

Abstract
The adaptive immune system generates CD8 cytotoxic T lymphocytes (CTLs) as a major component of the protective response against viruses. Knowledge regarding the nature of the peptide sequences presented by HLA class I molecules and recognized by CTLs is thus important for understanding host-pathogen interactions. In this study, we focused on identification of a CTL epitope generated from coxsackievirus B4 (CVB4), a member of the enterovirus group responsible for several inflammatory diseases in humans and often implicated in the triggering and/or acceleration of the autoimmune disease type 1 diabetes. We identified a 9-mer peptide epitope that can be generated from the P2C nonstructural protein of CVB4 (P2C1137-1145) and from whole virus by antigen-presenting cells and presented by HLA-A2.1. This epitope is recognized by effector memory (gamma interferon [IFN-γ]-producing) CD8 T cells in the peripheral blood at a frequency of responders that suggests that it is a major focus of the anti-CVB4 response. Short-term CD8 T-cell lines generated against P2C1137-1145 are cytotoxic against peptide-loaded target cells. Of particular interest, the epitope lies within a region of viral homology with the diabetes-related autoantigen, glutamic acid decarboxylase-65 (GAD65). However, P2C1137-1145-specific cytotoxic T lymphocyte (CTL) lines were not activated to produce IFN-γ by the GAD65 peptide homologue and did not show cytotoxic activity in the presence of appropriately labeled targets. These results describe the first CD8 T-cell epitope of CVB4 that will prove useful in the study of CVB4-associated disease.
Human enteroviruses (EVs) constitute a group of antigenically variable infectious agents belonging to the Picornaviridae family, which includes at least 67 serotypes, including poliovirus (3 serotypes), coxsackievirus A (23 serotypes), coxsackievirus B (CVB) (6 serotypes), echovirus (31 serotypes), and EV 68-71. Most EVs replicate initially in the gastrointestinal tract, and during the course of the infection there is involvement of other organs, notably the pancreas, heart, and central nervous system (21).
EV infections by nonpoliovirus agents are associated with a wide variety of clinical syndromes (21). The most common infections in humans are asymptomatic or result in mild respiratory illness. However, more severe clinical infections may result in meningitis, encephalitis, or acute and chronic myocarditis (3, 29, 52). Infections peak during summer and early autumn in temperate climates, and transmission occurs from person to person by the fecal-oral route, by contact with infected nose and throat discharges, and by droplet spread.
Although B- and T-lymphocyte responses during EV infections have a role in virus clearance (12, 14), there are also indications that they may lead to disease, and an immune-mediated pathological process is proposed in the context of EV-induced chronic dilated cardiomyopathy, chronic myositis, and diabetes (18, 27). Recent studies have demonstrated that in susceptible mouse strains lacking both CD4 and CD8 T cells, there is a decrease in mortality and cardiac tissue inflammation after infection with CVB3, when compared to mice lacking either CD4 or CD8 T cells alone (33). In some mouse strains, CD8 rather than CD4 T-cell responses appear to play a dominant role; in others, the CD8 T-cell response may be protective (15), suggesting that T cells recruited during EV infection are necessary for either protection from or induction of immunopathology, the net result depending upon the genetic background of the host.
In addition to cardiac inflammation, there is a considerable body of evidence linking viruses of the EV genus to the initiation and/or acceleration of events that lead to the inflammatory autoimmune disease type 1 diabetes mellitus (T1DM). This includes the isolation of CVB4 from the pancreas of a child at diagnosis of T1DM (51) and epidemiological, prospective, and case-control studies associating the disease with laboratory-derived evidence of EV infections (2, 9, 11, 16, 19, 24, 25, 30). More recently, we have shown that CD4 T cells from newly diagnosed T1DM patients up-regulate CD69 and produce the proinflammatory cytokine gamma interferon (IFN-γ) in response to in vitro challenge with CVB4 antigens more readily than matched control subjects (44, 46), supporting the contention that effector memory anti-EV responses are associated with the development of the disease.
Despite these data, there remains uncertainty regarding the mechanisms through which EVs could precipitate islet autoimmunity and β-cell destruction. It is known that the cellular infiltrate into islets during the development of T1DM is dominated by CD8 T cells and accompanied by hyperexpression of HLA class I molecules on islet cells themselves (6). Moreover, recent evidence links HLA class I alleles such as A*0201 with diabetes susceptibility (37). There is thus a pressing need for the identification of the molecular targets of these CD8 T cells, to facilitate functional studies in this arena. In the present study, we identify a CVB4 epitope that is frequently recognized by CD8 T cells and derives from a region of homology between CVB4 and a diabetes-related autoantigen, glutamic acid decarboxylase-65 (GAD65).
MATERIALS AND METHODS
Subjects.
We obtained blood from six healthy volunteer subjects (Table 1) and separated peripheral blood mononuclear cells (PBMCs) by density gradient centrifugation. This study was approved by the local research ethics committee.
TABLE 1.
Age, sex, and HLA class I A, B, and C locus genotypes in subjects studied for CD8 T-cell peptide-specific responsesa
| Subject no. | Sex | Age (yr) | HLA alleles
|
||
|---|---|---|---|---|---|
| A locus | B locus | C locus | |||
| 1 | M | 25 | 02, 01 | 08, 18 | 0501, 0701/6 |
| 2 | M | 34 | 02, 02 | 18, 51 | 04/18, 0701/6 |
| 3 | F | 34 | 02, 02 | 27, 51 | 0102/3/4, 0303 |
| 4 | F | 34 | 24, 24 | 51, 51 | 15, 15 |
| 5 | M | 40 | 01, 11 | 07, 35 | 04/18, 0702 |
| 6 | M | 37 | 02, 33 | 35, 44 | 04/18, 0501/3/4 |
All A*02-positive subjects were subtyped and shown to be HLA-A*0201 (A2.1).
Peptides.
Peptides used in this study were obtained from Interactiva (Ulm, Germany) and were more than 95% pure by reverse-phase high-performance liquid chromatography. The peptides were quantified by area-under-the-curve analysis at A210, dissolved in 20% dimethyl sulfoxide, and stored at −20°C until use. The sequences of the peptides used were as follows: CVB4 P2C protein, spanning residues 1137 to 1145 (EVKEKHEFL) and (referred to as P2C1137-1145, and GAD65, spanning residues 261 to 269 (EVKEKGMAA) and referred as GAD261-269. As a negative control peptide, predicted not to bind to either HLA-A*0201 or B*0801 (henceforth designated A2.1 and B8, respectively) we used GAD65, spanning residues 2 to 10 (ASPGSGFWS) and referred to as GAD2-10). As positive control peptides for ex vivo detection of CD8 T-cell responses, we used the HLA-A2.1 restricted influenza matrix 1 protein, spanning residues 58 to 66 (GILGFVFTL); B8-restricted influenza nucleoprotein, spanning residues 380 to 388 (ELRSRYWAI); A2-restricted human cytomegalovirus (CMV) pp65, spanning residues 495 to 503 (NLVPMVATV); and A2-restricted Epstein Barr virus (EBV) BMLF1, spanning positions 259 to 267 (GLCTLVAML) (10). In some experiments, these positive control peptides were pooled for use.
HLA stabilization assay.
Binding of the P2C1137-1145 and GAD261-269 peptides to HLA-A2.1 was examined by a conventional HLA stabilization assay (31). In this, HLA-A2.1-expressing T2 cells lacking the transporter associated with processing are pulsed with test peptide. In the absence of the transporter associated with processing and any exogenous peptides capable of A2.1 binding, stable HLA A2.1 molecules are not formed and there is no surface A2.1 expression. Addition of peptides with high affinity for A2.1 results in stable A2.1 generation and surface expression, which is detected by flow cytometry. Briefly, T2 cells (105) were washed twice with serum-free AIM-V medium (Sigma Chemical Company, Poole, Dorset, United Kingdom) and incubated in 50 μl of AIM-V medium containing 5 μg of β2-microglobulin (Sigma)/ml and each of the test peptides at concentrations varying from 100 to 0.78 μM at 37°C for 16 h in a CO2 incubator. After the incubation, surface HLA-A2.1 molecules were detected with anti-A2.1 monoclonal antibody (MAb) (BB7.2; BD Biosciences, Cowley, United Kingdom) and a FACSCalibur flow cytometer (BD Biosciences). Results are expressed as the mean (and standard deviation [SD]) of four separate determinations of the mean fluorescence intensity (MFI), which is a measure of the surface concentration of stable A2.1 molecules.
Peptide-specific CD8 T-cell line generation.
We selected a single volunteer on the basis of expression of both class I HLA types of interest (A2.1 and B8) from whom samples were taken to generate CVB4 peptide- and GAD65 peptide-specific CD8 T-cell lines. Dendritic cells (DCs) were generated as previously described (13) with monocyte enrichment from PBMCs after Percoll density centrifugation, followed by a 6-day culture in Iscove's modified Dulbecco's medium (Gibco BRL, Paisley, United Kingdom) supplemented with 250 U of interleukin-4 [IL-4]/ml (Strathmann Biotec AG, Hannover, Germany) and granulocyte-macrophage colony-stimulating factor (500 U/ml; Schering-Plough, Ltd., Hertfordshire, United Kingdom). Lymphocytes remaining after monocyte removal were cryopreserved for later use. After 6 days, immature DCs were matured by the addition of lipopolysaccharide (100 ng/ml; Sigma), IL-1β (10 ng/ml), and tumor necrosis factor alpha (25 ng/ml) (both from Strathmann Biotec AG) for 2 days, with the addition of P2C1137-1145 or GAD261-269 for the last 24 h. After maturation, the peptide-pulsed DCs were washed twice in Iscove's modified Dulbecco's medium containing 10% fetal calf serum (FCS) (Invitrogen, Ltd., Paisley, United Kingdom), counted, and plated at 200,000 cells/well on a 24-well plate. Autologous lymphocytes were removed from the liquid nitrogen and plated with peptide-pulsed DCs at a ratio of 10:1 in a total volume of 1 ml of RPMI containing 10% FCS (R10) supplemented with IL-7 (10 IU/ml; R&D Systems, Abingdon, United Kingdom). After 3 to 4 days half of the medium was replaced with fresh R10 containing IL-2 (proleukin, at a final concentration of 20 IU/ml; Chiron Corporation, Emeryville, Calif.), and cells were cultured for a further 10 days, being split and fed as necessary (typically, every 2 to 3 days).
Weekly restimulation of the T-cell line was carried out with cell blasts prepared by stimulation of PBMCs with phytohemagglutinin-L (PHA-L). Briefly, 5 days before restimulation, autologous PBMCs were stimulated with PHA-L (1 μg/ml; Biostat Diagnostic Systems, Stockport, United Kingdom) in R10 for 24 h; after which, IL-2 was added at a final concentration of 20 IU/ml and the medium was replenished. PHA-L lymphocyte blasts were harvested, counted, and pulsed with peptide at a 100 μM concentration in a final volume of 100 μl of R10 for 1 h with occasional agitation. After being pulsed, the cell blasts were washed three times, resuspended at 1 × 106/ml in R10, irradiated (3,000 rads), and plated at 1 × 106 cells/well on a 24-well plate together with 2 × 106 cells of the CD8 T-cell line. The medium was complemented with IL-7 (10 IU/ml) on the day of restimulation and with IL-2 (20 IU/ml) every 2 to 3 days thereafter.
To increase the proportion of peptide-specific CD8 T cells, the IFN-γ-producing cells were immunomagnetically enriched after a 6-h restimulation with the IFN-γ Secretion assay kit (Miltenyi Biotec, Bisley, United Kingdom), following the manufacturer's instructions. The positively selected cells were expanded in R10 medium for 14 days with autologous irradiated PBMCs and the addition of IL-7 (10 IU/ml), IL-2 (20 IU/ml), and IL-15 (0.1 ng/ml; Peprotech EC Ltd, London, United Kingdom) on the first day and with replenishment of the medium every 2 to 3 days with fresh R10 containing IL-2 and IL-15.
Intracellular cytokine staining.
After stimulation of fresh PBMCs from the donors or CD8 T-cell lines for 6 h in the presence of test peptide or peptide pools (10 μM final peptide concentration) at 37°C in R10 medium, cells were harvested into flow cytometry tubes and washed twice (500 × g for 6 min at 4°C) in phosphate-buffered saline (PBS). Cells were fixed in 2% formaldehyde in hypertonic PBS (1.2 times) for 15 min at room temperature. After fixation, cells were washed once with PBS, resuspended in flow buffer (PBS containing 2% FCS, 1% human AB serum, 1% bovine serum albumin, and 0.1% sodium azide), and stored at 4°C until the next day.
For staining, fixed cells were washed twice with 0.1% saponin in PBS (containing 1% FCS) and stained with titrated amounts of MAbs in 0.1% saponin buffer for 30 min on ice in the dark. The antibodies used were anti-CD3-APC (clone UCHT1), CD8-PerCP (clone SK1), IFN-γ (clone 25723.11), and the corresponding isotype control MAbs (all from BD Biosciences). After being stained, cells were washed once with saponin buffer, resuspended in PBS, and acquired with a FACSCalibur flow cytometer (BD Biosciences). Samples were analyzed with CellQuest software and a logical gate set using a dot plot of side-scatter versus CD3 staining was used to examine CD3+ T cells for the coexpression of CD8 and intracellular IFN-γ. At least 50,000 CD3+ CD8+ T cells were acquired for each analysis.
To determine restriction elements for CD8 T-cell lines, these were stimulated for 6 h with antigen-presenting cells (APCs) (represented by PHA-L lymphocyte blasts generated from different donors) and pulsed with peptide, followed by intracellular staining for IFN-γ as described above.
Natural processing and presentation of CVB4 epitope.
In order to examine whether professional APCs can process and present a defined viral epitope from whole protein and/or whole virus to CD8 T cells, DCs were generated as described above from the same source as the CD8 T-cell line (autologous; HLA-A2.1, B8) and from matched (HLA-A2.1) and unmatched (HLA-A1, A11, B7, and B35) donors. Whole P2C protein from the CVB4 E2 strain was generated as previously described (45) as a fusion protein with maltose binding protein (P2C-MBP). CVB4 (strain JVB)-infected viral lysates (Institute Virion, Zurich, Switzerland) were used as a source of CVB4 virus, as previously described (44, 46). Immature DCs from each donor were matured as above in the presence of peptide (100 μM), whole P2C protein (10 μg/ml), CVB4 lysate (0.2 μg/ml), or the relevant control (PBS, MBP, or uninfected cell lysate) and then cocultured for 6 h with the CD8 T-cell line, followed by intracellular staining for IFN-γ as described above.
Cytotoxicity assay.
Cells of an autologous EBV-transformed B-cell line were labeled with the dissociation-enhanced time-resolved fluoroimmunoassay (DELFIA) BATDA cytotoxicity assay kit (Perkin-Elmer Life Sciences, Cambridge, United Kingdom), following the indications of the manufacturer. Cells (2 × 106) were resuspended at a concentration of 1 × 106/ml in R10 and labeled with 3 μl of the DELFIA BATDA reagent for 20 min at 37°C. After being labeled, cells were centrifuged (400 × g for 5 min at room temperature). The supernatant was carefully taken out, and cells were washed three times in wash buffer (R10 containing 5 mM sulfinpyrazone [Sigma] to reduce the spontaneous label release). After being washed, cells were resuspended at 105/ml in R10 containing 5 mM sulfinpyrazone, and 10,000 cells were added per 96-well V-bottomed plate together with different amounts of cells from the CD8 T-cell line in triplicate, giving an effector:target (E:T) ratio ranging from 10:1 to 40:1. Wells with only medium from target cells (background), with only target cells (spontaneous), and with target cells lysed with water containing 5% NP-40 (maximum) were used to quantify the cytotoxicity of the CTL lines. Incubation was allowed for 2 h at 37°C, after which the plate was centrifuged for 5 min at 500× g and 20 μl of the supernatant was transferred to a flat-bottomed 96-well plate together with 200 μl of Europium solution. The plate was incubated for 15 min, and then fluorescence was read with a DELFIA research fluorimeter (Perkin-Elmer Life Sciences). The specific release was calculated as a percentage according to the formula (variables are of cell counts):
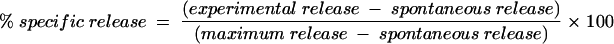 |
RESULTS
CD8 epitope prediction and binding motif.
We analyzed the P2C protein region from the diabetogenic E2 strain of CVB4 (accession number S76772) (20) using three different programs that predict binding affinities for HLA class I molecules,to identify potential 9-mer CD8 T-cell epitopes. The programs used were HLA Ligand/Motif Database (http://hlaligand.ouhsc.edu/LigandDB/servlet/GenerateFormServlet?form_type=index)(38), the HLA Peptide Binding Predictions Web site from the BioInformatics and Molecular Analysis Section (BIMAS) (http://bimas.dcrt.nih.gov/molbio/hla_bind/) (34), and the SYFPEITHI database (http://syfpeithi.bmi-heidelberg.com/scripts/MHCServer.dll/home.htm) (36). We focused our search on HLA molecules that are common in the general population and also associated with T1DM; HLA-A2.1 is frequently present on the HLA-DR4 haplotype among Caucasians (28) and is linked to the disease through epidemiological studies (37) and mechanistic research in the animal model of spontaneous diabetes, the nonobese diabetic mouse (26). HLA-B8 is also of interest, being present on the ancestral haplotype HLA-B8/DR3/DQ2 and associated with T1DM and autoimmunity in general (35).
We noted that the peptide sequence 1137-EVKEKHEFL-1145 in P2C is predicted to bind with high affinity to HLA-B8 and with intermediate affinity to -A2.1, characteristics also shared by the sequence 261-EVKEKGMAA-269 in GAD65 (see Tables 2 and 3). These two sequences had the highest cumulative predicted binding scores for B8 and A2.1 in the PEVKEK region. Indeed, the EVKEKHEFL region of P2C has the highest predicted binding scores for the whole of the CVB4 polyprotein for HLA-B8, with a score even higher than that predicted for known CD8 T-cell epitopes such as the B8-restricted influenza nucleoprotein epitope (380-ELRSRYWAI-388; score, 115) (42). Although 1137-EVKEKHEFL-1145 has a lower predicted score for the HLA-A2.1 molecule, it is in a similar range when compared with other known A2.1-restricted CD8 T-cell epitopes, such as that from the smallpox gene 018L (89-KVDDTFYYV-97; score, 89) (43), or the hepatitis C virus nonstructural protein 3 epitope (1073-CINGVCWTV-1081; score, 91) (49). Flanking 9-mer peptide sequences from the PEVKEK region are also predicted to bind to these HLA molecules (Table 3) but with lower affinity. In the light of these predictions, we focused on examining CD8 T-cell reactivity to the EVKEKHEFL sequence from P2C.
TABLE 2.
Sequence comparison between P2C protein of CVB4 E2 and GAD65, showing that in a 19-amino-acid sequence there is 52% identity and 73% similarity between the two proteinsa
| Antigen | Position | Sequencea |
|---|---|---|
| CVB4-E2 polyprotein | 1131-1149 | KVKILPEVKEKHEF-LNRL |
| GAD65 | 255-273 | RFKMFPEVKEKGMAALPRL |
The PEVKEK amino acid sequence is shown in boldface. Amino acid identity is shown by single underlining; amino acid similarity is shown by double underlining.
TABLE 3.
Motif-based CD8 T-cell epitope prediction for the PEVKEK region of CVB4 E2 and homologous region of GAD65a
| Antigen | Sequence no. | Sequence | Binding score
|
|
|---|---|---|---|---|
| A2.1 | B8 | |||
| CVB4-E2 polyprotein | 1133 | KILPEVKEK | 58 | 55 |
| 1135 | LPEVKEKHE | 32 | 100 | |
| 1137 | EVKEKHEFL | 64 | 220 | |
| GAD65 | 257 | KMFPEVKEK | 52 | 50 |
| 259 | FPEVKEKGM | 33 | 95 | |
| 261 | EVKEKGMAA | 46 | 165 | |
Class I epitope prediction using the HLA Ligand/Motif database (http://hlaligand.ouhsc.edu/LigandDB/servlet/GenerateFormServlet?form_type=index), is shown. For each peptide, the binding score represents the sum of each of the individual scores of the nine amino acid positions of the peptide binding groove. For each position of the peptide binding groove of a certain HLA allele, the percentage of all the amino acids is recorded, and based on this percentage, all the residues are scored for each position. Similar results were obtained with the SYFPEITHI database (http://www.uni-tuebingen.de/uni/kxi/).
Ex vivo detection of P2C1137-1145-specific CD8 T cells.
We reasoned that if the P2C1137-1145 peptide represents a CD8 T-cell epitope in the anti-CVB4 immune response, spontaneous effector memory CD8 T-cell reactivity against this sequence should be readily detectable in the general population. Infection with EVs such as CVB4 is common, and previous studies have shown that the P2C protein is a major viral target of the CD4 T-cell immune response (44, 45). We therefore examined direct ex vivo IFN-γ production by CD8 T cells in response to P2C1137-1145 and control peptides in five donors.
A representative set of results of these analyses is shown in Fig. 1 and data for all subjects are given in Table 4. As a validation of this approach for the detection of virus-specific CD8 T-cell responses, we first examined IFN-γ production by CD8 T cells after culture with a pool of peptides known to represent dominant A2.1 and B8 restricted epitopes of common viruses. These were the HLA-A2.1 restricted influenza matrix 1 protein, spanning positions 58 to 66 (GILGFVFTL); the B8-restricted influenza nucleoprotein, spanning positions 380 to 388 (ELRSRYWAI); A2-restricted human CMV pp65, spanning positions 495 to 503 (NLVPMVATV); and the A2-restricted EBVBMLF1, spanning positions 259 to 267 (GLCTLVAML) (10). Three of five subjects showed a detectable IFN-γ response to this positive peptide pool (0.06, 0.05, and 0.19%) (Fig. 1C and Table 4). Each of these three responding subjects was HLA-A2.1 positive and one (subject 1) was also HLA-B8 positive (see Table 1). In two subjects (subjects 2 and 3), responses to one of the peptides present in the pool, the HLA-A2.1-restricted influenza matrix 1 protein, were observed. This gives an indication of the frequency of CD8 T-cell responses to a single peptide epitope. For subject 3, response to this peptide represented 10.5% of the total pool response (0.02% of IFN-γ-secreting CD8 T cells compared to 0.19% for the whole pool) (Table 4). For subject 2, response to this peptide represented 40% of the total pool response (0.02% for the peptide compared with 0.05% for the whole pool, Table 4, Fig. 1D). These responses were clearly peptide and CD8 T-cell specific, since no IFN-γ response could be detected in CD3+ CD8− cells or when a negative control peptide was used (Table 4). No response to the HLA-A2.1-restricted influenza matrix 1 protein peptide could be detected in subjects 4 or 5, who also showed no response to the peptide pool (Table 4).
FIG. 1.

Ex vivo detection of CVB4 PEVKEK-specific CD8 T cells. Graph shows representative flow cytometry dot plots of staining of peripheral blood from a single healthy donor (subject 2) after stimulation directly ex vivo for 6 h with different peptide antigens. After the stimulation period, PBMCs were stained as described and analyzed by flow cytometry. CD3+ T cells were gated using anti-CD3 staining and side scatter (A) and then interrogated for expression of CD8 and intracellular IFN-γ (panels B to F, x axis and y axis, respectively). A very low level of IFN-γ production was detected when no antigen is added (B). Stimulation with a pool of HLA-A2.1- and B8-restricted CD8 epitopes from influenza virus, CMV, and EBV (see Materials and Methods) reveals the presence of virus-specific CD8 T cells (C) and responses to a single peptide from this pool (influenza matrix58-66) (D) indicate the typical frequency of CD8 T cells specific for a single virus epitope. A comparable response is seen to the CVB4-derived epitope P2C1137-1145 (E). No response was detected against the homologous peptide of GAD65 (F). Results for the remaining donors are shown in Table 4. Staining of IFN-γ+ CD3+ CD8− cells (unlabeled panel, upper-left quadrant) was typically ≪0.01%.
TABLE 4.
Percentages of IFN-γ-positive CD3+ CD8+ T cells from different donors after stimulation of cells with different antigens ex vivoa
| Subject no. | Antigen
|
||||
|---|---|---|---|---|---|
| Medium alone | Positive peptide pool | Influenza matrix 1 protein58-66 | P2C1137-1145 | GAD261-269 | |
| 1 | 0.01 | 0.06 | ND | 0.03 | 0.01 |
| 2 | 0 | 0.05 | 0.02 | 0.03 | 0 |
| 3 | 0 | 0.19 | 0.02 | 0 | 0 |
| 4 | 0 | 0 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 | 0 | 0 |
Data presented as percentages of CD3+ CD8+ cells. In all cases for all stimulation conditions, the percentages of IFN-γ+ CD3+ CD8− T cells were ≪0.01%. The positive peptide pool contains peptides representing known dominant EBV and CMV epitopes restricted by HLA A2.1 or B8 (see Materials and Methods for sequences). ND, not done.
Since the ex vivo detection system appeared capable of identifying peptide CD8 T-cell responses in an HLA class I-restricted manner, we next used the same approach to examine responses to P2C1137-1145 peptide. Samples from two subjects (numbers 1 and 2) showed an effector memory response to this peptide of 0.03% CD8+ IFN-γ+ T cells in both cases (Fig. 1E and Table 4), while in the other three subject specimens tested, no response to this peptide could be detected (Table 4).
P2C1137-1145 peptide-specific CD8 T cells recognize peptide presented by the HLA-A2.1 molecule and have cytotoxic potential against peptide-pulsed targets.
To examine the HLA molecule used by P2C1137-1145-specific CD8 T cells and the cytotoxic potential of the response, we selected samples from subject 1, who is both HLA-A2.1 and HLA-B8 positive, to generate a CD8 T-cell line specific for the peptide of interest. After initial lymphocyte stimulation with peptide-pulsed DCs and 10 days of in vitro expansion, the responding cells were restimulated weekly with peptide-pulsed autologous APCs. As shown in Fig. 2, after the first stimulation approximately 2% of the CD8 T cells were P2C peptide specific (Fig. 2D), a percentage that remained constant with successive restimulations (data not shown). This CD8 T-cell line was then stained for surface IFN-γ with phycoerythrin-labeled antibodies and subsequently enriched using paramagnetic beads coated with anti-phycoerythrin antibodies. By this means, we were able to enrich the CD8 T-cell line to contain approximately 11% responder cells specific for P2C1137-1145 peptide (Fig. 2F). We next examined the cytotoxic potential of this CTL line using autologous targets loaded with the P2C1137-1145 peptide. Results in Fig. 3 show that the P2C epitope-specific CD8 T-cell line very efficiently killed target cells loaded with the virus peptide (67% above background at a 10:1 E:T ratio).
FIG. 2.

Generation and enrichment of a P2C1137-1145-specific CD8 T-cell line. (A to C) A CD8 T-cell line from subject 1 was raised against the PEVKEK epitope P2C1137-1145 derived from CVB4. Graph shows flow cytometry dot plots of CD8 (x axis) versus isotype control (upper panels, y axis) or IFN-γ staining (lower panels, y axis) gated on CD3+ T cells after stimulation as shown above each column. (D) Approximately 2% of the original CD8+ T-cell line produced IFN-γ in response to the CVB4 peptide. (E and F) The same T-cell line after immunomagnetic enrichment of the IFN-γ+ cells. Percentages represent numbers of IFN-γ positive CD8 T cells. (G and H) Reactivity of the P2C epitope-specific CD8 T-cell line in the absence of peptide and in the presence of GAD261-269, respectively. No cross-reactivity against GAD was detected.
FIG. 3.

Cytotoxic activity of the P2C1137-1145-specific CTL line. As targets, homologous EBV-transformed B cells were fluorescently labeled and loaded with peptides for 1 h. After being washed, target cells were incubated with the CTL line at different E:T ratios, and killing was determined after 2 h of incubation. Cells that have were pulsed with the CVB4-derived peptide were completely killed by the CTLs (solid squares), while those pulsed with the GAD65-derived homologue (GAD261-269; open squares) showed no significant level of killing when compared to cells loaded with a peptide derived from GAD65 not predicted to bind to either A2.1 or B8 (GAD2-10; solid triangles) or nonpulsed target cells (results not shown). The horizontal line represents background release (no CTL cells).
To determine the HLA class I molecule capable of presenting peptide to this P2C1137-1145-specific CTL line, we obtained APCs from the different subjects shown in Table 1 and incubated these with P2C1137-1145 peptide. Only APCs from three of the subjects could present the P2C1137-1145 peptide and stimulate the CD8 T-cell line to make IFN-γ to a level comparable to that obtained with autologous APCs (approximately 10%) (Fig. 4). The HLA class I molecule in common between these donors was HLA A2.1, rather than HLA-B8, despite the fact that this peptide is predicted to bind with a much higher affinity to HLA-B8 (Table 3). Binding of P2C1137-1145 to HLA-A2.1 in the range of 25 to 100 μM peptide was then confirmed by an HLA stabilization assay (Fig. 5).
FIG. 4.

HLA molecule involved in presentation of P2C1137-1145 to the CD8 T-cell line. The CVB4 peptide-specific CTL line was incubated for 6 h with peptide-pulsed cells obtained from several individuals (subjects 1 to 6). The graphs show flow cytometry dot plot analyses of gated CD3+ T cells stained for CD8 (x axis) and IFN-γ (y axis). Subject number and HLA-A and HLA-B locus typing are shown for each subject above panels B to G. (A) Representative dot plot from one subject (subject number 1) showing that little or no IFN-γ could be detected when the CTL line was incubated with cells not pulsed with the peptide. In the presence of P2C peptide, only cells from three subjects (numbers 2, 3, and 6) (E, C, and F) could stimulate the line to a level similar to that seen in the presence of autologous cells (subject 1) (B). Since the common HLA class I molecule among these subjects was A2.1, this confirms that P2C1137-1145 is presented by this molecule. Values in the upper-right quadrant represent percentages of CD8+ T cells that are IFN-γ positive.
FIG. 5.

HLA stabilization assay to measure peptide binding to HLA-A2.1. The graph shows MFI (y axis) versus peptide concentration (x axis) for P2C1137-1145 (open diamonds) and GAD261-269 (solid squares). Symbols represent the means and SD of four determinations. Solid circle and error bars represent the mean MFIs and SD of T2 cells incubated in the absence of exogenous peptide to indicate background A2.1 expression levels. In this assay, P2C1137-1145 peptide shows binding to HLA-A2.1 at concentrations of >12.5 μM, while GAD261-269 fails to bind.
The P2C1137-1145 epitope is processed from whole P2C protein and CVB4-infected cell lysates and presented to CD8 T cells by HLA-A2.1.
It was important to determine whether this peptide region is immunologically processed by APCs in vivo and presented as a CD8 epitope by HLA-A2.1, which would imply its importance in natural infection. This phenomenon of cross-presentation is a critical feature of DC function and of major importance in the establishment of the anti-viral CD8 T-cell response (1). To examine this, monocyte-derived DCs were generated from the same donor as the PEVKEK1137-1145-specific CD8 T-cell line from an HLA-A2.1 matched donor and a mismatched (non-A2.1) donor. During DC maturation, cells were pulsed with whole recombinant P2C or with lysates of CVB4-infected Vero cells. The PEVKEK1137-1145-specific CD8 T-cell line responded by IFN-γ production to both autologous and HLA-A2.1 DCs pulsed with P2C protein or CVB4-infected lysates, but not to mismatched DCs, indicating natural processing and cross-presentation of the PEVKEK1137-1145 epitope by professional APCs (Fig. 6).
FIG. 6.

P2C1137-1145 is a naturally processed epitope presented by HLA-A2.1. The graph shows the percentages of the P2C1137-1145-specific CD8 T-cell line staining for IFN-γ after coculture with DCs from different donors, pulsed with either P2C (open bars), CVB4 lysates (solid bars), or P2C1137-1145 peptide (hatched bars). Autologous DCs and those from an HLA-A2.1 donor were able to process and present P2C1137-1145 from whole P2C and from CVB4-infected cell lysates. The graph shows representative results from two experiments; background staining with control preparations for P2C, CVB4, and peptide P2C1137-1145 (MBP, uninfected cell lysate, and PBS, respectively) has been subtracted. Staining of non-CD8 T cells with anti-IFN-γ was typically ≪0.1%.
P2C1137-1145-specific CD8 T cells do not cross-react with the homologous region from GAD65.
Having identified the first CD8 T-cell epitope from CVB4, and confirmed its natural presentation by HLA-A2.1, we went on to investigate the potential for cross-reactivity of T cells targeting the viral peptide with the peptide of similar sequence from GAD65. As described above, when the immunomagnetically enriched CTLs were stimulated in the presence of APCs preloaded with the P2C1137-1145 peptide, up to 11% of the CD8+ cells produced IFN-γ (Fig. 2F). However, when the same CTLs were cocultured with APCs preloaded with the homologous peptide GAD261-269, IFN-γ production by CD8 T cells was similar to that of the background (Fig. 2G and H). Moreover, we were unable to detect any direct ex vivo IFN-γ response against the GAD261-269 peptide in samples from any of the six healthy donors examined in this study (Fig. 1F and Table 4).
To exclude the possibility that cross-recognition of the homologous GAD65 peptide by the P2C1137-1145-specific CTL line was present but did not elicit IFN-γ production, we also examined the cytotoxic potential of this CTL line using autologous target cells loaded with either the P2C or GAD65 peptide. Only background levels of cytotoxicity were detected against targets loaded with the mimicry GAD261-269 peptide (approximately 7%), similar to that seen with targets loaded with the negative control peptide GAD2-10 (9%) (Fig. 3). Indeed, GAD261-269 peptide did not stabilise HLA-A2.1 formation in the stabilization assay, suggesting that binding to this molecule is weak or absent (Fig. 5).
DISCUSSION
Members of the EV genus of viruses, such as the CVB group, are implicated in the pathogenesis of a number of inflammatory conditions in humans, including chronic dilated myocarditis, chronic myositis, pancreatitis, and meningitis. Strong epidemiological and laboratory data also link these viruses with the autoimmune disease T1DM. A common feature of these disorders is that the immune response to the virus and associated inflammation may be major factors contributing to the extent of tissue damage. In the case of T1DM, several possible immunopathological mechanisms could be at play, most notably molecular mimicry between virus and host or proinflammatory bystander effects of virus-reactive immune cells. To further our understanding of these processes, it is important to characterize the cellular immune response to viruses in this group, which in turn requires the identification of the peptide targets and HLA restriction of virus-reactive T cells.
In the present study, we identify the first-known CD8 T-cell epitope of any of the coxsackievirus group. The number of IFN-γ-producing CD8 T cells detected following brief ex vivo culture with the P2C1137-1145 epitope was similar to that seen for the highly immunodominant influenza matrix epitope GILGFVFTL (41). This strongly implies that P2C1137-1145 represents an immunodominant A2.1-restricted epitope, despite its apparent intermediate affinity for this restriction element. Given the potential dominance of this epitope, it may prove useful in monitoring CVB4 infections, using the technologies described here (intracellular flow cytometry) or with fluorescent HLA-A2.1 tetramers loaded with P2C1137-1145 peptide.
It is clear from the epitope prediction programs interrogated in our study that the P2C peptide region spanning positions 1132 to 1145 contains peptide sequences with the potential to be presented by several HLA class I molecules. Indeed, one peptide, P2C1137-1145, is one of the best predicted HLA-B8 binders in the whole CVB4 polyprotein. We were able to demonstrate that CD8 effector memory T cells responding to this peptide epitope are present in the peripheral blood of healthy subjects, at levels that are typical of single-epitope responses associated with viral diseases. This suggests that these responses may be important in host protection and provides circumstantial evidence that this epitope is generated naturally through intracellular processing of viral P2C by APCs during CVB4 infection. The generation of this epitope from intact P2C- and CVB4-infected cells by professional APCs, involved in priming the anti-viral immune response, was confirmed with DCs matured in the presence of the relevant antigens. Taken together, these data support the proposal that P2C1137-1145 is an epitope generated during CVB4 infection, but further studies on the responses of naïve subjects to live virus will be required to show this with certainty. It is also of interest that despite the fact that the P2C1137-1145 sequence has a predicted 3.4-fold higher-affinity for HLA-B8 than A2.1, the CD8 T-cell line generated in our study from an HLA-A2.1 B8 individual is HLA-A2.1 restricted, and only samples from HLA-A2.1 individuals made responses to this epitope. The capacity for coexpressed HLA molecules to influence CD8 T-cell epitope restriction has been recognized in other studies, and it has been speculated that this may be due to either processing constraints or the influence of regulatory T-cell populations (5, 22).
The relationship between a peptide's affinity for major histocompatibility complex class I (MHC-I) molecules and its recruitment as a CD8 T-cell epitope is complex. For example, it has been determined in animal studies (40) that more than 80% of the peptide epitopes from a given protein antigen predicted to bind with high or intermediate affinity to their corresponding HLA allele (dissociation constant [Kd] < 0.05 μM) trigger a CD8 T-cell response. In contrast, in humans the situation is less clear; CD8 T-cell responses against peptides derived from hepatitis B virus in A2.1 subjects acutely infected with hepatitis B virus were directed against 45% of the high-affinity, 14% of the intermediate-affinity, and 6% of the low-affinity regions (40). Subsequent studies have indicated that the major determinant of CD8 T-cell epitope dominance is most likely to be the relative abundance of peptide-MHC complexes on the APC surface, which in turn is a function of both affinity and processing efficiency (7, 8, 47, 48). Taken together, these data lead us to speculate that the P2C1137-1145 CD8 T-cell epitope identified in our study may be immunodominant as a result of the combination of modest affinity for A2.1 balanced by efficient generation by the proteasome. It is noteworthy that previous studies also give prominence to the nonstructural P2C protein as an important antigen in the cellular immune response to the coxsackievirus group (44), making it an attractive target for the design of peptide vaccines due to its highly conserved protein sequence among the different EV members.
Having demonstrated CD8 T-cell responses to a CVB4 peptide in the PEVKEK region, we went on to address possible cross-reactivity between this and the homologous GAD65-derived peptide. The concept of molecular mimicry (32), in which T cells activated in response to microbial epitopes cross-react with sequences of an autoantigen, is perhaps the most frequently cited etiopathological mechanism in autoimmune disease. In T1DM, the most interest in this regard has focused on a sequence homology between the islet cell autoantigen GAD65 and P2C, in which a six-amino-acid sequence (PEVKEK) is shared and several flanking residues show conserved substitution (4). Thus far, studies examining immunological cross-reactivity in this region have focused on either antibodies (17, 23) or CD4 T cells, and evidence both for (4) and against (39) a functional mimicry has been presented. Our results show that T cells from healthy nondiabetic individuals responding to the P2C viral peptide epitope and restricted by HLA-A2.1 do not cross-react with the homologous region from GAD65, at least not at a level detectable by our in vitro assays.
The peptide binding motif for HLA-A2.1 predicts as major peptide anchor positions P2 and P9, with leucine (L) and methionine (M) at P2 as preferred residues and valine (V) and leucine (L) at P9. Accessory positions are P4 glutamic acid (E) and lysine (K) as preferred residues, P6 (V) and P8 (K) (http://syfpeithi.bmi-heidelberg.com/scripts/MHCServer.dll/home.htm)(36). Both the P2C1137-1145 and GAD261-269 peptides show lower predicted affinities for the A2.1 molecule than the well-characterized influenza matrix epitope known to have high affinity for A2.1. However, in HLA stabilization assays, P2C1137-1145 has measurable binding to A2.1, while the GAD261-269 peptide shows no appreciable binding. Both peptides have a V at position 2; at the other important anchor residue at position 9 the P2C1137-1145 epitope has an L, while GAD65261-269 has an A, which could explain the difference in binding affinity we observed. The HLA stabilization assay is relatively insensitive, and the relatively low affinity of GAD65261-269 does not exclude it per se from being a CD8 T-cell target. On the contrary, it is more likely that disease-related epitopes of autoantigens will have a relatively low affinity for self-MHC molecules, resulting in a low abundance of thymic peptide-MHC complexes and allowing autoreactive T cells to escape negative selection. This concept receives support from studies with the nonobese diabetic mouse, in which the affinity of the major disease-related autoreactive CD8 T-cell epitope in the insulin molecule B15-23, presented by Kd, is very low when measured by a conventional MHC stabilization assay (50).
The identification of the first CD8 epitope of CVB4 and its presence in the region of virus homology with the diabetes-related autoantigen GAD65 beg the question of the relevance of our findings to T1DM. Previous studies focusing on responses by CD4 T cells (4, 39) have provided conflicting results as to whether this homology region is targeted in patients with T1DM. In the work reported by Atkinson et al. (4), it was shown that peripheral blood lymphocytes from subjects at risk of T1DM development (autoantibody-positive, first-degree relatives of a T1DM subject) or newly diagnosed T1DM patients responded to two contiguous peptides in the PEVKEK region of GAD65, but healthy donors did not. Three T1DM subjects with cells that had responses to GAD65 peptides in the homologous region also responded to P2C peptides containing the PEVKEK motif, but this implies rather than proves cross-reactivity. Systematic analysis of responses to this region by Schloot et al. (39) failed to show cross-reactivity with CD4 T-cell lines and clones generated from T1DM patients by T-cell proliferation assays, but it is noteworthy that two of three CD4 T-cell clones produced IFN-γ in response to both the P2C- and GAD65-derived peptides. Our study shows that there is a CD8 T-cell epitope contiguous with this region of interest, and it remains a possibility that functional mimicry exists in patients with T1DM, perhaps arising as a result of ineffective central or peripheral deletion of cross-reactive T cells. Knowledge of the P2C epitope should facilitate further studies in T1DM, to establish its relevance to disease.
In conclusion, we have identified the first CD8 T-cell epitope from CVB4. The epitope is naturally generated by APCs, lies in the region of P2C with autoantigen homology, and is restricted by the diabetes-associated HLA-A2.1 molecule, but it does not appear to generate cross-reactive CD8 T-cell responses with the homologous region of GAD65, at least not in healthy nondiabetic donors.
Acknowledgments
M.P. is a Diabetes United Kingdom Senior Clinical Research Fellow. R.V.-C. was supported by the Wellcome Trust during this study.
REFERENCES
- 1.Ackerman, A. L., and P. Cresswell. 2004. Cellular mechanisms governing cross-presentation of exogenous antigens. Nat. Immunol. 5:678-684. [DOI] [PubMed] [Google Scholar]
- 2.Andreoletti, L., D. Hober, C. Hober-Vandenberghe, S. Belaich, M. C. Vantyghem, J. Lefebvre, and P. Wattre. 1997. Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J. Med. Virol. 52:121-127. [DOI] [PubMed] [Google Scholar]
- 3.Archard, L. C., M. A. Khan, B. A. Soteriou, H. Zhang, H. J. Why, N. M. Robinson, and P. J. Richardson. 1998. Characterization of coxsackie B virus RNA in myocardium from patients with dilated cardiomyopathy by nucleotide sequencing of reverse transcription-nested polymerase chain reaction products. Hum. Pathol. 29:578-584. [DOI] [PubMed] [Google Scholar]
- 4.Atkinson, M. A., M. A. Bowman, L. Campbell, B. L. Darrow, D. L. Kaufman, and N. K. Maclaren. 1994. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J. Clin. Investig. 94:2125-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betts, M. R., J. P. Casazza, B. A. Patterson, S. Waldrop, W. Trigona, T. M. Fu, F. Kern, L. J. Picker, and R. A. Koup. 2000. Putative immunodominant human immunodeficiency virus-specific CD8+ T-cell responses cannot be predicted by major histocompatibility complex class I haplotype. J. Virol. 74:9144-9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bottazzo, G. F., B. M. Dean, J. M. McNally, E. H. MacKay, P. G. Swift, and D. R. Gamble. 1985. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N. Engl. J. Med. 313:353-360. [DOI] [PubMed] [Google Scholar]
- 7.Busch, D. H., and E. G. Pamer. 1998. MHC class I/peptide stability: implications for immunodominance, in vitro proliferation, and diversity of responding CTL. J. Immunol. 160:4441-4448. [PubMed] [Google Scholar]
- 8.Busch, D. H., I. M. Pilip, S. Vijh, and E. G. Pamer. 1998. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity 8:353-362. [DOI] [PubMed] [Google Scholar]
- 9.Clements, G. B., D. N. Galbraith, and K. W. Taylor. 1995. Coxsackie B virus infection and onset of childhood diabetes. Lancet 346:221-223. [DOI] [PubMed] [Google Scholar]
- 10.Currier, J. R., E. G. Kuta, E. Turk, L. B. Earhart, L. Loomis-Price, S. Janetzki, G. Ferrari, D. L. Birx, and J. H. Cox. 2002. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J. Immunol. Methods 260:157-172. [DOI] [PubMed] [Google Scholar]
- 11.Dahlquist, G., G. Frisk, S. A. Ivarsson, L. Svanberg, M. Forsgren, and H. Diderholm. 1995. Indications that maternal coxsackie B virus infection during pregnancy is a risk factor for childhood-onset IDDM. Diabetologia 38:1371-1373. [DOI] [PubMed] [Google Scholar]
- 12.Davis, L. E., D. Bodian, D. Price, I. J. Butler, and J. H. Vickers. 1977. Chronic progressive poliomyelitis secondary to vaccination of an immunodeficient child. N. Engl. J. Med. 297:241-245. [DOI] [PubMed] [Google Scholar]
- 13.de Jong, E. C., P. L. Vieira, P. Kalinski, J. H. Schuitemaker, Y. Tanaka, E. A. Wierenga, M. Yazdanbakhsh, and M. L. Kapsenberg. 2002. Microbial compounds selectively induce Th1 cell-promoting or Th2 cell-promoting dendritic cells in vitro with diverse th cell-polarizing signals. J. Immunol. 168:1704-1709. [DOI] [PubMed] [Google Scholar]
- 14.Halliday, E., J. Winkelstein, and A. D. Webster. 2003. Enteroviral infections in primary immunodeficiency (PID): a survey of morbidity and mortality. J. Infect. 46:1-8. [DOI] [PubMed] [Google Scholar]
- 15.Henke, A., S. Huber, A. Stelzner, and J. L. Whitton. 1995. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J. Virol. 69:6720-6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiltunen, M., H. Hyoty, M. Knip, J. Ilonen, H. Reijonen, P. Vahasalo, M. Roivainen, M. Lonnrot, P. Leinikki, T. Hovi, H. K. Akerblom, et al. 1997. Islet cell antibody seroconversion in children is temporally associated with enterovirus infections. J. Infect. Dis. 175:554-560. [DOI] [PubMed] [Google Scholar]
- 17.Hou, J., C. Said, D. Franchi, P. Dockstader, and N. K. Chatterjee. 1994. Antibodies to glutamic acid decarboxylase and P2-C peptides in sera from coxsackie virus B4-infected mice and IDDM patients. Diabetes 43:1260-1266. [DOI] [PubMed] [Google Scholar]
- 18.Hyoty, H. 2002. Enterovirus infections and type 1 iabetes. Ann. Med. 34:138-147. [PubMed] [Google Scholar]
- 19.Hyoty, H., M. Hiltunen, M. Knip, M. Laakkonen, P. Vahasalo, J. Karjalainen, P. Koskela, M. Roivainen, P. Leinikki, T. Hovi, et al. 1995. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Diabetes 44:652-657. [DOI] [PubMed] [Google Scholar]
- 20.Kang, Y., N. K. Chatterjee, M. J. Nodwell, and J. W. Yoon. 1994. Complete nucleotide sequence of a strain of coxsackie B4 virus of human origin that induces diabetes in mice and its comparison with nondiabetogenic coxsackie B4 JBV strain. J. Med. Virol. 44:353-361. [DOI] [PubMed] [Google Scholar]
- 21.Knipe, D. M., P. M. Howley, D. E. Griffin, S. R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus. 2001. Fields Virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 22.Lacey, S. F., M. C. Villacres, C. La Rosa, Z. Wang, J. Longmate, J. Martinez, J. C. Brewer, S. Mekhoubad, R. Maas, J. M. Leedom, S. J. Forman, J. A. Zaia, and D. J. Diamond. 2003. Relative dominance of HLA-B*07 restricted CD8(+) T-Lymphocyte immune responses to human cytomegalovirus pp65 in persons sharing HLA-A*02 and HLA-B*07 alleles. Hum. Immunol. 64:440-452. [DOI] [PubMed] [Google Scholar]
- 23.Lonnrot, M., H. Hyoty, M. Knip, M. Roivainen, P. Kulmala, P. Leinikki, H. K. Akerblom, et al.. 1996. Antibody cross-reactivity induced by the homologous regions in glutamic acid decarboxylase (GAD65) and 2C protein of coxsackievirus B4. Clin. Exp. Immunol. 104:398-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lonnrot, M., K. Korpela, M. Knip, J. Ilonen, O. Simell, S. Korhonen, K. Savola, P. Muona, T. Simell, P. Koskela, and H. Hyoty. 2000. Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: the Finnish Diabetes Prediction and Prevention Study. Diabetes 49:1314-1318. [DOI] [PubMed] [Google Scholar]
- 25.Lonnrot, M., K. Salminen, M. Knip, K. Savola, P. Kulmala, P. Leinikki, T. Hyypia, H. K. Akerblom, H. Hyoty, et al. 2000. Enterovirus RNA in serum is a risk factor for beta-cell autoimmunity and clinical type 1 diabetes: a prospective study. J. Med. Virol. 61:214-220. [PubMed] [Google Scholar]
- 26.Marron, M. P., R. T. Graser, H. D. Chapman, and D. V. Serreze. 2002. Functional evidence for the mediation of diabetogenic T cell responses by HLA-A2.1 MHC class I molecules through transgenic expression in NOD mice. Proc. Natl. Acad. Sci. USA 99:13753-13758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martino, T. A., P. Liu, and M. J. Sole. 1994. Viral infection and the pathogenesis of dilated cardiomyopathy. Circ. Res. 74:182-188. [DOI] [PubMed] [Google Scholar]
- 28.Mori, M., P. G. Beatty, M. Graves, K. M. Boucher, and E. L. Milford. 1997. HLA gene and haplotype frequencies in the North American population: the National Marrow Donor Program Donor Registry. Transplantation. 64:1017-1027. [DOI] [PubMed] [Google Scholar]
- 29.Muir, P., and A. M. van Loon. 1997. Enterovirus infections of the central nervous system. Intervirology 40:153-166. [DOI] [PubMed] [Google Scholar]
- 30.Nairn, C., D. N. Galbraith, K. W. Taylor, and G. B. Clements. 1999. Enterovirus variants in the serum of children at the onset of type 1 diabetes mellitus. Diabet. Med. 16:509-513. [DOI] [PubMed] [Google Scholar]
- 31.Nijman, H. W., J. G. Houbiers, M. P. Vierboom, S. H. van der Burg, J. W. Drijfhout, J. D'Amaro, P. Kenemans, C. J. Melief, and W. M. Kast. 1993. Identification of peptide sequences that potentially trigger HLA-A2.1-restricted cytotoxic T lymphocytes. Eur. J. Immunol. 23:1215-1219. [DOI] [PubMed] [Google Scholar]
- 32.Oldstone, M. B. 1998. Molecular mimicry and immune-mediated diseases. FASEB J. 12:1255-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Opavsky, M. A., J. Penninger, K. Aitken, W. H. Wen, F. Dawood, T. Mak, and P. Liu. 1999. Susceptibility to myocarditis is dependent on the response of αβ T lymphocytes to coxsackieviral infection. Circ. Res. 85:551-558. [DOI] [PubMed] [Google Scholar]
- 34.Parker, K. C., M. A. Bednarek, and J. E. Coligan. 1994. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 152:163-175. [PubMed] [Google Scholar]
- 35.Price, P., C. Witt, R. Allcock, D. Sayer, M. Garlepp, C. C. Kok, M. French, S. Mallal, and F. Christiansen. 1999. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol. Rev. 167:257-274. [DOI] [PubMed] [Google Scholar]
- 36.Rammensee, H., J. Bachmann, N. P. Emmerich, O. A. Bachor, and S. Stevanovic. 1999. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213-219. [DOI] [PubMed] [Google Scholar]
- 37.Robles, D. T., G. S. Eisenbarth, T. Wang, H. A. Erlich, T. L. Bugawan, S. R. Babu, K. Barriga, J. M. Norris, M. Hoffman, G. Klingensmith, L. Yu, and M. Rewers. 2002. Millennium award recipient contribution. Identification of children with early onset and high incidence of anti-islet autoantibodies. Clin. Immunol. 102:217-224. [DOI] [PubMed] [Google Scholar]
- 38.Sathiamurthy, M., H. D. Hickman, J. W. Cavett, A. Zahoor, K. Prilliman, S. Metcalf, M. Fernandez Vina, and W. H. Hildebrand. 2003. Population of the HLA ligand database. Tissue Antigens 61:12-19. [DOI] [PubMed] [Google Scholar]
- 39.Schloot, N. C., S. J. Willemen, G. Duinkerken, J. W. Drijfhout, R. R. de Vries, and B. O. Roep. 2001. Molecular mimicry in type 1 diabetes mellitus revisited: T-cell clones to GAD65 peptides with sequence homology to Coxsackie or proinsulin peptides do not crossreact with homologous counterpart. Hum. Immunol. 62:299-309. [DOI] [PubMed] [Google Scholar]
- 40.Sette, A., A. Vitiello, B. Reherman, P. Fowler, R. Nayersina, W. M. Kast, C. J. Melief, C. Oseroff, L. Yuan, J. Ruppert, et al. 1994. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. 153:5586-5592. [PubMed] [Google Scholar]
- 41.Stewart-Jones, G. B., A. J. McMichael, J. I. Bell, D. I. Stuart, and E. Y. Jones. 2003. A structural basis for immunodominant human T cell receptor recognition. Nat. Immunol. 4:657-663. [DOI] [PubMed] [Google Scholar]
- 42.Suhrbier, A., C. Schmidt, and A. Fernan. 1993. Prediction of an HLA B8-restricted influenza epitope by motif. Immunology 79:171-173. [PMC free article] [PubMed] [Google Scholar]
- 43.Terajima, M., J. Cruz, G. Raines, E. D. Kilpatrick, J. S. Kennedy, A. L. Rothman, and F. A. Ennis. 2003. Quantitation of CD8+ T cell responses to newly identified HLA-A*0201-restricted T cell epitopes conserved among vaccinia and variola (smallpox) viruses. J. Exp. Med. 197:927-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varela-Calvino, R., R. Ellis, G. Sgarbi, C. M. Dayan, and M. Peakman. 2002. Characterization of the T-cell response to coxsackievirus B4: evidence that effector memory cells predominate in patients with type 1 diabetes. Diabetes 51:1745-1753. [DOI] [PubMed] [Google Scholar]
- 45.Varela-Calvino, R., G. Sgarbi, S. Arif, and M. Peakman. 2000. T-cell reactivity to the P2C nonstructural protein of a diabetogenic strain of coxsackievirus B4. Virology 274:56-64. [DOI] [PubMed] [Google Scholar]
- 46.Varela-Calvino, R., G. Sgarbi, L. R. Wedderburn, C. M. Dayan, J. Tremble, and M. Peakman. 2001. T cell activation by coxsackievirus B4 antigens in type 1 diabetes mellitus: evidence for selective TCR Vβ usage without superantigenic activity. J. Immunol. 167:3513-3520. [DOI] [PubMed] [Google Scholar]
- 47.Vijh, S., and E. G. Pamer. 1997. Immunodominant and subdominant CTL responses to Listeria monocytogenes infection. J. Immunol. 158:3366-3371. [PubMed] [Google Scholar]
- 48.Villanueva, M. S., P. Fischer, K. Feen, and E. G. Pamer. 1994. Efficiency of MHC class I antigen processing: a quantitative analysis. Immunity 1:479-489. [DOI] [PubMed] [Google Scholar]
- 49.Wertheimer, A. M., C. Miner, D. M. Lewinsohn, A. W. Sasaki, E. Kaufman, and H. R. Rosen. 2003. Novel CD4+ and CD8+ T-cell determinants within the NS3 protein in subjects with spontaneously resolved HCV infection. Hepatology 37:577-589. [DOI] [PubMed] [Google Scholar]
- 50.Wong, F. S., A. K. Moustakas, L. Wen, G. K. Papadopoulos, and C. A. Janeway, Jr. 2002. Analysis of structure and function relationships of an autoantigenic peptide of insulin bound to H-2K(d) that stimulates CD8 T cells in insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. USA 99:5551-5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoon, J. W., M. Austin, T. Onodera, and A. L. Notkins. 1979. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 300:1173-1179. [DOI] [PubMed] [Google Scholar]
- 52.Zaoutis, T., and J. D. Klein. 1998. Enterovirus infections. Pediatr. Rev. 19:183-191. [DOI] [PubMed] [Google Scholar]