

Abstract
Objective(s):
Oxidative stress has been established as a key cause of alcohol-induced hepatotoxicity. Licochalcone B, an extract of licorice root, has shown antioxidative properties. This study was to investigate the effects and mechanisms of licochalcone B in ethanol-induced hepatic injury in an in vitro study.
Materials and Methods:
An in vitro model of Ethanol-induced cytotoxicity in BRL cells was used in this study. Cell injury was assessed using WST-1 assay and lactate dehydrogenase, alanine transaminase, and aspartate aminotransferase release assay. Cell apoptosis were quantified by flow cytometric analysis. The intracellular oxidative level was evaluated by reactive oxidative species, malondialdehyde and glutathione detection. Furthermore, the expression level of Erk, p-Erk, Nrf-2 were assessed using Western blot.
Results:
Treatment with ethanol induced marked cell injury and cell apoptosis in BRL cells. Licochalcone B significantly attenuated ethanol-induced cell injury, and inhibited cell apoptosis. Furthermore, licochalcone B significantly inhibited ethanol-induced intracellular oxidative level, upregulated the expression of p-Erk, and promoted nuclear localization of Nrf2. Additionally, this hepatoprotective role was significantly abolished by inhibition of Erk signaling. However, no apparent effects of Erk inhibition were observed on ethanol-induced hepatotoxicity.
Conclusion:
This study demonstrates that licochalcone B protects hepatocyte from alcohol-induced cell injury, and this hepatoprotective role might be attributable to apoptosis reduction, inhibition of oxidative stress, and upregulation of Erk–Nrf2. Therefore, licochalcone B might possess potential as a novel therapeutic drug candidate for alcohol-related liver disorders.
Keywords: Alcohol, BRL cells, Erk, Hepatotoxicity, Nrf, Oxidative stress
Introduction
Excessive alcohol intake is a major public health challenge worldwide (1, 2). As the liver is the main organ involved in metabolism of alcohol, alcohol abuse causes various liver disorders, such as alcoholic hepatitis, alcoholic fatty liver, alcoholic cirrhosis, and even hepatocellular carcinoma, posing substantial economic and social burdens worldwide (3-5). Previous investigations have established that oxidative stress is an important factor associated with alcohol-related liver disorders (6, 7). Both increased production of ROS and compromised antioxidative defenses have been observed after ethanol exposure. Ethanol induces mitochondria dysfunction, expression of CYP2E1, and expression of NAD(P)H oxidase (Nox), all of which lead to the increased production of ROS (7). In addition, decline of non-enzymatic antioxidant (like GSH) and enzymatic antioxidant (like SOD, catalase and GPx) has
been also reported in ethanol-related animal models (8, 9). Based on this evidence, drugs targeting antioxidative stress have been extensively investigated (10). However, although some drugs may show a potential hepatoprotective role, limited efficiency and lack of favorable therapeutic measures have remained huge challenges for both investigators and clinics. Therefore, it is critical to search for novel liver-protecting drugs and investigate the hepatoprotective mechanisms involved.
Licorice root, an ancient Chinese Medicine, has shown effective roles in liver disease in Chinese Traditional Medicine. Licochalcone is an extract of licorice root, and belongs to the flavonoid family, including licochalcone A and licochalcone B (11). Previous studies have thoroughly investigated the effects of licochalcone A, especially its notable antitumor effects via induction of apoptosis in hepatocellular carcinoma (12), gastric cancer (13), and colon cancer (14). However, no apparent liver-protecting role was observed for licochalcone A. A few studies have found that licochalcone B has potential as an antioxidant or free-radical scavenger (11, 15). Moreover, a licochalcone B derivative compound shows some anti-inflammatory effect in RAW264.7 cells (16). Han et al reported that licochalcone B protected isolated rat hearts from ischemia/reperfusion injury (17). However, in view of the potential antioxidative and anti-inflammatory role of licochalcone B, whether licochalcone B protects against alcohol-induced liver injury remains unexplored.
Signaling via the Erk pathway is known to mediate survival signals in hepatocytes (18). The hepatopro-tective role of Erk signaling has established in conditions of oxidant-mediated hepatocyte injury by H2O2 (19) as well as ethanol (20, 21). Nuclear factor erythroid 2 p45-related factor 2 (Nrf2), is a critical transcription factor regulating expression of various antioxidant and cytoprotective genes, and is known to protect hepatocytes from various types of oxidative injuries (7). Additionally, some studies have also identified Erk-Nrf2 signaling as a critical hepatopro-tective signaling in oxidant-induced hepatocyte injury (20, 22). However, whether Erk-Nrf2 signaling regulates the role of licochalcone B in ethanol-induced hepatocyte injury need to be elucidated.
In the present study, by using an ethanol-induced hepatocyte injury model in vitro, we investigated the potentially hepatoprotective role of licochalcone B and explored the mechanisms in which Erk-Nrf2 might be involved. The identification of the role of licochalcone B in ethanol-induced hepatocyte injury may lead to the development of novel drugs for alcohol-related liver disorders.
Materials and Methods
Cell culture
A rat hepatocyte cell line, BRL cells (purchased from the cell bank of Chinese Academy of Science, Shanghai, China), was cultured in complete medium (90% Dulbecco’s modified Eagle’s medium and 10% fetal bovine serum) in a humidified incubator (Thermo Scientific, Waltham, MA, USA) in 95% air and 5% CO2 at 37 °C.
Cell viability assay
After various treatments as indicated, the cell viability of BRL cells was evaluated by WST-1 assay (Roche, Mannheim, Germany), based on the manufacturer’s instructions. Briefly, after the medium was refreshed, the WST-1 solution was added at a concentration of 10 μl/well and, then, the cells were incubated for 3 hr in the incubator. Absorbance was determined using a microplate reader (BD, San Diego, CA, USA) at 450 nm. After subtracting the background value, cell viability was calculated as a percentage of the control group.
LDH, ALT, and AST release detection
For detecting cell injury, levels of lactate dehydrogenase (LDH; Roche, Mannheim, Germany), alanine transaminase (ALT), and aspartate aminotransferase (AST; Randox Laboratories, Antrim, UK) in the medium were assayed by following the manufacturer’s protocol. After respective treatments as indicated, the supernatant was collected for LDH, ALT, and AST assays. LDH release was expressed as a percentage of Max release, and the ALT and AST levels as folds of levels for the control group.
Flow cytometric analysis
Cell apoptosis was evaluated by Annexin V (AV)/propidium iodide (PI) staining (Roche, Mannheim, Germany) and flow cytometric analysis as previously described (23). Briefly, BRL cells were harvested and re-suspended in a binding buffer. Then, after double-staining with fluorescein isothiocyanate (FITC)-AV and PI dye, cells were analyzed by flow cytometer (FC 500 MCL; Beckman-Coulter, CA, USA) at 530 and 575 nm. Cells expressing AV+/PI-/+ staining were considered apoptotic, and the apoptosis rate was calculated as the apoptotic cell number/total number of cells.
Measurement of reactive oxygen species
After the medium was removed, BRL cells were rinsed 3 times with phosphate-buffered saline (PBS) and incubated with a reactive oxygen series (ROS) indicator (DCF-HA 10 μM in HBSS) for 30 min in a light resistant incubator at 37 °C. Relative fluorescence values were determined by a fluorescence microplate (BD, San Diego, CA, USA) at 488/525 nm.
Detection of lipid peroxidation and glutathione
Lipid peroxidation was measured via detection of malondialdehyde (MDA) levels in the cell lysate using a commercialized kit (Beyotime, Shanghai, China) as specified by the manufacturer. Briefly, 100 μl MDA work buffer was added to 50 μl cell lysate supernatant; 15 min after the reaction, absorbance values were detected on a fluorescence microplate at 532 nm. Glutathione (GSH) levels was assayed using a GSH kit (Beyotime, Shanghai, China) as per manufacturer’s instructions. After removal of protein with a protein removal buffer, the non-protein supernatant was used for GSH detection using a GSH work buffer; 25 min after the reaction, absorbance values were detected on a fluorescence microplate at 412 nm.
Protein extraction and Western blot analysis
The proteins were harvested for Western blot (WB) assay using radioimmunoprecipitation assay (RIPA) lysis supplemented with a protease and phosphatase inhibitor cocktail (Roche, Mannheim, Germany), after the respective treatments. Nucleoprotein was extracted using NE-PER™ Nuclear Extraction Reagents (Thermo Scientific, Waltham, MA, USA) in accordance with the manufacturer’s instructions, and protein concentration was determined. Then, 30 μg of protein was separated by electrophoresis with 10–12% SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane. After blocking with 5% skimmed milk, the membrane was incubated with the primary antibodies: Nrf-2 (1:500), Histone H3 (1:1000), p-ERK (1:1,000), ERK1/2 (1:1000), and β-actin (1:2,000) (Cell Signaling Technology, Danvers, MA, USA) overnight on a rotator at 4 °C. Then, the membrane was washed and incubated with appropriate secondary antibodies for 1 hr. Immunoreactivity of the membrane was then detected using Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA). Optical densities were quantified using Image J (NIH, Bethesda, MD, USA).
Statistical analysis
Statistical analysis was performed using GraphPad prism 5 (GraphPad, San Diego, CA, USA). All data are expressed as means±standard errors of the means (SEMs) and analyzed using analysis of variance (ANOVA), followed by Bonferroni’s multiple comparisons or unpaired t-tests. When the P-value was <0.05, the difference was considered statistically significant.
Results
Ethanol-induced dose-dependent cytotoxicity and cell apoptosis in BRL cells
To determine the appropriate concentration of ethanol in our study, BRL cells were treated with different ethanol concentrations (10, 50, 75, 100, 150, and 200 mM); then, cytotoxicity was determined by cell viability assay and LDH release detection. As shown in Figure 1A, ethanol induced a dose-dependent cell-viability decrease 24 or 48 hr after treatment. Similar results were also obtained for LDH release detection, evidenced by a concentration-dependent increase in LDH release after ethanol treatment (Figure 1B). Compared with the control group, ethanol at 150 mM caused notable decrease in cell viability by nearly 50%. Additionally, apoptosis assay by flow cytometric detection showed that a 150-mM ethanol insult significantly increased cell apoptosis compared with that in the control group (Figure 1C and D). Therefore, this concentration was used in the subsequent experiments.
Figure 1.
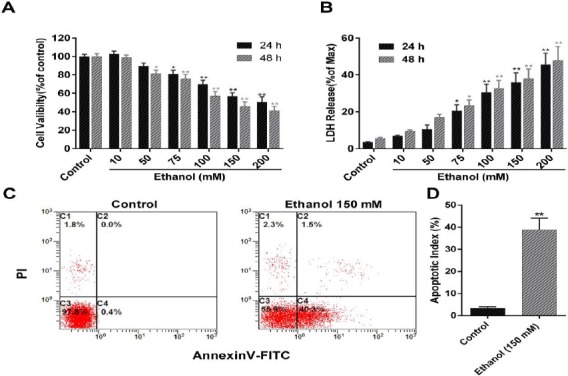
Ethanol-induced dose-dependent cytotoxicity and cell apoptosis in BRL cells. BRL cells were subjected to ethanol at concentrations of 10, 50, 75, 100, 150, or 200 mM for 24 or 48 hr. Cell viability was subsequently assessed using WST-1 assay and shown as a percentage of the control group (A). LDH release in the medium was also detected and expressed as a percentage of max release (B); 24 hr after the BRL cells were treated with ethanol 150 mM, cell apoptosis was analyzed using flow cytometry. Cells in C2 (AV+/PI+) and C4 (AV+/PI-) quadrant was considered apoptotic cells. The rate of apoptosis was calculated as apoptotic cell number/total number. Data represents means±SEMs, n=4 per group; *, P<0.05, **, P<0.01, vs. the control group
Effect of licochalcone B on ethanol-induced cytotoxicity and cell apoptosis in BRL cells
Proceeding to the hepatoprotective studies, we firstly analyzed whether licochalcone B affected cell viability in BRL cells. Twenty-four hours after treatment with different concentrations of licochalcone B, cytotoxicity was evaluated by WST-1. As shown in Figure 2A, no significant changes were observed in these groups treated with licochalcone B at a concentration less than 50 μM. However, treatment with licochalcone B at 50 μM provoked a slight but significant decrease of cell viability (Figure 2A). These data suggested that licochalcone B had no cytotoxicity at low concentrations. We further evaluated the hepatoprotective role of licochalcone B. Compared with the ethanol-alone group, significantly increased cell viability (Figure 2B) and decreased LDH release (Figure 2C) were identified in these groups cotreated with licochalcone B at 5, 10, 15, and 20 μM but not at 2.5 and 50 mM. Then, this hepatoprotective role was further evaluated by ALT and AST release. In line with a previous study, ethanol treatment induced an apparent increase of ALT and AST release in the medium (Figure 2D). This increase of ALT and AST was significant inhibited by treatment with licochalcone B at 5, 10, 15, and 20 μM (Figure 2D). Consistent with these results, treatment with licochalcone B 10 μM also significantly alleviated ethanol-induced cell apoptosis, compared with the ethanol-alone group (Figure 2E and F). Taken together, licochalcone B could alleviate ethanol-induced cytotoxicity and cell apoptosis.
Figure 2.
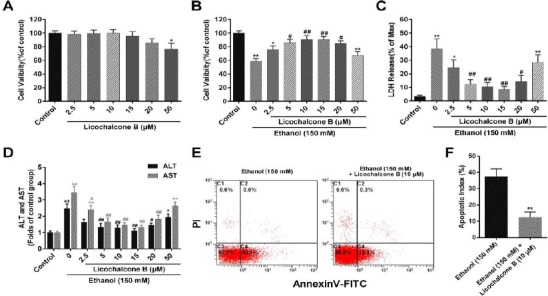
Effect of Licochalcone B on ethanol-induced cytotoxicity and cell apoptosis in BRL cells. BRL cells were subjected to licochalcone B at concentrations of 2.5, 5, 10, 15, 20, or 50 μM for 24 hr. Cell viability was subsequently assessed using WST-1 assay (A). BRL cells were subjected to ethanol (150 mM) and cotreated with licochalcone B at a concentration of 2.5, 5, 10, 15, 20, or 50 μM for 24 hr. Cytotoxicity was evaluated by cell viability assay (B), and LDH (C), ALT, and AST (D) release detection in the medium; 24 hr after BRL cells were treated with ethanol (150 mM) or cotreated with licochalcone B (10 μM), cell apoptosis was analyzed using flow cytometry and shown as the apoptosis rate (apoptotic cell number (AV+/PI-/+) total cell number × 100%). Data represents means±SEMs, n=4 per group; *,P<0.05, **, P<0.01, vs. the control group; #, P<0.05, ##, P<0.01, vs. the ethanol-alone group
Effect of licochalcone B on ethanol-induced oxidative stress in BRL cells
To evaluate the crucial roles of oxidative stress in ethanol-mediated hepatocyte injury, we evaluated the potentially antioxidative role of licochalcone B.
Cell oxidative levels were monitored by DCF-HA, a widely used reactive oxygen species (ROS) indictor. Twenty-four hours after exposure to ethanol 150 mM, a significant increase in the intracellular ROS levels was observed, compared with the control group (Figure 3A). Cotreatment with licochalcone B 10 μM significantly abolished the ethanol-induced increase in ROS levels. Furthermore, lipid peroxidation and GSH depletion were used to evaluate the antioxidative role of licochalcone B against ethanol. As shown in Figure 3B, the ethanol-induced increase of MDA in BRL cells was significantly inhibited by cotreatment with licochalcone B. Similarly, the ethanol-induced depletion of GSH was effectively ameliorated by cotreatment with licochalcone B (Figure 3C). Nrf-2, an important transcription factor regulating antioxidant genes, is involved in ethanol-induced oxidative stress. Our study found that cotreatment with licochalcone B significantly increased localization of the Nrf-2 protein in the cell nucleus (Figure 3D and F), but had no apparent effects on the total Nrf-2 protein level (Figure 3D and E). No significant change of Nrf-2 protein level was observed in the ethanol-alone group, compared with the control group.
Figure 3.
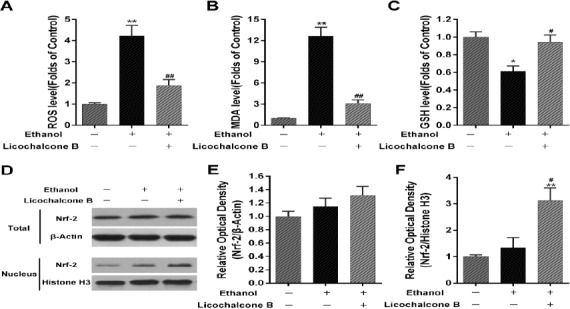
Effect of licochalcone B on ethanol-induced oxidative stress in BRL cells. After treatment with ethanol (150 mM) or cotreatment with licochalcone B (10 μM) for 24 hr, intracellular oxidative levels were evaluated using DCF-HA and shown as folds of control group. Cell lysates were harvested to detect intracellular MDA (B) and GSH (C) levels; 24 hr after treatment with ethanol (150 mM) alone or cotreatment with licochalcone B (10 μM), total proteins and nucleoproteins were harvested for Western blot assay. Representative bands are shown (D). The relative expression of Nrf-2 in total proteins (E) and in the nucleus (F) was calculated as folds of expression reported for the control group. β-Actin for total proteins and Histone H3 for nucleoproteins was used as an internal control. BRL cells not subjected to ethanol or licochalcone B were used as controls. Data are presented as mean ± SEMs; n=4 per group; *, P<0.05, **, P<0.01, vs. the control group; #, P<0.05, ##, P<0.01, vs. the ethanol-alone group
Involvement of Erk signaling in licochalcone B-mediated hepatoprotective role
A previous study found that Erk signaling was extensively involved in ethanol-induced oxidative stress (24). As shown in Figure 4A, the level of phosphorylation of Erk (p-Erk) was increased slightly 24 hr after ethanol treatment, but this change was not significant. However, cotreatment with licochalcone B significantly upregulated the p-Erk level 24 hr after ethanol treatment. No significant change of Erk protein level was observed. To further elucidate the role of Erk signaling in licochalcone B-induced hepatoprotective role against ethanol, an Erk inhibitor, PD98059, was applied to prevent Erk activation. As shown in Figure 4B, PD98059 significantly inhibited ethanol-induced upregulation of p-Erk, but had no significant effects on ethanol-induced hepatocyte injury, as evidenced by the cell viability (Figure 4C) and LDH-release assay (Figure 4D). In contrast, as compared with the ethanol+licochalcone B group, PD98059 significantly but partially abolished the licochalcone B-mediated hepatoprotective role against ethanol, evidenced by decreasing cell viability (Figure 4C) and increasing LDH release (Figure 4D). Taken together, licochalcone B might protect the BRL cell from ethanol partially via activation of Erk.
Figure 4.
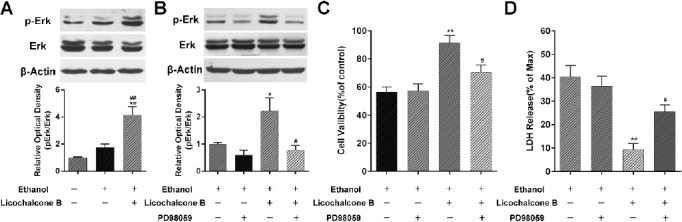
Involvement of Erk signaling in licochalcone B-mediated hepatoprotective role. Twenty-four hours after treatment with ethanol (150 mM) alone or cotreatment with licochalcone B (10 μM), the expression of p-Erk and Erk was detected by Western blot assay (A). BRL cells not subjected to ethanol or licochalcone B treatment were used as the control. **, P<0.01, vs. the control group; #, P<0.05, ##, P<0.01, vs. the ethanol-alone group. After pretreatment with an Erk inhibitor (PD98059, 20 μM), BRL cells were subsequently treated with ethanol (150 mM) alone or cotreated with licochalcone B (10 μM). The expression of p-Erk and Erk was detected by Western blot assay (B); cell cytotoxicity was evaluated by WST-1 assay (C) and LDH release detection (D). *, P<0.05, **, P<0.01, vs. the ethanol alone group; #, P<0.05, ##, P<0.01, vs. the ethanol + licochalcone B group. Data are represented as mean ± SEMs; n=4 per group
Discussion
Previous investigations have adequately established that alcohol produces a broad spectrum of hepatocyte injury and causes alcohol-related liver disorders. Further, ethanol-induced hepatocyte injury has been widely used to investigate the mechanisms involved in and to explore potentially efficient drugs against alcohol-related liver conditions (10, 25). In this study, ethanol-induced hepatotoxicity in the BRL cell, a rat hepatocyte cell line, was used. Treatment with ethanol induced a dose-dependent cell viability decrease and LDH increase. Moreover, a significant increase of apoptosis was observed after ethanol insult at a concentration of 150 mM. These results are in agreement with those reported from other studies in HepG2 or BRL 3A cells (26, 27).
Licochalcone B, which is an extract of licorice root, has shown some pharmacological properties, including antioxidant, free-radical scavenging, and anti-inflammatory effects (11, 15, 16). Therefore, we investigated the role of licochalcone B in ethanol-induced hepatotoxicity, and found that licochalcone B, at low concentrations (<50 μM), protects hepatocytes from ethanol-mediated injury, as evidenced by increased cell viability and decreased LDH and ALT/AST leakage. However, a high dose of licochalcone B (50 μM) did not show any hepatoprotective properties. This hepatoprotective role might partially be due to inhibition of apoptosis. A similar protective role against ischemia/reperfusion has been obtained in isolated rat hearts (17). Previous evidences have demonstrated the crucial role of oxidative stress in the pathogenesis of alcohol-induced hepatocyte injury (28-30). Alcohol is mainly metabolized in hepatocytes to form acetaldehyde, together with generation of ROS. Further, ROS overproduction could provoke oxidative stress, cause lipid peroxidation, and even lead to the production of reactive aldehydes with potent proinflammatory and profibrotic properties (31). Some drugs targeting alcohol-induced oxidative stress have shown a hepatoprotective role via an increase in endogenous antioxidant elements or removal of ROS, including cannabidiol (10) and diallyl trisulfide (32). Therefore, we further evaluated the antioxidative effects of licochalcone B in hepatoprotection. Our study found that licochalcone B notably inhibited ethanol-induced increase in oxidative levels (ROS and MDA), and restored downregulated GSH levels. Additionally, Nrf2, an important transcriptional factor, was intensively involved in, and protected from, ethanol-induced hepatocyte oxidative injury via induction of heme oxygenase 1 (HO-1) expression (20, 29, 33). Similarly as these studies, we found that licochalcone B promoted nuclear localization of Nrf2, but did not have a significant impact on the total level of Nrf2 protein. Taken together, licochalcone B alleviated ethanol-induced hepatotoxicity, and this role might be attributable to the inhibition of oxidative stress and promotion of Nrf2 nuclear location.
Erk signaling, a classic intracellular signaling, strongly participates in hepatocyte injury (20, 34). Our study found that Erk signaling was slightly activated after ethanol treatment, partially consistent with data on different hepatocytes (24, 35). Previous studies have demonstrated that the Erk–Nrf2–HO-1 pathway is intensively involved in hepatoprotective mechanisms, such as through chitooligosaccharides (21), antro-quinonol (36), and quercetin (20). Therefore, we further delineated the role of Erk signaling in licochalcone B-mediated hepatoprotective effects. After cotreatment with licochalcone B and ethanol, a significant increase of p-Erk expression was observed. Further, inhibition of Erk activation with an Erk inhibitor, PD98059, mitigated the licochalcone B-induced hepatoprotective role against ethanol insult, which is consistent with a study reported by Yao et al (20). However, inhibition of Erk activation with PD98059 had no apparent effect on ethanol-induced hepatotoxicity. Taken together, Erk–Nrf2 signaling may comprise downstream signaling of licochalcone B.
Conclusion
Licochalcone B inhibits ethanol-induced oxidative stress and protects hepatocyte from ethanol-induced injury. This hepatoprotective role may be related to the activation of Erk signaling and promotion of Nrf2 nuclear location. Additional experiments in vivo are need to further verify this hepatoprotective role. Despite these current limitations, these novel findings might provide a potential therapeutic candidate for alcohol-related liver disorders.
Acknowledgment
We would like to thank all the members in Department of General Surgery, Xi’an Central Hospital, Xi’an Jiaotong University.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Rusyn I, Bataller R. Alcohol and toxicity. J Hepatol. 2013;59:387–388. doi: 10.1016/j.jhep.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parry CD, Patra J, Rehm J. Alcohol consumption and non-communicable diseases: epidemiology and policy implications. Addiction. 2011;106:1718–1724. doi: 10.1111/j.1360-0443.2011.03605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouchery EE, Harwood HJ, Sacks JJ, Simon CJ, Brewer RD. Economic costs of excessive alcohol consumption in the U.S 2006. Am J Prev Med. 2011;41:516–524. doi: 10.1016/j.amepre.2011.06.045. [DOI] [PubMed] [Google Scholar]
- 4.McKillop IH, Schrum LW. Role of alcohol in liver carcinogenesis. Semin Liver Dis. 2009;29:222–232. doi: 10.1055/s-0029-1214377. [DOI] [PubMed] [Google Scholar]
- 5.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sid B, Verrax J, Calderon PB. Role of oxidative stress in the pathogenesis of alcohol-induced liver disease. Free Radic Res. 2013;47:894–904. doi: 10.3109/10715762.2013.819428. [DOI] [PubMed] [Google Scholar]
- 7.Zhu H, Jia Z, Misra H, Li YR. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: updated experimental and clinical evidence. J Dig Dis. 2012;13:133–142. doi: 10.1111/j.1751-2980.2011.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A, Miranda M, Mari M, et al. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol. 1997;273:G7–17. doi: 10.1152/ajpgi.1997.273.1.G7. [DOI] [PubMed] [Google Scholar]
- 9.Polavarapu R, Spitz DR, Sim JE, Follansbee MH, Oberley LW, Rahemtulla A, et al. Increased lipid peroxidation and impaired antioxidant enzyme function is associated with pathological liver injury in experimental alcoholic liver disease in rats fed diets high in corn oil and fish oil. Hepatology. 1998;27:1317–1323. doi: 10.1002/hep.510270518. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Rozenfeld R, Wu D, Devi LA, Zhang Z, Cederbaum A. Cannabidiol protects liver from binge alcohol-induced steatosis by mechanisms including inhibition of oxidative stress and increase in autophagy. Free Radic Biol Med. 2014;68:260–267. doi: 10.1016/j.freeradbiomed.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Y, Chen J, Li YJ, Zheng YF, Li P. Antioxidant and anti-inflammatory activities of six flavonoids separated from licorice. Food Chem. 2013;141:1063–1071. doi: 10.1016/j.foodchem.2013.03.089. [DOI] [PubMed] [Google Scholar]
- 12.Choi AY, Choi JH, Hwang KY, Jeong YJ, Choe W, Yoon KS, et al. Licochalcone A induces apoptosis through endoplasmic reticulum stress via a phospholipase Cgamma1-, Ca(2+)-, and reactive oxygen species-dependent pathway in HepG2 human hepatocellular carcinoma cells. Apoptosis. 2014;19:682–697. doi: 10.1007/s10495-013-0955-y. [DOI] [PubMed] [Google Scholar]
- 13.Xiao XY, Hao M, Yang XY, Ba Q, Li M, Ni SJ, et al. Licochalcone A inhibits growth of gastric cancer cells by arresting cell cycle progression and inducing apoptosis. Cancer Lett. 2011;302:69–75. doi: 10.1016/j.canlet.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 14.Kim JK, Shin EK, Park JH, Kim YH, Park JH. Antitumor and antimetastatic effects of licochalcone A in mouse models. J Mol Med (Berl) 2010;88:829–838. doi: 10.1007/s00109-010-0625-2. [DOI] [PubMed] [Google Scholar]
- 15.Furusawa J, Funakoshi-Tago M, Mashino T, Tago K, Inoue H, Sonoda Y, et al. Glycyrrhiza inflata-derived chalcones, Licochalcone A, Licochalcone B and Licochalcone D, inhibit phosphorylation of NF-kappaB p65 in LPS signaling pathway. Int Immunopharmacol. 2009;9:499–507. doi: 10.1016/j.intimp.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 16.Park JH, Jun JG, Kim JK. (E)-3-(3,4-dihydroxy-2-methoxyphenyl)-1-(2,4-dihydroxyphenyl)prop-2-en-1-one, a novel licochalcone B derivative compound, suppresses lipopolysaccharide-stimulated infla-mmatory reactions in RAW264.7 cells and endotoxin shock in mice. Chem Biol Interact. 2014;224C:142–148. doi: 10.1016/j.cbi.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 17.Han J, Wang D, Yu B, Wang Y, Ren H, Zhang B, et al. Cardioprotection against ischemia/reperfusion by licochalcone B in isolated rat hearts. Oxid Med Cell Longev. 2014;2014:134862. doi: 10.1155/2014/134862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coutant A, Rescan C, Gilot D, Loyer P, Guguen-Guillouzo C, Baffet G. PI3K-FRAP/mTOR pathway is critical for hepatocyte proliferation whereas MEK/ERK supports both proliferation and survival. Hepatology. 2002;36:1079–1088. doi: 10.1053/jhep.2002.36160. [DOI] [PubMed] [Google Scholar]
- 19.Rosseland CM, Wierod L, Oksvold MP, Werner H, Ostvold AC, Thoresen GH, et al. Cytoplasmic retention of peroxide-activated ERK provides survival in primary cultures of rat hepatocytes. Hepatology. 2005;42:200–207. doi: 10.1002/hep.20762. [DOI] [PubMed] [Google Scholar]
- 20.Yao P, Nussler A, Liu L, Hao L, Song F, Schirmeier A, et al. Quercetin protects human hepatocytes from ethanol-derived oxidative stress by inducing heme oxygenase-1 via the MAPK/Nrf2 pathways. J Hepatol. 2007;47:253–261. doi: 10.1016/j.jhep.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Luo Z, Dong X, Ke Q, Duan Q, Shen L. Chitooligosaccharides inhibit ethanol-induced oxidative stress via activation of Nrf2 and reduction of MAPK phosphorylation. Oncol Rep. 2014;32:2215–2222. doi: 10.3892/or.2014.3463. [DOI] [PubMed] [Google Scholar]
- 22.Choi HY, Lee JH, Jegal KH, Cho IJ, Kim YW, Kim SC. Oxyresveratrol abrogates oxidative stress by activating ERK-Nrf2 pathway in the liver. Chem Biol Interact. 2016;245:110–121. doi: 10.1016/j.cbi.2015.06.024. [DOI] [PubMed] [Google Scholar]
- 23.Toledo FD, Perez LM, Basiglio CL, Ochoa JE, Sanchez Pozzi EJ, Roma MG. The Ca(2)(+)-calmodulin-Ca(2)(+)/calmodulin-dependent protein kinase II signaling pathway is involved in oxidative stress-induced mitochondrial permeability transition and apoptosis in isolated rat hepatocytes. Arch Toxicol. 2014;88:1695–1709. doi: 10.1007/s00204-014-1219-5. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Gao C, Shi Y, Tang Y, Liu L, Xiong T, et al. Carbon monoxide alleviates ethanol-induced oxidative damage and inflammatory stress through activating p38 MAPK pathway. Toxicol Appl Pharmacol. 2013;273:53–58. doi: 10.1016/j.taap.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 25.Kannarkat GT, Tuma DJ, Tuma PL. Microtubules are more stable and more highly acetylated in ethanol-treated hepatic cells. J Hepatol. 2006;44:963–970. doi: 10.1016/j.jhep.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, Jia X, Luo Z, Guan H, Jiang H, Li X, et al. Inhibition of store-operated Ca(2+) channels prevent ethanol-induced intracellular Ca(2+) increase and cell injury in a human hepatoma cell line. Toxicol Lett. 2012;208:254–261. doi: 10.1016/j.toxlet.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 27.Cui R, Yan L, Luo Z, Guo X, Yan M. Blockade of store-operated calcium entry alleviates ethanol-induced hepatotoxicity via inhibiting apoptosis. Toxicol Appl Pharmacol. 2015;287:52–66. doi: 10.1016/j.taap.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 28.Albano E, Clot P, Morimoto M, Tomasi A, Ingelman-Sundberg M, French SW. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology. 1996;23:155–163. doi: 10.1002/hep.510230121. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Dou X, Li S, Zhang X, Sun X, Zhou Z, et al. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology. 2014;59:1381–1392. doi: 10.1002/hep.26912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meagher EA, Barry OP, Burke A, Lucey MR, Lawson JA, Rokach J, et al. Alcohol-induced generation of lipid peroxidation products in humans. J Clin Invest. 1999;104:805–813. doi: 10.1172/JCI5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tilg H, Moschen AR, Kaneider NC. Pathways of liver injury in alcoholic liver disease. J Hepatol. 2011;55:1159–1161. doi: 10.1016/j.jhep.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 32.Chen LY, Chen Q, Cheng YF, Jin HH, Kong DS, Zhang F, et al. Diallyl trisulfide attenuates ethanol-induced hepatic steatosis by inhibiting oxidative stress and apoptosis. Biomed Pharmacother. 2016;79:35–43. doi: 10.1016/j.biopha.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Lamle J, Marhenke S, Borlak J, von Wasielewski R, Eriksson CJ, Geffers R, et al. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology. 2008;134:1159–1168. doi: 10.1053/j.gastro.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 34.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park HM, Kim SJ, Mun AR, Go HK, Kim GB, Kim SZ, et al. Korean red ginseng and its primary ginsenosides inhibit ethanol-induced oxidative injury by suppression of the MAPK pathway in TIB-73 cells. J Ethnopharmacol. 2012;141:1071–1076. doi: 10.1016/j.jep.2012.03.038. [DOI] [PubMed] [Google Scholar]
- 36.Kumar KJ, Chu FH, Hsieh HW, Liao JW, Li WH, Lin JC, et al. Antroquinonol from ethanolic extract of mycelium of Antrodia cinnamomea protects hepatic cells from ethanol-induced oxidative stress through Nrf-2 activation. J Ethnopharmacol. 2011;136:168–177. doi: 10.1016/j.jep.2011.04.030. [DOI] [PubMed] [Google Scholar]