

Abstract
mid1p is a key factor for the central positioning of the cytokinetic ring in Schizosaccharomyces pombe. In interphase and early mitosis, mid1p forms a medial cortical band overlying the nucleus, which may represent a landmark for cytokinetic ring assembly. It compacts before anaphase into a tight ring with other cytokinetic ring components. We show here that mid1p binds to the medial cortex by at least two independent means. First, mid1p C-terminus association with the cortex requires a putative amphipathic helix adjacent to mid1p nuclear localization sequence (NLS), which is predicted to insert directly into the lipid bilayer. This association is stabilized by the polybasic NLS. mid1p mutated within the helix and the NLS forms abnormal filaments in early mitosis that are not properly anchored to the medial cortex. Misplaced rings assemble in late mitosis, indicating that mid1p C-terminus binding to membranes stabilizes cytokinetic ring position. Second, the N terminus of mid1p has the ability to associate faintly with the medial cortex and is sufficient to form tight rings. In addition, we show that mid1p oligomerizes. We propose that membrane-bound oligomers of mid1p assemble recruitment “platforms” for cytokinetic ring components at the medial cortex and stabilize the ring position during its compaction.
Cytokinesis is the last event of the cell division cycle, which allows the physical separation of the two sister cells. One early and critical step in this process is the definition of the orientation of the plane of division, since a proper orientation, perpendicular to the spindle elongation axis, is crucial for a faithful segregation of chromosomes between sister cells. In addition, division plane orientation also controls cell shape, tissue organization, or cell fate during development. For instance, during Caenorhabditis elegans early development, reorientation of the plane of division by rotation of the mitotic spindle in P cells allows the formation of the germ cell lineage by inheritance of specific determinants concentrated at the posterior pole of the embryo (18).
The fission yeast Schizosaccharomyces pombe offers a simple system for studying cytokinesis and division plane specification. Fission yeast cells have a well-defined rod shape and a stereotyped medial division to form symmetric daughter cells. The position of the division plane always coincides with the position of the nucleus, at the center in wild-type cells, or off the center in mutants with abnormal microtubules that are defective in nuclear positioning (12, 35, 41). This suggests that the nucleus or the microtubules play a major role in positioning the division plane.
As in higher eukaryotes, cell division depends on the assembly of a cytokinetic ring containing actin and type II myosin, as well as many other components involved in the nucleation of F-actin and the shaping F-actin and myosin II filaments into a compact ring (for reviews, see references 16, 19, and 20).
Several phases have been described during ring formation (44). Ten minutes before SPB separation, myosin II (myo2p heavy chain and cdc4p and rlc1p, two light chains) (24, 25, 28, 29, 31) is recruited to the cell cortex in a broad band overlying the nucleus, shortly followed by the IQGAP protein rng2 (14). When duplicated SPBs separate, actin nucleation factors cdc12p (a formin) (11) and cdc15p (a PSTPIP protein) (15) are in turn recruited in a central spot that may initiate the nucleation of an F-actin meshwork at the cell center (2, 9, 10). Tropomyosin (5) and alpha-actinin (43) are recruited a few minutes later. Compaction of all components into a tight ring then starts and is completed 10 min later by anaphase B onset.
One important question that remains to be understood is what spatial cues defining the future position of the division plane allow the recruitment of ring components to the central cortex. One key factor is mid1/dmf1p since mid1 mutants harbor specific defects in cytokinetic ring positioning (12, 37). mid1 protein actively shuttles during interphase between the nucleus and a central cortical band overlying the nucleus. The position of mid1p cortical band is coupled to the position of the nucleus and its formation in early G2, well before the entry into mitosis (33), is consistent with a role for mid1p as a cortical landmark for cytokinetic ring assembly. In early mitosis, mid1p cortical band is reinforced as the nuclear pool disappears in a plo1p-dependent manner (3). mid1p then compacts into a tight ring, together with other components of the cytokinetic ring. In late anaphase, it dissociates from the ring as contraction starts. Recent data suggest that mid1p may directly mediate myosin II recruitment to the central cortex (30, 44).
The mechanisms that control mid1p specific association with the central cortex and the coupling between the position of the nucleus and of mid1p cortical band are not understood. We have performed experiments to address how mid1p associates with the cell cortex. We report that mid1p N- and C-terminal domains both contain cortex-targeting motifs. In particular, mid1p C terminus contains an amphipathic helix that may act as a direct membrane-binding domain that stabilizes the position of the cytokinetic ring in early mitosis. In addition, we report that mid1p can form oligomers and that both N- and C-terminal domains can self-assemble. A model for how mid1p may associate with the cell cortex is proposed.
MATERIALS AND METHODS
Yeast strains and genetic methods.
S. pombe strains used in the present study are listed in Table 1. All strains are isogenic to 972. Standard S. pombe genetic techniques and cultures were performed as described at http://www.sanger.ac.uk/PostGenomics/S_pombe/docs/nurse_lab_manual.pdf.
TABLE 1.
Strains used in the study
| Strain | Genotype | Source or reference |
|---|---|---|
| FC418 | ade6-M210 ura4-D18 leu1-32 h− | F. Chang lab |
| AP490 | Same as FC418 + pDS473a | This study |
| AP761 | Same as FC418 + pAP183 (pnmt GST-mid1, ura4+) | This study |
| AP765 | Same as FC418 + pAP184 (pnmt Cter-GST, ura4+) | This study |
| AP810 | Same as FC418 + pAP187 (pnmt GST-Nter, ura4+) | This study |
| AP1383 | Same as FC418 + pSM78 (pnmt GFP-Helix-NLS, ura4+) | This study |
| AP1387 | Same as FC418 + pSM85 (pnmt GFP-(Helix-NLS)2, ura4+) | This study |
| SP1601 | ade6-M216 leu1-32 ura4-D18 dmf1::ura4+ h− | 37 |
| AP85 | Same as SP1601 + pAP97 integrated (pmid mid1-506, leu1+) | This study |
| AP616 | Same as SP1601 + pAP144 integrated (pmid Cter-GFP, leu1+) | This study |
| AP621 | Same as SP1601 + pAP144 integrated (pmid Cter-GFP, leu1+) | This study |
| AP528 | Same as SP1601 + pAP146 integrated (pmid mid1-GFP, leu1+) | This study |
| AP583 | Same as SP1601 + pAP159 integrated (pmid Helix* mid1-GFP, leu1+) | This study |
| AP630 | Same as SP1601 + pAP163 integrated (pmid Helix* NLS* mid1-GFP, leu1+) | This study |
| AP734 | Same as SP1601 + pAP167 integrated (pmid NLS* mid1-GFP, leu1+) | This study |
| AP826 | Same as SP1601 + pAP188 integrated (pmid Helix* NLS* Cter-GFP, leu1+) | This study |
| AP829 | Same as SP1601 + pAP189 integrated (pmid Helix* Cter-GFP, leu1+) | This study |
| AP1111 | Same as SP1601 + pAP200 integrated (pmid NLS* Cter-GFP, leu1+) | This study |
| AP998 | Same as SP1601 + pSM 26 integrated (pmid GFP-Nter, leu1+) | This study |
| AP1072 | ade6-M216 leu1-32 ura4-D18 dmf1::KanMX4 h− | This study |
| AP1055 | Same as AP1072 + pSM31 (pnmt** GST-mid1, LEU2) | This study |
| AP1059 | Same as AP1072 + pSM32 (pnmt** Cter-GST, LEU2) | This study |
| AP1061 | Same as AP1072 + pSM33 (pnmt** GST-Nter, LEU2) | This study |
| AP1063 | Same as AP1072 + pSM34 (pnmt** mid1-GFP, ura4+) | This study |
| AP1066 | Same as AP1072 + pSM35 (pnmt** Cter-GFP, ura4+) | This study |
| AP1069 | Same as AP1072 + pSM36 (pnmt** GFP-Nter, ura4+) | This study |
| AP1160 | Same as AP1072 + pSM31 + pSM34 | This study |
| AP1161 | Same as AP1072 + pSM31 + pSM36 | This study |
| AP1084 | Same as AP1072 + pSM31 + pSM35 | This study |
| AP1092 | Same as AP1072 + pSM32 + pSM36 | This study |
| AP1088 | Same as AP1072 + pSM33 + pSM35 | This study |
| AP1094 | Same as AP1072 + pSM33 + pSM36 | This study |
| YDM602 | mid1-13myc::ura4+ ura4-D18 leul-32 ade6-210 h+ | 3,4 |
| AP599 | Same as YDM602 + pAP146 integrated (pmid mid1-GFP, leu1+) | This study |
| MBY498 | myo2GFP::ura4+ ura4-D18 leu1-32 h− | M. Balasubramanian |
| FC630 | 41nmt-cdc12-GFP::ura4+ ura4-D18 h− | 10 |
| AP1178 | dmf1::KanMX4 41nmt-cdc12-GFP::ura4+ ura4-D18 leu1-32 ade6-M216 h− + pAP210 integrated (pmid Helix* NLS* mid1-mRFP, leu1+) | This study |
| AP1192 | dmf1::KanMX4 myo2GFP::ura4+ leu1-32 ura4-D18 h+ + pAP210 integrated (pmid Helix* NLS* mid1-mRFP, leu1+) | This study |
| HU0375 | pom152-GFP::kanMX leu1-32 ura4D18 ade6 M210 h− | 6 |
Yeast transformations were performed by using either a lithium acetate-dimethyl sulfoxide method (4) or electroporation (23).
Production of mid1 constructs.
PCR was performed by using PCR high-fidelity system (Roche Diagnostics, Mannheim, Germany), except when fragments were amplified from genomic DNA, in which case Tfl polymerase (Promega, Madison, Wis.) was used.
mid1-green fluorescent protein (GFP).
A XhoI-NotI fragment from pAP33 (33) containing the mid1 open reading frame (ORF) was subcloned into pSGP572a (generous gift from S. Forsburg) to create pAP119 (pnmt mid1-GFP, ura4+).
Cter-GFP.
A XhoI-NotI fragment corresponding to amino acids 507 to 920 of mid1p was amplified by PCR from pAP33 and inserted at XhoI and NotI sites in pSGP572a to create pAP120 (pnmt Cter-GFP, ura4+).
GFP-Nter.
To create a fusion between GFP and amino acids 1 to 506 of mid1p, a BglII-SacI fragment from pAP187 (see below) was inserted into pAP73 (33) to create pSM24.
To place these constructs under the control of mid1 endogenous promoter in an integrative vector, a XhoI-SalI fragment of genomic DNA containing 1,060 bp upstream of mid1 ORF was amplified by PCR and inserted at SalI site of the integrative vector pJK148 to create pAP140.
Then, a XhoI-SacI fragment from pAP119, pAP120 and pSM24 was introduced into pAP140, to create pAP146 (pmid1 mid1-GFP, leu1+), pAP144 (pmid1 Cter-GFP, leu1+), and pSM26 (pmid GFP-Nter, leu1+).
Helix* mid1-GFP, Helix* NLS* mid1-GFP and Helix* NLS* mid1-mRFP.
To produce Helix* mid1 and Helix* NLS* mid1 constructs, in which amino acids 681 to 688 (RKFFDKLF) were replaced by RKAADKAA, mutagenesis was performed on pAP32 containing wild-type mid1 and pAP59 containing NLS* mid1 (33) with QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, Calif.) with the forward oligonucleotide 5′-CGAAATCCAAAGCTAGGAAAGCTGCTGACAAGGCTGCCAACCGAC and the reverse oligonucleotide 5′-GTCGGTTGGCAGCCTTGTCAGCAGCTTTCCTAGCTTTGGATTTCG to create pAP141 and pAP142, respectively. A NarI-PstI fragment from pAP141 was subcloned into pAP32 to create pAP152. Finally, a BglII-XbaI fragment from pAP152 or pAP142 was introduced into pAP146 to produce pAP159 (pmid Helix* mid1-GFP, leu1+) and pAP163 (pmid Helix*NLS* mid1-GFP, leu1+). To produce pAP210 (pmid Helix*NLS* mid1-mRFP), a NotI-SalI fragment from pAP163 was replaced by a NotI-SalI fragment containing mRFP generated by PCR from pRSET B mRFP (a generous gift from R. Tsien [8]). Finally, a NarI-XbaI fragment from pAP142 and pAP152 was subcloned in pAP144, to create, respectively, pAP188 (pmid Helix*NLS* Cter-GFP, leu1+) and pAP189 (pmid Helix* Cter-GFP, leu1+).
All integrative plasmids were targeted by a NruI digest in the leu1 gene of pJK148 before transformation into the mid1Δ strain SP1601.
To obtain AP1178 and AP1192 strains, pAP210 was first integrated into AP1072 strain (dmf1Δ::kanMX4 ade6-M216 leu1-32 ura4-D18 h−; see below). The resulting strain was then crossed with AP1119 (41nmt-cdc12-GFP::ura4+ ura4-D18 leu1-32 h+) or AP1120 (myo2GFP::ura4+ leu1-32 ura4-D18 h+), themselves issued from a cross between FC630 and MBY498 strains (see Table 1) and a h+ ade6-M216 ura4-D18 leu1-32 strain.
GFP and GST-tagged mid1p expression vectors.
To create pAP183 (pnmt-GST-mid1), a NotI-SacI fragment from pAP34 containing mid1 ORF (33) was inserted into pDS473a (a generous gift from Susan Forsburg).
To produce pAP184 (pnmt Cter-GST), a XhoI-NotI fragment from pAP120 was introduced into pDS472a (a generous gift from Susan Forsburg).
To create pAP187 (pnmt GST-Nter), first a BamHI-SalI fragment corresponding to amino acids 1 to 300 of mid1p was inserted into pDS473a (pAP185), and then a BglII-SacI fragment from pAP60 (33) containing amino acids 74 to 500 of mid1p was subcloned in pAP185. These plasmids were transformed into FC418 strain.
To create pSM31 (pnmt** GST-mid1, LEU2), pSM32 (pnmt** Cter-GST, LEU2), and pSM33 (pnmt** GST-Nter, LEU2), XhoI-SacI fragments from pAP183, pAP184, and pAP187, respectively, were subcloned into pREP81x. Similarly, XhoI-SacI fragments from pAP119, pAP120, and pSM26 were subcloned into pSLF373 to produce pSM34 (pnmt** mid1-GFP, ura4+), pSM35 (pnmt** Cter-GFP, ura4+), and pSM36 (pnmt** GFP-Nter, ura4+).
These plasmids were transformed into AP1072 strain. This strain was obtained by transforming strain SP1601 (mid1Δ::ura4+, generous gift from V. Simanis) by SacII-linearized pSM29, a pBluescript plasmid containing ura4+ ORF (insertion at HindIII site) inside which the KanMX4 cassette from pFa6KanMX4 (BglII-SacI blunted fragment) had been inserted between BsgI and AvrII blunted sites.
Finally, to create pSM78 (pnmt GFP-Helix-NLS, ura4+), a 46-pb linker encoding the Helix-NLS peptide (RKFFDKLFNRRKKRK) was ligated between SalI and SmaI sites of pSGP573 (generous gift from Susan Forsburg), creating a linker of 8 amino acids (ACGRSRST) between GFP and Helix-NLS peptide. To create pSM85 [pnmt GFP-(Helix-NLS)2, ura4+], a second linker of 46 pb encoding the Helix-NLS peptide was inserted between the XhoI and NotI sites in pSM78, removing GFP ORF. Then, a XhoI-SalI fragment from pSGP573 containing GFP was reintroduced at the XhoI site. In this construct, the first copy of the Helix-NLS peptide was preceded by eight amino acids (ACGRSRSM) and the second copy by a seven amino acids (GGRSRST). These plasmids were transformed into FC418.
Protein extracts and immunoblotting.
Total extracts were prepared as described in reference 32 except that cells were broken by two cycles of 20 s of vortexing at maximum speed in a FastPrep FP120A Instrument (Qbiogene, Inc., Carlsbad, Calif.).
Determination of protein concentration and blotting were performed as described in reference 32. Mid1p was revealed by using anti-mid1p affinity-purified immunoglobulins (1/150) or an anti-GFP MAb (Roche; 1/500), peroxidase-coupled anti-rabbit or anti-mouse immunoglobulins (1/10,000; Jackson Immunoresearch, West Grove, Pa.), and a chemiluminescence kit (Pierce, Rockford, Ill.). Tubulin (loading control) was revealed by using monoclonal antibody (MAb) TAT1 (1/2,000; a generous gift from K. Gull).
Soluble extracts were prepared as follows. S. pombe cells grown at an optical density at 595 nm (OD595) of 0.5 were harvested, washed in STOP buffer (36), and broken in NP-40 buffer (0.6 ml per 109 cells, 50 mM HEPES [pH 7.5], 100 mM NaCl, 1 mM EDTA, 1% NP-40, 20 mM glycerophosphate, 50 mM NaF, 0.1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, EDTA-free complete protease inhibitor tablets [Roche]) by using a FastPrep FP120A instrument (Qbiogene; two cycles of 20 s at maximum speed). Lysates were then spun at 10,000 × g for 10 min at 4°C, and supernatants were recovered.
Immunoprecipitation experiments.
Immunoprecipitation experiments were performed by using magnetic M-280 sheep anti-mouse IgG Dynabeads (Dynal Biotech, Oslo, Norway). Anti-GFP MAb (6 μg) was coupled to the beads according to the manufacturer's instructions. First, 500 μl of soluble extract was incubated with the beads for 2 h at 4°C; then, the mixture was washed five times with NP-40 buffer, and the beads were resuspended in 2× sample buffer. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting were performed with anti-GFP MAb (1/500), anti-myc MAb 9E10 (1/500; a generous gift from Lucien Cabanié), and anti-GST immunoglobulins produced in rabbits against a glutathione S-transferase (GST) fusion protein (GST-pep3) and affinity purified on a GST column (1/1,000) (7).
Sucrose gradients.
A total of 150 μl of soluble extract was loaded on a 20 to 35% (wt/wt) linear sucrose gradient (500 μl) in UltraClear 5-by-41-mm centrifugal tubes (Beckman) and centrifuged at 100,000 × g during 16 h at 4°C. Molecular weight standards (Roche) were sedimented in a parallel gradient. Fractions of 20 μl were collected from top to bottom and analyzed by Coomassie blue staining for molecular weight standards or Western blotting with anti-mid1p immunoglobulin (1/150) or anti-GFP MAb (1/500).
Purification of overexpressed GST fusion proteins.
A total of 6 ml of soluble extract was mixed with glutathione-Sepharose beads (Amersham Biosciences, Uppsala, Sweden) according to the manufacturer's instructions for 2 h at 4°C. Beads were washed five times with NP-40 buffer, and GST proteins were eluted from the beads by incubation in elution buffer (50 mM Tris, 10 mM reduced glutathione; pH 8) for 45 min at room temperature. Eluates were directly subjected to SDS-PAGE, and gels were stained with colloidal blue (GST, Cter-GFP; Invitrogen, Carlsbad, Calif.) or loaded on 20 to 35% sucrose gradients. Fractions containing >450-kDa (GST-mid1) or 240-kDa (GST-Nter) complexes were pooled. Trichloroacetic acid (25%) was added, followed by a 30-min incubation on ice with periodic shaking. Tubes were centrifuged at 18,000 × g for 30 min at 4°C. Pellets were washed once with ice-cold acetone containing 0.05 N HCl and once with ice-cold acetone. Finally, pellets were dried and resuspended in sample buffer. Pellets were analyzed by SDS-PAGE, and gels were stained with colloidal blue.
Cell fractionation.
Spheroplasts were first produced as follows: 100 ml of cells grown in YE5S at an OD595 nm of 0.5 were harvested by centrifugation. The cell pellet was resuspended in 5 ml of buffer 1 (20 mM citrate, 20 mM Na2HPO4, 40 mM EDTA). Cells were pelleted again, resuspended in 2.5 ml of buffer 2 (50 mM citrate, 50 mM Na2HPO4, 1.2 M sorbitol) containing 12.5 mg of lysing enzymes (L1412; Sigma), and incubated at 37°C until cell wall digestion was completed in 90% of cells (about 1 h, as judged by rounding up and lysis of an aliquot of cells in H2O). Then, 17.5 ml of ice-cold buffer 3 was added (50 mM HEPES [pH 7.5], 50 mM NaCl, 1.2 M sorbitol). Spheroplasts were centrifuged at 850 × g for 5 min at 4°C and washed two times in 40 ml of ice-cold buffer 3. Spheroplasts were then lysed in 1 ml of hypotonic lysis buffer (50 mM HEPES [pH 7.5], 100 mM NaCl, 1 mM EDTA, 20 mM β-glycerophosphate, 50 mM NaF, 0.1 mM Na3VO4, 1 mM PMSF, EDTA-free complete protease inhibitor tablets) for 5 min at 4°C. Lysate was split into four tubes and centrifuged at 20,000 × g for 20 min at 4°C, and supernatants (S0) were collected and mixed with 8× sample buffer. Pellets were resuspended in lysis buffer (S1-P1) or lysis buffer supplemented with 1 M NaCl (S2-P2), 1% NP-40 (S3-P3), or both (S4-P4) for 5 min at 4°C and then centrifuged at 20,000 × g for 20 min at 4°C. Supernatants and pellets were finally mixed with 8× and 1× sample buffer, respectively, and subjected to SDS-PAGE and immunoblotting with anti-mid1p immunoglobulin (1/150), anti-GFP MAb (1/500), MAb TAT1 for tubulin (1/2,000), or anti-BIP serum (1/20,000; a generous gift from A. Pidoux and J. Armstrong).
Fluorescence microscopy and time-lapse video microscopy.
For GFP fluorescence, differential interference contrast (DIC) imaging, and time-lapse video microscopy, cells were grown in YE5S liquid medium at room temperature without shaking. Then, 2 μl of cells was mounted directly between the slide and coverslip or mounted on 2% EMM agar pads as described previously (42). Cells overexpressing GFP-Helix-NLS (AP1383) and GFP-(Helix-NLS)2 (AP1387) were grown for 20 h in absence of thiamine in liquid Edinburgh minimum medium (EMM) supplemented with adenine and leucine. Cells expressing Helix* NLS* mid1-mRFP were grown overnight at 30°C in patches on YE5S plates (AP1192) or EMM plates supplemented with adenine (AP1178) and then placed at 20°C for 6 h before acquisition. Cells were resuspended in liquid medium, mounted as before, and imaged immediately.
Three-dimensional deconvolution video microscopy was performed by using an upright motorized microscope (DM RXA2; Leica Microsystems, Mannheim, Germany) equipped with an oil immersion ×100/N.A. 1.4 Plan-Apochromat objective lens and a cooled interline charge-coupled device detector (Roper Coolsnap HQ). Z positioning was accomplished by using a piezoelectric driver (LVDT; Physik Instruments, Waldbronn, Germany) mounted underneath the objective lens. The whole system was steered by using Metamorph software (Universal Imaging Corp., Downingtown, Pa.). Deconvolution was performed by using the Metamorph PSF-based iterative constrained algorithm.
GFP fluorescence was captured for 2 s (gain 1, binning 1) after focusing on the DIC image except for AP1383 and AP1387 cells (200-ms exposure). For AP1178 and AP1192 strains, mRFP images were captured first with a 2-s exposure (gain 2, binning 1), and GFP images were acquired with a 1-s exposure (gain 2, binning 1). For time-lapse video microscopy, acquisition was performed for 500 ms (gain 2, binning 2). z series (0.2-μm intervals) were collected every 2 min.
All images were acquired and analyzed by using Metamorph imaging software.
Septa were stained as described in reference 32.
FRAP experiments.
For fluorescence recovery after photobleaching (FRAP) experiments, cells were grown and mounted as described above. The results shown in Fig. 3 were obtained from experiments with AP616 strain in which the Cter-GFP expression level is twofold higher than in AP621 (S. Celton-Morizur and A. Paoletti, unpublished results) because GFP fluorescence intensity at the central cortex in AP616 better matched that of AP1111 strain (NLS* Cter-GFP). Similar results were obtained with the AP621 strain (A. Paoletti and P. T. Tran, unpublished results).
FIG. 3.
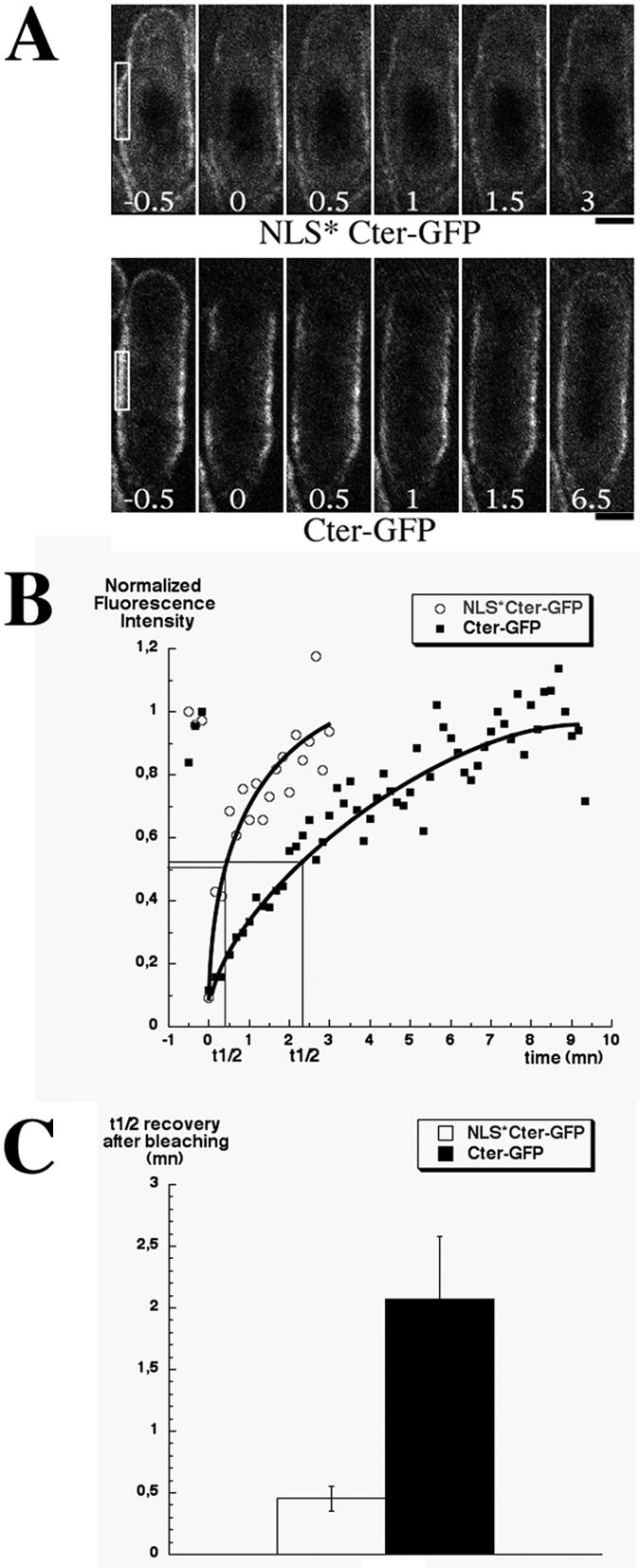
NLS mutations accelerate the exchange rate of cortical Cter-GFP. (A) Micrographs of NLS* Cter-GFP (top, strain AP1111) and Cter-GFP (bottom, strain AP616) cells before and after photobleaching (t = 0). Photobleached regions are boxed. Numbers indicate time elapsed after photobleaching in minutes. Upper cell tips were also subjected to bleaching and showed similar recovery rates. Bar, 2 μm. (B) Normalized fluorescence recovery after photobleaching in the regions boxed in A. (C) Mean half time recovery for NLS* Cter-GFP (n = 7) and Cter-GFP (n = 6). Error bars indicate the standard deviation.
FRAP was performed on a LSM510 Meta-Confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with a ×63/1.4 N.A. Plan-Apochromat objective lens. An argon-krypton continuous wave laser with about 10 mW of line 488 nm was chosen for both GFP imaging (2% power) and photobleaching (100% power). The fluorescence intensities of cells before and after photobleaching were measured by using the linescan tool of the MetaMorph imaging software. We corrected for background fluorescence by deducing background signals from measured intensities and for bleaching during imaging by using an unbleached region of the cortex to calculate the bleaching rate. The t1/2 recovery was determined graphically from the bleach recovery plots as the time corresponding to 50% intensity recovery.
RESULTS
The C terminus of mid1p contains cortex-targeting motifs.
During interphase, mid1p is partly associated with the central cell cortex (33; see also Fig. 4A). At the entry into mitosis, mid1p cortical staining is reinforced as mid1p exits the nucleus. To gain insight into how mid1p associates with the cell cortex, we decided to first determine which domain of mid1p mediates cortical anchoring. Our previous study (33) suggested that determinants for cortical anchoring were located in the C terminus of the protein. To test this hypothesis, we fused the C-terminal domain (amino acids 507 to 920) to GFP (Cter-GFP). The protein was expressed in mid1Δ strain from a genome-integrated construct under the control of mid1 promoter (see Materials and Methods). A Western blot showed that the concentration of this construct in total extracts was about 10-fold higher than the concentration of endogenous mid1p (Celton-Morizur and Paoletti, unpublished), suggesting that Cter-GFP, which lacks the PEST domains located in the N terminus of mid1p, is much more stable than full-length mid1p. In this strain (AP621), >95% of septa were aberrant (Table 2), indicating that mid1p C terminus is not functional. During interphase, Cter-GFP was strongly associated with the cell cortex and was ∼3-fold more concentrated in the medial region compared to the cell tips (Fig. 1A and C). Cter-GFP was also faintly associated with the nuclear envelope. During mitosis, the distribution at the central cortex became broader (Fig. 1B, time zero) and then split into two bands as shown by time-lapse video microscopy (Fig. 1B, 4 to 12 min). This indicates that Cter-GFP is unable to compact into a tight ring during mitosis and suggest that it may not interact with other components of the contractile ring. Cter-GFP was then observed in the region of septum formation (Fig. 1B, 16 to 52 min), where it was probably associated with the ingressing plasma membrane of the cleavage furrow. Finally, Cter-GFP remained more concentrated at the new tip compared to the old tip in short cells after sister cell separation (Fig. 1B, 60 min, and C, left panel).
FIG. 4.

Helix mutations perturb tight ring formation in early mitosis. (A) Localization of mid1-GFP (lane a, AP528), NLS* mid1-GFP (lane b, AP734), Helix* mid1-GFP (lane c, AP583), and Helix* NLS* mid1-GFP (lane d, AP630) in cells grown at 20°C. mid1-GFP and NLS*mid1-GFP form a broad band at the medial cortex during interphase and assemble a medial tight ring during mitosis (arrowheads). Helix* mid1-GFP is nuclear in interphase, whereas Helix* NLS* mid1-GFP is mostly cytoplasmic but is also present in the nucleus and forms faint patches at the cell cortex (long arrows). Both form disorganized filaments in the medial region in early mitosis (short arrows). Bar, 5 μm. (B) Expression levels of mid1-GFP (a), NLS* mid1-GFP (b), Helix* mid1-GFP (c), and Helix* NLS* mid1-GFP (d) at 30°C and 36°C. Western blot with anti-mid1p antibody and MAb TAT1 for tubulin (loading control). WT, wild-type cells.
TABLE 2.
Position of septa in mid1p helix mutants
| Straina | % Misplaced septab (level of septum displacement)c at:
|
||
|---|---|---|---|
| 20°C | 30°C | 36°C | |
| Wild type | 8 (moderate) | 5 (moderate) | 5 (moderate) |
| mid1Δ mutant | ND (severe) | 98 (severe) | 99 (severe) |
| Cter-GFP | >95 (severe) | >95 (severe) | >95 (severe) |
| Helix* Cter-GFP | >95 (severe) | >95 (severe) | >95 (severe) |
| NLS* Cter-GFP | >95 (severe) | >95 (severe) | >95 (severe) |
| Helix*NLS* Cter-GFP | >95 (severe) | >95 (severe) | >95 (severe) |
| Helix* mid1-GFP | 62 (moderate) | 27 (moderate) | >95 (severe)d |
| NLS* Helix* mid1-GFP | 50 (moderate) | 33 (moderate) | 47 (moderate) |
| GFP-Nter | 49 (moderate) | 29 (moderate) | 80 (moderate) |
Wild-type strain, mid1Δ strain, and mid1Δ strains expressing Cter-GFP, Helix* Cter-GFP, Helix*NLS* Cter-GFP, Helix* mid1-GFP, Helix*NLS* mid1-GFP, and GFP-Nter correspond, respectively, to FC418, SP1601, AP621, AP829, AP826, AP583, AP630, and AP998 (see Table 1).
Percentage of misplaced septa was determined on 200 cells (two independent experiments) after septum staining with Fluostain I.
Septum displacement was much weaker in full-length helix mutants and GFP-Nter mutant than in mid1Δ or Cter-GFP mutants.
This phenoype is due to Helix* mid1-GFP instability at 36°C (see Fig. 4B).
FIG. 1.

Localization of mid1p C terminus at the central cortex. (A) GFP fluorescence (left) and DIC images (right) of cells expressing mid1p C-terminal domain tagged with GFP (Cter-GFP; strain AP621) grown at 20°C. Cter-GFP is more concentrated at the central cortex than at the cell tips. Note the faint staining of the nuclear envelope. Bar, 5 μm. (B) Time-lapse movie of a Cter-GFP expressing cell during mitosis. A medial focal plane is shown. Numbers in white correspond to elapsed time in minutes. No tight ring is observed in early mitosis or mid-mitosis (t = 0 to 12). A staining of the ingressing plasma membrane is observed in late mitosis during septum formation (t = 16 to 52). (C) Cter-GFP fluorescence profile along the cell cortex. Fluorescence levels were recorded on the upper cortex of the two cells shown on the bottom from the left to the right cell tip. (Left) Very short G2 cell presenting a stronger staining at the new end compared to the old end; (middle) G2 cell presenting a strong staining at the medial cortex; (right) mean fluorescence intensity at the medial cortex and at the cell tips in cells presenting a symmetrical tip staining as in the middle panel (n = 8). Error bars indicate the standard deviation.
These data demonstrate that the C-terminal domain of mid1p contains determinants that mediate a preferential anchorage to the central cortex. An association with the cell tips and septum region was never observed for wild-type mid1p and may either result from the truncation of mid1p or from the higher concentration of this construct.
Role of an amphipathic helix in cortical anchorage.
The C-terminal domain possesses a basic region (amino acids 681 to 710) (37), which contains mid1p nuclear localization sequence (NLS; amino acids 691 to 695) (33). The sequence right upstream of the NLS (RKFFDKLF; amino acids 681 to 688) is typical of an amphipathic helix (Fig. 2A). Since amphipathic helices can directly insert into lipid bilayers (1, 17, 22, 26, 38, 39, 40, 46), we assayed the role of mid1p putative helix in cortical anchorage by substituting hydrophobic amino acids (F and L) by alanine (Fig. 2A). Since polybasic sequences such as mid1p NLS can also play a role in cortical anchorage (21, 27), helix mutations were also combined with NLS mutations. Cter-GFP mutated in the helix (Helix* Cter-GFP) was concentrated in the nucleus, even during mitosis (arrow in Fig. 2C). When mutations in the helix were combined with mutations in the NLS (Helix* NLS* Cter-GFP), Cter-GFP was found in the cytoplasm. As expected from previous work on full-length mid1p (33), NLS mutations alone (NLS* Cter-GFP) impaired nuclear localization but did not affect cortical anchorage. Thus, mid1p amphipathic helix plays a major role in cortical anchorage, and the polybasic NLS is dispensable. In addition, the fact that Helix* Cter-GFP is concentrated in the nucleus, whereas Cter-GFP is mostly found at the cell cortex, indicates that membrane anchorage competes with Cter-GFP import into the nucleus. Accordingly, Cter-GFP slowly accumulated in the nucleus when nuclear export was blocked upon leptomycin B treatment (Celton-Morizur and Paoletti, unpublished).
FIG. 2.

An amphipathic helix anchors mid1p C terminus to the cortex. (A) Schematic representation of mid1p C terminus tagged with GFP and mutations introduced by site-directed mutagenesis within an amphipathic helix and mid1p NLS, located inside the basic domain. (B) Expression levels of Cter-GFP (lane a, strain AP621), NLS* Cter-GFP (lane b, strain AP1111), Helix* Cter-GFP (lane c, strain AP829), and Helix*NLS* Cter-GFP (lane d, strain AP826). Western blotting with anti-GFP MAb and MAb TAT1 for tubulin (loading control). (C) Localization of C-terminal mutant constructs described in panel A in cells grown at 20°C. Helix mutations abolish recruitment to the cell cortex and induce a relocalization into the nucleus or the cytoplasm when the NLS is also mutated. An arrow indicates binucleated mitotic cell. Bar, 5 μm. (D) Biochemical analysis of Cter-GFP association with membranes. Spheroplasts from wild-type cells (AP240) and cells expressing Cter-GFP (AP621), mutated Cter-GFP (AP1111, -829, and -826), or pom152-GFP (HU0375) were processed as described on the left (see Materials and Methods). Pellets and supernatants were analyzed by Western blotting with anti-mid1p antibody (mid1p), anti-GFP MAb (Cter-GFP,pom152-GFP), MAb TAT1 for tubulin, and an anti-BIP antibody. A fraction of mid1p or Cter-GFP is solubilized by nonionic detergents (S3), indicating an association with membranes. Note instead the presence of a soluble pool in absence of detergents (S0) when the helix is mutated. (E) Cortical targeting of GFP fused to one or two copies of mid1p helix and NLS. Overexpressed GFP-Helix-NLS (strain AP1383 grown for 20 h in absence of thiamine) is only partially associated with the plasma membrane during interphase. An important fraction is targeted to the nucleus. Overexpressed GFP-(Helix-NLS)2 (strain AP1387 grown for 20 h in absence of thiamine) is strongly targeted to the plasma membrane and barely detected in the nucleus. Note that the two proteins are not restricted to the central cortex and associate with the ingressing plasma membrane during septation (arrows). Bar, 5 μm.
These results were confirmed by fractionation experiments (Fig. 2D). Spheroplasts obtained from wild-type cells and from cells expressing Cter-GFP, Helix* Cter-GFP, NLS* Cter-GFP, and Helix* NLS* Cter-GFP were subjected to hypotonic lysis and centrifuged at 20,000 × g. Due to a rupture of the integrity of the nuclear envelope during hypotonic lysis, both cytosolic and nucleoplasmic soluble proteins are released in S0, as evidenced in a strain expressing the nuclear reporter GFP-NLS-lacZ (45; Celton-Morizur and Paoletti, unpublished). Collected pellets were then extracted with 1% NP-40, 1 M NaCl, or both. mid1p and mid1p C terminus and NLS*-Cter-GFP were partially solubilized in presence of NP-40 (Fig. 2D, fraction S3), indicating that they are associated with membranes. In contrast, no detergent-soluble pool was observed for Helix* Cter-GFP and Helix* NLS* Cter-GFP, but a fraction of these proteins was directly soluble in the absence of detergents (Fig. 2D, fraction S0). All constructs were partially solubilized by 1 M NaCl (Fig. 2D, fraction S2), whereas a combination of NaCl and NP-40 (Fig. 2D, fraction S4) specifically enhanced the extraction of mid1p, Cter-GFP and NLS*-Cter-GFP. This indicates that these membrane-associated constructs are also engaged in high-salt-labile structures. The quality of our fractionation was checked by using a strain expressing the transmembrane nuclear pore protein pom152-GFP (6). As expected, a fraction of pom152-GFP was solubilized in presence of NP-40. The addition of 1 M NaCl was necessary to complete extraction. Conversely, soluble tubulin was fully released upon hypotonic lysis, whereas the endoplasmic reticulum protein BIP (34) required NP-40 for solubilization.
We conclude that mid1p and Cter-GFP are associated with membranes and that the amphipathic helix located in the basic domain plays a crucial role in membrane binding.
mid1p NLS may modulate the affinity for membranes.
We have shown that the NLS is dispensable for Cter-GFP association with the cell cortex. In order to determine whether the NLS could however modulate membrane anchorage, we analyzed the mobility of Cter-GFP and NLS* Cter-GFP by FRAP analysis (Fig. 3). We observed in half-bleached cells that fluorescence recovery at the cortex was not due to lateral mobility along the cortex (Paoletti and Tran, unpublished). Fluorescence recovery was considerably accelerated by the lack of NLS (Fig. 3B): fluorescence recovery was four times faster for NLS* Cter-GFP mutant than for Cter-GFP (t1/2 recovery in 27 s ± 6, [n = 7] versus 124 ± 30 [n = 6]; Fig. 3C).
This result indicates that cortical Cter-GFP exchanges faster in absence of NLS and suggests that the basic residues of this sequence may increase the affinity of the C-terminal domain for membranes by interacting with acidic phospholipids.
mid1p helix and NLS are sufficient to target GFP to the cortex.
Finally, to determine whether mid1p helix and NLS motifs were sufficient for membrane binding, we fused them to the C terminus of GFP (RKLFDKLFNRRKKRK peptide after a seven-amino-acid linker; see Materials and Methods). The fusion protein (GFP-Helix-NLS), overexpressed in wild-type cells from a multicopy plasmid, was partially associated with the plasma membrane (Fig. 2E) but was not concentrated in the medial region such as Cter-GFP. It was also present in the cytoplasm, and a fraction was targeted to the nucleus (Fig. 2E). In late mitosis, it was strongly associated with the plasma membrane in the region of septum formation (Fig. 2E, arrow). Targeting to the plasma membrane of GFP-helix-NLS seemed therefore weaker than that of Cter-GFP.
Anchoring to membranes of low-affinity membrane-targeting domains such as amphipathic helices is often enhanced by dimerization (39). Since we have evidence that mid1p may form oligomers (see below), we tested whether GFP targeting to the cortex could be enhanced by fusing two copies of mid1p helix and NLS peptide in tandem to the C terminus of GFP. The fusion protein [GFP-(Helix-NLS)2] overexpressed in wild-type cells from a multicopy plasmid was strongly targeted to the cortex and hardly detected in the nucleus, indicating that, as in the case of Cter-GFP, binding to the membrane competes with nuclear import. Finally, like GFP-Helix-NLS, GFP-(Helix-NLS)2 was strongly associated with the plasma membrane in the region of septum formation.
In conclusion, mid1p helix and NLS peptide is sufficient to target GFP to membranes. Targeting is enhanced in the presence of two copies of the peptide, which may indicate that mid1p oligomerization could facilitate membrane targeting in vivo.
Helix mutations in full-length mid1p perturb cortical anchorage in early mitosis and affect tight ring position.
In order to analyze the effects of helix mutations on the localization and function of mid1p, we produced full-length constructs encoding helix-mutated and helix and NLS-mutated mid1-GFP. Constructs were expressed in mid1Δ cells from genome-integrated constructs under the control of mid1 promoter. Western blot analysis showed that these constructs were ∼2-fold more expressed than wild-type mid1p at 30°C (Fig. 4B). At 36°C all GFP-tagged constructs except NLS*-mid1-GFP displayed reduced expression levels. These levels were, however, sufficient to carry out mid1 function, as judged by the ability of mid1-GFP to fully rescue the mid1Δ strain at this temperature, except for Helix*mid1-GFP, which was hardly detected (fivefold reduction compared to wild-type mid1p). During interphase, Helix* mid1-GFP was concentrated in the nucleus (Fig. 4Ac) and absent from the medial cortex where mid1-GFP and NLS* mid1-GFP were easily detected (Fig. 4Aa to b). Helix* NLS* mid1-GFP was essentially cytoplasmic but was also present in the nucleus and formed faint cortical patches (Fig. 4Ad). In early mitosis, the two helix mutants formed disorganized filaments at the cell center (Fig. 4Ac and d). Tight rings did form in late mitosis but were frequently slightly tilted or offset. Accordingly, septa were frequently mislocalized in these cells (62 and 50%, respectively, at 20°C and ∼30% at 30°C; Table 2).
In order to better understand the behavior of the filaments observed in early mitosis, we performed three-dimensional time-lapse video microscopy. We observed that the filaments formed by Helix* mid1-GFP or Helix* NLS* mid1-GFP were very dynamic. Projections at 90° of deconvolved z series also showed that the filaments were not properly anchored to the cell cortex (Fig. 5b and c). A stable association with the cortex was achieved in late mitosis when tight rings formed, but these rings were frequently misplaced. Helix* NLS* mid1-GFP remained faintly associated with the rings during their contraction (Fig. 5d and e). As expected, NLS* mid1-GFP always remained closely associated with the cell cortex from early to late mitosis (Fig. 5a).
FIG. 5.

The amphipathic helix anchors mid1p filaments to the cell cortex in early mitosis and stabilizes tight ring position. Time-lapse images of cells expressing NLS* mid1-GFP (a), Helix* mid1-GFP (b), and Helix* NLS* mid1-GFP (c to e). For each movie, maximum projections of deconvolved z series (top) and projections at 90° (bottom) are shown. DIC image of the cell at the end of the movie is shown on the right. Time elapsed between frames, 2 min. Bars, 4 μm. Note the presence of filaments not anchored to the cell cortex before anaphase in Helix* mid1-GFP cells (b, 90° projection) and in Helix*NLS*mid1-GFP cells (c, 90° projection). For this mutant, a faint staining of offset rings was observed during contraction (d and e).
In conclusion, the amphipathic helix plays a critical role in mid1p anchoring at the central cortex in early mitosis and stabilizes the position of the tight ring at the cell center.
Cdc12p and Myo2p colocalize with Helix* NLS* mid1-mRFP filaments in early mitosis.
Myosin II and formin cdc12p, an actin nucleation factor, are sequentially recruited to the medial cortex at the onset of mitosis (44). We wondered whether these proteins would be associated with the filaments formed by mid1p helix mutants in early mitosis. We performed microscopy on cells expressing Helix* NLS* mid1-monomeric red fluorescent protein (mRFP) (8) and either myo2-GFP or cdc12-GFP (strains AP1192 and AP1178). Helix* NLS* mid1-mRFP presented the same localization as Helix* NLS* mid1-GFP. mid1Δ (cells expressing these two proteins had the same phenotype (Celton-Morizur and Paoletti, unpublished). AP1192 cells had a more severe phenotype (Celton-Morizur and Paoletti, unpublished), suggesting that there may be a moderate synthetic effect between mid1p mutations or tagging and C-terminal tagging of myo2p with GFP. AP1178 cells had the same phenotype as Helix* NLS* mid1-GFP cells.
During interphase, myo2-GFP was diffuse in the cytoplasm and did not colocalize with Helix* NLS* mid1-mRFP patches at the central cortex (Celton-Morizur and Paoletti, unpublished). In early mitosis, as soon as Helix* NLS* mid1-mRFP nuclear staining disappeared and cortical staining reinforced, the two proteins colocalized in filaments at the cell center. In late mitosis, myo2-GFP formed misshaped rings, whereas Helix* NLS* mid1-mRFP cortical staining became fainter and nuclear staining reappeared.
Similarly, Helix* NLS* mid1-mRFP colocalized with cdc12-GFP in early mitosis (Fig. 6B) but not in interphase, during which Helix* NLS* mid1-mRFP was absent from cdc12p spots and cdc12-GFP did not colocalize with Helix* NLS* mid1-mRFP at the central cortex.
FIG. 6.

Filaments formed by Helix*NLS* mid1-mRFP in early mitosis contain myo2-GFP and cdc12-GFP. Shown is localization of myo2-GFP (strain AP1192) (A) or cdc12-GFP (strain AP1178) (B) in cells expressing Helix*NLS* mid1-mRFP grown at 20°C. DIC image of the same cells is shown on the bottom. Bar, 2 μm. Cdc12-GFP and Myo2-GFP colocalize with Helix*NLS* mid1-mRFP in early mitosis. Note also that Helix*NLS* mid1-mRFP is absent from cdc12-GFP spots before mitosis and has already reassociated with the central cortex after cell separation.
These observations indicate that Helix* NLS* mid1-mRFP colocalize with myo2-GFP and cdc12-GFP in early mitosis. This suggests that in wild-type cells mid1p mediates anchorage of myo2p and cdc12p to the central cortex in early mitosis.
The N-terminal domain of mid1p forms faint patches at the central cortex.
Since helix mutations in full-length mid1p did not totally abolish cortical anchorage, whereas they did abolish Cter-GFP anchorage, we suspected that the N-terminal domain of mid1p was also able to interact with the cortex. Such a cortical localization had not been observed in the mid1 1-506 strain by immunofluorescence previously (33) but could have been lost during cell fixation. Thus, we fused the N-terminal domain to GFP (GFP-Nter), expressed it under mid1 promoter control in a mid1Δ strain from a genome-integrated construct, and observed its localization in vivo (Fig. 7A). This construct was expressed at levels similar to those of the wild-type mid1p (Fig. 7C). In interphase, GFP-Nter was found not only in the cytoplasm but also in the nucleus and formed faint cortical patches similar to those observed in the Helix* NLS* mid1-GFP mutant (Fig. 7A). The patches were mainly located in the medial region (arrow in Fig. 7A; see also Fig. 7D), except in very short cells where some patches were associated with the new end (arrowheads in Fig. 7A; see also Fig. 7D). Moreover, GFP-Nter was able to assemble faint tight rings in late mitosis as observed by three-dimensional video microscopy (Fig. 7B). These rings were often slightly offset or tilted. Accordingly, septa were often misplaced as in helix mutant strains (50% at 20°C and 30% at 30°C; see Table 2). Rings contracted during septation (Fig. 7Bb and c).
FIG. 7.

The N-terminal domain of mid1p forms faint cortical patches and is sufficient to form tight rings during mitosis. (A) In vivo localization of mid1p N terminus tagged with GFP (GFP-Nter, strain AP998, top) and corresponding DIC images (bottom). Cells were grown at 20°C. GFP-Nter localizes in the cytoplasm, in the nucleus, and forms faint cortical patches in interphase, at the cell tips (arrowheads) or at the medial cortex (arrow). In the right-hand panel is shown a ×1.5 magnification. Bar, 5 μm. (B) Time-lapse images of cells expressing GFP-Nter. Maximum projections of deconvolved z series are shown. Numbers correspond to the time elapsed in minutes. During mitosis, GFP-Nter forms tight rings (a and b) that are often slightly tilted or offset (b). The rings constrict during septum formation (b and c). Bar, 2 μm. (C) Expression levels at 30 and 36°C of mid1p (lanes 1), mid1-GFP (lanes 2), and GFP-Nter (lanes 3). The loading control is tubulin (MAb TAT1). (D) Correlation between the distribution of cortical patches along the cell cortex and cell length. The distribution of GFP-Nter cortical patches was analyzed in 70 cells and correlated to cell length. Patches are prominently located in the central region, except in very short cells that recently cleaved their septa.
These results indicate that mid1p N-terminal domain can bind to the cortex independently of mid1p C-terminal helix. Its ability to form a tight ring also suggests that it may interact with other components of the cytokinetic ring.
mid1p forms oligomers.
Several pieces of evidence suggest that mid1p could recruit directly cytokinetic ring components at the medial cortex in early mitosis (33, 44). To test this idea, we tried to purify proteins interacting with mid1p from cell extracts.
First, we analyzed the sedimentation of mid1p and N- and C-terminal domains of mid1p on linear sucrose gradients (Fig. 8A). Detergent-soluble extracts prepared from asynchronous wild-type cells or cells expressing mid1-GFP, Cter-GFP, or mid1p N-terminal domain (Nter) were sedimented at 100,000 × g for 16 h on 20 to 35% sucrose gradients. We found that mid1p formed two high-molecular-mass complexes of more than 450 kDa. mid1-GFP behaved in a similar way, except that an additional 240-kDa complex was observed. Cter-GFP formed a single high-molecular-mass complex of >450 kDa. The N-terminal domain formed a soluble pool, together with degradation products, and a low-molecular-mass complex of ∼240 kDa.
FIG. 8.

Sedimentation and oligomerization of mid1p. (A) Sedimentation of mid1p and mid1p N- and C-terminal domains on sucrose gradients. Extracts prepared from asynchronous wild-type cells or cells expressing mid1-GFP, Cter-GFP and mid1p N-terminal domain (Nter) were run on 20 to 35% linear sucrose gradients. Gradient fractions (top on the left) were analyzed by Western blotting with anti-mid1p antibody (mid1p, mid1-GFP, and Nter) or anti-GFP MAb (Cter-GFP). The positions of peaks formed by bovine serum albumin (68 kDa), aldolase (160 kDa), catalase (240 kDa), and ferritin (450 kDa) run on a control gradient are shown at the bottom. (B) SDS-PAGE and colloidal blue staining of purified complexes. For GST-mid1p and Nter-GST, purified proteins were run on sucrose gradients as in panel B, and fractions corresponding to high-molecular-mass complexes were pooled before PAGE. Black dots correspond to major contaminants found in the GST control. (C) Coimmunoprecipitation of mid1-13myc with mid1-GFP. Detergent-soluble 10,000 × g extracts prepared from cells expressing mid1-13myc and mid1-GFP at endogenous levels (strain AP599) were submitted to immunoprecipitation with anti-GFP MAb. Supernatants (lanes S) and immunoprecipitation pellets (lanes IP) were subjected to Western blotting with anti-myc and anti-GFP MAbs. Mid1-GFP and mid1-13myc are indicated by arrows. Lower bands correspond to degradation products. (D) Coimmunoprecipitation of GST-tagged and GFP-tagged mid1p domains. Anti-GFP immunoprecipitations were performed on extracts from mid1Δ cells expressing GST-mid1p (left panel), GST-Nter (middle panel), or Cter-GST (right panel) alone (control) or in addition to mid1-GFP, GFP-Nter, or Cter-GFP. Constructs were expressed from plasmids under the control of pnmt** (low-strength promoter). Immunoprecipitation pellets were subjected to Western blotting with anti-GFP MAb and anti-GST purified immunoglobulins. GFP and GST-tagged N-terminal domains were coimmunoprecipitated. Similarly, GFP and GST-tagged C-terminal domains were coimmunoprecipitated. No association between N- and C-terminal domains was detected.
In order to purify these complexes, GST tags were fused to mid1p and mid1p N- and C-terminal domains (GST-mid1p, Cter-GST, and GST-Nter). These constructs were overexpressed in wild-type cells from plasmids (full strength nmt1 promoter). GST fusion proteins were purified on glutathione beads. Analysis of beads eluates on linear sucrose gradients showed that the purified proteins formed high-molecular-mass complexes like proteins expressed at endogenous levels (Celton-Morizur and Paoletti, unpublished).
SDS-PAGE analysis and colloidal blue staining of the high-molecular-mass fractions (Fig. 8B) showed that some polypeptides cosedimented with GST-mid1p or GST-Nter. Most of them were also found in a GST eluate used as a control. Mass spectrometry analysis revealed that GST-mid1p eluate contained the kinase plo1p, a known partner of mid1p (3), and a series of proteins highly expressed in cells such as ribosomal proteins, which may represent extensive contaminants. No other known component of the cytokinetic ring was detected in this fraction (S. Celton-Morizur, W. Faigle, D. Loew, and A. Paoletti, unpublished results). Cter-GST eluate did not contain any other major polypeptide. Since Cter-GST did form a high-molecular-mass complex (Celton-Morizur and Paoletti, unpublished), these data suggested to us that the C-terminal domain and possibly full-length mid1p were able to oligomerize.
To investigate this possibility, we performed anti-GFP immunoprecipitation in extracts from cells expressing mid1-13myc from its endogenous promoter and mid1-GFP from the same promoter, from a construct integrated at leu1+ locus. A large amount of mid1-13myc coimmunoprecipitated with mid1-GFP, indicating a strong interaction between the two tagged proteins expressed at endogenous levels (Fig. 8C). We then tested the role of mid1p N- and C-terminal domains in oligomerization. We performed anti-GFP immunoprecipitation on extracts from mid1Δ cells expressing GST-mid1p, GST-Nter, or Cter-GST and mid1-GFP, GFP-Nter or Cter-GFP from plasmids (low-strength nmt1 promoter; Fig. 8D). This experiment confirmed the interaction of full-length mid1p with itself and also demonstrated that the N- and C-terminal domains interact strongly with themselves. In contrast, no interaction was detected between the N- and C-terminal domains.
Together, these data indicate that mid1p is able to form oligomers in which molecules may adopt a parallel configuration.
DISCUSSION
A dual cortex binding mechanism.
Our data demonstrate that mid1p binds to the cell cortex by two independent means.
First, the mid1p C terminus binds to membranes. Binding involves an amphipathic helix and a polybasic sequence, previously described as the mid1p NLS, which are sufficient to target GFP to membranes.
Similar membrane-binding motifs have been described in a number of peripheral membrane proteins. For instance, binding of the small G protein Arf1 to membranes is mediated by a N-terminal amphipathic helix that is released from an hydrophobic pocket upon switch to the GTP state and can interact with membrane phospholipids (1, 26). More strikingly, targeting of MinD, a bacterial protein involved, like mid1p, in the spatial regulation of cytokinesis, relies on a C-terminal amphipathic helix (22, 40). This helix is highly similar in sequence to mid1p helix (KKGFLKRLF in Escherichia coli) and has been shown to bind to lipids by direct insertion of hydrophobic residues into the lipid bilayer (39, 46). Moreover, it has been proposed that ATP-regulated accessibility of this helix may account for MinD reversible membrane association during its pole-to-pole oscillation cycles (39). Interestingly, EcMinD helix fused to GFP is not sufficient for membrane targeting unless it is fused to a dimerization motif or present in two copies in a tandem array (39). This suggests that the low affinity of short amphipathic helices for lipids is either reinforced by additional lipid targeting motifs (as in Arf1) or by oligomerization (as in EcMinD). In the case of mid1p, both mechanisms may operate since we found evidence for both oligomerization properties for mid1p and a role for the polybasic NLS adjacent to the amphipathic helix in stabilizing mid1p C terminus binding to the cortex. Like the polybasic domain of ARNO (a nucleotide exchange factor for Arf1) (13), which acts in combination with a pleckstrin homology (PH) domain that provides a specific phosphoinositide-binding capacity (27), mid1p NLS may interact electrostatically with acidic phospholipids such as phosphatidyl serine, a common phospholipid from the cytoplasmic leaflet of membranes.
We thus propose a model in which mid1p amphipathic helix may directly insert inside the lipid bilayer: hydrophobic lateral chains of phenylalanines 683, 684, and 687 and leucine 688 may establish hydrophobic interactions inside one leaflet of the lipid bilayer and induce penetration of the helix in the membrane. In addition, the basic residues of mid1p NLS sequence (amino acids 690 to 695) may reinforce the affinity for membranes by establishing electrostatic interactions with head groups of acidic phospholipids (Fig. 9A).
FIG. 9.

Model for mid1p anchoring to the cortex and function. (A) Schematic association of mid1p amphipathic helix and NLS with phospholipids of the plasma membrane. Hydrophobic lateral chains of F and L residues could establish hydrophobic interactions inside one leaflet of the lipid bilayer, whereas basic lateral chains of R and K residues could interact electrostatically with head groups of acidic phospholipids. (B) Functional model for mid1p. Oligomers of mid1p that assemble in the cytoplasm or in the nucleus are recruited to the plasma membrane (or the nuclear envelope) by interactions between the C-terminal domain and phospholipids. Additional recruitment to membranes may be mediated by mid1p N-terminal domain, possibly by interacting with other cortical cues. In early mitosis, mid1p oligomers activated by plo1p phosphorylation form a “platform” recruiting ring components to initiate cytokinetic ring formation at the medial cortex. During ring compaction, anchorage of mid1 C terminus to membranes ensures a precise positioning of the contractile ring.
Mid1p also contains a PH domain at the C terminus. However, its deletion did not affect mid1p localization or function (33). It could, however, play a nonessential role in membrane binding like mid1p NLS. The possibility that it provides a specific phosphoinositide-binding capacity needs to be studied.
The mid1p polybasic domain has a dual function: as an NLS and as a membrane-anchoring domain. Similarly, a polybasic domain in Xenopus lamin 3b located next to a C-terminal prenylation motif was shown to serve as an NLS and as a membrane-targeting motif (21). This dual function is particularly interesting in light of the cell cycle regulation of mid1p localization. During interphase, mid1p is mostly found in the nucleus and shuttles in and out of the nucleus, suggesting that the polybasic sequence serves as an active NLS. The fact that Helix*mid1-GFP and Helix*Cter-GFP are much more concentrated in the nucleus than is mid1-GFP or Cter-GFP indicates that it is membrane anchorage rather than nuclear import/export rates that regulates the nucleus/cytoplasm ratio of mid1p by preventing mid1p NLS association with importins. One important question is how plo1p kinase induces mid1p relocalization to the cell cortex upon entry into mitosis. It was postulated that plo1p could trigger exportin-dependent nuclear export of mid1p (3, 33). In light of our experiments, we can propose alternatively that plo1p could reinforce mid1p binding to the cell cortex. The possibility that mid1p anchorage could be modulated by regulating the helix accessibility also needs to be explored.
Analysis of Nter-GFP localization shows that the N-terminal fragment of mid1p can also bind to the cortex. The proportion of N terminus bound to the cortex is not easy to quantify but seems very low compared to the C terminus. This suggests that the affinity of the N terminus for the cortex may be much weaker and may explain why it was not seen by immunofluorescence in the mid1 1-506 strain (33). Accordingly, we found that cortex binding via the N terminus is sufficient to initiate ring components recruitment, since myo2-GFP and cdc12-GFP colocalized with Helix*NLS*mid1-mRFP at the central cortex in early mitosis. However, it is not sufficient to properly maintain their anchorage to the cell cortex.
Strikingly, the anchorage defects observed in cells expressing full-length mid1p helix mutants seem to be compensated for in late mitosis since we observed properly anchored rings during anaphase. Two possibilities may explain this phenomenon. First, Nter-GFP cortex-binding activity could be reinforced by posttranslational modifications during late mitosis. Another possibility is that other components of the cytokinetic ring could mediate cortex anchorage at this stage. The second possibility is more likely since, in wild-type cells, ring anchorage to the cortex is maintained during ring contraction, although mid1p is no longer associated with the ring at this stage (37). However, the positioning of the anaphase rings was abnormal in Nter-GFP-expressing cells, indicating that the precise positioning of the ring requires a tight anchorage of ring components to the central cortex by mid1p during ring compaction.
The nature of the cortex-binding motif in mid1p N terminus remains to be identified. This interaction could be mediated by another cortical protein. The localization of Nter-GFP in the nucleus also indicates that the N terminus contains a nuclear-targeting motif in addition to mid1 NLS or that it interacts with another protein possessing a NLS. This may explain why Cter-GFP is less abundant in the nucleus compared to full-length mid1p.
One striking observation is that cortex-anchored mid1p C-terminal and N-terminal domains are, like full-length mid1p, more concentrated in the central region of the cell than at the cell tip. This feature seems essential to ensure that mid1p will function properly at the entry into mitosis to recruit cytokinetic ring components in the middle of the cell. The mechanism that enables this gradient of mid1p to be established on the cortex is unknown. We demonstrated that the position of mid1p cortical band is coupled to the position of the nucleus (33). However, how this coupling is achieved is not clear. Neither NLS mutation, which effectively blocks the entry of Cter-GFP into the nucleus, nor leptmomycin B treatment of Cter-GFP-expressing cells (which blocks nuclear exit), did affect the distribution of Cter-GFP or NLS*-Cter-GFP on the cortex (Celton-Morizur and Paoletti, unpublished). In addition, depolymerization of microtubules by a 30-min treatment with MBC (methyl 2-benzyimidaolecarbamate) did not affect the distribution of cortical Cter-GFP (Celton-Morizur and Paoletti, unpublished). Since Cter-GFP associated with the cell cortex exchanges in a few minutes according to our FRAP experiments, this indicates that neither nuclear shuttling nor microtubules are required to maintain Cter-GFP gradient on the cortex.
Oligomerization and function of mid1p in early mitosis.
Our results also demonstrate that mid1p can form oligomers. Since both the N terminus and the C terminus can oligomerize, we propose that in oligomers mid1p molecules adopt a parallel configuration (Fig. 9B).
Since mid1p is not extracted from cells in the absence of detergents (see Fig. 2D), the fraction of mid1p that was analyzed in sedimentation and immunoprecipitation experiments (detergent-soluble fraction) corresponds to a membrane-bound pool. This indicates that mid1p oligomers may form when mid1p associates with the membrane. However, the fact that Helix*mid1-GFP and Helix*NLS*mid1-GFP, which are mostly found in the nucleus and in the cytoplasm, respectively, also sedimented as high-molecular-mass complexes (Celton-Morizur and Paoletti, unpublished) suggests that oligomerization can also take place away from membranes.
What is the function of oligomerization? One possibility is that it enables the formation of “platforms” in which the presence of multiple membrane-binding domains in tandem reinforces cortex binding. This idea is strongly supported by the enhanced cortical targeting of GFP fused to two copies compared to one copy of mid1p helix and NLS peptide. The presence of closely packed binding domains for ring components could also help recruiting ring components at the entry into mitosis.
Recently, myosin II heavy-chain myo2 has been identified as one of the ring components recruited by mid1p (30). Interestingly, interaction between myo2p and mid1p was shown to be negatively regulated by a phosphorylation event in myo2 C terminus rather than by activation of mid1p upon entry into mitosis. It will now be of great interest to determine whether myo2p recruitment is the only function of mid1p or whether mid1p is also responsible for the recruitment of other ring components involved in F-actin nucleation, such as cdc12p and cdc15p.
In conclusion, our study has demonstrated that mid1p possesses a dual cortex binding mechanism. Cortical anchorage by the N terminus is sufficient to initiate ring components recruitment at the central cortex, whereas C-terminal anchorage is critical to maintain an association with the cortex during ring compaction and ensures the precise positioning of the ring in the middle of the cell.
Acknowledgments
We thank P. Chardin for pointing out to us the amphipathic helix in mid1p sequence and for very helpful discussions. We thank W. Faigle and D. Loew for mass spectrometry; M. Balasubramanian, F. Chang, D. McCollum, V. Simanis, and K. Ekwall for strains; S. Forsburg, J. Bähler, R. Tsien, and S. Sazer for vectors; L. Cabanié, K. Gull, A. Pidoux, and J. Armstrong for antibodies; and M. Bornens, F. Chang, I. Loïodice, and A.-M. Tassin for critical reading of the manuscript.
This study was supported by a grant from ARC to A.P. (subvention no. 4489) and a fellowship from the Ministère de la Recherche (France) to S.C.-M.
REFERENCES
- 1.Antonny, B., S. Beraud-Dufour, P. Chardin, and M. Chabre. 1997. N-terminal hydrophobic residues of the G-protein ADP-ribosylation factor-1 insert into membrane phospholipids upon GDP to GTP exchange. Biochemistry 36:4675-4684. [DOI] [PubMed] [Google Scholar]
- 2.Arai, R., and I. Mabuchi. 2002. F-actin ring formation and the role of F-actin cables in the fission yeast Schizosaccharomyces pombe. J. Cell Sci. 115:887-898. [DOI] [PubMed] [Google Scholar]
- 3.Bahler, J., A. B. Steever, S. Wheatley, Y. Wang, J. R. Pringle, K. L. Gould, and D. McCollum. 1998. Role of polo kinase and mid1p in determining the site of cell division in fission yeast. J. Cell Biol. 143:1603-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahler, J., J. Q. Wu, M. S. Longtine, N. G. Shah, A. R. McKenzie, A. B. Steever, A. Wach, P. Philippsen, and J. R. Pringle. 1998. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14:943-951. [DOI] [PubMed] [Google Scholar]
- 5.Balasubramanian, M. K., D. M. Helfman, and S. M. Hemmingsen. 1992. A new tropomyosin essential for cytokinesis in the fission yeast Schizosaccharomyces pombe. Nature 360:84-87. [DOI] [PubMed] [Google Scholar]
- 6.Bjerling, P., R. A. Silverstein, G. Thon, A. Caudy, S. Grewal, and K. Ekwall. 2002. Functional divergence between histone deacetylases in fission yeast by distinct cellular localization and in vivo specificity. Mol. Cell. Biol. 22:2170-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouckson-Castaing, V., M. Moudjou, D. J. Ferguson, S. Mucklow, Y. Belkaid, G. Milon, and P. R. Crocker. 1996. Molecular characterization of ninein, a new coiled-coil protein of the centrosome. J. Cell Sci. 109:179-190. [DOI] [PubMed] [Google Scholar]
- 8.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 99:7877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carnahan, R. H., and K. L. Gould. 2003. The PCH family protein, Cdc15p, recruits two F-actin nucleation pathways to coordinate cytokinetic actin ring formation in Schizosaccharomyces pombe. J. Cell Biol. 162:851-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, F. 1999. Movement of a cytokinesis factor cdc12p to the site of cell division. Curr. Biol. 9:849-852. [DOI] [PubMed] [Google Scholar]
- 11.Chang, F., D. Drubin, and P. Nurse. 1997. cdc12p, a protein required for cytokinesis in fission yeast, is a component of the cell division ring and interacts with profilin. J. Cell Biol. 137:169-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang, F., and P. Nurse. 1996. How fission yeast fission in the middle. Cell 84:191-194. [DOI] [PubMed] [Google Scholar]
- 13.Chardin, P., S. Paris, B. Antonny, S. Robineau, S. Beraud-Dufour, C. L. Jackson, and M. Chabre. 1996. A human exchange factor for ARF contains Sec7- and pleckstrin-homology domains. Nature 384:481-484. [DOI] [PubMed] [Google Scholar]
- 14.Eng, K., N. I. Naqvi, K. C. Wong, and M. K. Balasubramanian. 1998. Rng2p, a protein required for cytokinesis in fission yeast, is a component of the actomyosin ring and the spindle pole body. Curr. Biol. 8:611-621. [DOI] [PubMed] [Google Scholar]
- 15.Fankhauser, C., A. Reymond, L. Cerutti, S. Utzig, K. Hofmann, and V. Simanis. 1995. The Schizosaccharomyces pombe cdc15 gene is a key element in the reorganization of F-actin at mitosis. Cell 82:435-444. [DOI] [PubMed] [Google Scholar]
- 16.Feierbach, B., and F. Chang. 2001. Cytokinesis and the contractile ring in fission yeast. Curr. Opin. Microbiol. 4:713-719. [DOI] [PubMed] [Google Scholar]
- 17.Ford, M. G., I. G. Mills, B. J. Peter, Y. Vallis, G. J. Praefcke, P. R. Evans, and H. T. McMahon. 2002. Curvature of clathrin-coated pits driven by epsin. Nature 419:361-366. [DOI] [PubMed] [Google Scholar]
- 18.Gönczy, P., and A. A. Hyman. 1996. Cortical domains and the mechanisms of asymmetric cell division. Trends Cell Biol. 6:382-387. [DOI] [PubMed] [Google Scholar]
- 19.Gould, K. L., and V. Simanis. 1997. The control of septum formation in fission yeast. Genes Dev. 11:2939-2951. [DOI] [PubMed] [Google Scholar]
- 20.Guertin, D. A., S. Trautmann, and D. McCollum. 2002. Cytokinesis in eukaryotes. Microbiol. Mol. Biol. Rev. 66:155-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hofemeister, H., K. Weber, and R. Stick. 2000. Association of prenylated proteins with the plasma membrane and the inner nuclear membrane is mediated by the same membrane-targeting motifs. Mol. Biol. Cell 11:3233-3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu, Z., and J. Lutkenhaus. 2003. A conserved sequence at the C terminus of MinD is required for binding to the membrane and targeting MinC to the septum. Mol. Microbiol. 47:345-355. [DOI] [PubMed] [Google Scholar]
- 23.Kelly, T. J., G. S. Martin, S. L. Forsburg, R. J. Stephen, A. Russo, and P. Nurse. 1993. The fission yeast cdc18+ gene product couples S-phase to START and mitosis. Cell 74:371-382. [DOI] [PubMed] [Google Scholar]
- 24.Kitayama, C., A. Sugimoto, and M. Yamamoto. 1997. Type II myosin heavy chain encoded by the myo2 gene composes the contractile ring during cytokinesis in Schizosaccharomyces pombe. J. Cell Biol. 137:1309-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Goff, X., F. Motegi, E. Salimova, I. Mabuchi, and V. Simanis. 2000. The S. pombe rlc1 gene encodes a putative myosin regulatory light chain that binds the type II myosins myo3p and myo2p. J. Cell Sci. 113(Pt. 23):4157-4163. [DOI] [PubMed] [Google Scholar]
- 26.Losonczi, J. A., and J. H. Prestegard. 1998. Nuclear magnetic resonance characterization of the myristoylated, N-terminal fragment of ADP-ribosylation factor 1 in a magnetically oriented membrane array. Biochemistry 37:706-716. [DOI] [PubMed] [Google Scholar]
- 27.Macia, E., S. Paris, and M. Chabre. 2000. Binding of the PH and polybasic C-terminal domains of ARNO to phosphoinositides and to acidic lipids. Biochemistry 39:5893-5901. [DOI] [PubMed] [Google Scholar]
- 28.May, K. M., F. Z. Watts, N. Jones, and J. S. Hyams. 1997. Type II myosin involved in cytokinesis in the fission yeast, Schizosaccharomyces pombe. Cell Motil. Cytoskeleton 38:385-396. [DOI] [PubMed] [Google Scholar]
- 29.McCollum, D., M. Balasubramanian, and K. Gould. 1995. Schizosaccharomyces pombe cdc4+ gene encodes a novel EF-hand protein essential for cytokinesis. J. Cell Biol. 130:651-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motegi, F., M. Mishra, M. K. Balasubramanian, and I. Mabuchi. 2004. Myosin II reorganization during mitosis is controlled temporally by its dephosphorylation and spatially by Mid1 in fission yeast. J. Cell Biol. 165:685-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naqvi, N. I., K. C. Wong, X. Tang, and M. K. Balasubramanian. 2000. Type II myosin regulatory light chain relieves auto-inhibition of myosin heavy-chain function. Nat. Cell Biol. 2:855-858. [DOI] [PubMed] [Google Scholar]
- 32.Paoletti, A., N. Bordes, R. Haddad, C. L. Schwartz, F. Chang, and M. Bornens. 2003. Fission yeast cdc31p is a component of the half-bridge and controls SPB duplication. Mol. Biol. Cell 14:2793-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paoletti, A., and F. Chang. 2000. Analysis of mid1p, a protein required for placement of the cell division site, reveals a link between the nucleus and the cell surface in fission yeast. Mol. Biol. Cell 11:2757-2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pidoux, A. L., and J. Armstrong. 1993. The BiP protein and the endoplasmic reticulum of Schizosaccharomyces pombe: fate of the nuclear envelope during cell division. J. Cell Sci. 105:1115-1120. [DOI] [PubMed] [Google Scholar]
- 35.Radcliffe, P., D. Hirata, D. Childs, L. Vardy, and T. Toda. 1998. Identification of novel temperature-sensitive lethal alleles in essential beta-tubulin and nonessential alpha 2-tubulin genes as fission yeast polarity mutants. Mol. Biol. Cell 9:1757-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simanis, V., and P. Nurse. 1986. The cell cycle control gene cdc2+ of fission yeast encodes a protein kinase potentially regulated by phosphorylation. Cell 45:261-268. [DOI] [PubMed] [Google Scholar]
- 37.Sohrmann, M., C. Fankhauser, C. Brodbeck., and V. Simanis. 1996. The dmf1/mid1 gene is essential for correct positioning of the division septum in fission yeast. Genes Dev. 10:2707-2719. [DOI] [PubMed] [Google Scholar]
- 38.Stahelin, R. V., F. Long, B. J. Peter, D. Murray, P. De Camilli, H. T. McMahon, and W. Cho. 2003. Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J. Biol. Chem. 278:28993-28999. [DOI] [PubMed] [Google Scholar]
- 39.Szeto, T. H., S. L. Rowland, C. L. Habrukowich, and G. F. King. 2003. The MinD membrane targeting sequence is a transplantable lipid-binding helix. J. Biol. Chem. 278:40050-40056. [DOI] [PubMed] [Google Scholar]
- 40.Szeto, T. H., S. L. Rowland, L. I. Rothfield, and G. F. King. 2002. Membrane localization of MinD is mediated by a C-terminal motif that is conserved across eubacteria, archaea, and chloroplasts. Proc. Natl. Acad. Sci. USA 99:15693-15698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tran, P. T., L. Marsh, V. Doye, S. Inoue, and F. Chang. 2001. A mechanism for nuclear positioning in fission yeast based on microtubule pushing. J. Cell Biol. 153:397-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran, P. T., A. Paoletti, and F. Chang. 2004. Imaging green fluorescent protein fusions in living fission yeast cells. Methods 33:220-225. [DOI] [PubMed] [Google Scholar]
- 43.Wu, J. Q., J. Bahler, and J. R. Pringle. 2001. Roles of a fimbrin and an alpha-actinin-like protein in fission yeast cell polarization and cytokinesis. Mol. Biol. Cell 12:1061-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu, J. Q., J. R. Kuhn, D. R. Kovar, and T. D. Pollard. 2003. Spatial and temporal pathway for assembly and constriction of the contractile ring in fission yeast cytokinesis. Dev. Cell 5:723-734. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida, M., and S. Sazer. 2004. Nucleocytoplasmic transport and nuclear envelope integrity in the fission yeast Schizosaccharomyces pombe. Methods 33:226-238. [DOI] [PubMed] [Google Scholar]
- 46.Zhou, H., and J. Lutkenhaus. 2003. Membrane binding by MinD involves insertion of hydrophobic residues within the C-terminal amphipathic helix into the bilayer. J. Bacteriol. 185:4326-4335. [DOI] [PMC free article] [PubMed] [Google Scholar]