

Abstract
Brk (for breast tumor kinase) is a nonreceptor tyrosine kinase containing SH3, SH2, and tyrosine kinase catalytic domains. Brk was originally identified from a human metastatic breast tumor, and its overexpression is frequently observed in breast cancer and several other cancer types. However, the molecular mechanism by which this kinase participates in tumorigenesis remains poorly characterized. In the present study, we not only identified paxillin as the binding partner and substrate of Brk but also discovered a novel signaling pathway by which Brk mediates epidermal growth factor (EGF)-induced paxillin phosphorylation. We show that EGF stimulation activates the catalytic activity of Brk, which in turn phosphorylates paxillin at Y31 and Y118. These phosphorylation events promote the activation of small GTPase Rac1 via the function of CrkII. Through this pathway, Brk is capable of promoting cell motility and invasion and functions as a mediator of EGF-induced migration and invasion. In accordance with these functional roles, Brk translocates to membrane ruffles, where it colocalizes with paxillin during cell migration. Together, our findings identify novel signaling and biological roles of Brk and indicate the first potential link between Brk and metastatic malignancy.
Unraveling the signaling pathways responsible for the establishment of a metastatic phenotype in carcinoma cells is of crucial importance for the understanding of the pathology of cancer. The process of metastasis includes several components, such as the ability to invade through acquisition of cell motility, degradation of extracellular matrix and basement membrane, cell proliferation, and survival signaling. Aberrant tyrosine kinase signaling via stimulation of growth factor receptors or intracellular tyrosine kinases has been shown to contribute to various steps of tumor development and progression, including metastasis (6). Brk is an intracellular tyrosine kinase that was identified in a study for screening kinases expressed in human metastatic breast tumors (36). In addition to a typical tyrosine kinase domain, Brk possesses both SH3 and SH2 domains and thus is related to Src family kinases (36). However, unlike Src family kinases, Brk lacks an N-terminal consensus sequence for myristoylation and membrane association (36). Its genomic structure is also distinct from that of Src family kinases, suggesting that Brk has diverged significantly from Src kinases in evolution (37). The expression pattern and subcellular localization of Brk have suggested its role in tumorigenesis. In normal tissues, the expression of Brk or its mouse ortholog Sik is restricted to differentiating epithelial cells of the skin and gastrointestinal tract (27). However, it is highly expressed in many breast carcinoma cell lines and a significant portion of breast tumor tissues but not in human mammary epithelial cells (3, 34, 36) and mouse mammary glands at various developmental stages (27). Elevated expression of Brk has also been detected in metastatic melanoma cell lines (13) and in some colon tumors (27). In prostate cancers, although the expression of Brk is not significantly altered, Brk translocates from the nucleus to the cytoplasm during the progression of tumors (10).
Little is known about the signal transduction pathway in which Brk is involved. Brk expression sensitizes the mammary epithelial cells to the mitogenic response of epidermal growth factor (EGF) and potentiates their anchorage-independent growth (20). Accordingly, Brk was found to associate with EGF receptor and enhance EGF-dependent phosphorylation of erbB3, which subsequently leads to an increased recruitment of phosphoinositide 3-kinase (PI 3-kinase) and activation of Akt (19). This finding links Brk to the EGF-induced activation of PI 3-kinase/Akt pathway and may provide a mechanistic insight into Brk-dependent mitogenic sensitization. However, whether Brk is involved in other pathways induced by EGF and whether the catalytic activity of Brk is regulated by EGF signaling have not been explored.
Since the identification of Brk from metastatic breast carcinoma, it remains unclear whether this kinase contributes to the metastatic malignancy. To date, two substrates of Brk/Sik have been identified, i.e., the adaptor-like protein BKS (38) and the nuclear RNA-binding protein Sam68 (11). BKS possesses a PH-like domain, followed by an SH2 domain, and has recently been found to play a regulatory role in STAT3 activation (35). The physiological significance of BKS phosphorylation by Brk, however, is unclear. Another substrate is Sam68, which belongs to a member of the STAR family of RNA-binding proteins that regulate RNA metabolism (59). Phosphorylation of Sam68 by Brk attenuates its RNA-binding ability and Brk colocalizes with Sam68 in the nucleus (11). These findings point out a physiological role of Brk in regulating RNA function, although it remains largely elusive whether and how this effect contributes to the oncogenic function of Brk.
Paxillin is a multidomain protein that is recruited to the leading edges of cells upon the initiation of migration. It primarily functions as a molecular scaffold that provides multiple docking sites at the plasma membrane for an array of signaling, adaptor, and structural proteins (56). The motifs and domains present in paxillin include LIM domains, leucine-rich motifs (termed LD repeats), proline-rich sequences, and phosphotyrosine binding sites (46, 56). The LD repeats serve as binding regions for the focal adhesion proteins FAK, vinculin (7), the FAK-related kinase Pyk2 (57), and the ARF GAP, Git/PKL/CAT (57), whereas the LIM domains are crucial for focal adhesion targeting of paxillin (7), presumably through its association with the β-integrin tails. Importantly, many of the paxillin interacting proteins are involved in the regulation of actin cytoskeleton organization, which is necessary for cell motility events associated with diverse biological responses, such as embryonic development, wound repair, and tumor metastasis (46). Accordingly, paxillin-null mice die at embryonic day 9.5 and display impaired development of heart and somites (16). Furthermore, paxillin-null fibroblasts show abnormalities in focal adhesion structure and cortical actin cytoskeleton and have reduced rates of cell spreading and migration (16).
Paxillin undergoes tyrosine phosphorylation in response to various physiological stimuli and integrin-mediated cell adhesion events (46). Paxillin has four major tyrosine phosphorylation sites: Y31, Y40, Y118, and Y181 (40). Among them, phosphorylation of Y31/118 is highly augmented during cell adhesion and migration (40). These two phosphorylated residues serve as docking sites for the recruitment of downstream effectors such as Crk (42, 47) and p120RasGAP (55), thereby regulating cell motility and adhesion, respectively. In Nara Bladder Tumor II cells, replacement of Y31 and/or Y118 with phenylalanine greatly reduces CrkII association and cell motility (42), even though other reports demonstrated that the downstream events of paxillin tyrosine phosphorylation are cell type specific and could be influenced by the presence of other Crk-associated proteins, such as p130Cas (61). Notably, although a large number of stimuli induce tyrosine phosphorylation of paxillin, only a few tyrosine kinases have been reported to phosphorylate paxillin (46). FAK and its related protein Pyk2 interact and colocalize with paxillin, and evidence from both in vitro and in vivo studies indicates paxillin as a substrate of these kinases (5, 17, 47). Src family kinases have also been implicated in regulating tyrosine phosphorylation of paxillin since such phosphorylation is enhanced in fibroblasts derived from mice lacking Csk (an Src inhibitory kinase) (53) and decreased in SYF cells, which lacks Src, Fyn, and Yes (22). Furthermore, a recent study demonstrated a direct phosphorylation of paxillin by Src (18). In addition to FAK and Src, the proto-oncogene c-Abl was shown to associate with paxillin upon cell adhesion and to phosphorylate paxillin in vitro (26).
We report here the identification of a novel paxillin tyrosine kinase, Brk. Brk directly binds and phosphorylates paxillin both in vitro and in vivo and functions as a crucial mediator of EGF-induced paxillin phosphorylation at Y31 and Y118. As a consequence, Brk promotes the activation of Rac1, thereby stimulating cell migration and invasion in response to EGF. Our findings not only reveal a role of Brk in EGF-induced cell motility but also provide the first potential link between Brk and metastatic malignancy.
MATERIALS AND METHODS
Cell culture, transfection, and retroviral infection.
293T, HeLa, Cos-1, HaCaT, and A431 cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS). MDA-MB231 cells were cultured in DMEM-F-12 medium with 10% FCS. Transfection of 293T and Cos-1 cells was performed by the calcium phosphate method, and transfection of A431 cells was done by using Lipofectamine 2000 reagent (Invitrogen). Recombinant retroviruses were generated according to the procedures described previously (54). For infection of A431 and MDA-MB231 cells, the viral stock was supplemented with 8 and 6 μg of Polybrene/ml, respectively, and incubated with cells for 18 h. Infected cells were selected by using puromycin or hygromycin.
cDNA cloning and plasmid constructions.
The cDNA of Brk was generated by reverse transcription-PCR with RNA purified from T-47D human breast carcinoma cells, and its sequence was verified by sequencing analysis. This cDNA fragment was cloned into pRK5M and pRK5F to generate C-terminally myc- and Flag-tagged Brk, respectively. The BrkKM mutant was generated by PCR amplification with the primers 5′-ACAAGATCTGGCGGCGTGCC-3′ and 5′-TTGTCTCGAGAAATCACCATAATGGCCACC-3′ and the Advantage-GC 2 PCR kit (Clontech). The resulting fragment was digested by BglII and XhoI and was used to replace the BglII-XhoI fragment of pRK5M-Brk. The BrkΔSH2 mutant was constructed by a similar PCR-based strategy, whereas the BrkΔSH3 mutant was made by in vitro mutagenesis with a QuikChange site-directed mutagenesis kit. To obtain the full-length cDNA sequence of paxillin, two expressed sequence tag clones, BM561570 and BQ218095, covering the 5′ and 3′ of paxillin, respectively, were purchased from Open Biosystems. These two fragments were ligated by using the overlapping SacI site and cloned into pBluescript. The resulting full-length paxillin cDNA was cloned to pRK5F or pBabePuro3 or used as a template to generate its N-terminal and C-terminal deletion mutants by a PCR approach. The paxillinDYF mutant, in which the tyrosine 31 and tyrosine 118 were both replaced by phenylalanine, was constructed by site-directed mutagenesis and then subcloned to pBabeHygro. CrkII R38K and CrkII W170K mutants were generated by site-directed mutagenesis with the wild-type cDNA as a template.
Antibodies and reagents.
Antibodies to phosphotyrosine (4G10) and Rac1 (23A8), Src, and p418 Src were from UBI, whereas anti-Brk and anti-myc (9E10) were from Santa Cruz. The paxillin antibody was from BD Biosciences, and the antibodies specifically recognizing Y31- and Y118-phosphorylated paxillin were from Biosource. Anti-Flag M2 antibody was from Sigma, whereas anti-FAK was from Transduction Laboratories. EGF was purchased from UBI.
GST fusion proteins.
The cDNA fragments for paxillin and its mutant were cloned to pGEX-4T vector to generate glutathione S-transferase (GST) fusion proteins. Production and purification of the GST fusion proteins were performed as described previously (54). For purification of GST-PAK-CRIB, bacteria were lysed in buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 1% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride (PMSF) by vortexing the mixture for 15 min to 1 h at 4°C, followed by sonication two to three times at 4°C. Cell debris was removed by centrifugation at 10,000 rpm for 10 min, and the supernatant was incubated with glutathione-Sepharose beads for 1.5 h at 4°C. The beads were washed five to eight times with lysis buffer and then resuspended in lysis buffer containing 5% glycerol.
Production of baculovirus.
The cDNA for Brk or BrkKM containing a Flag tag was cloned to pVL1392 vector. These plasmids, together with the linearized baculovirus DNA (BaculoGold; Pharmingen), were introduced into monolayers of Sf9 cells cultured in Grace's medium (Life Technologies) supplemented with 10% FCS. The recombinant virus was harvested, amplified, and then used to infect monolayers of Sf9 cells in TMN-FH medium. After a 4-day incubation, the Flag-tagged Brk was purified by using the anti-Flag M2 agarose (Sigma).
Immunoprecipitations.
Cells were lysed in radioimmunoprecipitation assay buffer containing 50 mM Tris (pH 8.0), 0.15 M NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate (SDS), 1 mM PMSF, 1 μg of aprotinin/ml, 1 μg of leupeptin/ml, 1 mM sodium vanadate, 4 mM sodium pyrophosphate, and 20 mM NaF. Lysates containing equal amount of proteins were subjected to immunoprecipitation as described previously (54).
In vitro kinase assay.
293 or Sf9 cells expressing Brk or BrkKM were lysed in Brk lysis buffer containing 50 mM HEPES (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 1 mM PMSF, 1 μg of aprotonin/ml, 1 μg of leupeptin/ml, 1 mM sodium vanadate, 4 mM sodium pyrophosphate, and 20 mM NaF. Brk or BrkKM was precipitated from cell lysates by anti-Flag antibody and then incubated in 30 μl of kinase buffer containing 50 mM HEPES (pH 7.5), 10 mM MgCl2, 10 mM MnCl2, 1 mM DTT, 10 μM ATP, and 10 μCi of [γ-32P]ATP in the presence or absence of 2 μg of GST-paxillin or GST-paxillinDYF mutant at 30°C for 10 min. The reaction was stopped by boiling in SDS sample buffer, separated by SDS-polyacrylamide gel electrophoresis (PAGE), and analyzed by autoradiography, Coomassie blue staining, or Western blotting. To determine the stoichiometry of phosphorylation of paxillin by Brk, 0.8 μg of Flag-tagged Brk immobilized on protein G beads was incubated with 0.05 μg of GST-paxillin in 10 μl of kinase reaction mixture as described above. The reaction was allowed to proceed for 0 to 60 min and then stopped by boiling in SDS sample buffer. The reaction products were separated by SDS-PAGE, and the protein band corresponding to GST-paxillin was excised. 32P incorporation was determined by scintillation counting to calculate the number of moles of phosphate transferred per moles of recombinant GST-paxillin.
Assay for GTP-bound Rac.
Detection and quantitation of GTP-bound Rac1 was performed essentially as described previously (45). Cells were lysed in Rac lysis buffer containing 25 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10% glycerol, 1 mM PMSF, 1 μg of aprotinin/ml, 1 μg of leupeptin/ml, 1 mM sodium vanadate, 4 mM sodium pyrophosphate, and 20 mM NaF. Then, 300 μl of cell lysate was incubated with 10 μg of GST-PAK-CRIB at 4°C for 1.5 h. The pull-down complexes were washed with Rac lysis buffer three times and then subjected to Western blotting to detect the bound Rac.
Immunofluorescence and confocal studies.
A431 cells were fixed with 3.7% paraformaldehyde in phosphate-buffered saline at 4°C overnight, followed by permeabilization with extraction buffer containing 50 mM NaCl, 300 mM sucrose, 10 mM PIPES (pH 6.8), 3 mM MgCl2, and 0.5% Triton X-100 for 7 min. Cells were blocked with phosphate-buffered saline supplemented with 10% goat serum, 1% bovine serum albumin, and 50 mM NH4Cl for 1 h and then incubated with anti-Brk antibody for 2 h, followed by incubation with anti-paxillin antibody for 1.5 h. Cells were then washed and incubated with Alexa Fluor 488- and Texas red-conjugated secondary antibodies. Cells were then washed, mounted, and examined with a Leica DM RA epifluorescence microscope or a Leica TCS SP2 confocal laser-scanning microscope with a ×63 or ×100 oil objective lens. Fluorescent images were captured by using the MetaMorph Image System and then processed by using Adobe Photoshop software.
Cell migration assays.
For chemotaxis assays, Transwell chamber filters (8-μm pore size; Costar) were coated with 15 μg of collagen/ml on both sides. Serum-starved cells (104) resuspended in DMEM with 1% bovine serum albumin were added to the upper chamber. The same medium supplemented with 0.15 to 15 ng of EGF/ml as a chemoattractant was added to the lower chamber. After incubation at 37°C for 3 to 5 h, the cells on the upper side of the filter were removed by wiping it with a cotton swab, and migratory cells on the lower membrane surface were then counted. When transiently transfected cells were used for this assay, green fluorescent protein (GFP)-positive cells migrated to the underside of the filter were counted under a fluorescence microscope. Each assay was set up in triplicate, and at least five random fields were analyzed for each filter.
For wound-healing migration assay by time-lapse video microscopy, confluent cells were wounded by using disposable plastic tips and washed with growth medium. Dishes were placed in an open chamber with atmospheric and temperature controls, and cell movement was viewed by using a Zeiss Axiovert 100 TV microscope equipped with a cooled charge-coupled device video camera. Phase-contrast images obtained at 5-min intervals were collected. In some cases, fluorescence images were also obtained during the recording period. Images were analyzed with the Metamorph program and processed by using Adobe Photoshop software.
Cell invasion assays.
Cells were serum starved for 6 h and then seeded at a density of 104 per well in the upper chamber of Transwells precoated with 54 μg of Matrigel (BD Biosciences). DMEM containing 10 ng of EGF/ml was added to the lower chamber. After incubation for 48 h, cells that appeared on the lower side of filter were counted as described for the Transwell cell migration assay.
Reduction of endogenous Brk expression with siRNA.
A 19-nucleotide small interfering RNA (siRNA) duplex with 3′ TT overhangs corresponding to the Brk mRNA positions 645 to 663 (AAGGUGGCCAUUAAGGUGAUU) and a control siRNA were synthesized by Dharmacon. siRNA transfections were performed by using the RNAi Shuttle Transfection Kit (Orbigen) according to the manufacturer's protocol. To achieve a high efficiency of transfection, cells were transfected three times at 24-h intervals. After transfection, cells were harvested for Western blot analysis or assayed for migration.
RESULTS
Brk induces paxillin phosphorylation at Y31 and Y118.
In an attempt to identify cellular substrates for Brk, we infected HaCaT cells with recombinant retrovirus carrying wild-type Brk or BrkKM (a kinase-defective mutant in which the ATP binding Lys 219 residue was substituted with a Met), together with the puromycin resistance gene. After selection with puromycin, pools of infected cells were generated and then used for analyzing tyrosine-phosphorylated proteins in cell lysates with the anti-phosphotyrosine antibody 4G10. Several tyrosine-phosphorylated bands at ∼68 kDa were significantly induced in cells overexpressing Brk compared to those carrying BrkKM or a control vector (Fig. 1A). A similar induction of the phosphorylation of proteins ∼68 kDa by Brk was observed in 293T cells and Cos-1 cells (data not shown). In searching for cellular proteins with a molecular mass of ∼68 kDa, we noticed that the 68-kDa protein paxillin serves as a substrate for certain Src family kinases (18, 22, 53). Furthermore, paxillin is an abundant cellular protein, which is coincident with the broad, strong band revealed by our Western blots. In addition, overexpression of Brk in certain epithelial cells induced biological and morphological effects that resembled those induced by paxillin phosphorylation (see below). We therefore investigated whether Brk could promote paxillin phosphorylation. Indeed, overexpression of Brk in quiescent Cos-1 cells led to a marked enhancement of paxillin tyrosine phosphorylation, as determined by immunoprecipitation with anti-paxillin antibody followed by Western blotting with the anti-phosphotyrosine antibody. This induction of paxillin phosphorylation was not observed in cells expressing BrkKM (Fig. 1B), indicating a requirement of Brk catalytic activity. Similar induction of paxillin tyrosine phosphorylation by Brk, but not BrkKM, was observed in 293T, HaCaT (Fig. 1B), and HeLa cells (data not shown). In contrast, Brk did not promote FAK tyrosine phosphorylation (Fig. 1C), indicating that the induction of paxillin tyrosine phosphorylation in cells overexpressing Brk is independent of FAK. To determine the tyrosine residues that are involved in this phosphorylation event, we utilized phosphorylation-specific paxillin antibodies. This analysis revealed that phosphorylation at both Y31 and Y118 was increased in cells expressing Brk (Fig. 1D). To investigate whether these two residues are the major phosphorylation sites targeted by Brk, we generated a Flag-tagged paxillin mutant (paxillinDYF), in which the Y31 and Y118 were both replaced by phenylalanine. When this mutant and Brk were coexpressed in Cos-1 cells, we found that Brk could not phosphorylate the paxillinDYF mutant (Fig. 1E). Altogether, our data indicate that Brk promotes paxillin phosphorylation at Y31 and Y118 in vivo.
FIG. 1.

Brk promotes paxillin phosphorylation in vivo. (A) HaCaT cells stably expressing Brk or BrkKM or a control vector were lysed, and tyrosine phosphorylated proteins were detected by Western blotting with anti-phosphotyrosine antibody (upper panel). The expressions of endogenous and exogenous Brk (myc-Brk) are shown in the bottom panel. The positions of protein bands ∼68 kDa and the tyrosine phosphorylated Brk are indicated by an arrowhead and an arrow, respectively. (B) Cos-1 and 293T cells transfected with myc-Brk, myc-BrkKM, or a control vector or HaCaT cells infected with retroviruses carrying Brk or control vector were serum starved and lysed. Cell lysates were subjected to immunoprecipitation with antipaxillin (Pax) antibody, followed by Western blotting with the antibodies indicated. The expression of overexpressed Brk is shown on the bottom. (C) Cos-1 cells transfected and cultured as in panel B were lysed for immunoprecipitation with anti-FAK antibody, followed by Western blotting with the indicated antibodies. The expression of overexpressed Brk is shown on the bottom. (D) Cos-1 cells transfected and cultured as in panel B were assayed for paxillin tyrosine phosphorylation at Y31 and Y118. The immunocomplexes or cell lysates were analyzed by Western blotting with the indicated antibodies. (E) Cos-1 cells were transfected with myc-Brk, Flag-paxillin or their mutants as indicated. Serum-starved cells were lysed and subjected to immunoprecipitation with anti-Flag antibody, followed by Western blotting with antibody to phosphotyrosine (pTyr) (upper panel) or Flag (middle panel). The expressions of myc-Brk and myc-BrkKM are shown in the bottom panel.
Brk interacts with paxillin in vivo.
The induction of paxillin tyrosine phosphorylation by Brk raises a possibility that paxillin is an in vivo substrate of Brk. If so, these two proteins should be able to interact in vivo. Indeed, immunoprecipitation with anti-paxillin antibody from lysates of Cos-1 cells transfected with Flag-Brk revealed the coprecipitation of the Flag-Brk (Fig. 2A, top panels). Furthermore, this coprecipitation of Flag-Brk with paxillin was also detected by a reverse immunoprecipitation with anti-Flag antibody, followed by Western blotting with the antipaxillin antibody (Fig. 2A, middle panels). To demonstrate an association between endogenous Brk and endogenous paxillin, we utilized the lysates of A431 cells, which express a relatively high level of endogenous Brk (see below). Indeed, the endogenous Brk was readily coprecipitated by the antipaxillin antibody but not by a control antibody (Fig. 2B). These results indicate a specific interaction of Brk and paxillin in vivo. To determine the domain requirement of this association, we generated BrkΔSH2 and BrkΔSH3 mutants in which the SH2 and SH3 domains, respectively, were deleted. Notably, deletion of either the SH2 or the SH3 domain of Brk almost completely abolished its interaction with paxillin (Fig. 2C), indicating that both domains are important for this interaction. Next, we mapped the region in paxillin involved in Brk interaction. Flag-tagged full-length paxillin, its N-terminal LD motif-containing region (amino acids 1 to 315), or the C-terminal LIM domain-containing region (amino acids 316 to 557), together with myc-tagged Brk, was coexpressed in Cos-1 cells, and cell lysates were used for immunoprecipitation analysis. We found that both the N-terminal and C-terminal segments of paxillin displayed weak interactions with Brk compared to the full-length protein (Fig. 2D). Together, these results reveal a complex interaction mode of the two proteins and suggest the involvement of multiple regions in their interaction.
FIG. 2.

Brk interacts with paxillin. (A) Interaction of Brk and paxillin in vivo. Cos-1 cells transfected with or without (−) Flag-Brk were subjected to immunoprecipitation with antipaxillin antibody, anti-Flag antibody, or control antibody as indicated. The immunocomplexes and cell lysates were resolved by SDS-PAGE and analyzed by Western blots with antibodies as indicated. (B) Brk and paxillin interact endogenously. A431 cells were lysed and subjected to immunoprecipitation with antipaxillin antibody or a control immunoglobulin G. The immunocomplexes and cell lysate were analyzed by Western blotting with anti-Brk or antipaxillin antibody as indicated. (C) The SH2 and SH3 domains of Brk are both involved in paxillin binding. Cos-1 cellstransfected with various Flag-tagged Brk constructs or a control vector were subjected to coimmunoprecipitation analysis as in panel B. The expressions of Brk and its deletion mutants in transfected cells are shown on the bottom. (D) Both N-terminal and C-terminal segments of paxillin are involved in Brk binding. Cos-1 cells were transfected with Flag-tagged full-length paxillin, its N-terminal segment (PaxLD), or C-terminal segment (PaxLIM), together with myc-tagged Brk. Cell lysates were used for immunoprecipitations with anti-Flag antibody, followed by Western blotting with anti-Flag or anti-Brk. The expressions of Brk and Flag-tagged paxillin or its deletion mutants are shown on the bottom. The bands corresponding to Flag-paxillin, Flag-PaxLIM, and Flag-PaxLD are indicated by asterisks.
Paxillin is a direct substrate of Brk.
The induction of paxillin tyrosine phosphorylation by Brk in vivo, combined with the physical interaction between the two proteins, strongly suggest that paxillin is a direct substrate of Brk. To substantiate this notion, we tested whether Brk could phosphorylate paxillin in an in vitro kinase reaction, in which the purified, baculovirus-expressed Brk was incubated with purified, bacterially expressed GST-paxillin. This analysis revealed that Brk could phosphorylate GST-paxillin in a dose-dependent manner (Fig. 3A). The phosphorylation observed on paxillin was specific to Brk, since the kinase-dead BrkKM mutant did not result in any phosphorylation (Fig. 3B). Furthermore, when GST-paxillinDYF mutant was introduced as a substrate, Brk-induced phosphorylation was greatly reduced. Stoichiometry analysis revealed that Brk catalyzed ∼1.61 mol of phosphate incorporation per mol of GST-paxillin (Fig. 3C), a finding consistent with two phosphorylation sites. Taken together, these results not only identify paxillin as a direct substrate of Brk but also indicate that Y31 and Y118 are the principal sites phosphorylated by Brk.
FIG. 3.
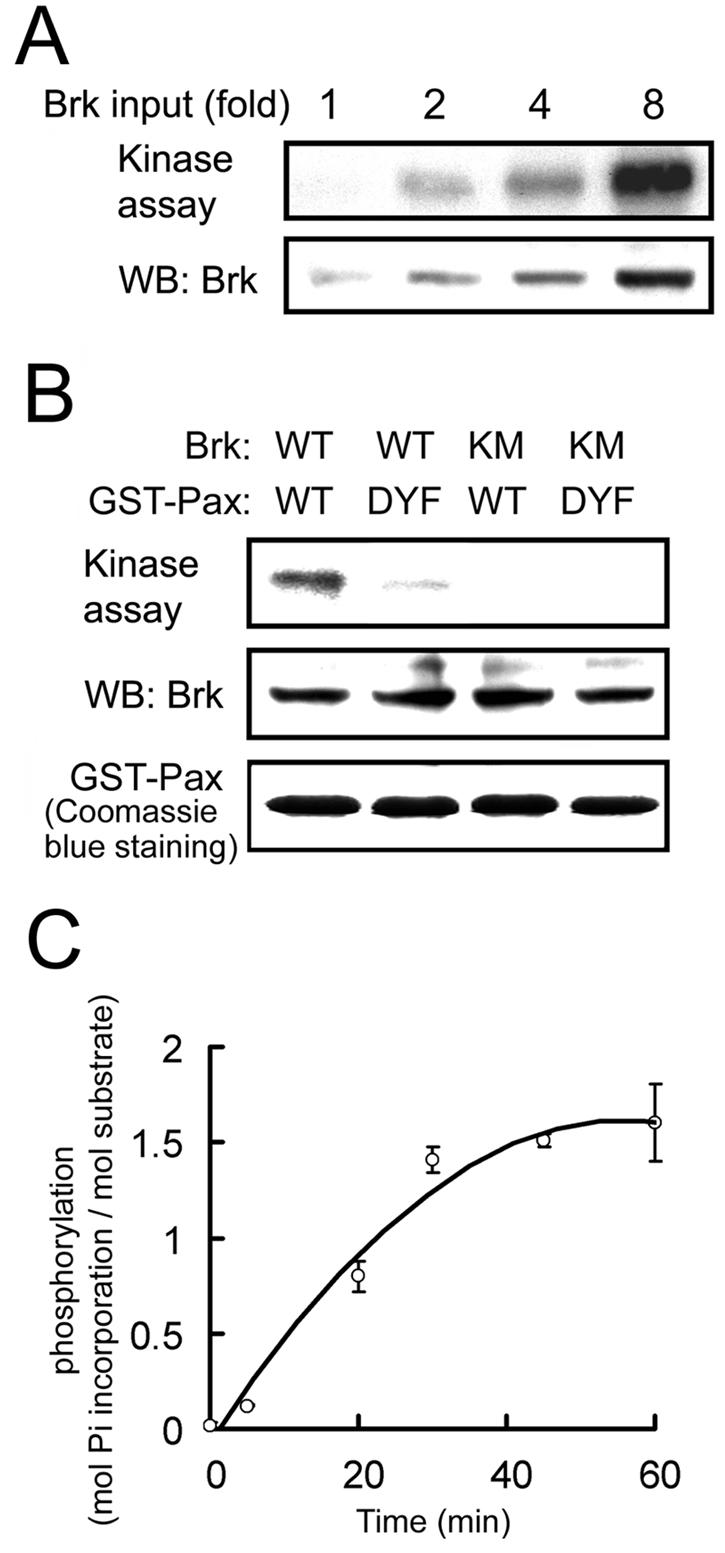
Brk directly phosphorylates paxillin at Y31 and Y118. (A) Equal amounts of GST-paxillin purified from bacteria were incubated with increasing amounts of purified baculovirus-expressed Brk in in vitro kinase reactions. The reaction products were analyzed by autoradiography to detect GST-paxillin phosphorylation (upper panel) or by Western blotting with anti-Brk antibody (bottom panel). (B) GST-paxillin or GST-paxillinDYF mutant was phosphorylated by Brk or BrkKM as described for panel A. The equal input of GST fusion proteins is shown on the bottom. (C) The stoichiometry of GST-paxillin phosphorylation by Brk was determined as described in Materials and Methods. The data shown are means ± the standard deviations from three independent experiments.
Brk promotes Rac1 activation.
Phosphorylation of paxillin at Y31 and Y118 was reported to create binding sites for the SH2 domain of adaptor protein CrkII (42, 47), thereby promoting cell migration (42). CrkII mediates adhesion and growth factor-induced lamellipodia formation and cell migration through a Rac1-dependent manner (12, 15, 21), and a CrkII effector, DOCK180, was recently identified as a novel guanine nucleotide exchange factor for Rac1 (9). On the basis of these previous studies, together with our finding that Brk is a paxillin Y31/118 kinase, we sought to investigate whether Brk could promote Rac1 activation and, if so, whether this Rac1 activation involves paxillin phosphorylation at Y31 and Y118. We found that overexpression of Brk, but not BrkKM, in Cos-1 cells indeed induced a 2.3-fold elevation of Rac1 activity, as determined by GST-PAK-CRIB pull-down analysis (Fig. 4A). This activation of Rac1, however, was completely abolished by coexpression of Brk with the paxillinDYF mutant but not with the wild-type paxillin, indicating that this mutant could inhibit Brk-induced Rac1 activity in a dominant-negative fashion. To further investigate the involvement of CrkII in this pathway, two dominant-negative mutants of CrkII were generated. The CrkIIR38K is an SH2 domain mutant that interrupts the binding to its SH2 domain partner, including Y31/118-phosphorylated paxillin (32, 42), whereas the SH3-domain mutant CrkIIW170K interferes with the binding of CrkII to its SH3 domain effector molecules (14). As shown in Fig. 4B, introduction of either mutant was capable of attenuating Brk-induced Rac1 activation. The smaller effect elicited by the W170K mutant might be due to its lower expression level than the CrkIIR38K mutant. Together, our data revealed that Brk promotes Rac1 activation through its phosphorylation of paxillin and subsequent recruitment of CrkII.
FIG. 4.
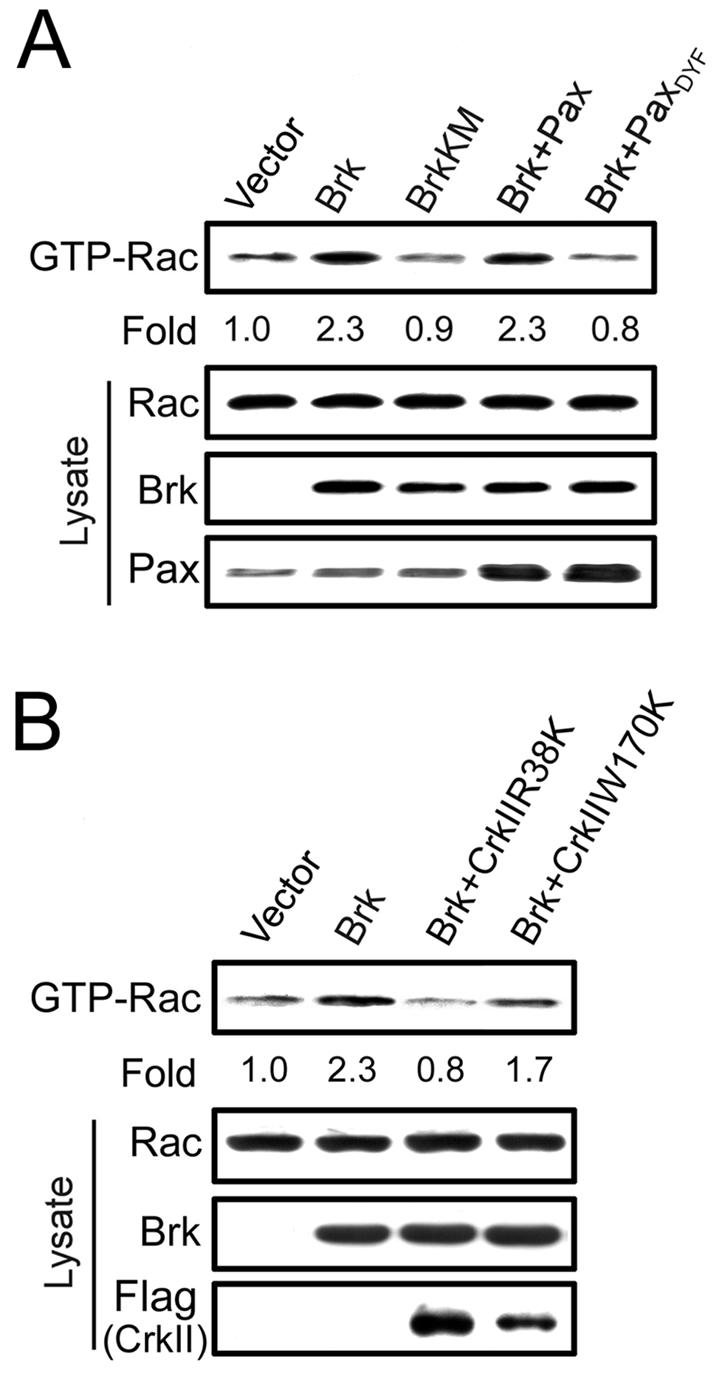
Brk promotes Rac1 activation through paxillin phosphorylation and CrkII adaptor function. (A and B) Cos-1 cells transfected with myc-Brk, myc-BrkKM, paxillin, paxillinDYF, and/or Flag-CrkII mutant constructs as indicated were serum starved and then lysed. The amount of GTP-bound Rac1 in cell lysates was determined by GST-PAK-CRIB pull-down analysis, followed by Western blotting with anti-Rac1 antibody. The expression levels of Rac1, Brk, paxillin, or their mutants in cell lysates were determined by Western blotting (bottom panels). The amounts of GTP-bound Rac1 were normalized by using those of total Rac1 protein present in cell lysates and are expressed as the fold induction relative to cells transfected with the control vector.
Brk is activated by EGF signaling.
Having identified a downstream signaling pathway for Brk, we next explored its upstream activator. Since Brk was reported to associate with EGF receptor and potentiate certain EGF-elicited signaling pathway and biological effects (19, 20), we investigated whether the kinase activity of Brk could be stimulated by EGF. Brk activity was determined by its autophosphorylation in the in vitro kinase assay, which was previously demonstrated as an eligible method for measuring Brk catalytic activity (43). When quiescent Cos-1 cells transfected with Flag-Brk were stimulated with EGF, we observed a time-dependent activation of this Flag-Brk. The catalytic activity of Flag-Brk was highest at 15 min after the addition of EGF and then gradually declined (Fig. 5A). The kinase-dead BrkKM did not show any autophosphorylation signal, indicating that the phosphorylation signal observed was specific to Brk. In contrast to EGF, integrin signaling did not significantly affect Brk catalytic activity, as determined by replating quiescent Cos-1 cells on fibronectin under serum-starved conditions (Fig. 5B). Thus, our results demonstrate that EGF, but not integrin, acted as an upstream activating signal for Brk.
FIG. 5.

Brk is activated by EGF signaling and is a mediator of EGF-induced paxillin phosphorylation and Rac1 activation. (A) Cos-1 cells transfected with Flag-Brk or Flag-BrkKM were serum starved and then stimulated with 15 ng of EGF/ml for various time points. The Flag-Brk or Flag-BrkKM was precipitated from cell lysates and subjected to in vitro kinase assays to detect Brk autophosphorylation. An equal input of Brk is shown in the bottom panel. (B) Cos-1 cells transfected with Flag-Brk were serum starved and then reseeded on plates coated with 1 μg of fibronectin/ml for various times as indicated. Cells were harvested to determine Brk catalytic activity as described for panel A. (C) EGF induces paxillin tyrosine phosphorylation in Cos-1 cells reseeded on fibronectin. Serum-starved Cos-1 cells were detached from culture plates, kept in suspension for 30 min, and then reseeded on plates coated with 1 μg of fibronectin/ml for 3 h. The cells were then treated with (+) or without (−) 15 ng of EGF/ml for various times as indicated. Paxillin tyrosine phosphorylation was analyzed as described for Fig. 1. (D and E) Brk activity is critical for EGF-induced paxillin tyrosine phosphorylation. Cos-1 cells transfected with BrkKM or a control vector were serum starved and reseeded on fibronectin as described for panel C and then treated with 15 ng of EGF/ml for 15 min. Paxillin tyrosine phosphorylation (D) and phosphorylation at Y31 and Y118 (E) were detected as described in Fig. 1. (F) Brk and paxillin phosphorylation are important for EGF-induced Rac1 activation. Cos-1 cells transfected with BrkKM, paxillin, paxillinDYF mutant, or a control vector were starved, reseeded, and stimulated with EGF as in panel D. GTP-bound Rac1 was detected and then calculated as the induction relative to unstimulated cells as described for Fig. 4. The expression levels of various proteins in cell lysates were determined by Western blotting (bottom panels).
Brk mediates EGF-induced paxillin phosphorylation and Rac1 activation.
We next explored the possible connection between the upstream activator (EGF) and downstream signaling molecules (paxillin and Rac1) of Brk. Despite the activation of Rac1 upon EGF stimulation is well documented (58), the role of EGF in paxillin tyrosine phosphorylation is controversial. Both induction (25) and reduction (28) of tyrosine phosphorylation on paxillin have been reported upon EGF stimulation. In the latter case, dephosphorylation of paxillin is attributed to the rapid downregulation of FAK activity, which can be overcome by replating cells on fibronectin (28). To avoid the complications resulting from FAK downregulation, we studied the role of EGF in paxillin tyrosine phosphorylation on reseeded cells. When Cos-1 cells reseeded on fibronectin for 3 h were treated with EGF, we observed a time-dependent induction of paxillin tyrosine phosphorylation (Fig. 5C). This increase occurred within 5 min and reached the maximal level at 15 min after the addition of EGF. In the control population reseeded on fibronectin but without receiving EGF, the paxillin phosphorylation level was not altered, indicating that the induction of paxillin phosphorylation observed in EGF-treated cells was due to the effect of EGF. This EGF-induced paxillin phosphorylation, however, was completely inhibited by expression of the BrkKM mutant (Fig. 5D). Using the phosphorylation site-specific antibodies, we found that both Y31 and Y118 phosphorylation were induced by EGF and that expression of BrkKM significantly blocked such effect of EGF (Fig. 5E). We also noted that BrkKM did not nonspecifically block the kinase activity of related Src kinase, as determined by Src autophosphorylation at Y418 (unpublished data), indicating that the results obtained with BrkKM are due to its inhibition of endogenous Brk. Thus, our data revealed a novel role of Brk in EGF-induced paxillin phosphorylation at Y31 and Y118, which prompted us to further investigate the role of Brk in EGF-induced Rac1 activation. EGF treatment of reseeded Cos-1 cells led to a 3.3-fold increase in Rac1 activity. However, in cells overexpressing BrkKM or paxillinDYF mutant, but not wild-type paxillin, this EGF-induced Rac1 activity was greatly downregulated (Fig. 5F). Together, our results indicate that Brk is a mediator of EGF-induced paxillin phosphorylation and Rac1 activation.
Brk is colocalized with paxillin at the leading cell edges of migratory cells.
The identification of Brk as a paxillin kinase and a mediator of EGF-induced paxillin phosphorylation prompted us to examine whether Brk could colocalize with paxillin in any subcellular compartment. Paxillin is a focal adhesion protein and recruited to the leading cell edges upon the initiation of migration (24, 46). Furthermore, the phosphorylation of paxillin at Y31 and Y118 is highly augmented during cell migration, and this phosphorylated protein is present at the leading cell edges of the migrating epithelial cells (41). To monitor the subcellular localization of Brk during cell migration, confluent A431 cells were induced for migration by scrapping to generate a cell-free zone, as used in the “wound healing” motility assay. Immediately after wounding, endogenous paxillin was mainly localized in the cytoplasm, whereas endogenous Brk was distributed in both the cytoplasm and the nucleus (Fig. 6A). At 2 h after wounding, membrane ruffles prevailed at the leading edges, indicating the highly migratory behavior of the cells. Importantly, at this time we observed an accumulation of Brk to the membrane ruffles, where it was almost completely colocalized with paxillin. The Brk image also partially merged with that of Y31/118-phosphorylated paxillin at the cell periphery at this time point (Fig. 6B). At 6 h after wounding, certain cells formed prominent focal adhesions at the cell periphery. Notably, Brk was relocalized back to the cytoplasm and nucleus and was virtually absent at these focal adhesions (Fig. 6A). This dynamic distribution of Brk and its colocalization with paxillin not only further strengthens the relationship between the two proteins but also implies a possible role of Brk in cell migration.
FIG. 6.

Brk and paxillin colocalize at membrane ruffles but not at focal adhesions in migratory cells. (A) Confluent A431 cells were scratched to induce wounds and fixed at various times after wounding. Endogenous Brk or paxillin was detected by immunofluorescence staining with anti-Brk (green) or antipaxillin (red) antibody. Green, red, and merged images are shown. At 2 h after wounding, cells displayed membrane ruffles, whereas certain cells at 6 h after wounding formed prominent focal adhesions (arrowheads). Confocal images are shown for cells at 0 and 2 h after wounding, whereas cells at 6 h after wounding were examined by fluorescence microscopy to visualize focal adhesions. The large arrows indicate the direction of the wounds. (B) A431 cells stably expressing myc-Brk were scratched to induce migration as for panel A and then double stained with anti-myc and anti-pY31/118 paxillin (1:1 mixture of the pY31-paxillin and pY118-paxillin antibodies). Confocal images were captured and analyzed as in panel A. (C) Serum-starved A431 cells were stimulated with EGF for indicated times. The locations of endogenous paxillin and Brk were detected by immunostaining as in panel A and examined by confocal microscopy.
Next, we investigated the subcellular localization of Brk during EGF-induced cell motility. To address this issue, the human epidermoid carcinoma A431 cells were used, since such cells are highly responsive to EGF-stimulated chemotactic migration (29) and express a high level of Brk (Fig. 7B.) In quiescent A431 cells, paxillin was mainly localized in the cytoplasm, whereas Brk was diffusively distributed in the nucleus and cytoplasm (Fig. 6C). EGF treatment rapidly induced the formation of lamellipodia and membrane ruffles (Fig. 6C), characteristics of migratory cells. Under this circumstance, Brk and paxillin could both be found at ruffles and colocalized with each other. This finding suggests a role of Brk in phosphorylating paxillin at membrane ruffles during EGF-stimulated motility event.
FIG. 7.

Brk promotes cell migration and mediates EGF-induced motility through phosphorylating paxillin at Y31 and Y118. (A) Cos-1 cells cotransfected with Brk, BrkKM, or a control vector, together with GFP (10:1 ratio), were plated onto Transwell chambers and assayed for migration into lower chamber containing 0 (−EGF) or 15 ng of EGF/ml (left panel) or different concentrations of EGF as indicated (right panel). After 4 h for migration, the GFP-positive cells that migrated to the lower side of the chamber were counted. For each well, five randomly selected fields were analyzed, and the data shown are means ± standard deviations of triplicate experiments (∗, P < 0.005; ∗∗, P < 0.0005 [compared to the control vector]). (B) The expression levels of endogenous Brk in A431 and MDA-MB231 cells were assessed by Western blotting. (C and D) A431 (C) and MDA-MB231 (D) cells were infected with various recombinant retroviruses as indicated. Infected cells were selected by using puromycin and/or hygromycin and then assayed for basal migration (−EGF) or chemotaxis migration toward 1.5 ng/ml (for A431 cells) or 15 ng/ml (for MDA-MB231 cells) of EGF. After 3 h (for A431 cells) or 5 h (for MDA-MB231 cells), the cells that migrated to the lower side of the chamber were counted, and values were calculated as for panel A. The expression levels of endogenous and exogenous Brk, Flag-paxillin, and their mutants are shown in the bottom panels.
Brk promotes cell migration and invasion and mediates a EGF-induced migratory event.
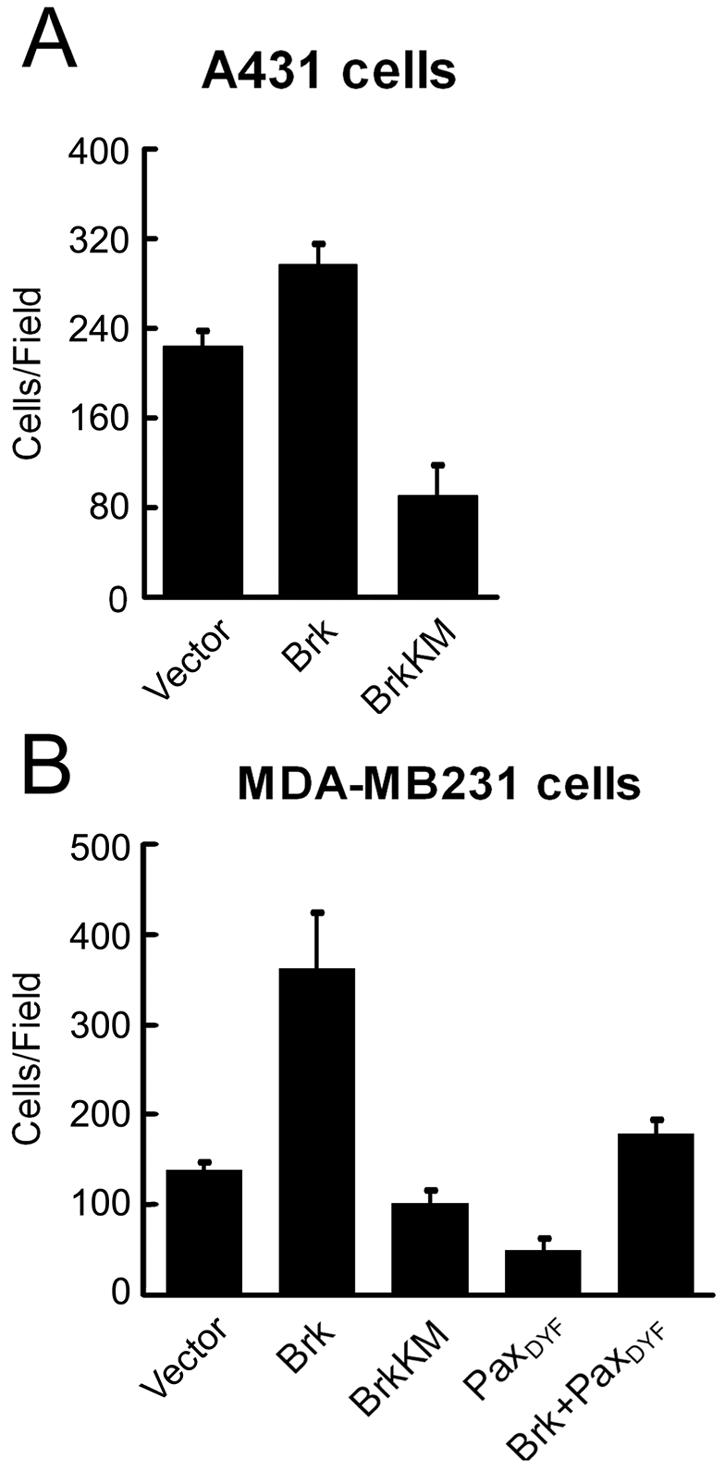
The finding that Brk promotes Rac1 activation by phosphorylating paxillin, combined with its recruitment to membrane ruffles in migratory cells, points out that it may function as a migration-promoting factor. Furthermore, the ability of Brk to mediate EGF-induced paxillin phosphorylation and Rac1 activation predicts an involvement of Brk in EGF-induced motility. To assess these possibilities, Cos-1 cells were cotransfected with Brk (or BrkKM) and GFP, and the chemotaxis effect of the GFP-positive transfectants toward EGF was monitored by Transwell analysis. As shown in Fig. 7A (left panel), BrkKM greatly impaired EGF-induced motility without affecting basal motility, whereas overexpression of Brk promoted chemotaxis toward EGF. In fact, the ability of Brk to enhance chemotaxis was observed at all EGF concentrations tested (from 0.15 to 15 ng/ml), whereas BrkKM displayed an inhibitory role in EGF-induced motility, which was most evident at a high concentration of EGF (Fig. 7A, right panel). To demonstrate that the role of Brk in EGF-induced motility is not a cell-type-specific effect, we used the human epidermoid carcinoma A431 cells. Brk and BrkKM were stably introduced into A431 cells by retrovirus-mediated gene transfer. Transwell motility assay revealed that expression of BrkKM in this cell line significantly reduced chemotaxis toward EGF (Fig. 7C). However, overexpression of Brk only modestly promoted the motility of A431 cells, most likely due to the high endogenous level of Brk (Fig. 7B). To demonstrate a greater effect of Brk ectopic expression on cell motility, we sought to express Brk in cells that do not contain a high level of endogenous Brk. The breast carcinoma cell line MDA-MB231, which expresses a low level of Brk (Fig. 7B) (3, 34), was infected with recombinant retrovirus expressing Brk, paxillin, and/or their mutants. Overexpression of Brk into these cells indeed markedly induced chemotaxis motility toward EGF and, furthermore, this induction of cell migration was efficiently blocked by coexpression of the paxillinDYF mutant (Fig. 7D). Together, these results indicate that Brk is able to promote cell migration through phosphorylating paxillin and is responsible for EGF-induced migratory effect.
In addition to Transwell motility analysis, a wound-healing assay was performed as an alternative method for monitoring cell migration. In this assay, Cos-1 cells cotransfected with GFP and BrkKM (or a control vector) were grown to confluent and then wounded by scratching. Cell migration toward the wound was recorded by time-lapse video microscopy, and cells expressing BrkKM or the control vector were traced by monitoring their GFP fluorescence. At 16 h after wounding, Cos-1 cells transfected with a control vector were capable of moving into the center of the wound. However, cells expressing BrkKM failed to migrate into the center of the wound at this time point (Fig. 8). A similar reduction in wound-induced migratory event was seen in HeLa cells expressing BrkKM (data not shown). To demonstrate a physiological role of Brk in cell migration, we assessed whether knockdown of the endogenous Brk would impair motility. Using a Brk-specific siRNA, but not a control siRNA, we were able to reduce the expression of Brk in HeLa cells to 15% of the endogenous level (Fig. 9A). When these siRNA-treated cells and parental HeLa cells were tested for EGF-induced chemotaxis by using Transwell assays, we observed a nearly complete inhibition of EGF-induced motility in cells receiving Brk siRNA compared to those treated with a control siRNA or without siRNA (Fig. 9B). In addition, these Brk siRNA-treated cells displayed a significant reduction in wound-induced migration (Fig. 9C). Together, the results not only provide additional evidence for the role of Brk in motility but also demonstrate a physiological significance of this effect of Brk.
FIG. 8.

Time-lapse monitoring of the wound-healing migration of cells expressing BrkKM. Cos-1 cells cotransfected with GFP and BrkKM or a control vector (ratio of 1:10) were cultured to confluence in growth medium and then scratched. Cell migration into wounds was monitored by time-lapse videomicroscopy. Still images were captured at the indicated time after wounding. Cells carrying BrkKM or control vector were indicated by their GFP fluorescence. The time after scratching is shown on the left.
FIG. 9.

siRNA-mediated reduction of Brk interferes with cell migration. (A) HeLa cells transfected with or without Brk siRNA or a control siRNA were lysed at 72 h after transfection and then assayed for the expression of Brk by Western blotting. (B) Cells as described for panel A were assayed for chemotaxis migration toward 15 ng of EGF/ml. After 7 h, cells that migrated to the lower side of the chamber were counted and calculated. (C) Cells as in panel A were assayed for wound-healing migration as described for Fig. 8. The percentages of wound closure at 24 h after wounding are indicated on the bottom.
The capability of Brk to stimulate cell migration predicts a role in promoting cell invasion. Indeed, in the presence of EGF, invasion of A431 cells through Matrigel was markedly reduced by expression of BrkKM, whereas the ectopic expression of Brk modestly enhanced invasion (Fig. 10A). Furthermore, similar to what was observed with the migration assay, expression of Brk in MDA-MB231 cells greatly promoted invasion, and this effect was blocked by the coexpression of paxillinDYF mutant (Fig. 10B). The present study thus identifies a novel function of Brk in promoting cell migration and invasion, which likely plays a significant role in the malignant progression.
FIG. 10.
Brk promotes cell invasion. A431 (A) and MDA-MB231 (B) cells stably expressing various proteins as indicated were assayed for invasion through Matrigel as described in Materials and Methods. The data shown are means ± standard deviations of triplicate experiments.
DISCUSSION
We have identified here not only the upstream regulator and downstream effector for the nonreceptor tyrosine kinase Brk but also a novel signaling pathway through which Brk mediates EGF-induced phosphorylation of paxillin. Since both EGF and Y31/118-phosphorylated paxillin display a migration-promoting effect (39, 42), our findings suggest a role for Brk in migration. Indeed, Brk is capable of stimulating migration and invasion and is critical for mediating EGF-induced cell motility. Based on the findings presented here, we postulated a model that depicts the signaling pathway through which Brk mediates EGF-induced cell motility and invasion (Fig. 11). Upon EGF stimulation, Brk is recruited to the EGF receptor (19), and this event activates the kinase activity of Brk, presumably due to a conformational change. The activated Brk in turn phosphorylates paxillin at Y31 and Y118 to create docking sites for the adaptor protein CrkII. CrkII subsequently activates Rac1 and promotes lamellipodium formation, cell motility, and invasion. Several lines of evidence presented here support a role for Brk in this signaling pathway. First, overexpression of Brk promotes paxillin phosphorylation and Rac1 activation in the absence of EGF and enhances EGF-induced cell migration and invasion. Second, overexpression of Brk dominant-negative mutant inhibits all of these downstream signaling events in response to EGF and prevents membrane protruding and ruffling during cell migration. Third, dominant-negative interference of either paxillin or CrkII blocks Brk-induced Rac1 activation. Fourth, siRNA-mediated downregulation of Brk leads to a great suppression in cell migration. Finally, Brk is recruited to the membrane ruffles in migratory cells, where both paxillin phosphorylation and Rac1 activation occurred (41). Our discovery of the participation of Brk in this EGF-induced motility pathway suggests an important role for this kinase in promoting the migration and invasion of tumors with an aberrant activation of the EGF pathway. Accordingly, Brk overexpression is frequently observed in breast cancer cells (3), in which aberrant activation of the EGF family receptor HER2 is prevalent (33, 51). We hypothesize that Brk overexpression in these tumors would synergize the EGF motility effect, thereby promoting tumor metastasis.
FIG. 11.
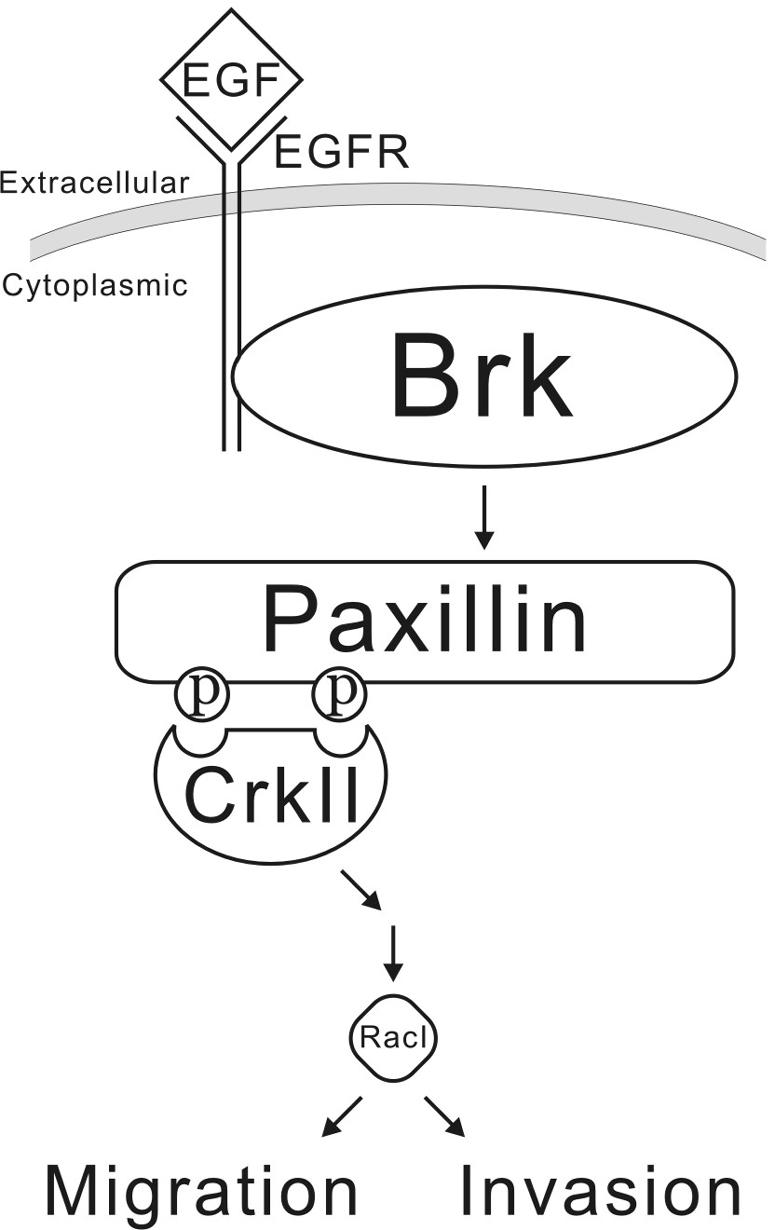
Model illustrating the signaling pathway through which Brk participates in EGF-induced cell migration and invasion.
The adaptor function of CrkII was reported to be critical for EGF-induced activation of Rac-JNK, since deletion of either SH2 or SH3 domain of CrkII blocks this pathway (12). CrkII promotes Rac1 activation, presumably through its SH3 domain binding partner DOCK180, which was recently identified as a two-component Rac1 exchange factor through its interaction with ELMO (15). In addition, the coupling of CrkII with phosphorylated paxillin promotes the assembly of paxillin/GIT2/β-PIX complex (23), in which β-PIX functions as a Rac1 exchange factor (2, 30). In the present study, we have demonstrated that Brk acts upstream of paxillin-CrkII complex in mediating EGF-induced Rac1 activation. However, many other signaling molecules and pathways have been demonstrated or proposed for the EGF-induced activation of Rac1 and also for the accompanying lamellipodium formation and membrane ruffling, such as Vav2, Ras, Abl-Sos-1, PLCγ, and PI 3-kinase (4, 31, 49, 50, 52). Therefore, it is likely that multiple pathways elicited by EGF converge on the Rac1 activation, thereby promoting cell morphogenesis and motility. Consistent with this notion, we show that blockage of Brk activity or paxillin phosphorylation significantly reduced but did not completely inhibit EGF-induced Rac1 activity.
Our study identified paxillin as a binding partner and a direct substrate of Brk. The association of Brk and paxillin is complex and involves the SH2 and SH3 domains of Brk and the N-terminal and C-terminal regions of paxillin. Notably, the SH2 and SH3 domains of Brk are also involved in the interaction with another Brk substrate, Sam68 (11), and similar substrate binding via SH2 and SH3 domains has been reported for Src family kinases (8). To date, only a few tyrosine kinases have been implicated in regulating the phosphorylation of paxillin (46). FAK and its related protein CAKβ/Pyk2 are the two major players controlling the tyrosine phosphorylation of paxillin (1, 47). Similar to Brk, these two kinases physically associate with paxillin and phosphorylate paxillin at Y31 and Y118 (1, 48). However, Brk differs from FAK in certain aspects. First, Brk is recruited to membrane ruffles but not focal adhesions in migratory cells, whereas FAK is a major component of focal adhesions. Second, FAK is activated in response to both growth factors and integrin signaling (48), whereas the catalytic activity of Brk is not significantly regulated by integrin-mediated cell adhesions. These divergences of Brk with FAK predict a more restricted role of Brk in regulating growth factor (e.g., EGF)-induced motility rather than integrin-mediated cell adhesions. Consistent with this notion, Brk overexpression did not significantly affect the adhesion of Cos-1 cell on fibronectin (data not shown).
Phosphorylation of paxillin at Y31 and Y118 is reported to promote migration (42) and affect focal adhesion dynamics (60). Mutation of these two tyrosine residues significantly decreases the disassembly rate of focal adhesions (60) and leads to the formation of large focal adhesion structures at the cell periphery (41). In addition to serving as a binding site for CrkII, Y31/118-phosphorylated paxillin is capable of recruiting p120RasGAP at the cell periphery, thereby releasing its binding partner p190RhoGAP to downregulate RhoA (55). This pathway is necessary for efficient membrane ruffling and spreading during cell migration (55). We hypothesize that Brk-induced paxillin phosphorylation might similarly decrease RhoA activity at the cell periphery, since inhibition of Brk activity in Cos-1 cells prevents membrane ruffling and extension in response to migratory cues (data not shown). The effect of Y31/118 phosphorylation on cell migration, however, seems to be context dependent (61). Since Y31/118-phosphorylated paxillin is present in both focal adhesions and leading cell edges, it has been proposed that this context-dependent migratory effect may have resulted from the distinct downstream signaling pathways engaged in the differentially localized phosphorylated paxillin (55). Consistent with this notion, the recruitment of p120RasGAP occurs only at leading cell edge but not at focal adhesions (55). In this regard, periphery-localized rather than focal-adhesion-localized Brk might selectively activate downstream pathways that promote cell migration by phosphorylating paxillin only at the cell periphery.
The details of the mechanism by which EGF induces the catalytic activity of Brk are currently unknown. Recent studies have revealed that, similar to Src family kinases, Brk activity is negatively regulated by intramolecular interactions, which involve the binding of SH2 and SH3 domains to the autophosphorylated Y447 residue and a proline-rich linker region, respectively (43, 44). Disruption of these interactions by binding to a synthetic SH2 or SH3 ligand leads to Brk activation (44). Since EGF stimulation induces the interaction of Brk with EGF receptor (19), presumably via the SH2 domain of Brk, this intermolecular interaction might relieve the intramolecular constraint, thereby activating Brk catalytic activity. Regardless of the specific mechanism, our finding that Brk is a downstream target of EGF is consistent with previous findings that Brk is able to enhance certain biological functions and signaling events of EGF (19, 20).
Overexpression of Brk has been found in >60% of breast carcinoma tissues and in several other cancer types (3, 13, 27, 34, 36), suggesting a role for Brk in tumorigenesis. Furthermore, during the progression of prostate cancers, Brk translocation from the nucleus to the cytoplasm was observed (10), implying that Brk might facilitate tumor progression by phosphorylating cytoplasmic proteins. Our finding that Brk phosphorylates the cytoplasm-localized paxillin, which in turn activates a signaling pathway to promote cell migration and invasion, correlates well with these expression data obtained from tumor cells and tissues. We hypothesize that Brk probably utilizes this pathway to participate in tumor progression and metastasis. In addition to promoting cell migration and invasion, Brk has also been reported to sensitize cells to the proliferation effect of EGF (20). Thus, Brk might elicit diverse biological effects, thereby participating in the distinct stages of tumorigenesis.
Acknowledgments
We thank Hsin-Ying Chen for contributions in the initial stage of this study, Tzuu-Shuh Jou for the GST-PAK-CRIB construct, and Tsun-Cheng Wang for the A431 cells.
This study was supported by grant NSC92-2312-B-002-005.
REFERENCES
- 1.Avraham, H., S. Y. Park, K. Schinkmann, and S. Avraham. 2000. RAFTK/Pyk2-mediated cellular signalling. Cell Signal 12:123-133. [DOI] [PubMed] [Google Scholar]
- 2.Bagrodia, S., S. J. Taylor, K. A. Jordon, L. Van Aelst, and R. A. Cerione. 1998. A novel regulator of p21-activated kinases. J. Biol. Chem. 273:23633-23636. [DOI] [PubMed] [Google Scholar]
- 3.Barker, K. T., L. E. Jackson, and M. R. Crompton. 1997. BRK tyrosine kinase expression in a high proportion of human breast carcinomas. Oncogene 15:799-805. [DOI] [PubMed] [Google Scholar]
- 4.Bar-Sagi, D., and A. Hall. 2000. Ras and Rho GTPases: a family reunion. Cell 103:227-238. [DOI] [PubMed] [Google Scholar]
- 5.Bellis, S. L., J. T. Miller, and C. E. Turner. 1995. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J. Biol. Chem. 270:17437-17441. [DOI] [PubMed] [Google Scholar]
- 6.Blume-Jensen, P., and T. Hunter. 2001. Oncogenic kinase signaling. Nature 411:355-365. [DOI] [PubMed] [Google Scholar]
- 7.Brown, M. C., J. A. Perrotta, and C. E. Turner. 1996. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J. Cell Biol. 135:1109-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown, M. T., and J. A. Cooper. 1996. Regulation, substrates and functions of src. Biochim. Biophys. Acta 1287:121-149. [DOI] [PubMed] [Google Scholar]
- 9.Brugnera, E., L. Haney, C. Grimsley, M. Lu, S. F. Walk, A. C. Tosello-Trampont, I. G. Macara, H. Madhani, G. R. Fink, and K. S. Ravichandran. 2002. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell Biol. 4:574-582. [DOI] [PubMed] [Google Scholar]
- 10.Derry, J. J., G. S. Prins, V. Ray, and A. L. Tyner. 2003. Altered localization and activity of the intracellular tyrosine kinase BRK/Sik in prostate tumor cells. Oncogene 22:4212-4220. [DOI] [PubMed] [Google Scholar]
- 11.Derry, J. J., S. Richard, Valderrama-Carvajal, H., X. Ye, V. Vasioukhin, A. W. Cochrane, T. Chen, and A. L. Tyner. 2000. Sik (BRK) phosphorylates Sam68 in the nucleus and negatively regulates its RNA binding ability. Mol. Cell. Biol. 20:6114-6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolfi, F., M. Garcia-Guzman, M. Ojaniemi, H. Nakamura, M. Matsuda, and K. Vuori. 1998. The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc. Natl. Acad. Sci. USA 95:15394-15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Easty, D. J., P. J. Mitchell, K. Patel, V. A. Florenes, R. A. Spritz, and D. C. Bennett. 1997. Loss of expression of receptor tyrosine kinase family genes PTK7 and SEK in metastatic melanoma. Int. J. Cancer 71:1061-1065. [DOI] [PubMed] [Google Scholar]
- 14.Feller, S. M. 2001. Crk family adaptors-signalling complex formation and biological roles. Oncogene 20:6348-6371. [DOI] [PubMed] [Google Scholar]
- 15.Gumienny, T. L., E. Brugnera, A. C. Tosello-Trampont, J. M. Kinchen, L. B. Haney, K. Nishiwaki, S. F. Walk, M. E. Nemergut, I. G. Macara, R. Francis, T. Schedl, Y. Qin, L. Van Aelst, M. O. Hengartner, and K. S. Ravichandran. 2001. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107:27-41. [DOI] [PubMed] [Google Scholar]
- 16.Hagel, M., E. L. George, A. Kim, R. Tamimi, S. L. Opitz, C. E. Turner, A. Imamoto, and S. M. Thomas. 2002. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell. Biol. 22:901-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanks, S. K., and T. R. Polte. 1997. Signaling through focal adhesion kinase. Bioessays 19:137-145. [DOI] [PubMed] [Google Scholar]
- 18.Ishibe, S., D. Joly, X. Zhu, and L. G. Cantley. 2003. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol. Cell 12:1275-1285. [DOI] [PubMed] [Google Scholar]
- 19.Kamalati, T., H. E. Jolin, M. J. Fry, and M. R. Crompton. 2000. Expression of the BRK tyrosine kinase in mammary epithelial cells enhances the coupling of EGF signalling to PI 3-kinase and Akt, via erbB3 phosphorylation. Oncogene 19:5471-5476. [DOI] [PubMed] [Google Scholar]
- 20.Kamalati, T., H. E. Jolin, P. J. Mitchell, K. T. Barker, L. E. Jackson, C. J. Dean, M. J. Page, B. A. Gusterson, and M. R. Crompton. 1996. Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. J. Biol. Chem. 271:30956-30963. [DOI] [PubMed] [Google Scholar]
- 21.Klemke, R. L., J. Leng, R. Molander, P. C. Brooks, K. Vuori, and D. A. Cheresh. 1998. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J. Cell Biol. 140:961-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klinghoffer, R. A., C. Sachsenmaier, J. A. Cooper, and P. Soriano. 1999. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18:2459-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamorte, L., S. Rodrigues, V. Sangwan, C. E. Turner, and M. Park. 2003. Crk associates with a multimolecular paxillin/GIT2/beta-PIX complex and promotes Rac-dependent relocalization of paxillin to focal contacts. Mol. Biol. Cell 14:2818-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laukaitis, C. M., D. J. Webb, K. Donais, and A. F. Horwitz. 2001. Differential dynamics of alpha 5 integrin, paxillin, and alpha-actinin during formation and disassembly of adhesions in migrating cells. J. Cell Biol. 153:1427-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leduc, I., and S. Meloche. 1995. Angiotensin II stimulates tyrosine phosphorylation of the focal adhesion-associated protein paxillin in aortic smooth muscle cells. J. Biol. Chem. 270:4401-4404. [DOI] [PubMed] [Google Scholar]
- 26.Lewis, J. M., and M. A. Schwartz. 1998. Integrins regulate the association and phosphorylation of paxillin by c-Abl. J. Biol. Chem. 273:14225-14230. [DOI] [PubMed] [Google Scholar]
- 27.Llor, X., M. S. Serfas, W. Bie, V. Vasioukhin, M. Polonskaia, J. Derry, C. M. Abbott, and A. L. Tyner. 1999. BRK/Sik expression in the gastrointestinal tract and in colon tumors. Clin. Cancer Res. 5:1767-1777. [PubMed] [Google Scholar]
- 28.Lu, Z., G. Jiang, P. Blume-Jensen, and T. Hunter. 2001. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell. Biol. 21:4016-4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malliri, A., M. Symons, R. F. Hennigan, A. F. Hurlstone, R. F. Lamb, T. Wheeler, and B. W. Ozanne. 1998. The transcription factor AP-1 is required for EGF-induced activation of rho-like GTPases, cytoskeletal rearrangements, motility, and in vitro invasion of A431 cells. J. Cell Biol. 143:1087-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manser, E., T. H. Loo, C. G. Koh, Z. S. Zhao, X. Q. Chen, L. Tan, I. Tan, T. Leung, and L. Lim. 1998. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell 1:183-192. [DOI] [PubMed] [Google Scholar]
- 31.Marcoux, N., and K. Vuori. 2003. EGF receptor mediates adhesion-dependent activation of the Rac GTPase: a role for phosphatidylinositol 3-kinase and Vav2. Oncogene 22:6100-6106. [DOI] [PubMed] [Google Scholar]
- 32.Matsuda, M., S. Nagata, S. Tanaka, K. Nagashima, and T. Kurata. 1993. Structural requirement of CRK SH2 region for binding to phosphotyrosine-containing proteins: evidence from reactivity to monoclonal antibodies. J. Biol. Chem. 268:4441-4446. [PubMed] [Google Scholar]
- 33.McCann, A. H., P. A. Dervan, M. O'Regan, M. B. Codd, W. J. Gullick, B. M. Tobin, and D. N. Carney. 1991. Prognostic significance of c-erbB-2 and estrogen receptor status in human breast cancer. Cancer Res. 51:3296-3303. [PubMed] [Google Scholar]
- 34.Meric, F., W. P. Lee, A. Sahin, H. Zhang, H. J. Kung, and M. C. Hung. 2002. Expression profile of tyrosine kinases in breast cancer. Clin. Cancer Res. 8:361-367. [PubMed] [Google Scholar]
- 35.Minoguchi, M., S. Minoguchi, D. Aki, A. Joo, T. Yamamoto, T. Yumioka, T. Matsuda, and A. Yoshimura. 2003. STAP-2/BKS, an adaptor/docking protein, modulates STAT3 activation in acute-phase response through its YXXQ motif. J. Biol. Chem. 278:11182-11189. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell, P. J., K. T. Barker, J. E. Martindale, T. Kamalati, P. N. Lowe, M. J. Page, B. A. Gusterson, and M. R. Crompton. 1994. Cloning and characterization of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene 9:2383-2390. [PubMed] [Google Scholar]
- 37.Mitchell, P. J., K. T. Barker, J. Shipley, and M. R. Crompton. 1997. Characterization and chromosome mapping of the human nonreceptor tyrosine kinase gene, brk. Oncogene 15:1497-1502. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell, P. J., E. A. Sara, and M. R. Crompton. 2000. A novel adaptor-like protein which is a substrate for the non-receptor tyrosine kinase, BRK. Oncogene 19:4273-4282. [DOI] [PubMed] [Google Scholar]
- 39.Moulin, V. 1995. Growth factors in skin wound healing. Eur. J. Cell Biol. 68:1-7. [PubMed] [Google Scholar]
- 40.Nakamura, H. K., Yano, E. Schaefer, and H. Sabe. 2001. Different modes and qualities of tyrosine phosphorylation of Fak and Pyk2 during epithelial-mesenchymal transdifferentiation and cell migration: analysis of specific phosphorylation events using site-directed antibodies. Oncogene 20:2626-2635. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura, K., H. Yano, H. Uchida, S. Hashimoto, E. Schaefer, and H. Sabe. 2000. Tyrosine phosphorylation of paxillin alpha is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J. Biol. Chem. 275:27155-27164. [DOI] [PubMed] [Google Scholar]
- 42.Petit, V., B. Boyer, D. Lentz, C. E. Turner, J. P. Thiery, and A. M. Valles. 2000. Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J. Cell Biol. 148:957-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu, H., and W. T. Miller. 2002. Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J. Biol. Chem. 277:34634-34641. [DOI] [PubMed] [Google Scholar]
- 44.Qiu, H., and W. Miller.T. 2004. Role of the Brk SH3 domain in substrate recognition. Oncogene 23:2216-2223. [DOI] [PubMed] [Google Scholar]
- 45.Sander, E. E., J. P. ten Klooster, S. van Delft, R. A. van der Kammen, and J. G. Collard. 1999. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 147:1009-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaller, M. D. 2001. Paxillin: a focal adhesion-associated adaptor protein. Oncogene 20:6459-6472. [DOI] [PubMed] [Google Scholar]
- 47.Schaller, M. D., and J. T. Parsons. 1995. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol. Cell. Biol. 15:2635-2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schlaepfer, D. D., C. R. Hauck, and D. J. Sieg. 1999. Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 71:435-478. [DOI] [PubMed] [Google Scholar]
- 49.Scita, G., P. Tenca, E. Frittoli, A. Tocchetti, M. Innocenti, G. Giardina, and P. P. Di Fiore. 2000. Signaling from Ras to Rac and beyond: not just a matter of GEFs. EMBO J. 19:2393-2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sini, P., A. Cannas, A. J. Koleske, P. P. Di Fiore, and G. Scita. 2004. Abl-dependent tyrosine phosphorylation of Sos-1 mediates growth-factor-induced Rac activation. Nature Cell Biol. 6:268-274. [DOI] [PubMed] [Google Scholar]
- 51.Slamon, D. J., G. M. Clark, S. G. Wong, W. J. Levin, A. Ullrich, and W. L. McGuire. 1987. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177-182. [DOI] [PubMed] [Google Scholar]
- 52.Tamas, P., Z. Solti, P. Bauer, A. Illes, S. Sipeki, A. Bauer, A. Farago, J. Downward, and L. Buday. 2003. Mechanism of epidermal growth factor regulation of Vav2, a guanine nucleotide exchange factor for Rac. J. Biol. Chem. 278:5163-5171. [DOI] [PubMed] [Google Scholar]
- 53.Thomas, S. M., P. Soriano, and A. Imamoto. 1995. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature 376:267-271. [DOI] [PubMed] [Google Scholar]
- 54.Tsai, Y. T., Y. H. Su, S. S. Fang, T. N. Huang, Y. Qiu, Y. S. Jou, H. M. Shih, H. J. Kung, and R. H. Chen. 2000. Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Mol. Cell. Biol. 20:2043-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsubouchi, A., J. Sakakura, R. Yagi, Y. Mazaki, E. Schaefer, H. Yano, and H. Sabe. 2002. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell Biol. 159:673-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turner, C. E. 2000. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2:E231-E236. [DOI] [PubMed] [Google Scholar]
- 57.Turner, C. E., M. C. Brown, J. A. Perrotta, M. C. Riedy, S. N. Nikolopoulos, A. R. McDonald, S. Bagrodia, S. Thomas, and P. S. Leventhal. 1999. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J. Cell Biol. 145:851-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Aelst, L., and C. D'Souza-Schorey. 1997. Rho GTPases and signaling networks. Genes Dev. 11:2295-2322. [DOI] [PubMed] [Google Scholar]
- 59.Vernet, C., and K. Artzt. 1997. STAR, a gene family involved in signal transduction and activation of RNA. Trends Genet. 13:479-484. [DOI] [PubMed] [Google Scholar]
- 60.Webb, D. J., K. Donais, L. A. Whitmore, S. M. Thomas, C. E. Turner, J. T. Parsons, and A. F. Horwitz. 2004. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6:154-161. [DOI] [PubMed] [Google Scholar]
- 61.Yano, H., H. Uchida, T. Iwasaki, M. Mukai, H. Akedo, K. Nakamura, S. Hashimoto, and H. Sabe. 2000. Paxillin alpha and Crk-associated substrate exert opposing effects on cell migration and contact inhibition of growth through tyrosine phosphorylation. Proc. Natl. Acad. Sci. USA 97:9076-9081. [DOI] [PMC free article] [PubMed] [Google Scholar]