] | |
| High dose UDCA | RCT | 126 | Improvement on FibroTest | [87] | |
| FXR agonist (obeticholic acid) | RCT | 141 | 45% vs. 21%, Fibrosis improvement (p = 0.002; 24-week F/U) | [88] | |
| Autoimmune hepatitis | Steroid | Non RCT | 87 | 53% Fibrosis improvement (57-month F/U) | [79] |
| Cyclosporine A and steroid | Non RCT | 19 | Mean fibrosis stage decreased from 4.53 to 2.16 (3.63-year F/U) | [80] | |
| PBC | UDCA | RCT | 146 | No fibrosis improvement | [89] |
| UDCA | RCT | 103 | Lower fibrosis progression rate (p < 0.002) | [90] |
RTC, randomized control trial; F/U, follow-up; ACE, angiotensin-converting enzyme; NASH, nonalcoholic steatohepatitis; PPAR-γ, peroxisome proliferator-activated receptor γ; TNF, tumor necrosis factor; UDCA, ursodeoxycholic acid; FXR, farnesoid X receptor; PBC, primary biliary cirrhosis.
Hepatitis B virus-related liver disease
Chronic hepatitis B (CHB) is a significant worldwide problem as CHB patients develop cirrhosis and hepatocellular carcinoma. Standard treatments include pegylated IFN-α and nucleos(t)ide analogues [91]. Several studies have demonstrated that hepatitis B virus (HBV) DNA suppression is associated with biochemical and histological responses. There is currently evidence that these surrogate markers correlate with improved long-term clinical outcome [92]. IFN has been used in the treatment of CHB since the 1980s. Peginterferon therapy has been shown to reduce fibrosis progression in hepatitis B envelop antigen (HBeAg)-positive patients, with a greater response seen in those who sustain HBeAg seroconversion, as well as in HBeAg negative patients with a sustained virologic/biochemical response [72,93]. In one experimental study, IFN-α therapy exhibited antifibrotic activity by inhibiting the production of TGF-β, reducing HSC activation, and stimulating HSC apoptosis in vitro [94]. Another study observed the antifibrotic effect that IFN-γ exerted in liver cells through STAT-1 phosphorylation and impaired TGF-β signaling [95]. Long-term therapy with nucleoside analogues has also been shown to improve liver fibrosis and disease progression. In a 3-year study of lamivudine for hepatitis B treatment, follow-up liver biopsies indicated reversal of cirrhosis in eight of 11 patients (73%) [73]. Entecavir, which is a more potent inhibitor of viral replication in CHB, improves liver fibrosis. In a recent study, 96% of patients had histological improvement after long-term treatment with entecavir. Ten of the 57 patients had advanced fibrosis or cirrhosis (Ishak score 4 to 6) at baseline. All 10 patients achieved at least a 1-point reduction in the Ishak fibrosis score after long-term entecavir therapy [74]. In a more recent 5-year study with tenofovir treatment for chronic HBV infection that included patients with liver cirrhosis at the start of the study, 74% demonstrated extensive histological improvement, such that they were no longer considered to be cirrhotic [75].
Recently, instead of liver biopsy, transient elastography (TE) has been applied for the clinical assessment of liver fibrosis [96,97]. A large prospective cohort study of 426 individuals reported a significant decline in TE values in CHB patients after 3 years of antiviral treatment. However, the significant reduction in TE values at follow-up, compared to those at baseline, was limited in patients who had initially elevated alanine transaminase (ALT) levels [96]. To exclude the confounding effect of high ALT, another study investigated changes in TE values during antiviral treatment in 41 patients with CHB exhibiting low ALT levels (≤ 2 × the upper limit of normal). After 1 to 2 years of antiviral treatment, TE values significantly decreased compared with baseline, whereas ALT levels remained unchanged [97]. Non-invasive serum fibrosis markers were also utilized for the assessment of changes in liver fibrosis. It was reported that fibrosis based on the four factors (FIB-4) and the aspartate aminotransferase-to-platelet ratio index (APRI) were significantly improved in 370 HBV-associated cirrhosis patients who received 2 years of entecavir therapy [98]. These results suggest that potential fibrosis regression could be possible with long-term antiviral treatment and that clinical monitoring using non-invasive methods is useful [99].
Hepatitis C virus-related liver disease
Patients with compensated cirrhosis and chronic hepatitis C (CHC) benefit from IFN-based antiviral treatment. Viral eradication can be achieved in up to 40% of patients with genotype 1, and in 70% of patients with genotypes 2 or 3, reducing the risk of developing cirrhosis, hepatic decompensation, and hepatocellular carcinoma [100]. In a 5-year follow-up study of CHC patients with stage 2 or greater fibrosis at baseline, 82% of patients who had achieved sustained virologic response (SVR) after IFN treatment had decreased fibrosis scores. Another study demonstrated a reduction in clinical events after SVR [76,101]. In addition to IFN, excellent efficacy has been reported with several new directly acting antiviral agents (DAA) for HCV. Although little data were reported on follow-up liver biopsy, a recent report showed the possibility of reversal of liver fibrosis and cirrhosis by indirect measurement. TE values as well as FIB-4 and APRI scores were evaluated prior to therapy and within 18 months after DAA therapy; patients who had achieved SVR after DAA therapy showed significant regression of TE values and improvement of FIB-4 and APRI scores [102]. Liver fibrosis and cirrhosis are expected to improve in these patients with resolution of HCV infection.
Alcoholic liver disease
Clinical evidence for regression of fibrosis in alcoholic liver disease is limited. Results from randomized controlled trials (RCTs) assessing the effects of pharmacological agents on alcoholic fibrosis and cirrhosis have been disappointing. A Cochrane Intervention Review assessing the effect of colchicine for alcoholic and non-alcoholic liver fibrosis and cirrhosis from 15 RCTs reported the absence of statistically significant improvements in any significant clinical outcome, including liver histology [103]. However, there is a slight effect of abstinence of alcohol on clinical outcome. In one study, 100 patients with alcoholic cirrhosis were followed for 7 years after their baseline histological assessment. Abstinence at 1 month post-biopsy was associated with a significant improvement in long-term survival. This article demonstrated that there were benefits of abstinence after longer follow-up, with statistically significant differences in 5-year survival rates between those who abstained and persistent alcoholic drinkers (75% and 50%, respectively; p < 0.002) [77].
Non-alcoholic fatty liver disease
There are currently no approved treatments for non-alcoholic fatty liver disease (NAFLD) and therapies are based on targeting risk factors. Although there have been several studies defining the benefits of various pharmacological agents for NAFLD, these studies have been limited by small study populations and short-term follow-up periods [104]. Weight reduction through lifestyle modification is the first treatment strategy in NAFLD patients. Weight reduction is often associated with beneficial effects on multiple components of metabolic syndrome. Histological improvements have also been observed, particularly with respect to steatosis, but evidence of fibrosis regression is controversial. A RCT assessing the effect of weight reduction through lifestyle modification in 31 NASH patients over a 48-week period demonstrated significant improvements in the NASH histological activity score following an average weight loss of 9.3%, but failed to show a significant change in fibrosis [105].
Supplementation with the natural form of vitamin E (800 IU/day) has beneficial effects in patients with NASH, but benefits of pioglitazone are less clear according to the latest findings from the Pioglitazone vs. Vitamin E vs. Placebo for the Treatment of Nondiabetic Patients with Nonalcoholic Steatohepatitis (PIVENS) trial. In this study, 247 adults with biopsy-confirmed NASH without diabetes mellitus were randomly assigned to one of the three following treatment groups: (1) 30 mg per day of pioglitazone in addition to placebo; (2) 800 IU of vitamin E per day in addition to placebo; and (3) two placebo tablets daily. The primary outcome was a composite of improvement in hepatocellular ballooning, no worsening of fibrosis, and improved activity scores for NASH. Twice as many patients treated with vitamin E achieved the primary outcome compared to those who received placebo (43% vs. 19%). Although more patients in the pioglitazone group than the placebo group fulfilled the primary outcome (34% vs. 19%), the difference was not statistically significant [78].
A recent placebo-controlled randomized trial demonstrated that obeticholic acid, a synthetic farnesoid X receptor agonist, was effective in patients with NASH. Patients were treated for 72 weeks and the primary end-point was improvement in histology, as measured by a two-point reduction in a composite activity histological score without worsening of fibrosis. The therapeutic phase of the trial was stopped early partly because a preplanned interim analysis revealed that more patients on obeticholic acid (45%, 50 of 110) than on placebo (21%, 23 of 109) reached the primary endpoint (relative risk, 1.9; 95% confidence interval, 1.3 to 2.8). Thirty-six of 102 obeticholic acid-treated patients (35%) demonstrated fibrosis regression by one stage or more compared to 19 of 98 placebo-treated patients (19%). Although the study was stopped after the interim analysis, the main result of this study demonstrates a clear improvement in all histological features of NASH, including steatosis, inflammation, and liver-cell injury, together with a reduction in aminotransferases, biochemical markers of hepatic damage [106].
Autoimmune hepatitis
Approximately 40% of patients with autoimmune hepatitis develop cirrhosis under current therapies. Although the course is variable depending on the period of observation, the annual occurrence of cirrhosis is estimated at 3% per year [107,108]. Several studies have not only demonstrated the antifibrotic effects of immunosuppressive therapy in autoimmune hepatitis but have also strengthened the association between the indices of hepatic inflammation and the progression of liver fibrosis [79,80]. Fibrosis scores using the Ishak system improved in 46 of 87 treated patients (53%) with autoimmune hepatitis during 63 months, and the histological activity index decreased concurrently [79]. They suggested that improvement of hepatic fibrosis is possible in the majority of treated patients with autoimmune hepatitis and that failure to suppress liver inflammation worsens fibrosis [109].
Primary biliary cirrhosis
The only clinically approved medical treatment for primary biliary cirrhosis (PBC) is ursodeoxycholic acid (UDCA) [110,111]. However, there are controversies regarding the interpretation of current evidence. In several studies with crossover from placebo or no UDCA treatment, the crossover patients’ conditions deteriorated despite using UDCA [112]. A Cochrane Review evaluating 16 RCTs using UDCA versus placebo revealed that almost half of these trials had a high risk of bias and concluded that UDCA did not significantly improve liver histology and had no demonstrable effect on improving mortality [113]. Nevertheless, UDCA may have benefits in early stage and asymptomatic PBC. In the asymptomatic PBC cohort described by Prince et al. [114], 45% of the patients taking UDCA did not develop liver-related symptoms during a median follow-up of 7.4 years.
The role of immunosuppressive agents in PBC remains controversial. A few studies evaluating methotrexate have presented conflicting results and some studies suggest that methotrexate may worsen mortality [115].
Obeticholic acid, a derivative of chenodeoxycholic acid, has, unlike UDCA, strong activating effects on the nuclear receptor farnesoid X receptor in phase II results [116]. In a recent clinical trial with obeticholic acid as therapy for PBC, favorable effects were also observed in PBC patients with an inadequate response to UDCA [117]. Alkaline phosphatase, γ glutamyl transpeptidase, and alanine aminotransferase levels were significantly improved in patients receiving obeticholic acid compared with those in the placebo group. In this study, the treatment period was only 3 months and liver biopsies were not obtained to identify histologic changes. Subjects were allowed to participate in an open-label extension trial, which demonstrated sustained decreases in liver enzyme levels over 12 months [117]. Future trials to determine the beneficial effects of obeticholic acid on hepatic fibrosis in patients with primary cirrhosis are warranted.
Hemochromatosis
Few studies have identified a regression of fibrosis and reversal of cirrhosis using liver biopsy following phlebotomy in patients with hereditary hemochromatosis [118]. The most recent study addressing reversibility of liver fibrosis or cirrhosis assessed histological outcome following venesection in 36 cases of C282Y homozygotes with documented F3 or F4 fibrosis on index biopsy. The 36 patients were enrolled from C282Y homozygotes with either severe fibrosis or cirrhosis (F3 or F4 fibrosis, staged according to the METAVIR grading system). When defining regression of fibrosis as a decrease of at least 2 METAVIR units, fibrosis regressed in nine of 13 patients (69%) with stage F3 and in eight of 23 patients (35%) with stage F4 fibrosis [119].
NEW ANTIFIBROTIC DRUGS AND EXPECTATIONS FOR CLINICAL USE
Recently, there has been a steady addition to the number of molecules and pathways that are targets for antifibrotic therapy (Table 3) [88,120-128]; TGF-β1 remains the most important of such molecules. However, systemic inhibition of TGF-β1 results in increased inflammation [129]. This has spurred the targeting of specific steps of TGF-β1 activation in a localized manner. Inhibition of integrin αvβ6, with reduction of TGF-β1 activation, promises to be a highly effective and localized antifibrotic approach [121]. Connective tissue growth factor (CTGF) amplifies TGF-β1 signaling, and a monoclonal antibody targeting CTGF has shown promise in animal models of organ fibrosis [122]. Attenuating the activated phenotype of myofibroblasts is an attractive approach due to their key role in ECM deposition. Inhibition of the CB1 reverses myofibroblast activation and attenuates experimental liver fibrosis [123]. This has passed the proof of principle state, and peripheral-acting CB1 antagonists that may circumvent adverse side effects on the central nervous system, such as depression, are being developed [130]. In fibrotic NASH, progression is intimately linked with insulin resistance/type 2 diabetes as well as lipotoxic hepatocyte death and intestinal dysbiosis, providing rational targets for both anti-inflammatory and antifibrotic therapy. Therapeutic strategies include reducing oxidative stress, improving insulin signaling, activating the farnesoid X receptor (e.g., with obeticholic acid), fibrosis-targeted inhibitors of hedgehog signaling, combined PPAR-α/δ agonists, or manipulation of altered gut microbiota using probiotics or microbiota transfer [120]. Although oxidative stress is an important cofactor in fibrosis, the use of antioxidants has proved disappointing. Activation of NADPH oxidases (NOX1, NOX 2, and NOX4) induces HSC activation. NOX4 can trigger apoptosis in hepatocytes [27,124]. NOX inhibitors have been studied for the prevention of liver fibrosis.
Table 3.
New antifibrotic targets and agents for clinical use
| Anti-fibrotic target | Agent | Effects | Reference |
|---|---|---|---|
| TGF-β1 inhibition | Anti-αvβ6 monoclonal antibody | Inhibition of integrin αvβ6 | [121] |
| Anti-CTGF monoclonal antibody | Inhibition of CTGF | [122] | |
| Apoptosis or quiescence of myofibroblast activation | CB1 antagonist | Inhibition of CB1 | [123] |
| FXR agonist | Activating the farnesoid X receptor | [88] | |
| miRNA-29 | Inhibition of the coreceptor smoothened, fibrosis-targeted inhibitors of hedgehog signaling | [120,128] | |
| CCR1/CCR5 antagonists | Inhibition of HSC activation and increase of hepatocyte survival | [127] | |
| Reduction of oxidative stress | NOX inhibitor | Reduction of reactive oxygen species production, the expression of fibrogenic markers, and hepatocyte apoptosis | [124] |
| Reduction of hepatic storage of triglycerides and fatty acid esters | Combined PPAR-α/δ agonists | Regulation of fatty acid transport and β-oxidation | [125] |
| Fatty acid-bile acid conjugates | Decrease in the synthesis of fatty acid and increase of the β-oxidation by inhibition of SCD1, which is a key enzyme for fatty acid metabolism | [126] |
TGF-β1, transforming growth factor β1; CTCF, connective tissue growth factor; CB1, cannabinoid receptor 1; FXR, farnesoid X receptor; CCR, C-C motif chemokine receptor; HSC, hepatic stellate cell; NOX, NADPH (nicotinamide adenine dinucleotide phosphate) oxidases; PPAR, peroxisome proliferator-activated receptor; SCD1, stearoyl-CoA desaturase 1.
In addition, several candidate molecules have been tested in steatohepatitis patients that have a strong preclinical rationale. These include the dual PPAR-α/δ (GFT505), CCR1 and CCR5 antagonists, antifibrotic agents (simtuzumab), Takeda G-protein coupled receptor 5 (TGR5) agonists or receptor agonists, and the fatty acid-bile acid conjugate aramchol. Most of these agents are already in advanced phase 2b and phase 3 clinical trials. The drug pipeline is slowly building to address the clinical needs of this silent but damaging liver disease [117,125-127,131]. Recently, MSC therapy has been suggested as an effective alternate approach for the treatment of hepatic fibrosis. MSCs have the potential to differentiate into hepatocytes and their therapeutic value lies in their immune-modulatory properties and secretion of trophic factors, such as growth factors and cytokines [132].
CONCLUSIONS
Hepatic fibrogenesis is a complex and regulated process that represents the balance between matrix production and degradation. HSCs are important factors in the fibrogenic process and are a promising target for antifibrotic therapies. Although the immune system and HSCs reciprocally regulate each other to propagate fibrogenesis, an antifibrotic pathway including NK and DCs also stimulates HSC apoptosis. Despite the advances in understanding the mechanisms underlying hepatic fibrosis, there is a significant lag in applying these to clinical treatments. Nevertheless, progress in the next decade is expected to uncover approaches to reverse fibrosis through further translational research.
Clinically, the most convincing evidence for the regression of liver fibrosis is derived from large-scale studies of antiviral therapies for the treatment of CHC and hepatitis B. Long-term follow-up studies indicate that regression of liver fibrosis is associated with improved clinical outcomes by strengthening perceived histological regression. Although fibrosis regression remains a controversial topic, we believe that regression of fibrosis could eventually be achieved by elucidating the multiple signaling pathways involved in HSC activation as well as through the application of potential new antifibrotic strategies.
Acknowledgments
The authors thank Dr. Young Sun Lee for his critical review of the manuscript.
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Baik SJ, Kim TH, Yoo K, et al. Decreased S100B expression in chronic liver diseases. Korean J Intern Med. 2016 Jun 3; doi: 10.3904/kjim.2015.296. [Epub]. http://doi.org/10.3904/kjim.2015.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ueno T, Sata M, Sakata R, et al. Hepatic stellate cells and intralobular innervation in human liver cirrhosis. Hum Pathol. 1997;28:953–959. doi: 10.1016/s0046-8177(97)90011-3. [DOI] [PubMed] [Google Scholar]
- 4.Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol. 1999;30:77–87. doi: 10.1016/s0168-8278(99)80010-5. [DOI] [PubMed] [Google Scholar]
- 5.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mormone E, George J, Nieto N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem Biol Interact. 2011;193:225–231. doi: 10.1016/j.cbi.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology. 2006;44:1487–1501. doi: 10.1002/hep.21427. [DOI] [PubMed] [Google Scholar]
- 9.Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22 Suppl 1:S73–S78. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- 10.Sanz-Cameno P, Medina J, Garcia-Buey L, et al. Enhanced intrahepatic inducible nitric oxide synthase expression and nitrotyrosine accumulation in primary biliary cirrhosis and autoimmune hepatitis. J Hepatol. 2002;37:723–729. doi: 10.1016/s0168-8278(02)00266-0. [DOI] [PubMed] [Google Scholar]
- 11.Zhan SS, Jiang JX, Wu J, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 12.Iwakiri Y. Nitric oxide in liver fibrosis: the role of inducible nitric oxide synthase. Clin Mol Hepatol. 2015;21:319–325. doi: 10.3350/cmh.2015.21.4.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Minicis S, Brenner DA. NOX in liver fibrosis. Arch Biochem Biophys. 2007;462:266–272. doi: 10.1016/j.abb.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayashi H, Sakai T. Biological significance of local TGF-β activation in liver diseases. Front Physiol. 2012;3:12. doi: 10.3389/fphys.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bataller R, Sancho-Bru P, Gines P, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology. 2003;125:117–125. doi: 10.1016/s0016-5085(03)00695-4. [DOI] [PubMed] [Google Scholar]
- 16.Wasmuth HE, Trautwein C. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Hepatology. 2007;45:543–544. doi: 10.1002/hep.21527. [DOI] [PubMed] [Google Scholar]
- 17.Julien B, Grenard P, Teixeira-Clerc F, et al. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology. 2005;128:742–755. doi: 10.1053/j.gastro.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 18.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 19.Bomble M, Tacke F, Rink L, Kovalenko E, Weiskirchen R. Analysis of antigen-presenting functionality of cultured rat hepatic stellate cells and transdifferentiated myofibroblasts. Biochem Biophys Res Commun. 2010;396:342–347. doi: 10.1016/j.bbrc.2010.04.094. [DOI] [PubMed] [Google Scholar]
- 20.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol. 2010;53:976–980. doi: 10.1016/j.jhep.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Muhanna N, Abu Tair L, Doron S, et al. Amelioration of hepatic fibrosis by NK cell activation. Gut. 2011;60:90–98. doi: 10.1136/gut.2010.211136. [DOI] [PubMed] [Google Scholar]
- 23.Tiggelman AM, Boers W, Linthorst C, Brand HS, Sala M, Chamuleau RA. Interleukin-6 production by human liver (myo)fibroblasts in culture: evidence for a regulatory role of LPS, IL-1 beta and TNF alpha. J Hepatol. 1995;23:295–306. [PubMed] [Google Scholar]
- 24.Schwabe RF, Bataller R, Brenner DA. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am J Physiol Gastrointest Liver Physiol. 2003;285:G949–G958. doi: 10.1152/ajpgi.00215.2003. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Yang S, Lin H, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 26.Wasmuth HE, Tacke F, Trautwein C. Chemokines in liver inflammation and fibrosis. Semin Liver Dis. 2010;30:215–225. doi: 10.1055/s-0030-1255351. [DOI] [PubMed] [Google Scholar]
- 27.Jiang JX, Venugopal S, Serizawa N, et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology. 2010;139:1375–1384. doi: 10.1053/j.gastro.2010.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang JX, Mikami K, Venugopal S, Li Y, Torok NJ. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. 2009;51:139–148. doi: 10.1016/j.jhep.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeong WI, Park O, Suh YG, et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology. 2011;53:1342–1351. doi: 10.1002/hep.24190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang SC, Tsukamoto H, Brenner DA, Rippe RA. Expression of intracellular adhesion molecule 1 by activated hepatic stellate cells. Hepatology. 1996;24:670–676. doi: 10.1002/hep.510240333. [DOI] [PubMed] [Google Scholar]
- 31.Knittel T, Dinter C, Kobold D, et al. Expression and regulation of cell adhesion molecules by hepatic stellate cells (HSC) of rat liver: involvement of HSC in recruitment of inflammatory cells during hepatic tissue repair. Am J Pathol. 1999;154:153–167. doi: 10.1016/s0002-9440(10)65262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wynn TA, Cheever AW, Jankovic D, et al. An IL-12-based vaccination method for preventing fibrosis induced by schistosome infection. Nature. 1995;376:594–596. doi: 10.1038/376594a0. [DOI] [PubMed] [Google Scholar]
- 35.Marra F, Aleffi S, Galastri S, Provenzano A. Mononuclear cells in liver fibrosis. Semin Immunopathol. 2009;31:345–358. doi: 10.1007/s00281-009-0169-0. [DOI] [PubMed] [Google Scholar]
- 36.Zimmermann HW, Seidler S, Nattermann J, et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS One. 2010;5:e11049. doi: 10.1371/journal.pone.0011049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1310–G1321. doi: 10.1152/ajpgi.00365.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kisseleva T, Brenner DA. The phenotypic fate and functional role for bone marrow-derived stem cells in liver fibrosis. J Hepatol. 2012;56:965–972. doi: 10.1016/j.jhep.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albillos A, Lario M, Alvarez-Mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol. 2014;61:1385–1396. doi: 10.1016/j.jhep.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 41.Ubeda M, Munoz L, Borrero MJ, et al. Critical role of the liver in the induction of systemic inflammation in rats with preascitic cirrhosis. Hepatology. 2010;52:2086–2095. doi: 10.1002/hep.23961. [DOI] [PubMed] [Google Scholar]
- 42.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 43.Song Y, Zhao Y, Wang F, Tao L, Xiao J, Yang C. Autophagy in hepatic fibrosis. Biomed Res Int. 2014;2014:436242. doi: 10.1155/2014/436242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Senoo H, Yoshikawa K, Morii M, Miura M, Imai K, Mezaki Y. Hepatic stellate cell (vitamin A-storing cell) and its relative: past, present and future. Cell Biol Int. 2010;34:1247–1272. doi: 10.1042/CBI20100321. [DOI] [PubMed] [Google Scholar]
- 45.Thoen LF, Guimaraes EL, Dolle L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–1360. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 46.Gressner AM, Polzar B, Lahme B, Mannherz HG. Induction of rat liver parenchymal cell apoptosis by hepatic myofibroblasts via transforming growth factor beta. Hepatology. 1996;23:571–581. doi: 10.1002/hep.510230324. [DOI] [PubMed] [Google Scholar]
- 47.Gressner AM, Bachem MG. Molecular mechanisms of liver fibrogenesis: a homage to the role of activated fat-storing cells. Digestion. 1995;56:335–346. doi: 10.1159/000201257. [DOI] [PubMed] [Google Scholar]
- 48.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 49.Latella G, Vetuschi A, Sferra R, et al. Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int. 2009;29:997–1009. doi: 10.1111/j.1478-3231.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- 50.Dooley S, Hamzavi J, Ciuclan L, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135:642–659. doi: 10.1053/j.gastro.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 51.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noetel A, Kwiecinski M, Elfimova N, Huang J, Odenthal M. MicroRNA are central players in anti- and profibrotic gene regulation during liver fibrosis. Front Physiol. 2012;3:49. doi: 10.3389/fphys.2012.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roderburg C, Urban GW, Bettermann K, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 54.Lakner AM, Steuerwald NM, Walling TL, et al. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology. 2012;56:300–310. doi: 10.1002/hep.25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guimaraes EL, Empsen C, Geerts A, van Grunsven LA. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol. 2010;52:389–397. doi: 10.1016/j.jhep.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 56.Benyon RC, Iredale JP, Goddard S, Winwood PJ, Arthur MJ. Expression of tissue inhibitor of metalloproteinases 1 and 2 is increased in fibrotic human liver. Gastroenterology. 1996;110:821–831. doi: 10.1053/gast.1996.v110.pm8608892. [DOI] [PubMed] [Google Scholar]
- 57.Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–549. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murphy FR, Issa R, Zhou X, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277:11069–11076. doi: 10.1074/jbc.M111490200. [DOI] [PubMed] [Google Scholar]
- 59.Iredale J. Defining therapeutic targets for liver fibrosis: exploiting the biology of inflammation and repair. Pharmacol Res. 2008;58:129–136. doi: 10.1016/j.phrs.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 60.Iredale JP, Benyon RC, Arthur MJ, et al. Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology. 1996;24:176–184. doi: 10.1002/hep.510240129. [DOI] [PubMed] [Google Scholar]
- 61.Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795–1808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 62.Parsons CJ, Bradford BU, Pan CQ, et al. Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology. 2004;40:1106–1115. doi: 10.1002/hep.20425. [DOI] [PubMed] [Google Scholar]
- 63.Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 64.Pellicoro A, Aucott RL, Ramachandran P, et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology. 2012;55:1965–1975. doi: 10.1002/hep.25567. [DOI] [PubMed] [Google Scholar]
- 65.Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60:1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 66.Jiao J, Sastre D, Fiel MI, et al. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology. 2012;55:244–255. doi: 10.1002/hep.24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mallat A, Lotersztajn S. Reversion of hepatic stellate cell to a quiescent phenotype: from myth to reality? J Hepatol. 2013;59:383–386. doi: 10.1016/j.jhep.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 68.Troeger JS, Mederacke I, Gwak GY, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073–1083. doi: 10.1053/j.gastro.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:9448–9453. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 71.Ellis EL, Mann DA. Clinical evidence for the regression of liver fibrosis. J Hepatol. 2012;56:1171–1180. doi: 10.1016/j.jhep.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 72.van Zonneveld M, Zondervan PE, Cakaloglu Y, et al. Peg-interferon improves liver histology in patients with HBeAg-positive chronic hepatitis B: no additional benefit of combination with lamivudine. Liver Int. 2006;26:399–405. doi: 10.1111/j.1478-3231.2006.01257.x. [DOI] [PubMed] [Google Scholar]
- 73.Dienstag JL, Goldin RD, Heathcote EJ, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124:105–117. doi: 10.1053/gast.2003.50013. [DOI] [PubMed] [Google Scholar]
- 74.Chang TT, Liaw YF, Wu SS, et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology. 2010;52:886–893. doi: 10.1002/hep.23785. [DOI] [PubMed] [Google Scholar]
- 75.Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381:468–475. doi: 10.1016/S0140-6736(12)61425-1. [DOI] [PubMed] [Google Scholar]
- 76.George SL, Bacon BR, Brunt EM, Mihindukulasuriya KL, Hoffmann J, Di Bisceglie AM. Clinical, virologic, histologic, and biochemical outcomes after successful HCV therapy: a 5-year follow-up of 150 patients. Hepatology. 2009;49:729–738. doi: 10.1002/hep.22694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Verrill C, Markham H, Templeton A, Carr NJ, Sheron N. Alcohol-related cirrhosis: early abstinence is a key factor in prognosis, even in the most severe cases. Addiction. 2009;104:768–774. doi: 10.1111/j.1360-0443.2009.02521.x. [DOI] [PubMed] [Google Scholar]
- 78.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Czaja AJ, Carpenter HA. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. J Hepatol. 2004;40:646–652. doi: 10.1016/j.jhep.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 80.Mohamadnejad M, Malekzadeh R, Nasseri-Moghaddam S, et al. Impact of immunosuppressive treatment on liver fibrosis in autoimmune hepatitis. Dig Dis Sci. 2005;50:547–551. doi: 10.1007/s10620-005-2472-5. [DOI] [PubMed] [Google Scholar]
- 81.Colmenero J, Bataller R, Sancho-Bru P, et al. Effects of losartan on hepatic expression of nonphagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am J Physiol Gastrointest Liver Physiol. 2009;297:G726–G734. doi: 10.1152/ajpgi.00162.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McHutchison J, Goodman Z, Patel K, et al. Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection. Gastroenterology. 2010;138:1365–1373. doi: 10.1053/j.gastro.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 83.Kim MY, Cho MY, Baik SK, et al. Beneficial effects of candesartan, an angiotensin-blocking agent, on compensated alcoholic liver fibrosis: a randomized open-label controlled study. Liver Int. 2012;32:977–987. doi: 10.1111/j.1478-3231.2012.02774.x. [DOI] [PubMed] [Google Scholar]
- 84.Aithal GP, Thomas JA, Kaye PV, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 85.Ratziu V, Charlotte F, Bernhardt C, et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51:445–453. doi: 10.1002/hep.23270. [DOI] [PubMed] [Google Scholar]
- 86.Zein CO, Yerian LM, Gogate P, et al. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology. 2011;54:1610–1619. doi: 10.1002/hep.24544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ratziu V, de Ledinghen V, Oberti F, et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54:1011–1019. doi: 10.1016/j.jhep.2010.08.030. [DOI] [PubMed] [Google Scholar]
- 88.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poupon RE, Poupon R, Balkau B. Ursodiol for the long-term treatment of primary biliary cirrhosis: the UDCA-PBC Study Group. N Engl J Med. 1994;330:1342–1327. doi: 10.1056/NEJM199405123301903. [DOI] [PubMed] [Google Scholar]
- 90.Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology. 2000;32:1196–1199. doi: 10.1053/jhep.2000.20240. [DOI] [PubMed] [Google Scholar]
- 91.Korean Association for the Study of the Liver. KASL clinical practice guidelines: management of chronic hepatitis B. Clin Mol Hepatol. 2016;22:18–75. doi: 10.3350/cmh.2016.22.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liaw YF. Reversal of cirrhosis: an achievable goal of hepatitis B antiviral therapy. J Hepatol. 2013;59:880–881. doi: 10.1016/j.jhep.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 93.Papatheodoridis GV, Petraki K, Cholongitas E, Kanta E, Ketikoglou I, Manesis EK. Impact of interferon-alpha therapy on liver fibrosis progression in patients with HBeAg-negative chronic hepatitis B. J Viral Hepat. 2005;12:199–206. doi: 10.1111/j.1365-2893.2005.00582.x. [DOI] [PubMed] [Google Scholar]
- 94.Chang XM, Chang Y, Jia A. Effects of interferon-alpha on expression of hepatic stellate cell and transforming growth factor-beta1 and alpha-smooth muscle actin in rats with hepatic fibrosis. World J Gastroenterol. 2005;11:2634–2636. doi: 10.3748/wjg.v11.i17.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weng H, Mertens PR, Gressner AM, Dooley S. IFN-gamma abrogates profibrogenic TGF-beta signaling in liver by targeting expression of inhibitory and receptor Smads. J Hepatol. 2007;46:295–303. doi: 10.1016/j.jhep.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 96.Fung J, Lai CL, Wong DK, Seto WK, Hung I, Yuen MF. Significant changes in liver stiffness measurements in patients with chronic hepatitis B: 3-year follow-up study. J Viral Hepat. 2011;18:e200. doi: 10.1111/j.1365-2893.2010.01428.x. [DOI] [PubMed] [Google Scholar]
- 97.Kim SU, Park JY, Kim DY, et al. Non-invasive assessment of changes in liver fibrosis via liver stiffness measurement in patients with chronic hepatitis B: impact of antiviral treatment on fibrosis regression. Hepatol Int. 2010;4:673–680. doi: 10.1007/s12072-010-9201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shin SK, Kim JH, Park H, et al. Improvement of liver function and non-invasive fibrosis markers in hepatitis B virus-associated cirrhosis: 2years of entecavir treatment. J Gastroenterol Hepatol. 2015;30:1775–1781. doi: 10.1111/jgh.13020. [DOI] [PubMed] [Google Scholar]
- 99.Yo IK, Kwon OS, Park JW, et al. The factors associated with longitudinal changes in liver stiffness in patients with chronic hepatitis B. Clin Mol Hepatol. 2015;21:32–40. doi: 10.3350/cmh.2015.21.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Everson GT. Management of cirrhosis due to chronic hepatitis C. J Hepatol. 2005;42 Suppl:S65–S74. doi: 10.1016/j.jhep.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 101.Veldt BJ, Heathcote EJ, Wedemeyer H, et al. Sustained virologic response and clinical outcomes in patients with chronic hepatitis C and advanced fibrosis. Ann Intern Med. 2007;147:677–684. doi: 10.7326/0003-4819-147-10-200711200-00003. [DOI] [PubMed] [Google Scholar]
- 102.Bachofner JA, Valli PV, Kroger A, et al. Direct antiviral agent treatment of chronic hepatitis C results in rapid regression of transient elastography and fibrosis markers fibrosis-4 score and aspartate aminotransferase-platelet ratio index. Liver Int. 2016 Sep 28; doi: 10.1111/liv.13256. [Epub]. http://doi.org/10.1111/liv.13256. [DOI] [PubMed]
- 103.Rambaldi A, Gluud C. Colchicine for alcoholic and non-alcoholic liver fibrosis and cirrhosis. Cochrane Database Syst Rev. 2005;(2):CD002148. doi: 10.1002/14651858.CD002148.pub2. [DOI] [PMC free article] [PubMed]
- 104.Musso G, Gambino R, Cassader M, Pagano G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology. 2010;52:79–104. doi: 10.1002/hep.23623. [DOI] [PubMed] [Google Scholar]
- 105.Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–129. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Neuschwander-Tetri BA. Targeting the FXR nuclear receptor to treat liver disease. Gastroenterology. 2015;148:704–706. doi: 10.1053/j.gastro.2015.02.037. [DOI] [PubMed] [Google Scholar]
- 107.Roberts SK, Therneau TM, Czaja AJ. Prognosis of histological cirrhosis in type 1 autoimmune hepatitis. Gastroenterology. 1996;110:848–857. doi: 10.1053/gast.1996.v110.pm8608895. [DOI] [PubMed] [Google Scholar]
- 108.Czaja AJ, Manns MP. Advances in the diagnosis, pathogenesis, and management of autoimmune hepatitis. Gastroenterology. 2010;139:58–72. doi: 10.1053/j.gastro.2010.04.053. [DOI] [PubMed] [Google Scholar]
- 109.Czaja AJ. Review article: the prevention and reversal of hepatic fibrosis in autoimmune hepatitis. Aliment Pharmacol Ther. 2014;39:385–406. doi: 10.1111/apt.12592. [DOI] [PubMed] [Google Scholar]
- 110.Karlsen TH, Vesterhus M, Boberg KM. Review article: controversies in the management of primary biliary cirrhosis and primary sclerosing cholangitis. Aliment Pharmacol Ther. 2014;39:282–301. doi: 10.1111/apt.12581. [DOI] [PubMed] [Google Scholar]
- 111.Lindor KD, Gershwin ME, Poupon R, et al. Primary biliary cirrhosis. Hepatology. 2009;50:291–308. doi: 10.1002/hep.22906. [DOI] [PubMed] [Google Scholar]
- 112.Goulis J, Leandro G, Burroughs AK. Randomised controlled trials of ursodeoxycholic-acid therapy for primary biliary cirrhosis: a meta-analysis. Lancet. 1999;354:1053–1060. doi: 10.1016/S0140-6736(98)11293-X. [DOI] [PubMed] [Google Scholar]
- 113.Gong Y, Huang ZB, Christensen E, Gluud C. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database Syst Rev. 2008;(3):CD000551. doi: 10.1002/14651858.CD000551.pub2. [DOI] [PubMed] [Google Scholar]
- 114.Prince MI, Chetwynd A, Craig WL, Metcalf JV, James OF. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53:865–870. doi: 10.1136/gut.2003.023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaplan MM, Cheng S, Price LL, Bonis PA. A randomized controlled trial of colchicine plus ursodiol versus methotrexate plus ursodiol in primary biliary cirrhosis: ten-year results. Hepatology. 2004;39:915–923. doi: 10.1002/hep.20103. [DOI] [PubMed] [Google Scholar]
- 116.Corpechot C. Primary biliary cirrhosis and bile acids. Clin Res Hepatol Gastroenterol. 2012;36 Suppl 1:S13–S20. doi: 10.1016/S2210-7401(12)70016-5. [DOI] [PubMed] [Google Scholar]
- 117.Hirschfield GM, Mason A, Luketic V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148:751–761. doi: 10.1053/j.gastro.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 118.Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS, American Association for the Study of Liver Diseases Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328–343. doi: 10.1002/hep.24330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Falize L, Guillygomarc’h A, Perrin M, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology. 2006;44:472–477. doi: 10.1002/hep.21260. [DOI] [PubMed] [Google Scholar]
- 120.Mehal WZ, Schuppan D. Antifibrotic therapies in the liver. Semin Liver Dis. 2015;35:184–198. doi: 10.1055/s-0035-1550055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology. 2008;135:660–670. doi: 10.1053/j.gastro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang Q, Usinger W, Nichols B, et al. Cooperative interaction of CTGF and TGF-β in animal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011;4:4. doi: 10.1186/1755-1536-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Giannone FA, Baldassarre M, Domenicali M, et al. Reversal of liver fibrosis by the antagonism of endocannabinoid CB1 receptor in a rat model of CCl(4)-induced advanced cirrhosis. Lab Invest. 2012;92:384–395. doi: 10.1038/labinvest.2011.191. [DOI] [PubMed] [Google Scholar]
- 124.Jiang JX, Chen X, Serizawa N, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53:289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Staels B, Rubenstrunk A, Noel B, et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2013;58:1941–1952. doi: 10.1002/hep.26461. [DOI] [PubMed] [Google Scholar]
- 126.Safadi R, Konikoff FM, Mahamid M, et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12:2085–2091. doi: 10.1016/j.cgh.2014.04.038. [DOI] [PubMed] [Google Scholar]
- 127.Gonzalez EO, Boix V, Deltoro MG, et al. The effects of Maraviroc on liver fibrosis in HIV/HCV co-infected patients. J Int AIDS Soc. 2014;17(4 Suppl 3):19643. doi: 10.7448/IAS.17.4.19643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang JJ, Tao H, Li J. Hedgehog signaling pathway as key player in liver fibrosis: new insights and perspectives. Expert Opin Ther Targets. 2014;18:1011–1021. doi: 10.1517/14728222.2014.927443. [DOI] [PubMed] [Google Scholar]
- 129.Samarakoon R, Overstreet JM, Higgins PJ. TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal. 2013;25:264–268. doi: 10.1016/j.cellsig.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tam J, Cinar R, Liu J, et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167–179. doi: 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Baghdasaryan A, Claudel T, Gumhold J, et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2-/- (Abcb4-/-) mouse cholangiopathy model by promoting biliary HCO– 3 output. Hepatology. 2011;54:1303–1312. doi: 10.1002/hep.24537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eom YW, Shim KY, Baik SK. Mesenchymal stem cell therapy for liver fibrosis. Korean J Intern Med. 2015;30:580–589. doi: 10.3904/kjim.2015.30.5.580. [DOI] [PMC free article] [PubMed] [Google Scholar]