

Abstract
The basic leucine zipper transcription factor, CCAAT/enhancer binding protein α (C/EBPα), is abundantly expressed in keratinocytes of the skin; however, its function in skin is poorly characterized. UVB radiation is responsible for the majority of human skin cancers. In response to UVB-induced DNA damage, keratinocytes activate cell cycle checkpoints that arrest cell cycle progression and prevent replication of damaged DNA, allowing time for DNA repair. We report here that UVB radiation is a potent inducer of C/EBPα in human and mouse keratinocytes, as well as in mouse skin in vivo. UVB irradiation of keratinocytes resulted in the transcriptional up-regulation of C/EBPα mRNA, producing a >70-fold increase in C/EBPα protein levels. N-Methyl-N′-nitro-N-nitrosoguanidine, etoposide, and bleomycin also induced C/EBPα. UVB-induced C/EBPα was accompanied by an increase in p53 protein and caffeine, an inhibitor of ataxia-telangiectasia-mutated kinase, and ataxia-telangiectasia-mutated and Rad3-related kinase inhibited UVB-induced increases in both C/EBPα and p53. UVB irradiation of p53-null or mutant p53-containing keratinocytes failed to induce C/EBPα. UVB irradiation of C/EBPα knockdown keratinocytes displayed a greatly diminished DNA damage G1 checkpoint, and this was associated with increased sensitivity to UVB-induced apoptosis. Our results uncover a novel role for C/EBPα as a p53-regulated DNA damage-inducible gene that has a critical function in the DNA damage G1 checkpoint response in keratinocytes.
CCAAT/enhancer binding protein α (C/EBPα) is a member of the basic leucine zipper class of transcription factors (for a review, see reference 40). C/EBPα plays a critical role in the regulation of differentiation in myeloid cells (39, 56), preadipocytes (30, 52), and hepatocytes (11). In these cell types, C/EBPα is involved in the regulation of mitotic growth arrest associated with terminal differentiation as well as the expression of genes associated with the differentiated phenotype. C/EBPα is also expressed in skin (27, 33, 47), intestine (1), adrenal gland (4), and ovary (37); however, its function in these tissues is poorly characterized. While the function of C/EBPα in skin has not been characterized, C/EBPβ is involved in the regulation of the early stages of squamous differentiation (61). C/EBPβ also plays a critical role in Ras-mediated mouse skin tumorigenesis and keratinocyte survival (62). Unlike C/EBPβ, C/EBPα does not cooperate with Ras to induce transformation of NIH 3T3 cells (62). However, forced expression of C/EBPα blocks epidermal keratinocyte proliferation, suggesting a cell cycle regulatory function for C/EBPα in skin (61).
The ability of cells to respond to DNA damage is essential to ensure the integrity of the genome. With regard to environmental DNA-damaging agents, sun- or UV-exposed areas of human skin represent a major site of DNA damage. UV radiation induces cyclobutane pyrimidine dimers, 6-4 photoproducts, cytosine photohydrates, DNA strand breaks, and DNA cross-links (for reviews, see references 6 and 10). UVB radiation is responsible for 1,000,000 non-melanoma skin cancer cases in the United States each year, accounting for 40% of all new cancer cases diagnosed annually in the United States (29). In response to UV-induced DNA damage, keratinocytes activate cell cycle checkpoints (28, 32, 53) that arrest cell cycle progression, prevent replication of damaged DNA, and allow time for DNA repair; if the damage is too extensive, the keratinocytes initiate apoptosis (63). Failure to repair UV-induced DNA damage in keratinocytes is linked to the development of nonmelanoma skin cancer (7). While the expression of C/EBPα and its antiproliferative function are regulated during development and differentiation, its antiproliferative function is not thought to be involved in the DNA damage response network. However, in view of the high potential of the skin for exposure to extrinsic factors such as UVB that induce DNA damage, it is possible that one function of C/EBPα in the skin is to participate in mitotic growth arrest in response to environmental stressors such as UVB-induced DNA damage. The results presented in this study are the first to identify a role for C/EBPα in the cellular response to DNA damage induced by extrinsic DNA-damaging agents. We demonstrate that the C/EBPα gene is a p53-regulated DNA damage-inducible gene in keratinocytes and that it is an important link between UVB-induced DNA damage and cell cycle arrest in epidermal keratinocytes.
MATERIALS AND METHODS
Cell lines and cell culture.
BALB/MK2 keratinocytes (B. Weissman, University of North Carolina) were cultured in Ca2+-free Eagle's minimal essential medium (EMEM; BioWhittaker) supplemented with 8% Chelex-treated fetal bovine serum (FBS; Invitrogen), 4 ng of human epidermal growth factor (hEGF)/ml (Invitrogen), and 0.05 mM calcium (keratinocyte medium). Mouse primary keratinocytes were isolated from the epidermis of newborn mice by overnight flotation in trypsin at 4°C (19). Isolated keratinocytes were plated in Ca2+-free EMEM supplemented with 10% non-Chelex-treated FBS and 10 ng of hEGF/ml. Four hours later, cultures were washed with Mg2+- and Ca2+-free phosphate-buffered saline (PBS) and refed with Ca2+-free EMEM supplemented with 4% Chelex-treated FBS, 10 ng of hEGF/ml, 100 U of penicillin/ml, 100 μg of streptomycin/ml, 250 ng of amphotericin B (Fungizone)/ml and 0.05 mM calcium. Normal human epidermal keratinocytes (NHEK) were purchased from Cambrex and cultured in KGM keratinocyte medium (Cambrex). HaCaT cells were cultured in Dulbecco's modified Eagle essential medium (DMEM) (Sigma) supplemented with 10% FBS, 100 U of penicillin/ml, and 100 μg of streptomycin/ml. NIH 3T3 cells and normal rat kidney (NRK) fibroblasts were cultured in DMEM supplemented with 10% calf serum. HepG2 cells were cultured in MEM supplemented with 10% FBS, 1.0 mM sodium pyruvate, and 0.1 mM nonessential amino acids.
Animals.
CD-1 (Charles River) and C57BL/6 and 129/SV mixed-strain mice were used for in vivo UVB irradiation. Wild-type and p53 mice were provided by J. French (National Institute of Environmental Health Sciences). Primers and PCR conditions were published previously (20). p53-deficient male mice were mated with heterozygous female mice to produce p53-deficient pups. C57BL/6 mice were mated to generate control subjects.
UVB irradiation and chemical treatment.
The UVB lamp (model EB 280C; Spectronics) emits wavelengths between 280 to 320 nm with a spectrum peak at 312 nm. The light intensity of the lamp was measured by the IL-1700 Research Radiometer (International Light) equipped with an SED 240 sensor. The UVB lamp was positioned 15 cm above the cells or mice. For cells in culture, the medium was removed, and the cells were washed with PBS and irradiated in the presence of PBS for the amount of time corresponding to the indicated UVB dose. After irradiation, PBS was removed and replaced with the specified medium. N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG; Aldrich), etoposide (Sigma), and actinomycin D (Sigma) were dissolved in dimethyl sulfoxide. Bleomycin A2 (Calbiochem) and caffeine (Sigma) were dissolved in water. For mouse studies, the hair of the dorsal skin of 6- to 7-week-old female mice was clipped; 2 days later, the mice were individually irradiated with UVB.
Preparation of cell lysates.
Nuclear extracts were prepared as previously described (43). For the preparation of whole-cell lysates, cells were washed with cold PBS, harvested by scraping, and collected by centrifugation. Cells were placed in hypotonic buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail [Roche], 1 mM sodium orthovanadate with 0.6% NP-40) and lysed by sonication, and then a 1/10 volume of 5 M NaCl was added. The cell lysates were vortexed, incubated for 15 min on ice, and centrifuged at 14,000 × g for 10 min. Supernatants were stored at −80°C until use. Protein concentration was determined with the Bio-Rad protein assay reagent.
Western blot analysis.
Equal amounts of protein were precipitated by the addition of an equal volume of 20% trichloroacetic acid and washed with cold acetone. Protein samples were dissolved in sodium dodecyl sulfate (SDS) sample buffer, boiled, and separated by SDS-polyacrylamide gel electrophoresis. The separated proteins were transferred to an Immobilon-P membrane (Millipore). Following incubation in blocking buffer, the membranes were probed with rabbit polyclonal immunoglobulin G (IgG) raised against C/EBPα (sc-61), C/EBPβ (sc-150), p53 (sc-6243), or p21 (sc-757) (Santa Cruz). The membranes were washed and then probed with a horseradish peroxidase-linked secondary antibody (Amersham). Detection was made with an enhanced chemiluminescence reagent (Amersham), followed by exposure of the membrane to film. Membranes were stained with Coomassie blue to confirm equal protein loading. Band intensity was measured by densitometry.
Electrophoretic mobility shift assay (EMSA) and supershift.
C/EBP consensus oligonucleotides (Santa Cruz) were labeled with [γ-32P]ATP by kinase reaction withT4 polynucleotide kinase. Radiolabeled oligonucleotides were incubated with 4 μg of nuclear extract at room temperature in binding buffer containing poly(dI-dC). For the supershift assay, samples were further incubated with anti-C/EBPα antibody (sc-61X; Santa Cruz). DNA-protein complexes were loaded onto 4% polyacrylamide gels and run in 0.25× Tris-borate-EDTA buffer. The gel was transferred to Whatman paper, dried, and exposed to film.
Northern blot analysis.
Total RNA was isolated from UVB-irradiated or nonirradiated BALB/MK2 cells in culture with acid guanidium thiocyanate-phenol-chloroform extraction. C/EBPα cDNA was labeled with [α-32P]dCTP with Ready-To-Go labeling beads (Amersham). RNA was electrophoresed on an agarose-formaldehyde gel, transferred to a Zeta-Probe GT membrane (Bio-Rad), and UV cross-linked. Membranes were incubated at 65°C in hybridization buffer (0.25 M Na2HPO4 [pH 7.2], 7% SDS) overnight, washed, exposed to film at −80°C, and developed.
Chromatin immunoprecipitation (ChIP) assay.
BALB/MK2 cells were plated in 100-mm culture plates and 24 h later were irradiated or not with UVB at the dose of 5 or 10 mJ/cm2. Twelve hours postirradiation, cells were cross-linked with 1% formaldehyde for 10 min at room temperature, and the ChIP assay was performed according to the manufacturer's instructions (Upstate Biotechnology). Samples were immunoprecipitated with rabbit polyclonal antibody against p53 (sc-6243) or rabbit IgG. Immunoprecipitated DNA was purified and amplified by PCR. Nine potential p53 binding sites in C/EBPα promoter were identified with the p53 Scanner program from the Human Cancer Genetics Bioinformatics Group, Ohio State University. The location of the nine sites relative to the transcriptional start site are as follows: −452 to −424, −2605 to −2571, −2963 to −2935, −5870 to −5844, −6587 to −6550, −7314 to −7287, −9657 to −9628, −10446 to −10417, and −13577 to −13544. Nine primer sets for the corresponding sites were synthesized, and primers for locations spanning −10446 to −10417 and the p21 promoter are as follows. For −10446 to −10417, the forward primer was 5′-CAGTTTTTCCAATGTCACCCCTAC-3′, and the reverse primer was 5′-TATCTTCATCAGCAGCCCCAGG-3′. For the p21 promoter, the forward priner was 5′-CCAGAGGATACCTTGCAAGGC-3′, and the reverse primer was 5′-TCTCTGTCTCCATTCATGCTCCTCC-3′.
Construction of C/EBPα promoter-reporter and luciferase assay.
The region between −11133 to −10171 of the C/EBPα promoter (P. Johnson, National Cancer Institute [NCI]) was amplified by PCR. The amplified product was inserted into the pGL3-promoter vector (Promega) using KpnI and NheI sites. Colonies were screened by restriction enzyme analysis. BALB/MK2 keratinocytes were plated and transfected 24 h later with Tfx-10 (Promega) with the pGL3-promoter-reporter (pGL3; Promega) or with the pGL3-promoter-reporter containing the C/EBPα promoter (C/EBPα-pGL3), the pCMV-p53 or pCMV control vector, and the phRL-CMV Renilla luciferase vector (Promega) as an internal control. Cells were harvested 24 h after transfection, and luciferase activity was determined with the luciferase assay kit (Promega).
Immunohistochemistry.
Normal and irradiated mouse skins were fixed in 10% neutral buffered formalin phosphate and embedded in paraffin. Tissue sections (5 μm) were subjected to antigen retrieval, followed by treatment with 0.1% H2O2 and blocking with normal goat serum. Sections were incubated with the anti-C/EBPα antibody or anti-p53 antibody at 4°C overnight and a biotinylated goat anti-rabbit IgG at room temperature for 30 min. Detection was made with the ABC kit (Vector Laboratories) and 3,3′-diaminobenzidine (BioGenex) as the chromogen, following the manufacturer's protocol.
Transfection and colony formation assay.
BALB/MK2 keratinocytes were plated and transfected 24 h later with 2 μg of pcDNA3 or pcDNA3-C/EBPα with Lipofectamine (Invitrogen) according to the manufacturer's protocol. Two days later, the cultures were split (1:3), and selection medium containing 300 μg of G418 (Sigma)/ml was added 24 h after the cultures were replated. On days 3, 5, and 7 after G418 selection, the number of colonies and the number of cells per colony in 30 random grid squares were counted.
Retroviral infection and thymidine incorporation assay.
φNX cells were grown in DMEM supplemented with 10% FBS and transfected with 15 μg of pWZL or pWZL-C/EBPα (P. Johnson, NCI) by the calcium phosphate precipitation method. C/EBPα nucleotide sequences were confirmed by DNA sequencing. Twenty-four hours after transfection, the culture medium was replaced with keratinocyte medium. The next day, medium containing virus was collected, filtered, and transferred onto the keratinocytes for infection with 5 μg of polybrene (Sigma)/ml. Fresh keratinocyte medium was added to the packaging cells. The infection procedure was repeated two more times at 4-h intervals. Infected keratinocytes were selected with 100 μg of hygromycin (Roche)/ml for 2 weeks, and colonies were pooled. Cells were pulse-labeled with 3 μCi of [3H-methyl]thymidine per ml (20 Ci/mmol) for 1 h before collection. Cells were collected by trypsinization, resuspended in 1 mM EDTA buffer, and sonicated; aliquots were collected onto glass fiber filters; and the filters were placed in a liquid scintillation cocktail and subjected to scintillation counting. For DNA quantitation, an aliquot of each sample and 5 μl of 0.1 mg of Hoechst 33258 solution/ml were mixed in 1 ml of 0.01 M Tris (pH 7.0)-0.1 M NaCl-0.01 M EDTA buffer. The fluorescence units were determined with a fluorometer (excitation at 365 nm and emission at 450 nm).
Generation of C/EBPα knockdown BALB/MK2 keratinocytes.
The siRNA target sequence for C/EBPα, 5′-907AAAGCCAAACAACGCAACGTG-3′, was selected following the manufacturer's protocol; the target sequence was analyzed by BLAST search to check significant sequence homology with other genes. Two DNA oligonucleotides were designed and cloned into the pSilencer 2.3-U6 hygro vector (Ambion), following the manufacturer's protocol. BALB/MK2 keratinocytes were transfected with these constructs as well as an empty vector and selected with hygromycin, and colonies were ring cloned. The colonies were expanded and screened for C/EBPα expression after UVB irradiation.
5-Bromo-2′-deoxyuridine (BrdU) labeling and fluorescence-activated cell sorting (FACS) analysis.
For the G1 checkpoint, C/EBPα knockdown keratinocytes or control keratinocytes were synchronized by serum and hEGF starvation (0.5% Chelex-treated FBS) at ∼25% confluence for 24 h and then released into the cell cycle by the addition of keratinocyte medium. Six hours later, cells were irradiated or not with UVB (5 mJ/cm2) and harvested at 18 and 24 h after release into the cell cycle. Cells were incubated with 10 μM BrdU for 2 h before the cells at each time point were harvested. Cells were trypsinized, pelleted, resuspended in 100 μl of cold PBS, and fixed in 70% ethanol. Cells were treated with 2 N HCl-Triton X-100 to denature DNA, followed by neutralization with Na2B4O7. Cells were pelleted, resuspended in 0.5% Tween 20-1% bovine serum albumin-PBS with anti-BrdU-fluorescein isothiocyanate antibody (1:50; Becton Dickinson) and 0.5 mg of RNase/ml, and incubated at 4°C overnight. Cells were pelleted and resuspended in PBS containing 5 mg of propidium iodide (PI)/ml and subjected to FACS analysis. Data were collected and presented on a scatter plot with BrdU intensity on the y axis and PI intensity on the x axis.
Caspase 3/7 assay.
C/EBPα knockdown keratinocytes or control keratinocytes were plated in fluorescence-readable 96-well plates and grown in keratinocyte medium. The Apo-ONE Homogeneous Caspase-3/7 assay (Promega) was used to measure apoptosis, following the manufacturer's protocol. Caspase 3/7 activity was normalized to the total amount of protein.
RESULTS
We examined the effect of UVB treatment on C/EBPα expression in BALB/MK2 keratinocytes. BALB/MK2 keratinocytes, a nontransformed immortalized mouse cell line, were irradiated with a single dose of UVB (10 mJ/cm2). As shown in Fig. 1A, UVB produced a very large increase in C/EBPα protein levels. At 6 h post-UVB treatment, there was approximately a 10-fold increase in C/EBPα protein; by 18 h, C/EBPα protein was increased >70 fold. Elevated levels of C/EBPα protein (two- to threefold) could be detected as early as 3 h post-UVB treatment (data not shown). By 48 h post-UVB treatment, C/EBPα protein levels began to return to baseline levels (Fig. 1A). These results demonstrate that UVB radiation is a potent and rapid inducer of the transient expression of C/EBPα protein in BALB/MK2 keratinocytes. While UVB is a potent inducer of C/EBPα, it had a comparatively much smaller effect on C/EBPβ protein levels (Fig. 1A). C/EBPδ (34) and C/EBPζ (CHOP/GADD 153) (13, 16, 58) can be induced by various cellular stressors; however, neither C/EBPδ protein nor C/EBPζ protein was induced by UVB at the doses and time points we used to examine C/EBPα (data not shown). EMSA analysis utilizing a C/EBP consensus sequence demonstrated that UVB treatment also produced a large increase in DNA binding activity (Fig. 1B). Supershift EMSA analysis with antibodies to C/EBPα revealed that the great majority of the UVB-induced increase in DNA binding was due to C/EBPα binding (Fig. 1C). Therefore, the increase in UVB-induced C/EBPα protein levels is accompanied by a concomitant increase in C/EBPα DNA binding. As shown in Fig. 1D, UVB induced C/EBPα in a dose-dependent manner. To determine whether the UVB induction of C/EBPα protein is a result of increased mRNA levels, we isolated total RNA from UVB-treated BALB/MK2 keratinocytes and conducted Northern analyses. As shown in Fig. 1E, UVB treatment produced a large increase in C/EBPα mRNA levels. Actinomycin D treatment completely blocked the UVB induction of C/EBPα protein levels (Fig. 1F). Actinomycin D also blocked a UVB-induced increase in C/EBPα mRNA, as determined by quantitative reverse transcription-PCR (data not shown). Collectively, these data indicate that UVB is a potent inducer of C/EBPα at the transcriptional level.
FIG. 1.

UVB is a potent inducer of C/EBPα. (A) BALB/MK2 cells were irradiated with UVB (10 mJ/cm2) and harvested at various time points. Whole-cell lysates were prepared, and equal amounts of protein were subjected to immunoblot analysis with rabbit polyclonal anti-C/EBPα or anti-C/EBPβ antibody. Nonspecific bands (NS) are shown to confirm equal loading. (B) BALB/MK2 cells were irradiated with UVB (10 mJ/cm2); 18 h later, nuclear extracts were prepared and EMSA and (C) a supershift assay with anti-C/EBPα antibody were conducted. (D) BALB/MK2 cells were irradiated with various doses of UVB; 18 h later, cell lysates were prepared and a Western blot analysis with anti-C/EBPα antibody was conducted. (E) Total RNA was isolated from UVB-irradiated BALB/MK2 cells (10 mJ/cm2) 14 h after irradiation. Northern blot analysis for C/EBPα was conducted. 18S RNA was shown for equal loading. (F) BALB/MK2 cells were preincubated with actinomycin D for 30 min before UVB irradiation and incubated further for 6 h. Cell lysates were prepared, and Western blot analysis was conducted for C/EBPα.
As shown in Fig. 2A, UVB induced C/EBPα protein levels in human primary keratinocytes (NHEK) as well as in mouse primary keratinocytes. UVB also induced C/EBPα in the epidermis of skin of mice treated in vivo with UVB (Fig. 2A). To determine whether other types of DNA damage and/or DNA-damaging agents could also induce C/EBPα, we treated BALB/MK2 keratinocytes with MNNG, a carcinogen that methylates DNA; etoposide, an inhibitor of topoisomerase II that produces double-strand DNA breaks; or bleomycin, an antineoplastic drug that produces both single- and double-strand breaks. As shown in Fig. 2B, all three DNA-damaging agents are potent inducers of C/EBPα protein expression. While human and mouse keratinocytes responded to UVB with the robust induction of C/EBPα, treatment of NIH 3T3 fibroblasts, human hepatoma cells (HepG2), and normal rat kidney fibroblasts (NRK cells) with various doses of UVB did not induce C/EBPα (Fig. 2C). UVB treatment did, however, increase p53 protein levels, indicating that these cells are capable of responding to UVB (Fig. 2C). Treatment of NIH 3T3, HepG2, and NRK cells with MNNG or etoposide did not induce C/EBPα (data not shown). These results indicate UVB induction of C/EBPα is cell type specific.
FIG. 2.

UVB induces C/EBPα in human and mouse primary keratinocytes and mouse skin in vivo, and induction of C/EBPα is a general DNA damage response. (A) NHEK, primary mouse keratinocytes, or 8-week-old mice were irradiated with indicated doses of UVB. Keratinocytes were collected at the indicated time points, cell lysates were prepared, and Western blots were probed with anti-C/EBPα antibody. (B) BALB/MK2 cells were treated with MNNG, etoposide, or bleomycin A2 for the indicated times and harvested. Cell lysates were prepared, and Western blot analysis was conducted for C/EBPα. (C) NIH 3T3, NRK, or HepG2 cells were irradiated with the indicated doses of UVB. Cells were collected 12 h after irradiation, cell lysates were prepared, and Western blotting was conducted with anti-C/EBPα or anti-p53 antibody.
As shown in Fig. 3A, UVB treatment of BALB/MK2 keratinocytes induced the accumulation of p53 protein as well as p21, a p53-inducible protein. The kinetics of the increase in C/EBPα protein was similar to p53 accumulation, suggesting a possible relationship between the two proteins (Fig. 3A). To begin to determine if p53 is involved in UVB induction of C/EBPα, we first examined the effect of caffeine (42), a pharmacological inhibitor of ataxia-telangiectasia-mutated (ATM) and ataxia-telangiectasia-mutated and Rad3-related (ATR) kinases on UVB induction of C/EBPα and p53 accumulation. ATR is activated by UVB-induced DNA damage and subsequently phosphorylates p53, contributing to the activation (9, 12) and accumulation of p53 protein (45). As shown in Fig. 3B, treatment of BALB/MK2 keratinocytes with caffeine blocked UVB-induced p53 accumulation as well as C/EBPα induction, suggesting that UVB-induced C/EBPα is ATM/ATR and/or p53 dependent. To determine whether the UVB induction of C/EBPα is p53 dependent, we examined the effects of UVB in HaCaT cells, a human keratinocyte cell line that contains inactive mutant p53 (24). While UVB is an effective inducer of C/EBPα in NHEK cells, UVB did not induce C/EBPα or p21 in HaCaT cells, indicating that functional p53 is required for C/EBPα induction (Fig. 3C). HaCaT cells displayed elevated levels of p53 in the absence of UVB treatment as well as a lack of increase in p21, both of which are indicative of the presence of inactive mutant p53. To provide additional direct evidence that p53 is required for UVB-induced C/EBPα expression, we examined the effects of UVB in p53-nullizygous and wild-type primary mouse keratinocytes. In primary mouse keratinocytes, maximal UVB induction of C/EBPα was achieved 6 h after irradiation (Fig. 2A). As shown in Fig. 3D, UVB failed to induce C/EBPα in p53-nullizygous mouse keratinocytes. Similar results were obtained at 12 and 18 h post-UVB treatment and at a lower dose (5 mJ/cm2) (data not shown). As shown in Fig. 3E, UVB treatment increased C/EBPα and p53 protein levels in the epidermis of the skin of wild-type mice but not in p53-nullizygous mice. These results demonstrate that p53 is required for UVB induction of C/EBPα in human and mouse keratinocytes and in mouse skin in vivo. We also observed that p53 is also required for C/EBPα induction in keratinocytes treated with other DNA-damaging agents including MNNG, etoposide, and bleomycin (data not shown). UVB increased C/EBPβ protein levels in the epidermis of the skin of both wild-type and p53-nullizygous mice, indicating that the moderate increase in UVB-induced C/EBPβ is p53 independent (data not shown).
FIG. 3.

UVB induction of C/EBPα requires p53. (A) Time course study of C/EBPα, p53, and p21 protein induction after UVB irradiation of BALB/MK2 cells. (B) BALB/MK2 cells were pretreated with the indicated concentrations of caffeine for 1 h before UVB irradiation. Cells were irradiated, incubated for 12 h in the presence of caffeine, and collected. Cell lysates were prepared, and a Western blot analysis was conducted with anti-p53 or anti-C/EBPα antibody. (C) NHEK or HaCaT cells were irradiated with UVB. At the indicated time points, nuclear extracts were prepared, and a Western blot analysis was conducted to determine p53, p21, and C/EBPα protein levels. (D) Primary keratinocytes were isolated from wild-type or p53-null newborn mice. Three plates of each genotype were irradiated with UVB (10 mJ/cm2), and three plates of each genotype were not treated. Whole-cell lysates were prepared 6 h after irradiation, and a Western blot analysis was conducted with anti-C/EBPα or anti-p53 antibody. (E) Adult wild-type (W) and p53 knockout (K) mice were treated with the indicated doses of UVB, epidermis was isolated 12 h post-UVB treatment, and whole cell lysates were prepared for Western blot analysis. (F) ChIP assay with p53 antibody. BALB/MK2 cells were irradiated with the indicated doses of UVB, and cross-linking was initiated at 12 h post-UVB treatment. The input DNA is the source DNA prior to immunoprecipitation; p53-immunoprecipitated DNA is designated IP p53 while IP control DNA was precipitated with rabbit IgG. Results presented are from a single experiment, which is representative of three independent experiments. (G) BALB/MK2 cells were transfected with C/EPBα-pGL3, which contains the region of C/EBPα promoter including the identified p53 site (−10446 to −10417), or empty pGL3 with or without the p53 expression vector as indicated. A luciferase assay was done in triplicate and normalized to the Renilla luciferase reporter (phRL-CMV). Another C/EBPα promoter-reporter construct prepared from an independent clone was tested and showed similar results. *, significantly different from other groups as determined by Student's t test (P < 0.05).
To begin to determine whether p53 regulates C/EBPα through a mechanism involving the direct binding of p53 to the C/EBPα promoter, we analyzed the C/EBPα promoter for p53 binding sites. Computer analysis of the initial 16 kb of the C/EBPα promoter revealed nine p53 consensus sites with high levels of p53 site sequence conservation. To determine if UVB treatment results in p53 binding to any of these sites, BALB/MK2 cells were UVB irradiated and a ChIP assay was conducted with a p53 antibody. PCR was conducted on the input DNA and p53-immunoprecipitated DNA with primers that flank each of the p53 sites and primers that flank the p53 site in the p21 promoter (positive control). With the use of input DNA, all nine sites produced the correctly sized PCR product; however, only one of the p53 sites (−10446 to −10417) in the C/EBPα promoter was positive in the p53 ChIP assay, as was the p53 site in the p21 promoter (Fig. 3F). These results suggest that p53 binding site located 10 kb upstream of the C/EBPα transcriptional start site is an active UVB-inducible p53 site involved in UVB induction of C/EBPα. To determine whether p53 can regulate the C/EBPα promoter through this site, we constructed a promoter-reporter construct that contained the portion of C/EBPα promoter with the identified p53 site (−10446 to −10417). As shown in Fig. 3G, p53 stimulated the C/EBPα promoter-reporter that contained the p53 site. These data, along with the p53 ChIP results, suggest that one mechanism of UVB induction of C/EBPα involves direct binding of p53 to the C/EBPα promoter.
The epidermis is primarily composed of basal and suprabasal keratinocytes. Basal keratinocytes are undifferentiated and capable of mitosis, while suprabasal keratinocytes are postmitotic and have entered the squamous differentiation program. Knowing the cellular location of the UVB-induced increase in C/EBPα within the epidermis could provide insight into the function of C/EBPα in the epidermal UVB response. Immunohistochemical staining for C/EBPα in UVB-irradiated mouse skin revealed increased staining of C/EBPα in the nucleus and cytoplasm of both the suprabasal and basal keratinocytes of the epidermis (Fig. 4A). Of particular significance was the observation that UVB produced a three- to fourfold increase in the number of basal keratinocytes expressing nuclear C/EBPα (Fig. 4B). Thus, UVB treatment not only increases the level of C/EBPα in keratinocytes previously expressing C/EBPα, but it induces the expression of C/EBPα in basal keratinocytes in which C/EBPα was previously not expressed or was undetectable. We also examined the localization of p53 and C/EBPα in wild-type and p53-nullizygous mice. In wild-type mice, p53 was not detectable in untreated mouse epidermis. However, UVB treatment induced p53 in the epidermis, and this increase occurred predominately in the basal keratinocytes (Fig. 4C). Neither p53 nor C/EBPα was induced in the p53-nullizygous mouse epidermis (Fig. 3E and data not shown). These results are consistent with our observation that in UVB-treated skin the number of basal keratinocytes expressing C/EBPα is preferentially increased when compared to suprabasal keratinocytes (Fig. 4B) and further strengthen the role of p53 in the regulation of C/EBPα expression in basal keratinocytes in UVB-treated mouse skin. The UVB-induced increase of C/EBPα in the basal keratinocytes, which represent the proliferating compartment of the epidermis, suggests that C/EBPα may have a role in UVB-induced inhibition of keratinocyte proliferation. In agreement with earlier studies (61), forced expression of C/EBPα in BALB/MK2 keratinocytes inhibited keratinocyte proliferation, as indicated by the decrease in the number of cells per colony and the total number of colonies. As shown in Fig. 5A, the number of cells per colony changed little over time in the C/EBPα-transfected cells, while the number of cells per colony in the control cells transfected with the empty vector increased over fourfold (Fig. 5A). C/EBPα also produced approximately a 75% decrease in the total number of colonies (data not shown).
FIG. 4.

C/EBPα protein is induced in the basal and suprabasal epidermal keratinocytes irradiated with UVB in vivo. (A) Dorsal area hair of 7-week-old CD-1 mice was clipped and irradiated with UVB (100 mJ/cm2). At the indicated times after UVB irradiation, skins were collected, processed, and embedded in paraffin. Tissue sections were stained with anti-C/EBPα antibody, and basal and suprabasal interfollicular keratinocytes with positive nuclear C/EBPα staining were scored separately and are shown in panel B. Photographs were taken at a magnification of ×400. B, basal; S, suprabasal. (B) Data are expressed as means ± standard errors from three mice. Three sections per mouse were processed. *, significantly different from untreated group as determined by Student's t test (P < 0.05). (C) Mice were irradiated with UVB (100 mJ/cm2). At 12 h after UVB irradiation, skins were collected, processed, and embedded in paraffin. Immunohistochemical staining was conducted with anti-p53 antibody, and photographs were taken at a magnification of ×400.
FIG. 5.
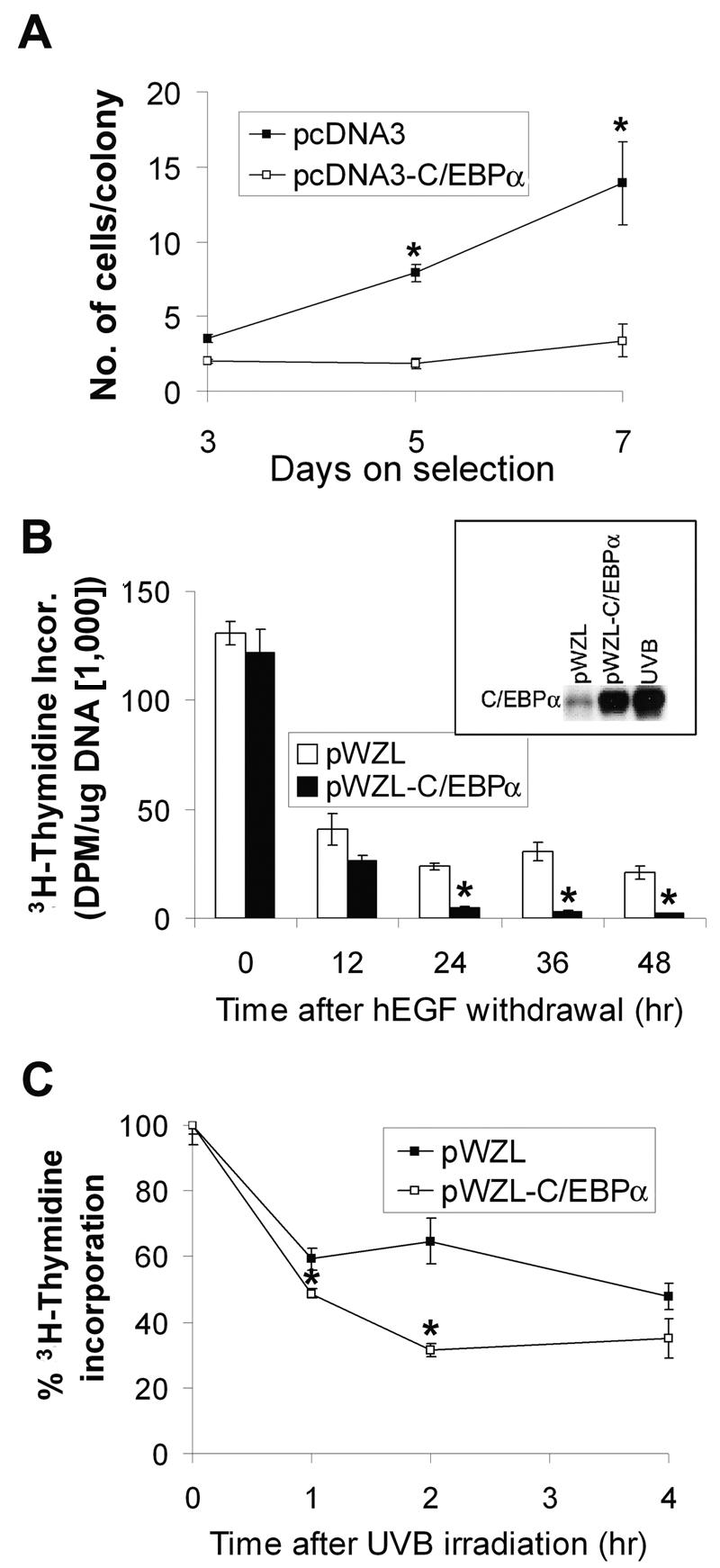
Enforced expression of C/EBPα inhibits proliferation of keratinocytes. (A) BALB/MK2 cells were transfected with pcDNA3 or pcDNA3-C/EBPα and subcultured in the presence of 300 μg of G418/ml. The number of cells per colony was counted on days 3, 5, and 7 after G418 selection. (B) hEGF was withdrawn from BALB/MK2-pWZL or BALB/MK2-pWZL-C/EBPα cells. Cells were pulse-labeled with [3H-methyl]thymidine for 1 h before each time point indicated, and [3H-methyl]thymidine incorporation into DNA was determined. The box shows the C/EBPα protein levels in BALB/MK2-pWZL, BALB/MK2-pWZL-C/EBPα, and BALB/MK2 cells irradiated with UVB (10 mJ/cm2) at the 18-h time point. (C) BALB/MK2-pWZL or BALB/MK2-pWZL-C/EBPα cells were irradiated with UVB (5 mJ/cm2) and pulse-labeled with [3H-methyl]thymidine for 1 h before each time point indicated; [3H-methyl]thymidine incorporated into DNA was determined. Data are expressed as the means ± standard error of a representative experiment done in triplicate. *, significantly different from similarly treated BALB/MK2-pWZL as determined by Student's t test (P < 0.05).
Transient transfection of BALB/MK2 cells with pcDNA3-C/EBPα produces supraphysiologic levels of C/EBPα, and transfection efficiency is less than 5%. To further study the effect of C/EBPα on keratinocyte proliferation, we infected BALB/MK2 keratinocytes with a retrovirus containing C/EBPα and selected the cells with hygromycin. Cells infected with the C/EBPα-containing retrovirus displayed elevated levels of C/EBPα that were comparable to the UVB-induced C/EBPα levels (Fig. 5B, insert). These elevated C/EBPα levels are at least an order of magnitude less than the cells transfected with pcDNA3-C/EBPα (data not shown). Unexpectedly, the keratinocytes infected with the retrovirus containing C/EBPα continued to proliferate, similar to the control cells infected with empty virus, as determined by [3H]thymidine incorporation into DNA and FACS analysis (data not shown). While overexpression of C/EBPα inhibits cell proliferation in many cells, this appears to be dependent on the degree of overexpression as well as the cell type (14, 25, 44). When hEGF was removed from the culture medium, cells infected with the C/EBPα-containing retrovirus displayed a 80 to 90% greater decrease in [3H]thymidine incorporation into DNA at 24 to 48 h compared to control cells infected with empty virus (Fig. 5B). We conducted caspase 3/7 assays and cell counts of C/EBPα-overexpressing keratinocytes and control keratinocytes upon hEGF withdrawal. We observed that apoptosis was not significantly increased and the cell numbers did not decrease after hEGF withdrawal, indicating that the decrease in [3H]thymidine incorporation into DNA was not due to apoptosis (data not shown). Conditioned medium from control cells did not alter the growth inhibitory response of the C/EBPα-expressing cells, nor did conditioned medium from the C/EBPα-expressing cells induce growth inhibition in the control cells (data not shown), indicating that the differential growth response of C/EBPα-overexpressing cells was not due to altered production of autocrine factors present in the medium. Collectively, these results suggest that hEGF in the culture medium antagonizes the antiproliferative function of C/EBPα under unstressed conditions.
Next, we examined the effect of UVB on cell proliferation in keratinocytes infected with either a retrovirus containing C/EBPα or an empty retrovirus. Since C/EBPα is rapidly induced by UVB, we examined proliferation at early time points post-UVB treatment before substantial levels of endogenous C/EBPα were induced in the control cells. UVB inhibited cell proliferation in both C/EBPα-overexpressing and control cells; however, cells overexpressing C/EBPα displayed approximately 50% greater inhibition in cell proliferation as determined by [3H]thymidine incorporation into DNA at 2 h post-UVB treatment (Fig. 5C). These results indicate that C/EBPα contributes to growth arrest in UVB-treated keratinocytes. Later time points (up to 24 h) did not show significant differences in [3H]thymidine incorporation into DNA in cells infected with the retrovirus containing C/EBPα, compared to cells infected with empty retrovirus (data not shown). This lack of effect at the later time points could be due to the induction of endogenous C/EBPα in the control cells, which complicates the interpretation of these C/EBPα overexpression studies.
To better define the role of C/EBPα in UVB-induced growth arrest, we utilized small-interference RNA-mediated knockdown of C/EBPα. C/EBPα small-interference RNA blocked C/EBPα induction by approximately 80% (Fig. 6B). To determine whether C/EBPα has a role in the UVB-induced DNA damage G1 checkpoint, keratinocytes were synchronized by serum and hEGF starvation, released from starvation, irradiated with UVB or not 6 h after release from starvation, and pulsed with BrdU 2 h prior to collection. At 6 h after the release from serum starvation, <1% of the untreated control cells and C/EBPα knockdown cells were in S phase (Fig. 6A). Similar results were observed at 8 h postrelease (data not shown). As shown in Fig. 6A, at 18 h postrelease, 13.5% of the nonirradiated control and 30.7% of the C/EBPα knockdown cells progressed into S phase. In contrast, 1.4% ± 0.26% of the irradiated control and 10.3% ± 0.33% of the irradiated C/EBPα knockdown cells progressed into S phase. Therefore, UVB inhibited by ∼90% the entry of control cells into S phase; however, entry into S phase was only inhibited by 67% in the C/EBPα knockdown cells (Fig. 6A). Similar results were obtained with another independently isolated C/EBPα knockdown clone and with UVB irradiation 4 h after release into the cell cycle (data not shown). At 36 h post-UVB irradiation, ∼30% of control cells entered into S phase (data not shown). These results demonstrate that the UVB-irradiated control cells have an effective G1 checkpoint, while C/EBPα knockdown cells have a greatly attenuated G1 checkpoint in response to UVB-induced DNA damage. We also conducted a BrdU pulse-chase experiment in asynchronously growing control and C/EBPα knockdown keratinocytes. Both UVB-irradiated control and C/EBPα knockdown cells arrested in S phase at 6 and 12 h post-UVB treatment to a similar degree, suggesting that C/EBPα is not involved in the S-phase checkpoint (data not shown). Attenuation of DNA damage checkpoints, including the G1 checkpoint, can result in increased apoptosis (5, 36). As shown in Fig. 6C, C/EBPα knockdown keratinocytes display an increased sensitivity to UVB-induced apoptosis, as determined by an increase in caspase 3/7 activity.
FIG. 6.

UVB irradiation of C/EBPα knockdown keratinocytes displays an attenuated DNA damage G1 checkpoint. (A) C/EBPα knockdown keratinocytes or control keratinocytes were synchronized by starvation as described above and either irradiated or not 6 h after release into the cell cycle. Cells were pulse-labeled with BrdU 2 h before collection, stained with anti-BrdU antibody and PI, and subjected to FACS analysis at the different time points indicated after release. The percentages of cells in G1 (lower left), S (top), and G2/M (lower right) phases of the cell cycle are shown. Values reported are from duplicates for nonirradiated samples (variance, <20%) and triplicates for irradiated samples (standard deviation < 20%). (B) C/EBPα expression level after indicated treatments. (C) C/EBPα knockdown keratinocytes or control keratinocytes were grown in regular medium and irradiated with UVB (10 mJ/cm2). At the indicated time points, caspase 3/7 activity was measured and corrected by total cell number. *, significantly different from UVB-irradiated control group as determined by Student's t test (P < 0.05).
DISCUSSION
We have demonstrated that C/EBPα is a p53-regulated DNA damage-inducible gene that has a role in the G1 checkpoint in epidermal keratinocytes. Our study is the first to characterize a function for C/EBPα in skin and it uncovers a previously unidentified role for C/EBPα in the cellular DNA damage response network, as well as a novel regulation of C/EBPα expression involving p53. Previous studies have demonstrated an important role for C/EBPα in growth arrest that is associated with differentiation and development. Our study expands the antiproliferative function of C/EBPα to now include growth arrest associated with DNA damage mediated by extrinsic factors. We observed that a variety of agents including UVB, MNNG, etoposide, and bleomycin, which produce different types of DNA damage, all induce C/EBPα in keratinocytes through a p53-dependent pathway. Thus, the induction of C/EBPα appears to be a general DNA damage response in keratinocytes. In contrast, UVB irradiation or MNNG or etoposide treatment of HepG2, NRK, or NIH 3T3 cells did not result in the induction of C/EBPα, suggesting that the induction of C/EBPα by DNA damage is cell type specific. Epidermal keratinocytes constitute the outer epithelial surface of the body; as such, these cells are exposed to a variety of extrinsic factors that have the potential to damage DNA. Accordingly, it is possible that keratinocytes have evolved additional mechanisms to deal with DNA damage in order to ensure the integrity of the genome. It will be of interest to examine whether DNA damage induces C/EBPα in other types of epithelial cells that are exposed to extrinsic DNA damaging agents, such as those cells that line the gastrointestinal, urinary, and respiratory tracts.
UVB treatment results in the posttranslational modification of the p53 protein and involves phosphorylation of multiple serine/threonine sites and acetylation sites located in the carboxy terminus of p53 (41). These complex and multiple modifications are important in stabilizing the p53 protein and enhancing the transcription activity of p53 and may also be involved in the determination of promoter specificity (2, 9). ATR is activated by UV-induced DNA damage and phosphorylates serine 15 of p53 (corresponding to serine 18 of mouse p53) (48). The phosphorylation of serine 15 has been reported to enhance p53 transcriptional activity through a mechanism involving acetylation (9) and to disrupt mdm2/p53 interaction, resulting in increased level of p53 (45). Our results showing that caffeine, a known ATM/ATR inhibitor, blocks UVB induction of C/EBPα and p53 accumulation support a role for ATR as one of the upstream kinases of p53 in the induction of C/EBPα. In addition, our observations that etoposide induces C/EBPα through a p53-dependent pathway suggest ATM may also participate in the induction of C/EBPα upstream of p53 in keratinocytes.
Multiple mechanisms can converge on DNA damage-induced cell cycle checkpoints. For example, p53-regulated proteins p21, 14-3-3σ, and GADD45 have roles in the G2/M checkpoint (8, 57). In contrast to the G2/M checkpoint in which numerous p53-regulated proteins have a role, p21 is considered the sole p53-dependent mediator of the G1 DNA damage checkpoint (54). However, we observed that C/EBPα knockdown keratinocytes have an attenuated G1 checkpoint after UV-induced DNA damage, indicating that C/EBPα is a novel p53-regulated mediator of the G1 checkpoint in keratinocytes. Thus, we have identified the C/EBPα gene as a second p53-regulated gene that participates in the G1 checkpoint. The antiproliferative function of C/EBPα is multifaceted and involves the up-regulation and activation of p21 (18, 51), interaction with retinoblastoma family proteins (49, 50), and repression of E2F (38, 46). Moreover, C/EBPα has been shown to block cell cycle progression independent of its DNA binding and transcriptional activity, through the formation of an inhibitory complex with cyclin-dependent kinase 4 (cdk4) and cdk2 (55). More recently, the antiproliferative activity of C/EBPα was shown to require a SWI/SNF chromatin remodeling complex, supporting a transcriptional basis for its antiproliferative activity (31). All of these mechanisms are consistent with our observation that C/EBPα has a role in the DNA damage-induced G1 checkpoint and indicate that C/EBPα regulates the G1 checkpoint through multiple mechanisms. However, it is unlikely that one of the mechanisms involves p21, as we observed that p21 levels were not decreased in UVB-treated C/EBPα knockdown cells (data not shown). Moreover, C/EBPα can inhibit cell cycle progression in p21-deficient cells (30), and p21 levels are not altered in livers of mice that are C/EBPα deficient (23).
The importance of C/EBPα in the regulation of growth arrest and differentiation is highlighted by recent studies in which C/EBPα was implicated as a human tumor suppressor gene. In human acute myeloid leukemia (AML), C/EBPα is inactivated by dominant negative mutation or through its association with oncoprotein AML-1-ETO (35, 59). The inactivation of C/EBPα is thought to result in a differentiation block of the granulocytic blasts. C/EBPα expression is reduced in hepatocellular carcinomas (60), lung cancer, and lung cancer cell lines (17), supporting a possible tumor suppressor function in these organs. We have observed that C/EBPα protein levels are greatly diminished or undetectable in mouse skin squamous cell carcinomas (SCCs) (33) and mouse skin SCC cell lines; reintroduction of C/EBPα in SCC cells inhibits cell proliferation (M. Shim and R. C. Smart, unpublished results). Taken together, our results suggest that the loss of C/EBPα expression may contribute to the altered growth characteristics of skin SCCs.
In terms of human skin cancer, more than 90% of SCCs and 50% of basal cell carcinomas (BCCs) contain UV signature mutations (C→T, CC→TT) in the p53 gene (15, 63). Clusters of mutant p53-containing preneoplastic keratinocytes have been identified in sun-exposed areas of human skin (21) and UVB-treated skin of mice (3). It has been proposed that additional multiple exposures to UVB allow these “initiated” cells to clonally expand because they are resistant to UVB-induced apoptosis (63). At the same time, UVB exposure of these mutant p53 keratinocytes would not result in p21 and C/EBPα induction; as such, the DNA damage G1 checkpoint would be compromised, allowing for the accumulation of additional critical mutations that contribute to SCC and BCC. Therefore, the loss of function of C/EBPα in the presence of UVB-induced DNA damage may contribute to the development of SCCs and BCCs that contain p53 mutations. It will also be of interest to examine C/EBPα for inactivating or dominant negative mutations in SCCs and BCCs that do not contain p53 mutations.
Previous studies have shown that p53 can modulate the transcription activity of C/EBPα and C/EBPβ (22, 26). Our results demonstrate that p53 is required for C/EBPα induction by MNNG, etoposide, bleomycin, and UVB in keratinocytes and that UVB treatment results in the binding of p53 to a p53 site in the C/EBPα distal promoter. This region of the C/EBPα promoter containing the p53 site can be stimulated by p53 in a promoter-reporter assay. Collectively, these results provide evidence that p53 directly stimulates the C/EBPα promoter; however, the fold stimulation is modest. It is therefore likely that there are other p53-dependent mechanisms that impinge upon UVB induction of C/EBPα. While further studies are required to determine if other mechanisms, either directly or indirectly involving p53, regulate the expression of C/EBPα, our study uncovers a previously unidentified role for the C/EBPα gene as a p53-regulated gene that links DNA damage to cell cycle arrest in keratinocytes. Our results suggest that the loss of UVB-induced C/EBPα expression may contribute to UVB-induced skin carcinogenesis.
Acknowledgments
We thank J. French (NIEHS) for the generous gift of the p53 newborn mice and Peter Johnson (NCI) for providing the C/EBPα constructs, φNX cells, and the C/EBPα promoter. We also thank T. Archer and H. Kinyama (NIEHS) for their help with the ChIP assay.
This work was supported by a grant (CA046637) from the National Cancer Institute.
REFERENCES
- 1.Antonson, P., and K. G. Xanthopoulos. 1995. Molecular cloning, sequence, and expression patterns of the human gene encoding CCAAT/enhancer binding protein alpha (C/EBP alpha). Biochem. Biophys. Res. Commun. 215:106-113. [DOI] [PubMed] [Google Scholar]
- 2.Appella, E., and C. W. Anderson. 2001. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 268:2764-2772. [DOI] [PubMed] [Google Scholar]
- 3.Berg, R. J., H. J. van Kranen, H. G. Rebel, A. de Vries, W. A. van Vloten, C. F. Van Kreijl, J. C. van der Leun, and F. R. de Gruijl. 1996. Early p53 alterations in mouse skin carcinogenesis by UVB radiation: immunohistochemical detection of mutant p53 protein in clusters of preneoplastic epidermal cells. Proc. Natl. Acad. Sci. USA 93:274-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birkenmeier, E. H., B. Gwynn, S. Howard, J. Jerry, J. I. Gordon, W. H. Landschulz, and S. L. McKinght. 1989. Tissue-specific expression, developmental regulation, and genetic mapping of the gene encoding CCAAT/enhancer binding protein. Genes Dev. 3:1146-1156. [DOI] [PubMed] [Google Scholar]
- 5.Bissonnette, N., and D. J. Hunting. 1998. p21-induced cycle arrest in G1 protects cells from apoptosis induced by UV-irradiation or RNA polymerase II blockage. Oncogene 16:3461-3469. [DOI] [PubMed] [Google Scholar]
- 6.Brash, D. E. 1997. Sunlight and the onset of skin cancer. Trends Genet. 13:410-414. [DOI] [PubMed] [Google Scholar]
- 7.Brash, D. E., J. A. Rudolph, J. A. Simon, A. Lin, G. J. McKenna, H. P. Baden, A. J. Halperin, and J. Ponten. 1991. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 88:10124-10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan, T. A., P. M. Hwang, H. Hermeking, K. W. Kinzler, and B. Vogelstein. 2000. Cooperative effects of genes controlling the G2/M checkpoint. Genes Dev. 14:1584-1588. [PMC free article] [PubMed] [Google Scholar]
- 9.Chao, C., M. Hergenhahn, M. D. Kaeser, Z. Wu, S. Saito, R. Iggo, M. Hollstein, E. Appella, and Y. Xu. 2003. Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J. Biol. Chem. 278:41028-41033. [DOI] [PubMed] [Google Scholar]
- 10.de Gruijl, F. R., H. J. van Kranen, and L. H. Mullenders. 2001. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J. Photochem. Photobiol. B 63:19-27. [DOI] [PubMed] [Google Scholar]
- 11.Diehl, A. M., D. C. Johns, S. Yang, H. Lin, M. Yin, L. A. Matelis, and J. H. Lawrence. 1996. Adenovirus-mediated transfer of CCAAT/enhancer-binding protein-α identifies a dominant antiproliferative role for this isoform in hepatocytes. J. Biol. Chem. 271:7343-7350. [DOI] [PubMed] [Google Scholar]
- 12.Dumaz, N., and D. W. Meek. 1999. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 18:7002-7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fornace, A. J., Jr., D. W. Nebert, M. C. Hollander, J. D. Luethy, M. Papathanasiou, J. Fargnoli, and N. J. Holbrook. 1989. Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol. Cell. Biol. 9:4196-4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freytag, S. O., D. L. Paielli, and J. D. Gilbert. 1994. Ectopic expression of the CCAAT/enhancer-binding protein α promotes the adipogenic program in a variety of mouse fibroblastic cells. Genes Dev. 8:1654-1663. [DOI] [PubMed] [Google Scholar]
- 15.Gailani, M. R., D. J. Leffell, A. Ziegler, E. G. Gross, D. E. Brash, and A. E. Bale. 1996. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J. Natl. Cancer Inst. 88:349-354. [DOI] [PubMed] [Google Scholar]
- 16.Garmyn, M., M. Yaar, N. Holbrook, and B. A. Gilchrest. 1991. Immediate and delayed molecular response of human keratinocytes to solar-simulated irradiation. Lab. Investig. 65:471-478. [PubMed] [Google Scholar]
- 17.Halmos, B., C. S. Huettner, O. Kocher, K. Ferenczi, D. D. Karp, and D. G. Tenen. 2002. Down-regulation and antiproliferative role of C/EBPα in lung cancer. Cancer Res. 62:528-534. [PubMed] [Google Scholar]
- 18.Harris, T. E., J. H. Albrecht, M. Nakanishi, and G. J. Darlington. 2001. CCAAT/enhancer-binding protein-α cooperates with p21 to inhibit cyclin-dependent kinase-2 activity and induces growth arrest independent of DNA binding. J. Biol. Chem. 276:29200-29209. [DOI] [PubMed] [Google Scholar]
- 19.Hennings, H., D. Michael, C. Cheng, P. Steinert, K. Holbrook, and S. H. Yuspa. 1980. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell 19:245-254. [DOI] [PubMed] [Google Scholar]
- 20.Hulla, J. E., J. E. French, and J. K. Dunnick. 2001. Chromosome 11 allelotypes reflect a mechanism of chemical carcinogenesis in heterozygous p53-deficient mice. Carcinogenesis 22:89-98. [DOI] [PubMed] [Google Scholar]
- 21.Jonason, A. S., S. Kunala, G. J. Price, R. J. Restifo, H. M. Spinelli, J. A. Persing, D. J. Leffell, R. E. Tarone, and D. E. Brash. 1996. Frequent clones of p53 keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 93:14025-14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubicka, S., F. Kuhnel, L. Zender, K. L. Rudolph, J. Plumpe, M. Manns, and C. Trautwein. 1999. p53 represses CAAT enhancer-binding protein (C/EBP)-dependent transcription of the albumin gene. A molecular mechanism involved in viral liver infection with implications for hepatocarcinogenesis. J. Biol. Chem. 274:32137-32144. [DOI] [PubMed] [Google Scholar]
- 23.Lee, Y. H., B. Sauer, P. F. Johnson, and F. J. Gonzalez. 1997. Disruption of the c/ebpα gene in adult mouse liver. Mol. Cell. Biol. 17:6014-6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehman, T. A., R. Modali, P. Boukamp, J. Stanek, W. P. Bennett, J. A. Welsh, R. A. Metcalf, M. R. Stampfer, N. Fusenig, E. M. Rogan, et al. 1993. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis 14:833-839. [DOI] [PubMed] [Google Scholar]
- 25.Lin, F. T., and M. D. Lane. 1994. CCAAT/enhancer binding protein alpha is sufficient to initiate the 3T3-L1 adipocyte differentiation program. Proc. Natl. Acad. Sci. USA 91:8757-8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margulies, L., and P. B. Sehgal. 1993. Modulation of the human interleukin-6 promoter (IL-6) and transcription factor C/EBP beta (NF-IL6) activity by p53 species. J. Biol. Chem. 268:15096-15100. [PubMed] [Google Scholar]
- 27.Maytin, E. V., and J. F. Habener. 1998. Transcription factors C/EBPα, C/EBPβ, and CHOP (Gadd153) expressed during the differentiation program of keratinocytes in vivo and in vitro. J. Investig. Dermatol. 110:238-246. [DOI] [PubMed] [Google Scholar]
- 28.Medrano, E. E., S. Im, F. Yang, and Z. A. Abdel-Malek. 1995. Ultraviolet B light induces G1 arrest in human melanocytes by prolonged inhibition of retinoblastoma protein phosphorylation associated with long-term expression of the p21Waf-1/SDI-1/Cip-1 protein. Cancer Res. 55:4047-4052. [PubMed] [Google Scholar]
- 29.Miller, D. L., and M. A. Weinstock. 1994. Nonmelanoma skin cancer in the United States: incidence. J. Am. Acad. Dermatol. 30:774-778. [DOI] [PubMed] [Google Scholar]
- 30.Muller, C., M. Alunni-Fabbroni, E. Kowenz-Leutz, X. Mo, M. Tommasino, and A. Leutz. 1999. Separation of C/EBPα-mediated proliferation arrest and differentiation pathways. Proc. Natl. Acad. Sci. USA 96:7276-7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller, C., C. F. Calkhoven, X. Sha, and A. Leutz. 2004. The CCAAT enhancer-binding protein alpha (C/EBPα) requires a SWI/SNF complex for proliferation arrest. J. Biol. Chem. 279:7353-7358. [DOI] [PubMed] [Google Scholar]
- 32.Neades, R., L. Cox, and J. C. Pelling. 1998. S-phase arrest in mouse keratinocytes exposed to multiple doses of ultraviolet B/A radiation. Mol. Carcinog. 23:159-167. [PubMed] [Google Scholar]
- 33.Oh, H.-S., and R. C. Smart. 1998. Expression of CCAAT/enhancer binding protein (C/EBP) is associated with squamous differentiation in epidermis and isolated primary keratinocytes and is altered in skin neoplasms. J. Investig. Dermatol. 110:939-945. [DOI] [PubMed] [Google Scholar]
- 34.O'Rourke, J., R. Yuan, and J. DeWille. 1997. CCAAT/enhancer-binding protein-delta (C/EBP-δ) is induced in growth-arrested mouse mammary epithelial cells. J. Biol. Chem. 272:6291-6296. [DOI] [PubMed] [Google Scholar]
- 35.Pabst, T., B. U. Mueller, P. Zhang, H. S. Radomska, S. Narravula, S. Schnittger, G. Behre, W. Hiddemann, and D. G. Tenen. 2001. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-α (C/EBPα), in acute myeloid leukemia. Nat. Genet. 27:263-270. [DOI] [PubMed] [Google Scholar]
- 36.Park, D. S., E. J. Morris, L. A. Greene, and H. M. Geller. 1997. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J. Neurosci. 17:1256-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piontkewitz, Y., S. Enerback, and L. Hedin. 1993. Expression and hormonal regulation of the CCAAT enhancer binding protein-alpha during differentiation of rat ovarian follicles. Endocrinology 133:2327-2333. [DOI] [PubMed] [Google Scholar]
- 38.Porse, B. T., T. A. Pedersen, X. Xu, B. Lindberg, U. M. Wewer, L. Friis-Hansen, and C. Nerlov. 2001. E2F repression by C/EBPα is required for adipogenesis and granulopoiesis in vivo. Cell 107:247-258. [DOI] [PubMed] [Google Scholar]
- 39.Radomska, H. S., C. S. Huettner, P. Zhang, T. Cheng, D. T. Scadden, and D. G. Tenen. 1998. CCAAT/enhancer binding protein α is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol. Cell. Biol. 18:4301-4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramji, D. P., and P. Foka. 2002. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem. J. 365:561-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saito, S., H. Yamaguchi, Y. Higashimoto, C. Chao, Y. Xu, A. J. Fornace, Jr., E. Appella, and C. W. Anderson. 2003. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J. Biol. Chem. 278:37536-37544. [DOI] [PubMed] [Google Scholar]
- 42.Sarkaria, J. N., E. C. Busby, R. S. Tibbetts, P. Roos, Y. Taya, L. M. Karnitz, and R. T. Abraham. 1999. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 59:4375-4382. [PubMed] [Google Scholar]
- 43.Schreiber, E., P. Matthias, M. M. Muller, and W. Schaffner. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts,’ prepared from a small number of cells. Nucleic Acids Res. 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott, L. M., C. I. Civin, P. Roth, and A. D. Friedman. 1992. A novel temporal expression pattern of three C/EBP family members in differentiating myelomonocytic cells. Blood 80:1725-1735. [PubMed] [Google Scholar]
- 45.Shieh, S. Y., M. Ikeda, Y. Taya, and C. Prives. 1997. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91:325-334. [DOI] [PubMed] [Google Scholar]
- 46.Slomiany, B. A., K. L. D'Arigo, M. M. Kelly, and D. T. Kurtz. 2000. C/EBPα inhibits cell growth via direct repression of E2F-DP-mediated transcription. Mol. Cell. Biol. 20:5986-5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swart, G. W. M., J. J. M. van Groningen, F. van Ruissen, M. Bergers, and J. Schalkwijk. 1997. Transcription factor C/EBPα: novel sites of expression and cloning of the human gene. Biol. Chem. 378:373-379. [DOI] [PubMed] [Google Scholar]
- 48.Tibbetts, R. S., K. M. Brumbaugh, J. M. Williams, J. N. Sarkaria, W. A. Cliby, S. Y. Shieh, Y. Taya, C. Prives, and R. T. Abraham. 1999. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13:152-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Timchenko, N. A., M. Wilde, and G. J. Darlington. 1999. C/EBPα regulates formation of S-phase-specific E2F-p107 complexes in livers of newborn mice. Mol. Cell. Biol. 19:2936-2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Timchenko, N. A., M. Wilde, P. Iakova, J. H. Albrecht, and G. J. Darlington. 1999. E2F/107 and E2F/p130 complexes are regulated by C/EBPα in 3T3-L1 adipocytes. Nucleic Acids Res. 27:3621-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Timchenko, N. A., M. Wilde, M. Nakanishi, J. R. Smith, and G. J. Darlington. 1996. CCAAT/enhancer-binding protein alpha (C/EBPα) inhibits cell proliferation through the p21 (WAF-1/CIP-1/SDI-1) protein. Genes Dev. 10:804-815. [DOI] [PubMed] [Google Scholar]
- 52.Umek, R. M., A. D. Friedman, and S. L. McKnight. 1991. CCAAT-enhancer binding protein: a component of a differentiation switch. Science 251:288-292. [DOI] [PubMed] [Google Scholar]
- 53.Vincent, F., G. Deplanque, J. Ceraline, B. Duclos, and J. P. Bergerat. 1999. p53-independent regulation of cyclin B1 in normal human fibroblasts during UV-induced G2-arrest. Biol. Cell 91:665-674. [PubMed] [Google Scholar]
- 54.Waldman, T., K. W. Kinzler, and B. Vogelstein. 1995. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 55:5187-5190. [PubMed] [Google Scholar]
- 55.Wang, H., P. Iakova, M. Wilde, A. Welm, T. Goode, W. J. Roesler, and N. A. Timchenko. 2001. C/EBPα arrests cell proliferation through direct inhibition of cdk2 and cdk4. Mol. Cell 8:817-828. [DOI] [PubMed] [Google Scholar]
- 56.Wang, X., E. Scott, C. L. Sawyers, and A. D. Friedman. 1999. C/EBPα bypasses granulocyte colony-stimulating factor signals to rapidly induce PU.1 gene expression, stimulate granulocytic differentiation, and limit proliferation in 32D cl3 myeloblasts. Blood 94:560-571. [PubMed] [Google Scholar]
- 57.Wang, X. W., Q. Zhan, J. D. Coursen, M. A. Khan, H. U. Kontny, L. Yu, M. C. Hollander, P. M. O'Connor, A. J. Fornace, Jr., and C. C. Harris. 1999. GADD45 induction of a G2/M cell cycle checkpoint. Proc. Natl. Acad. Sci. USA 96:3706-3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang, X. Z., B. Lawson, J. W. Brewer, H. Zinszner, A. Sanjay, L. J. Mi, R. Boorstein, G. Kreibich, L. M. Hendershot, and D. Ron. 1996. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 16:4273-4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Westendorf, J. J., C. M. Yamamoto, N. Lenny, J. R. Downing, M. E. Selsted, and S. W. Hiebert. 1998. The t(8;21) fusion protein, AML-1-ETO, associated with C/EBP-α, inhibits C/EBP-α-dependent transcription and blocks granulocytic differentiation. Mol. Cell. Biol. 18:322-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu, L., L. Hui, S. Wang, J. Gong, Y. Jin, Y. Wang, Y. Ji, X. Wu, Z. Han, and G. Hu. 2001. Expression profiling suggested a regulatory role of liver-enriched transcription factors in human hepatocellular carcinoma. Cancer Res. 61:3176-3181. [PubMed] [Google Scholar]
- 61.Zhu, S., H. S. Oh, M. Shim, E. Sterneck, P. F. Johnson, and R. C. Smart. 1999. C/EBPβ modulates the early events of keratinocyte differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol. Cell. Biol. 19:7181-7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu, S., K. Yoon, E. Sterneck, P. F. Johnson, and R. C. Smart. 2002. CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc. Natl. Acad. Sci. USA 99:207-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ziegler, A., A. S. Jonason, D. J. Leffell, J. A. Simon, H. W. Sharma, J. Kimmelman, L. Remington, T. Jacks, and D. E. Brash. 1994. Sunburn and p53 in the onset of skin cancer. Nature 372:773-776. [DOI] [PubMed] [Google Scholar]