

Abstract
Mutations of the tyrosine kinase-directed ubiquitin ligase CBL cause myeloid leukemias, but the molecular determinants of the dominant leukemogenic activity of mutant CBL oncogenes are unclear. Here, we first define a gain-of-function attribute of the most common leukemia-associated CBL mutant, Y371H, by demonstrating its ability to increase proliferation of hematopoietic stem/progenitor cells (HSPCs) derived from CBL-null and CBL/CBL-B-null mice. Next, we express second-site point/deletion mutants of CBL-Y371H in CBL/CBL-B-null HSPCs or the cytokine-dependent human leukemic cell line TF-1 to show that individual or combined Tyr → Phe mutations of established phosphotyrosine residues (Tyr-700, Tyr-731, and Tyr-774) had little impact on the activity of the CBL-Y371H mutant in HSPCs, and the triple Tyr → Phe mutant was only modestly impaired in TF-1 cells. In contrast, intact tyrosine kinase-binding (TKB) domain and proline-rich region (PRR) were critical in both cell models. PRR deletion reduced the stem cell factor (SCF)-induced hyper-phosphorylation of the CBL-Y371H mutant and the c-KIT receptor and eliminated the sustained p-ERK1/2 and p-AKT induction by SCF. GST fusion protein pulldowns followed by phospho-specific antibody array analysis identified distinct CBL TKB domains or PRR-binding proteins that are phosphorylated in CBL-Y371H-expressing TF-1 cells. Our results support a model of mutant CBL gain-of-function in which mutant CBL proteins effectively compete with the remaining wild type CBL-B and juxtapose TKB domain-associated PTKs with PRR-associated signaling proteins to hyper-activate signaling downstream of hematopoietic growth factor receptors. Elucidation of mutant CBL domains required for leukemogenesis should facilitate targeted therapy approaches for patients with mutant CBL-driven leukemias.
Keywords: E3 ubiquitin ligase, leukemia, mutagenesis, oncogene, receptor tyrosine kinase, CBL, proline-Rich region, tyrosine kinase binding domain
Introduction
The CBL family ubiquitin ligases (E3s) specifically interact with activated protein-tyrosine kinases (PTKs)6 and target them for ubiquitination followed by lysosomal or proteasomal degradation (1). Members of this protein family, which includes CBL, CBL-B, and CBL-C in mammals, share a highly conserved N-terminal tyrosine kinase-binding (TKB) domain, a short linker helical region (LHR), and a RING finger (RF) domain. The LHR and RF domains dictate the ubiquitin ligase (E3) activity of CBL family proteins by serving as a structural platform for optimal binding of a ubiquitin-conjugating enzyme (E2).
CBL and CBL-B, but not CBL-C, contain an extensive C-terminal region that includes a proline-rich region (PRR) for interactions with SH3 domain-containing proteins such as Grb2, Nck, CIN85, and Src family PTKs (2–6) and several tyrosine residues that undergo phosphorylation by associated PTKs to form binding sites for interactions with SH2 domain-containing signaling intermediates: Tyr(P)-700 (numbering corresponds to human CBL residues) mediates binding to Vav family of Rho/Rac/Cdc42 guanine nucleotide exchange factors; Tyr(P)-731 mediates binding to PI3-kinase via its p85 subunit, and Tyr(P)-774 mediates binding to Crk family adaptors, which in turn promote the interaction with C3G, a guanine nucleotide exchange factor for Ras-related small GTPases Rap1 (6–9). Near the C terminus, CBL and CBL-B contain a leucine zipper/ubiquitin-associated (UBA) domain involved in ubiquitin binding and dimerization of CBL proteins (10, 11).
Investigations in a variety of cell biological models have elucidated the basic biochemical mechanisms by which CBL proteins function as negative regulators of PTK signaling (1, 12). Following cell surface receptor stimulation and activation of intrinsic (for receptor tyrosine kinases (RTKs)) or non-covalently associated PTK, CBL proteins are recruited through the interaction of their TKB domains to specific phosphotyrosine (Tyr(P))-containing motifs on PTKs. Interaction with PTKs leads to phosphorylation of CBL on multiple residues, including a key regulatory tyrosine within the LHR (human CBL-Tyr-371). This phosphorylation event causes a conformational change in CBL that activates its E3 ubiquitin ligase activity by creating a more optimal E2 interaction interface on the LHR and RF domain and by positioning the bound E2 in closer proximity to the TKB domain-bound PTK target (13). Mutational studies have established that the conserved TKB domain, LHR and RF domains, are required for CBL to function as a negative regulator of PTKs (14–16). Interaction partners that associate with the PRR or Tyr(P) motifs also undergo ubiquitin-dependent negative regulation, although the role of degradation of these targets is less clear (17–19). Thus, CBL functions as an activation-induced negative feedback regulator of PTK-mediated receptor signaling. Consistent with this role, mice with deletion of CBL, CBL-B, or both exhibit hyperactive PTK signaling and associated biological phenotypes (1, 20–24).
A substantial number of recent studies (15, 25–31) have revealed that mutations in CBL, and less frequently CBL-B, are associated with various types of myeloid malignancies, accounting for about 3.5% of all hematological malignancies (COSMIC database). In particular, CBL mutations are found in up to 15% of patients within the category of including the juvenile myelomonocytic leukemia (JMML), chronic myelomonocytic leukemia, and atypical chronic myeloid leukemia cases. Notably, these mutations cluster in the LHR and RF domain, regions indispensable for the E3 activity of CBL proteins. The most common site of CBL mutations in myeloid leukemias involves the regulatory Tyr-371 residue in the LHR, accounting for nearly 10% of all CBL mutations in hematological malignancies. Moreover, this mutation is invariably found in JMML patients, most of whom represent germ line transmission of the mutant gene from a non-cancerous heterozygous parent with Noonan syndrome (27). Oncogenic mutations of CBL-Tyr-371 have been established to abrogate the E3 activity (27, 32, 33). Thus, clinical evidence strongly suggests that loss of E3 activity of CBL mutants is a critical feature leading to the conversion of a negative regulatory CBL protein into a dominant oncogene.
A key additional feature of CBL mutations found in myeloid leukemias is that the remaining wild type (WT) CBL allele is replaced by the mutant allele, resulting in the frequently observed acquired uniparental disomy at the 11q-23 locus where the CBL gene is located (15, 27, 30). This has led to suggestions that mutant CBL proteins may function through a gain-of-function mechanism and/or by competing with the remaining wild type CBL family member CBL-B (34), because CBL-C is not expressed in hematopoietic lineages (35). Consistent with this idea, mouse hematopoietic stem cell knock-out of CBL and CBL-B together (36, 37) or a CBL RF domain mutant knock-in on a CBL-null background (16) leads to a myeloproliferative disease similar to human leukemic disease caused by CBL/CBL-B mutations. How the leukemia-associated CBL mutants function as dominant oncogenes, however, remains unclear. This question is of obvious interest, as this information could help design therapeutic strategies to treat patient with mutant CBL-driven leukemia. Because mutant CBL proteins are expected to interact with cellular partners of normal CBL, and may also interact with other proteins, we undertook a structure-function analysis to delineate the structural regions in mutant CBL that are essential for its leukemogenic activity using in vitro cell models. By expressing the most common leukemia-associated CBL mutant (CBL-Y371H) in CBL-null or CBL/CBL-B-null hematopoietic stem/progenitor cells, we show that mutant CBL functions both through competition with endogenous CBL-B and an inherent gain-of-function. By engineering second-site mutations or deletions in key structural motifs/domains of the CBL-Y371H mutant to disable its protein-protein interactions, we identify a critical role of the proline-rich region in its ability to promote cytokine hypersensitivity in murine primary hematopoietic stem/progenitor cells (HSPCs), which we confirm and extend by expressing these engineered mutants in the TF-1 human leukemia cell line. Coupled with an essential requirement of an intact TKB domain of mutant CBL, our results provide a model for how mutant CBL functions as a dominant oncogene.
Results
Demonstration of a Gain-of-Function Phenotype of Leukemia-associated CBL Mutant, CBL-Y371H, through Expression in CBL-null and CBL/CBL-B-null Primary Mouse Hematopoietic Stem/Progenitor Cells
Clinical studies and experimental modeling in mice or in vitro (15, 27, 30) have demonstrated that loss of endogenous WT CBL expression is a prerequisite for mutant CBL-driven leukemogenesis. Furthermore, introduction of an oncogenic CBL mutant into murine HSPCs led to cytokine hyper-responsiveness, a feature of mutant CBL-associated human leukemias (15). These findings have promoted the idea that CBL mutants attain gain-of-function attributes (15, 34). Yet, deletion of both alleles of CBL or even two alleles of CBL and one allele of CBL-B in mouse HSPCs did not trigger myeloproliferative disease, whereas homozygous CBL/CBL-B deletion produced a rapidly lethal myeloproliferative disease (38). Thus, whether the gain-of-function of mutant CBL simply reflects the relative increase in the dosage of mutant CBL protein relative to that of CBL-B, the remaining wild type CBL family member expressed in hematopoietic stem cells or a true gain-of-function that can be observed in the absence of any wild type CBL and CBL-B expression is unknown. To address this issue, we introduced retroviruses that code for WT CBL or the leukemia-associated CBL-Y371H mutant (Fig. 1A) as well as GFP into primary mouse CBL-null or CBL/CBL-B-null HSPCs, selected the infected cells by FACS based on GFP expression (Fig. 1B), and assessed the extent of cytokine hyper-responsiveness imparted by mutant CBL expression. As reported previously (15), introduction of CBL-Y371H into CBL-null HSPCs led to a marked hyper-responsiveness to stem cell factor (SCF) or thrombopoietin (TPO), as assessed using cell counting as an index of proliferation (Fig. 1, C–E). Notably, although CBL-Y371H expression was not enough to promote cytokine independent proliferation, it enhanced the survival of CBL-null cells under cytokine-independent conditions (Fig. 1C). In contrast, WT CBL expression led to an expected small reduction in cytokine responsiveness (Fig. 1, C–E). Next, we expressed the WT CBL or CBL-Y371H mutant in CBL/CBL-B DKO HSPCs and assessed their responsiveness to SCF or thrombopoietin, as above. As we have shown previously (36, 37), CBL/CBL-B DKO HSPCs by themselves show a dramatically heightened response to cytokines (Fig. 1, F–H). In this setting, introduction of WT CBL led to a marked reduction in the level of basal and cytokine-induced cell proliferation (Fig. 1, F–H). Notably, despite the marked hyperproliferation of CBL/CBL-B DKO HSPCs, CBL-Y371H mutant expression led to a significant further increase in cytokine-induced proliferation; this was more clearly seen under cytokine-independent conditions and in TPO-treated groups (Fig. 1, F–H). These results show that mutant CBL proteins attain a true gain-of-function phenotype that can be seen even in the absence of any WT CBL family proteins.
FIGURE 1.

Analysis of gain-of-function of CBL-Y371H in CBL-null and CBL/CBL-B-null primary mouse HSPCs. A, schematic illustration of various second-site mutants of CBL-Y371H mutants used in this study. B, mouse primary HSPCs were isolated and infected with the indicated retroviruses as described under “Experimental Procedures.” Data shown are representative FACS plot of the GFP+ population from Cbl null and Cbl/Cbl-b DKO HSPCs. C–H, lineage-negative HSPCs were purified by immuno-depletion of lineage-positive mature cells from bone marrow mononuclear cells collected from CBL-null (C–E) or CBL/CBL-B-null (DKO) mice (F–H). The cells were retrovirally infected to express the indicated constructs carrying an IRES-GFP, and infected (GFP+) cells obtained by FACS sorting were assessed in three replicates for cytokine-independent (C and F), SCF-stimulated (D and G), or TPO-stimulated (E and H) proliferation as described under “Experimental Procedures.” For each in C–H, left is one representative experiment with six replicates (mean ± S.D.); right is pooled data of three independent experiments shown as percentage of uninfected control (mean ± S.E.). *, p < 0.05.
Deletion of the Proline-rich Region of CBL-Y371H Mutant Abrogates Its Ability to Promote Cytokine Hypersensitivity in HSPCs
The leukemia-associated oncogenic mutations of CBL cluster in the LHR and RF domain and inactivate the E3 activity (15), but the key protein-protein interaction motifs and domains, including the C-terminal proline-rich region and the tyrosine phosphorylation sites, are intact in the oncogenic mutant. To determine whether these domains/motifs of mutant CBL are required for leukemogenic activity, we engineered HA-CBL-Y371H mutant constructs in which second-site mutations or deletions were introduced (Fig. 1A) as follows: the Tyr → Phe mutations of Tyr-700, Tyr-731, and Tyr-774 to disable interactions with the corresponding SH2 domain-containing signaling proteins or deletion of amino acids 477–688 to create an internal deletion of the proline-rich region.
To examine the impact of the C-terminal phosphorylation sites on the oncogenic ability of CBL-Y371H mutant, DKO HSPCs were infected with retroviruses encoding CBL-Y371H with secondary Tyr → Phe mutations of Tyr-700, Tyr-731, and Tyr-774, individually or together. SCF-induced cell proliferation of GFP+ (mutant-expressing) cells was assessed to determine the effect of Tyr → Phe mutations on the ability of CBL-Tyr-371 mutant to promote SCF hyper-responsiveness. Notably, the enhancement of SCF-dependent cell proliferation conferred by the CBL-Tyr-371 mutant expression was unaffected by individual as well as combined Tyr → Phe mutations of Tyr-700, Tyr-731, and Tyr-774 (Fig. 2A).
FIGURE 2.

Proline-rich region of CBL-Y371H mutant is essential for its gain-of-function phenotype in HSPCs. A, CBL/CBL-B DKO mouse HSPCs were isolated, retrovirally infected with the indicated CBL constructs, and infected (GFP+) cells FACS-sorted as in Fig. 1, followed by analysis of SCF-induced proliferation (1 ng/ml) using the Cell TiterGlo assay. B–D, CBL-null mouse HSPCs were retrovirally infected with the indicated CBL constructs, and GFP+ cells were assessed in three replicates for cytokine-independent (B) or SCF-stimulated proliferation (C) (Cell TiterGlo), or for SCF-stimulated colony forming ability (D). Proliferation based on luminescence arbitrary unit in A–C was normalized to the vector-only control. Shown is pooled data of three independent experiments demonstrated as percentage to vector-only control (mean ± S.E.).
Because the marked cytokine hypersensitivity of CBL/CBL-B-null HSPCs results in a more modest further increment upon CBL-Y371H expression, the subsequent structure-function analyses were carried out using CBL-null HSPCs. Remarkably, the CBL-Y371H mutant with a secondary deletion of the PRR showed a drastically reduced ability to promote the cytokine-independent as well as the cytokine-dependent hyper-proliferation of CBL-null HSPCs, based on both cell counting and colony-forming assays of cell proliferation (Fig. 2, B–D). This finding demonstrated, for the first time, that the C-terminal sequences of mutant CBL distinct from the mutational hot spot region (LHR and RF), and in particular the PRR, are required for the oncogenic activity of leukemia-associated mutant CBL.
Proline-rich Region and an Intact TKB Domain Are Required for CBL-Y371H Mutant to Promote Cytokine Hypersensitivity and Cytokine-independent Survival of Cytokine-dependent TF-1 Leukemia Cell Line
To further establish the requirement of the PRR of mutant CBL in its gain-of-function phenotype and to begin to gain mechanistic insights, we carried out further analyses using the GM-CSF-dependent TF-1 human leukemic cell line model that has been used recently to demonstrate the signaling aberrations caused by leukemia-associated CBL mutants (32, 39, 40). Infection of TF-1 cells with retroviruses used above for HSPCs was carried out, and FACS-sorted GFP+ cells were propagated to generate stable cell lines expressing the HA epitope-tagged CBL-Y371H or HA-CBL-WT and a control line with empty vector alone. The expression of exogenous CBL proteins was verified using Western blotting with an anti-HA antibody (Fig. 3A). We next compared the effect of ectopic mutant versus WT CBL expression in TF-1 cells on cell survival under GM-CSF deprivation conditions. Confirming previous reports (32, 39, 40), CBL-Y371H-expressing TF-1 cells exhibited a modest but significant increase in viable cell numbers when grown in GM-CSF-free medium compared with empty vector or WT CBL-expressing cells (Fig. 3B). Next, we compared the control, CBL-WT-, and CBL-Y371H-expressing TF-1 cells for relative sensitivity to GM-CSF by measuring cell proliferation in response to increasing concentrations of GM-CSF. The CBL-Y371H-expressing cell line showed a moderate but significant shift in the dose-response curve toward the left compared with vector control and CBL-WT-expressing cell lines (Fig. 3C), and the difference between CBL-Y371H and CBL empty vector was significant.
FIGURE 3.

Expression of CBL-Y371H in TF-1 leukemic cell line enhances cytokine-independent as well as cytokine-dependent proliferation. A, TF-1 cells were retrovirally infected with the vector or the indicated HA-tagged CBL constructs, the GFP+ population sorted and maintained continuously in GM-CSF-containing media. The expression of ectopic CBL constructs was verified using immunoprecipitation of cell lysates with anti-HA antibody. Reactivity with the culture medium-derived IgG heavy chain bands in the lysates provides a loading control. B and C, TF-1 cell lines expressing the indicated CBL constructs were GM-CSF deprived for 24 h, followed by culture in medium without GM-CSF (B) or supplemented with the indicated concentrations of GM-CSF (C). D, TF-1 cell lines expressing the indicated CBL constructs were GM-CSF deprived for 24 h, followed by culture in medium supplemented with the indicated concentrations of SCF. Proliferation was assessed after 72 h, using the Cell TiterGlo assay. B–D, left is one representative experiment with six replicates (mean ± S.D.); right is pooled data of three independent experiment shown as percentage to vector only control (mean ± S.E.). *, p < 0.05.
Although the GM-CSF dose-response analysis indicated that the CBL-Y371H mutant expression led to GM-CSF hyper-responsiveness, the magnitude of this effect was modest. Our recent studies in murine hematopoietic stem cells have highlighted the importance of stem cell factor (SCF)/c-KIT receptor signaling as an important target of CBL proteins (37). Furthermore, aberrations of c-KIT signaling are well established in myeloid leukemia (41). Consistent with the low levels of surface c-KIT, stimulation with increasing concentrations of SCF (in the absence of GM-CSF) only elicited a modest increase in the proliferation of vector control and CBL-WT-expressing TF-1 cell lines (Fig. 3D). In contrast, CBL-Y371H-expressing TF-1 cells exhibited a robust and SCF dose-dependent increase in proliferation compared with that of WT CBL or empty vector expressing cell lines (Fig. 3D). The relatively low SCF-induced cell proliferation of control cell lines versus a robust proliferation of the CBL-Y371H-expressing cell line provided a sensitive measure of the ability of the CBL-Y371H mutant and its second-site mutants to up-regulate PTK signaling downstream of a hematopoietic RTK intimately associated with leukemogenesis.
To determine the importance of the protein-protein interaction domains/motifs of mutant CBL in the TF-1 cell model, we utilized selected second-site mutants of CBL-Y371H based on analyses of the HSPCs above: CBL-Y371H with Tyr → Phe mutations of Tyr-700, Tyr-731, and Tyr-774 together or an internal deletion of the PRR (amino acids 477–688). In addition, we utilized a second-site mutant with a truncation after amino acid 436 to delete all of the C-terminal motifs and a G306E point mutant to abrogate the ability of the TKB domain to bind to activated PTKs (Fig. 1A). The corresponding mutations were also engineered within a WT CBL construct as controls. These second-site mutants were retrovirally transduced into TF-1 cells, and the cells were selected based on GFP expression, and the expression of the introduced constructs was verified by Western blotting with an anti-HA antibody (Fig. 4A). The impact of the second-site mutations on the oncogenic activity of the CBL-Y371H mutant in TF-1 cells was assessed by measuring the SCF-induced cell proliferation (Fig. 4B).
FIGURE 4.

TKB domain and proline-rich region of CBL-Y371H mutant are essential for the enhanced cytokine-independent and cytokine-dependent proliferation of TF-1 cell lines. A, stable retroviral expression of the indicated CBL constructs was verified using Western blotting with anti-HA antibody, with IgG heavy chain serving as a loading control as in Fig. 3A. B and C, TF-1 cell lines expressing the indicated ectopic CBL constructs were analyzed for SCF-stimulated (B) or cytokine-independent (C) proliferation as in Fig. 3. Shown are pooled data from three independent repeats (mean ± S.E.). *, p < 0.05.
In contrast to the unmodified CBL-Y371H mutant, expression of the same mutant with a secondary G306E mutation eliminated the hypersensitivity of TF-1 cells to SCF stimulation (Fig. 4B), demonstrating that the ability of mutant CBL to bind to its PTK targets is essential for its oncogenic activity. As expected, the WT CBL or its G306E mutant had no significant impact on the SCF response of TF-1 cells. Notably, the CBL-Y371H mutant with an engineered internal PRR deletion (CBL-Y371H-ΔPRR) was drastically impaired in its ability to impart the SCF hypersensitivity, indicating that the PRR-mediated protein-protein interactions are indispensable for oncogenic activity of mutant CBL. Notably, the combined mutation of Y700F/Y731F/Y774F induced a modest but significant decrease in CBL-Tyr-371-induced SCF responsiveness, suggesting that the protein-protein interactions mediated by the phosphorylation sites within the C-terminal part of mutant CBL do play a positive role in the oncogenic activity of mutant CBL in the TF-1 model. Consistent with our results using the CBL-Y371H mutant with Tyr → Phe secondary mutations or PRR deletion, truncation of CBL-Y371H distal to the RF domain (CBL-Y371H-436-Stop), which excludes the proline-rich region as well as the phosphorylation sites, completely abrogated its ability to promote TF-1 cell hyper-proliferation in response to SCF, clearly establishing that the C-terminal motifs of mutant CBL are required for oncogenic activity.
We also tested TF-1 cells expressing selected second-site mutants of CBL-Y371H for cytokine-independent proliferation (Fig. 4C). Interestingly, TF-1 cells expressing the CBL-Y371H-Y700F/Y731F/Y774F mutant displayed cytokine-independent growth comparable with that of cells expressing the unmodified CBL-Y371H mutant. In contrast, TF-1 cells expressing the internal PRR deletion mutant of CBL-Y371H failed to exhibit the cytokine-independent growth advantage, further demonstrating the importance of an intact PRR of mutant CBL for its oncogenic activity. Altogether, these data reveal an essential role of the TKB domain and the PRR of mutant CBL for its oncogenic activity, with a positive but less critical role of tyrosine phosphorylation sites.
Proline-rich Region of Mutant CBL Is Critical for Its Ability to Enhance SCF-induced c-KIT Signaling
Studies using HSPCs derived from mouse models of mutant CBL leukemogenesis and leukemic cell lines have demonstrated that cytokine hypersensitivity promoted by mutant CBL proteins or by loss of CBL/CBL-B expression is associated with increased downstream signaling (16, 32, 37, 39, 40, 42–44). Given our findings that the PRR of mutant CBL is essential for its ability to promote cytokine hyper-responsiveness, we assessed the impact of PRR deletion on the ability of the CBL-Y371H mutant to promote the hyper-activation of signaling in response to SCF stimulation. TF-1 cells stably expressing CBL-Y371H or its PRR deletion construct were stimulated with SCF for various time point, and the levels of phosphorylated AKT and ERK1/2 were assessed using Western blotting of cell lysates (Fig. 5A). As expected (16, 37), SCF stimulation induced a higher and more sustained AKT and ERK1/2 phosphorylation in CBL-Y371H-expressing TF-1 cells compared with the empty vector or WT CBL-expressing control cells. Importantly, the TF-1 cells expressing the PRR-deleted CBL-Y371H mutant failed to exhibit the enhanced and sustained AKT and ERK1/2 phosphorylation (Fig. 5A).
FIGURE 5.

Proline-rich region of CBL-Y371H mutant essential for enhanced signaling downstream of SCF-induced c-KIT activation. TF-1 cell lines expressing the indicated CBL mutant constructs were GM-CSF-deprived and then either left unstimulated (time 0) or stimulated with SCF (100 ng/ml) for the indicated time points (minutes). A–C, cell lysates were subjected to Western blotting for the indicated proteins: p-ERK, total ERK, and p-AKT (A) and total c-KIT and p-c-KIT (B). Shown is a representative experiment of two independent repeats with similar results. C, phospho-c-KIT signals in Western blottings were quantified using ImageJ analysis of scanned images, and the signals were first normalized to HSC-70 and then expressed as phospho-c-KIT signals relative to those in vector-transfected cells at 5 min, which were assigned a value of 1. Shown are pooled data of three independent repeats (mean ± S.E.). Red asterisks (*) indicate significance between CBL-Y371H and CBL-Y371-ΔPRR, and black asterisks (*) show significance between CBL-Y371H and vector cells (D). The indicated live TF-1 cell lines were analyzed by FACS for surface levels of c-KIT at various points after SCF stimulation. Mean fluorescence intensity relative to unstimulated controls is plotted. Shown are pooled data of three independent repeats. *, p < 0.05. E, THP-1 and GDM-1 cell lines were serum-deprived for 24 h and then left unstimulated or stimulated with SCF (100 ng/ml) for the indicated time points (minutes). Cell lysates were Western-blotted for phospho-c-KIT, total c-KIT, phospho-CBL, total CBL, and total CBL-B with HSC-70 used as a loading control. Shown is a representative of two independent repeats with similar results.
We also examined the impact of the expression of CBL-Y371H or its PRR deletion mutant on the kinetics of the loss of c-KIT receptor upon SCF stimulation. As expected (37, 45), the total and phosphorylated c-KIT levels declined in a time-dependent manner following SCF stimulation of the vector control TF-1 cells (Fig. 5, B and C). In contrast, the down-regulation of phospho-c-KIT levels was delayed in SCF-stimulated TF-1 cells expressing the CBL-Y371H mutant compared with control TF-1 cells expressing the empty vector, consistent with the previously established role of CBL in the down-regulation of c-KIT (43, 46, 47) and other receptor tyrosine kinases (1). Notably, TF-1 cells expressing the CBL-Y371H-ΔPRR mutant showed a reduced level of phosphorylation of c-KIT compared with that observed in CBL-Y371H-expressing cells, although the kinetics of total c-KIT down-regulation did not differ substantially between the two cell lines (Fig. 5, B and C). Furthermore, FACS analysis of live cells revealed slower down-regulation of surface c-KIT upon SCF stimulation in TF-1 cell expression of CBL-Y371H compared with empty vector control, whereas TF-1 cells expressing the CBL-Y371H-ΔPRR mutant exhibited a c-KIT down-regulation kinetics comparable with that of the vector control cells (Fig. 5D). To exclude the possibility that altered SCF signaling seen upon CBL-Y371H mutant expression in TF-1 cells can also be observed in leukemia cell lines with endogenously expressed mutant CBL, we compared the SCF-induced c-KIT and CBL phosphorylation in THP-1 (wild type CBL and c-KIT) and GDM-1, which c-KIT harbors bi-allelic CBL-R420Q RING finger mutations (44). Currently, GDM1 cell line is the only reported myeloid leukemia cell line with a duplicated CBL mutation akin to that seen in leukemia patients with CBL mutations. Consistent with low levels of c-KIT expression in THP-1 cells, a relatively low level of c-KIT and CBL phosphorylation was seen upon SCF stimulation (Fig. 5E, longer exposure). In contrast, SCF stimulation of c-KIT in GDM-1 cells, which express high levels of c-KIT, exhibited high and sustained levels of c-KIT phosphorylation (Fig. 5E); notably, even though the levels of total CBL in the GDM1 cell line are lower than that in THP-1 cells, markedly higher and sustained phosphorylation of CBL was seen in these cells (Fig. 5E). These analyses further support our conclusion that mutant CBL promotes sustained c-KIT phosphorylation and signaling.
CBL-Y371H Mutant Is Hyper-phosphorylated Following SCF Stimulation and the Internal PRR Deletion Negates This Effect
One characteristic of oncogenic CBL mutants, observed in leukemic cells as well as upon ectopic expression, is their hyper-phosphorylation (14, 46, 48). We therefore examined the impact of PRR deletion on the hyper-phosphorylation of the CBL-Y371H mutant under basal conditions and upon SCF stimulation. Growth factor-deprived TF-1 cells expressing the HA-tagged CBL-Y371H mutant or its PPR deletion were either left unstimulated or stimulated with SCF for the indicated time points followed by immunoprecipitation of CBL and Western blotting with anti-Tyr(P), anti-phospho-CBL Tyr-700, or anti-phospho-CBL Tyr-774 antibodies to assess the level of phosphorylation on mutant CBL proteins. Notably, despite higher expression of the PRR-deleted CBL-Y371H mutant, it displayed lower levels of basal as well as SCF-induced tyrosine phosphorylation as detected by either anti-Tyr(P) or phosphotyrosine site-specific antibodies (Fig. 6A). A time course experiment revealed that while the SCF-induced phosphorylation of CBL-Y371H mutant was sustained for >90 min (the longest time point analyzed) that of the PRR-deleted mutant was transient, reaching basal levels by 30 min (Fig. 6A). These results suggest that interactions mediated by the PRR are required for the hyper-phosphorylation of mutant CBL.
FIGURE 6.

Proline-rich region of CBL-Y371H is required for hyper-activation of basal and SCF-induced signaling. A, TF-1 cell lines expressing indicated CBL mutant constructs were GM-CSF deprived and either left unstimulated or stimulated with SCF for the indicated time points. Anti-HA immunoprecipitations were carried out followed by anti-Tyr(P), anti-CBL-Tyr(P)-700, or anti-CBL-Tyr(P)-774 immunoblotting to visualize the phosphorylation of ectopic overall and site-specific phosphorylation of CBL proteins. Anti-HA blotting shows comparable loading. Shown is a representative experiment of two independent repeats with similar results. B, GM-CSF-deprived TF-1 cells expressing CBL-Y371H were stimulated with SCF for 15 min, and 5 mg of cell lysate protein aliquots used for pulldown with 50 μg of GST (lane 1, control), GST-CBL-N (lane 2), or GST-CBL-C (lane 3) fusion proteins non-covalently bound to glutathione-Sepharose beads. After washing, the bound proteins were visualized anti-Tyr(P) (4G10) immunoblotting, and the membrane was re-probed with an anti-phospho-c-KIT antibody. The whole cell lysate (25 μg) was concurrently resolved (lane 4). The molecular mass markers (in kilodaltons) are indicated on left. C, antibody array analysis of phosphoproteins in whole cell lysates. TF-1 cells expressing empty vector, CBL-Y371H, or CBL-Y371H-ΔPRR were GM-CSF-deprived and stimulated with SCF for 15 min. Cell lysates were collected in the antibody array lysis buffer supplied with the kit and incubated with antibody array filters and developed using enhanced chemiluminescence, followed by densitometry analysis of phosphoprotein signals. Signals were expressed relative to those in unstimulated vector control TF-1 cell lysates. D, antibody array analysis of preferential binding of cellular phosphoproteins with PRR-containing GST-CBL-C versus GST-CBL-N (TKB domain). TF-1 cells expressing CBL-Y371H were GM-CSF-deprived and stimulated with SCF for 15 min. 5-mg aliquots of cell lysate protein were incubated with 50 μg of glutathione-Sepharose-bound GST-CBL-N or GST-CBL-C fusion proteins. Bound proteins were eluted in 1% SDS-containing lysis buffer by a 5-min incubation at 95 °C, cooled to room temperature, diluted 10-fold in antibody array buffer, and analyzed for phosphoprotein signals using antibody arrays as in C. Data are presented as a ratio of GST-CBL-C pulldown signals over GST-CBL-N pulldown signals for each phosphoprotein. Boxes indicate preferential binding to GSCT-CBL-C or GST-CBL-N.
PRR of CBL-Y371H Mutant Is Required to Promote Hyper-phosphorylation of Certain Signaling Pathway Proteins Downstream of SCF and Shows Preferential Interaction with Some of These Proteins
To gain further insights into the requirements of the TKB domain and the PRR for the leukemogenic ability of CBL-Y371H mutant, we undertook a combination of GST fusion protein pulldown assays and a commercially available antibody array-based detection approach focused on phospho-proteins linked to normal as well as oncogenic signaling. We first assessed whether any of the proteins exhibiting hyper-phosphorylation on a tyrosine residue in the CBL-Y371H mutant-expressing TF-1 cells were potential TKB domain or PRR-binding partners. For this purpose, we used bacterially expressed GST fusion proteins of the N-terminal TKB domain (GST-CBL-N) or the C-terminal part of CBL (CBL-C), which includes the proline-rich region, to carry out a pulldown from CBL-Y371H-expressing cells that were stimulated with SCF. Of note, the bacterially expressed GST fusion proteins lack any phosphorylation on tyrosine residues, which are present within the CBL-C fusion protein, thereby eliminating any possibility of the SH2 domain-containing signaling proteins interacting with this probe via phosphotyrosine-containing motifs on the fusion protein. The only motif within this fusion protein that interacts with signaling proteins is the proline-rich region (1). Consistent with the primary function of the TKB domain of CBL proteins to interact with phosphorylated PTKs (1), including c-KIT (50), anti-Tyr(P) blotting of GST-CBL-N pulldown included prominent bands in the region where phospho-c-KIT is expected to migrate, and anti-phospho-c-KIT immunoblotting confirmed this (Fig. 6B). In contrast, fewer Tyr(P)-containing bands were detected in GST-CBL-C pulldowns, notably a band in the 55–60-kDa range, and only weak phospho-c-KIT signals were detected (Fig. 6B). Given that only few Tyr(P)-containing bands were seen with GST-CBL-C pulldown, we utilized an unbiased approach to query the pulldowns using a commercial antibody array of phosphoproteins linked to PTK signaling (phosphokinase array, R & D Systems).
First, GM-CSF-deprived TF-1 cells expressing the CBL-Y371H mutant or its PRR deletion as well as empty vector control cells were stimulated with SCF for 15 min, and their lysates were used to probe the relative level of phosphorylation of individual proteins represented in the antibody array. Lysates of unstimulated empty vector control TF-1 cells were used to normalize the level of phosphorylation for each individual protein. Albeit the differences were modest, higher p-AKT and p-ERK signals were seen in lysates of CBL-Y371H,-expressing cells compared with control lysates, and these signals were reduced in lysates of cells expressing the PRR-deletion mutant of CBL-Y371H (Fig. 6C), confirming the results obtained with traditional Western blotting (Fig. 5A). In addition, other cellular signaling proteins, such as STAT proteins, showed moderately elevated phosphorylation in CBL-Y371H mutant-expressing TF-1 cells, and this was reduced in cells expressing the PRR deletion mutant (Fig. 6C). These results further support a role of the mutant CBL PRR in the hyper-activation of cellular signaling.
Next, we carried out GST-CBL-N or GST-CBL-C pulldowns from lysates of SCF-stimulated CBL-Y371H mutant-expressing TF-1 cells. The GST fusion protein-bound proteins were eluted in 1% SDS-containing buffer and diluted in the antibody array sample buffer to reduce the SDS concentration to 0.1% followed by analysis of these samples using the phospho-protein antibody array to assess those signaling proteins that might have selectivity for binding to PRR or TKB domain. By expressing the signals of phosphoproteins as a ratio of signals in the GST-CBL-C pulldown over the GST-CBL-N pulldown, we observed moderate to high selective pulldown of Src family kinases (Lck, Lyn, Fyn, and Yes) and PLCγ1 with the PRR-containing GST-CBL-C fusion protein (Fig. 6D). However, signaling proteins involved in JAK-STAT signaling appeared modestly enriched in the CBL TKB domain pulldown (Fig. 6D). There is an overlap between the phospho-protein pulldowns with the TKB domain, and the PRR is consistent with previous findings that CBL can interact with its targets, such as the SRC family kinase Fyn, through multiple independent interactions (49). Collectively, these data support a critical role for the PRR and TKB domain of mutant CBL in hyper-activating hematopoietic growth factor receptor signaling.
Discussion
Mutations or deletions within the LHR and the RING finger domain of CBL, and rarely CBL-B, are now firmly established as a causal event in a subpopulation of leukemias within the myelodysplastic syndrome/myeloproliferative neoplasms (myelodysplastic/myeloproliferative disorders) category. How these E3 activity-nullifying point mutations or small deletions convert CBL from a negative regulator of PTK signaling and a tumor suppressor into a dominant oncogene is not fully understood. Previous work has suggested that mutant CBL proteins attain a gain-of-function, but the structural basis for this key attribute of mutant CBL proteins remains unclear. Using CBL-Y371H, the single most common leukemia-associated CBL mutant, as a representative example, we use genetic and structure-functional analyses to reveal novel insights into mechanisms of the gain-of-function phenotype of mutant CBL proteins.
As reported previously (15), ectopic expression of CBL-Y371H in CBL-null primary HSPCs markedly elevated the cytokine responsiveness, which reflects the ability of mutant CBL to compete with endogenous WT CBL-B (Fig. 1, C–E). Consistent with this idea, CBL/CBL-B DKO HSPCs exhibit a dramatically higher cytokine responsiveness compared with CBL-null HSPCs (Fig. 1, F–H), as noted previously (36, 37). Remarkably, however, the mutant CBL-Y371H expression in CBL/CBL-B DKO HSPCs consistently led to a further increase in cytokine-independent proliferation as well as cytokine-induced hyper-proliferation of these cells (Fig. 1, F–H). Thus, although competition with CBL-B clearly accounts for a major part of the gain-of-function phenotype of mutant CBL, our analyses show that oncogenic CBL mutants enhance receptor signaling beyond what can be attributed to just a lack of functional CBL and CBL-B proteins.
A plausible mechanism to account for the gain-of-function phenotype of oncogenic CBL mutants is that the intact protein-protein interaction domains/motifs in these mutants continue to engage the PTK signaling machinery components that are physiologically accessed by the wild type CBL/CBL-B. However, unlike WT CBL/CBL-B-associated partners, the PTKs and signaling proteins bound to E3 activity-deficient mutant CBL proteins cannot undergo ubiquitination and consequent degradation (50–52). Furthermore, unlike the autocatalytic ubiquitination and subsequent degradation of WT CBL/CBL-B (53, 54), mutant CBL proteins complexed with PTKs and signaling proteins are expected to remain stable. Consistent with this notion, the leukemia-associated CBL mutants, including the CBL-Y371H, lack E3 activity (27, 32, 33). We therefore posit that the combination of these processes transforms the mutant CBL protein into a stable adaptor to juxtapose the signaling proteins bound to mutant CBL's intact protein-protein interaction domains with the stabilized PTKs bound to its TKB domain (Fig. 7). This model predicts that an intact TKB domain, which helps recruit the mutant CBL to surface receptor-associated PTKs, and one or more of the C-terminal motifs, which mediate interactions with downstream signaling proteins and accessory PTKs, will be essential for the mutant CBL gain-of-function. Findings presented here support such a model and establish that an intact N-terminal TKB domain and the PRR within the C-terminal portion of mutant CBL are essential for the gain-of-function of an oncogenic CBL mutant (Figs. 4B and 5, A–D).
FIGURE 7.
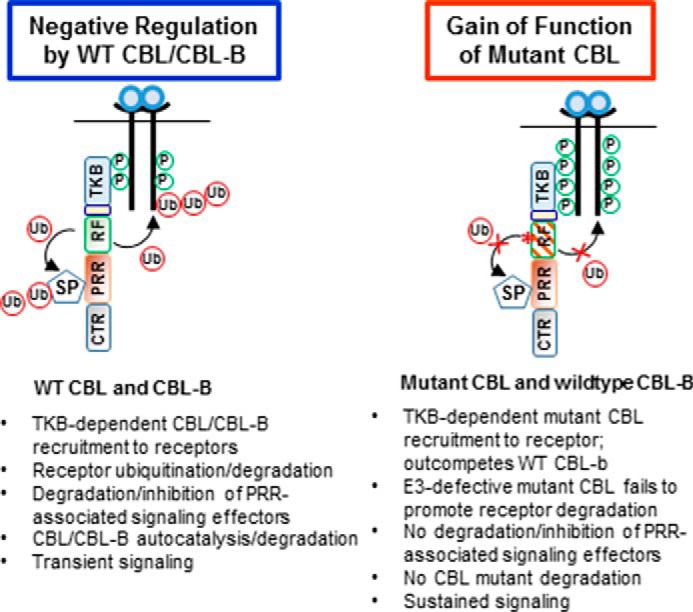
Schematic presentation of the model of mutant CBL gain-of-function based on present studies. TKB, tyrosine kinase-binding domain; RF, RING finger domain; PRR, proline-rich region; CTR, C-terminal region; Ub, Ubiquitin; P, phosphorylation.
A major mechanism of protein-protein interactions of CBL family proteins, which allows them to bind to and negatively regulate a number of signaling proteins, is through the induced tyrosine phosphorylation sites within the regions C-terminal to the RING finger domain (1). Biochemical, cell biological, and animal studies have validated the importance of the phosphorylation of Tyr-700, Tyr-731, and Tyr-774 of CBL in such protein-protein interactions (6, 7, 9, 55, 56). Therefore, we used the murine HSPC expression system to assess whether second-site Tyr → Phe mutations of these residues, singly or in combination, would abrogate the cytokine hyper-responsiveness imparted by the CBL-Y371H mutant (Fig. 2A). Surprisingly, neither single second-site mutations of the indicated tyrosine residues nor their combination had a significant impact on the cytokine hyper-responsive phenotype imparted by CBL-Y371H. This result suggested that either additional uncharacterized phosphorylation sites are responsible for the oncogenic phenotype or that a different protein-protein interaction mechanism is of primary importance in the mutant CBL gain-of-function. When the triple Tyr → Phe mutant of CBL-Y371H was expressed in the TF-1 leukemia cell line model, however, it partially reduced the SCF hyper-responsiveness (Fig. 4B). However, the cytokine-independent growth advantage conferred by CBL-Y371H was not seen with this construct (Fig. 4C). Overall, these results support the idea that the C-terminal phosphorylation sites of mutant CBL proteins are not essential, although it is likely that they contribute to oncogenesis especially under conditions where the mutant CBL gene is expressed under its physiological regulatory control. Future studies using mouse knock-in of Tyr → Phe mutations in the context of a mutant CBL allele should help address this issue categorically. Although the exact reason for the difference in the phenotype of the triple Tyr → Phe mutant of CBL-Y371H in the primary HSPC versus the TF-1 cell systems is not clear, it remains possible that the putative secondary phosphorylation sites play a less important role in the TF-1 cell system. Future proteomic studies will be needed to test this hypothesis.
The other protein-protein interaction motifs retained in CBL mutants, aside from the phosphorylation sites, are the PRR and the UBA domain. Because the PRR is extensive, we engineered an internal PRR deletion in CBL-Y371H mutant to test the role of the PRR-mediated protein-protein interactions. Notably, the PRR deletion essentially completely abrogated the gain-of-function phenotype imparted by CBL-Y371H expression in primary HSPCs as well as in TF-1 cells (Figs. 2B and 4B). Furthermore, truncation of CBL-Y371H mutant at residue 436, which results in deletion of the PRR, the UBA domain, and the intervening sequences, similarly abrogated the gain-of-function phenotype (Fig. 4B). Because the extent to which the gain-of-function attribute was reduced by the PRR deletion or truncation at residue 436 was comparable (Fig. 4B), we conclude that the PRR is the primary C-terminal motif that is required for the gain-of-function mechanism we have proposed (Fig. 7).
Further biochemical analyses carried out in the TF-1 model support the importance of the PRR in the gain-of-function phenotype of CBL mutants. Both the basal and the SCF-induced tyrosine phosphorylation of CBL-Y371H mutant was reduced by the PRR deletion (Fig. 6A). This result suggests that any role of the Tyr(P) motifs in mutant CBL gain-of-function is likely to be largely secondary to interactions mediated by the PRR. Furthermore, although CBL-Y371H expression led to more sustained phosphorylation of c-KIT and downstream signaling intermediates ERK and AKT (Fig. 5, A–C), these traits were lost upon PRR deletion. To ensure that these observations are not simply a reflection of the ectopic overexpression of the CBL mutant in TF-1 cells, we examined the only available myeloid leukemia cell line GDM-1 with a duplicated oncogenic mutation akin to those seen in human myeloid leukemia patients (44). These cells also exhibited SCF-induced sustained c-KIT phosphorylation and marked hyper-phosphorylation of the endogenously expressed mutant CBL (CBL-R420Q) expressed in these cells. By employing an antibody array approach to concurrently assess the levels of phosphorylation of a larger number of signaling proteins, we confirmed the hyper-phosphorylation of ERK and AKT proteins in mutant CBL-expressing TF-1 cells and also observed moderate hyper-activation of other signaling pathways such as members of the SRC family kinases, JAK-STAT pathway, focal adhesion kinase, and PLCγ1 (Fig. 6C). Importantly, hyper-phosphorylation of some of these signaling proteins, such as ERK, AKT, and STAT, appeared to be dependent on an intact PRR in the mutant CBL (Fig. 6C), although more sensitive methods and broader analyses of the phospho-proteome will be needed to put these initial findings on a robust footing. Using the phosphoprotein antibody array analysis together with pulldown with GST fusion proteins incorporating the CBL TKB or the PRR, we found modest to substantial selective binding of the PRR-containing CBL-C region to SRC family kinases Lck, Lyn, Fyn, and Yes as well as to PLCγ1, whereas the phosphorylated c-KIT receptor (Fig. 6B) and STAT proteins were more selectively pulled down with the TKB domain (Fig. 6D). Although these findings need to be further expanded using classical biochemical approaches, these initial findings further support the role of the TKB domain and the PRR in mutant CBL gain-of-function.
The pulldown results are consistent with previous work in which we and others have shown that CBL, via its proline-rich region, interacts with the SH3 domains of Src family kinases (6, 49, 57–60). The PLCγ1 SH3 domain is also known to bind to CBL (61). The observation that these proline-rich region-interacting partners are also pulled down at lower levels with the TKB domain is also consistent with our work in which we have shown the CBL TKB domain to bind to phosphorylated Src family kinase Fyn, separate from the SH3-PRR interaction (49). Furthermore, as signaling proteins are present in larger complexes, some indirect pulldown is expected. The pulldown analyses also confirmed the relatively preferential interaction of the CBL TKB domain with phospho-c-KIT (Fig. 6B), as expected (50).
Consistent with the key role of the PRR in mutant CBL gain-of-function phenotype identified here, several recent reports have shown a role for SFKs in mutant CBL-dependent functional traits in leukemic cell models. Dasatinib, a multikinase inhibitor whose targets include SFKs, was shown to preferentially reduce the proliferation of a leukemic cell line that harbors a duplicated copy of mutant CBL compared with a leukemic cell line without a CBL mutation (44). Introduction of Y371H or C384R mutants of CBL into TF-1 cells led to increased GM-CSF-induced activation of JAK2 and LYN, the latter an SFK member, as well as increased GM-CSF receptor phosphorylation, and the latter was inhibited by dasatinib (32). Another study showed that GM-CSF hypersensitivity imparted by the CBL-Y371H mutant in TF-1 or BAF-3 cells was associated with increased SRC and LYN phosphorylation and was reversed by dasatinib (40). Finally, a recent study in which all coding exons of CBL family proteins were analyzed in JAK2-V617F mutation-negative leukemia samples identified occasional PRR mutations which, like RING finger mutations, imparted IL3 hypersensitivity when introduced in the cytokine-dependent cell line 32D-FLT3 (62).
How might the PRR participate in promoting the gain-of-function phenotype of oncogenic CBL mutants? Our data and previous studies cited above collectively point to a role of SFKs and potentially of PLCγ1. Mechanistically, the PRR forms a primary determinant of the interaction of CBL family proteins with SFKs (59); however, as we have demonstrated using the SFK FYN (49), additional SFK SH2 domain binding to phospho-CBL and CBL-TKB domain binding to phospho-SFK are also involved. The PRR-recruited SFKs may promote mutant CBL gain-of-function in many ways. For example, their juxtaposition to upstream receptors and their associated intrinsic (e.g. c-KIT) or associated (e.g. JAK2) tyrosine kinases can lead to hyper-activation of signaling through pathways directly recruited both by SFKs (via their SH2 domain) and phosphorylated receptors/RTKs. Consistent with such an idea, recent studies indicate that mutant CBL-dependent AKT hyper-phosphorylation in TF-1 and BAF-3 cell lines is SFK-mediated (39). Mutant CBL PRR-recruited SFKs could also promote the hyper-phosphorylation of upstream receptors/RTKs, resulting in enhanced recruitment of signaling machinery components to these receptors, a suggestion consistent with increased pulldown of phosphorylated STAT proteins with CBL TKB domain (Fig. 6C). This mechanism is consistent with our results (Fig. 5A) and those of others (32) in the context of hematopoietic growth factor receptors and our previous studies demonstrating the hyper-phosphorylation of PDGF or EGF receptors and activation of their downstream signaling in non-hematopoietic cells expressing oncogenic CBL mutants (14, 64–66). It is also possible that SFKs promote the hyper-phosphorylation of mutant CBL, consistent with our results (Fig. 5A), although our studies suggest that this mechanism may be either dispensable (lack of an effect of mutating three major sites of phosphorylation) or involve redundant phosphorylation of other tyrosine residues.
Although the SFK interaction with mutant CBL PRR is of obvious interest, the PRR is known to mediate interactions with a variety of other adaptor (e.g. Grb2 and related adaptors, Nck, CBL-associated protein or CAP, CIN85, and others) or enzymatic proteins (e.g. PLCγ1, PI3K, and others) in PTK signaling pathways (1, 4–7, 19, 65, 67, 68), of which we show a PRR-dependent interaction of PLCγ1 (Fig. 6C). It remains possible that the PRR in mutant CBL is displayed differently compared with that in the WT protein, resulting in binding to novel proteins. Also, the spectrum of signaling proteins represented in the array we utilized was relatively narrow. Thus, it is likely that other unknown proteins interacting through the PRR region are more critical in the gain-of-function phenotype of mutant CBL. Future extension of the biochemical approaches used here together with unbiased proteomics will be needed to determine the extent to which one or more of these proposed mechanisms may be involved.
A particularly important downstream pathway exaggerated in mutant CBL-associated leukemias is the RAS pathway, and CBL mutations in JMML are mutually exclusive with other RAS pathway activating mutations such as those in PTPN11, NF1, or NRAS/KRAS, indicating a key role of this signaling pathway in leukemogenesis (28, 34). Notably, the prolonged ERK and AKT phosphorylation imparted by CBL-Y371H was abrogated by the PRR deletion (Fig. 5, A and C). Thus, we surmise that the PRR of mutant CBL may also be essential to engage the RAS signaling pathway, which is thought to be critical for leukemogenesis by CBL mutants based on clinical observations.
Our observations that the oncogenic phenotype of the CBL-Y371H mutant is abrogated by the PRR deletion or truncation stand in contrast to the impact of truncating the C terminus of WT CBL, which showed that the TKB and RING finger domains were necessary and sufficient for the ubiquitination and degradation of RTKs and non-receptor PTKs (50, 51). Mutations of the RING finger domain or the LHR abolish the E3 activity of CBL (49–52, 56, 65, 66, 69–72), and this has been demonstrated for several human leukemia-derived CBL mutants, including Y371H (15, 32, 73). As the E3-inactivating LHR mutation is still present in the second-site mutants of CBL-Y371H with PRR or entire C-terminal region deletion (Fig. 4, B and C), these results support the notion that loss of E3 activity is required but not sufficient for oncogenesis by CBL mutants. This notion is consistent with a previous analysis of introduced mutations of the LHR of CBL, which identified mutants that were non-transforming in rodent fibroblasts despite their loss of E3 activity (74). It is notable that viral CBL represents a vastly more truncated protein that encompasses only the TKB domain (residues 1–357) (75), and its ability to transform fibroblasts is dependent on its intact TKB domain (14). Yet, v-CBL sequences alone cause milder transformation of rodent fibroblasts compared with LHR mutants that retain the C-terminal region (48). When these sequences were expressed in a hematopoietic cell line, mild cytokine hyper-responsiveness was seen, although a direct comparison with mutant CBL proteins that retain the C-terminal region was not performed (76). Yet, truncation mutants of CBL akin to the v-CBL oncogene are not observed in human leukemias. Together, these findings further support our model that effective leukemogenic ability of mutant CBL proteins involves a gain-of-function that requires an intact C-terminal region, particularly the PRR of mutant CBL, in addition to an N-terminal TKB domain.
In conclusion, our second-site mutational analyses of the most common leukemia-associated CBL oncogene reveal the structural basis for its gain-of-function and identify an essential role of the TKB domain and the PRR of mutant CBL in oncogenesis. Further identification and characterization of partners of mutant CBL's TKB domain and the PRR should provide a fruitful approach to further elucidate mutant CBL oncogenesis and reveal potential strategies to treat leukemias associated with CBL mutations.
Experimental Procedures
Reagents and Antibodies
Anti-hemagglutinin (HA) epitope monoclonal antibody (clone 12CA5) was purified i-house as described (77). Anti-phospho-ERK1/2 (pERK1/2), anti-ERK1/2 (ERK1/2), anti-phospho-AKT (pAKT), anti-AKT (tAKT), anti-phospho-c-KIT (p-c-KIT), anti-c-KIT anti-phospho-CBL (Tyr-700), and anti-phospho-CBL (Tyr-774) were obtained from Cell Signaling Technologies. Goat anti-mouse and anti-rabbit HRP-conjugated secondary antibodies were obtained from Invitrogen. Glutathione-Sepharose beads were from GE Healthcare. Biochemicals were from Sigma or Life Technologies, Inc., unless indicated.
Vectors and Cloning
WT HA-CBL cDNA sequences obtained from the MSCV promoter-driven pGCDNsam retrovirus constructs containing an IRES-EGFP have been described (15). HA-CBL sequences were released by BamHI digestion of previously described pAlterMax2 mammalian expression constructs (77) and cloned non-directionally into the BamHI site of pGCDNsam-IRES-EGFP followed by screening for clones with desired directionality using AgeI/PvuII double digestion.
Site-directed Mutagenesis
All mutagenesis was performed in the pAlterMax2 vector before cloning the cDNAs fragments into retroviral vector to minimize potential deleterious effects of undesired mutations in the retroviral vector backbone. Complementary forward and reverse primers containing the desired mutations were designed using the QuikChange Primer Design Program (sequences are shown in Table 1). These were used for mutagenesis using the QuikChange mutagenesis kit according to the supplier's recommendation (Agilent Technologies). The presence of introduced mutations and lack of any undesired PCR-introduced changes were validated by sequencing the entire HA-CBL cDNA within the construct (sequencing primer sequences listed in Table 2).
TABLE 1.
Primer sequences used to generate various second-site mutants of CBL-Y371H used in this study
F means forward, and R means reverse.
| Mutagenesis oligonucleotide sequences | |
|---|---|
| Mutation | Sequence (5′–3′) |
| Y371H F | GAACAATATGAATTACACTGTGAGATGGGCTCCACA |
| Y371H R | TGTGGAGCCCATCTCACAGTGTAATTCATATTGTTC |
| Y700F F | GAAGAGGACACAGAGTTCATGACTCCCTCTTCCAGG |
| Y700F R | CCTGGAAGAGGGAGTCATGAACTCTGTGTCCTCTTC |
| Y731F F | ATTGATAGCTGTACGTTTGAAGCAATGTATAATATT |
| Y731F R | AATATTATACATTGCTTCAAACGTACAGCTATCAAT |
| Y774F F | AATGAGGATGATGGGTTTGATGTCCCAAAGCCACCT |
| Y774F R | AGGTGGCTTTGGGACATCATACCCATCATCCTCATT |
| 436STOP F | GGTAGATCCGTTTGATCCTTGATAGAGTGGCAGCCTGTTGAGG |
| 436STOP R | CCTCAACAGGCTGCCACTCTATCAAGGATCAAACGGATCTACC |
| ΔPRR F | CAATGTGAGGGTGAAGAGGACAC |
| ΔPRR R | CCGTTCCACCTTGGCACCAG |
TABLE 2.
Sequencing primers used to verify correct sequences of engineered second site mutants of CBL-Y371H
Fwd means forward, and Rev means reverse.
| CBL sequencing primers | ||
|---|---|---|
| Primer | Sequence (5′–3′) | Start site |
| Fwd 1 | TGTTTATGGAGAATTTGATGAAGAA | 350 |
| Fwd 2 | CCTCTTTGCTCAGGAATTGG | 755 |
| Fwd 3 | TGTGCTGAAAATGATAAGGATG | 1150 |
| Fwd 4 | TCTAAGGCTGCTTCTGGCTC | 1558 |
| Fwd 5 | GCAGCCCATTAGTAGGTCCA | 1952 |
| Fwd 6 | CCGCCGAACTCTCTCAGAT | 2358 |
| Rev 1 | TTCGCCTAGGCTGAGAAT | 448 |
| Rev 2 | CCGAGCTTTCACTTCGTCAT | 840 |
| Rev 3 | CTGACCTTCTGATTCCTGCC | 1242 |
| Rev 4 | AGGCGGTGGTGGTGGAAG | 1641 |
| Rev 5 | CACAGGAAGAGGTCTGGCA | 2049 |
| Rev 6 | TGACATTTGTTGTAGGATCACCA | 2440 |
Mice
Cbl−/− (CBL-null) and MMTV-Cre; Cblflox/flox; Cbl-b−/− (CBL/CBL-B-null) mice on a C57BL/6 background (36, 37) were housed under specific pathogen-free conditions at the Center for Comparative Medicine of the University of Nebraska Medical Center. All mouse experiments were approved by the Institutional Animal Care and Use Committee.
Culture of Established Cell Lines
TF-1 human erythroleukemic cell line (ATCC) was cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (HyClone) and 2 ng/ml recombinant human GM-CSF (PeproTech). THP-1 human monocytic leukemic cell line (ATCC) and GDM-1 human myelomonoblastic leukemic cell line (ATCC) were cultured in RPMI 1640 medium (HyClone) supplemented with 10% fetal bovine serum. Phoenix-Eco and Phoenix-Ampho packaging cell lines were obtained from ATCC and grown in DMEM supplemented with 10% fetal bovine serum.
Mouse Bone Marrow-derived HSPC Isolation
Primary bone marrow cells harvested from femurs and tibiae of mice and lineage marker-negative cells, representing the HSPCs, were MACS-purified (Miltenyi) using a lineage depletion kit (Miltenyi) according to the manufacturer's instructions. HSPCs were cultured in α-minimal essential medium supplemented with 10% fetal bovine serum, 25 ng/ml recombinant murine SCF, 10 ng/ml recombinant murine TPO, and 10 ng/ml recombinant murine FLT3 ligand (all from PeproTech). All cells were grown in 37 °C incubators with 5% CO2.
Retrovirus Production and Infection
Phoenix-Eco or Phoenix-Ampho packaging cell lines were used to produce viruses for infection of mouse or human cells, respectively. The cells were plated in 10-cm plates and transfected with 20 μg of retroviral vector and 7 μg of the appropriate envelope vector (pCL-Eco for Phoenix-Eco and pIK for Phoenix-Ampho, respectively) (Phoenix-Eco and Phoenix-Ampho were from ATCC; pCL-Eco was a gift from Dr. Inder Verma, Salk Institute; plK was a gift from Dr. David Root, Broad Institute) using the calcium phosphate method (72). Supernatants were collected at 48, 60, or 72 h post-transfection and filtered through a 0.45-μm cellulose acetate syringe filter (WWR International). For TF-1 infections, supernatants were directly added to the cell as a 1:1 mixture with regular TF-1 cell growth media along with 4 μg/ml Polybrene. Cells were collected 2 days after infection and sorted for GFP expression. For primary HSPC infections, supernatants were concentrated 100-fold using the Retro-X Concentrator (Takara), according to the manufacturer's instructions. 24-Well non-tissue culture treated plates were coated with Retronectin (Takara) to allow virus coating, according to manufacturer's instructions. 125,000 HSPCs were added to each well, 12 h after culturing with SCF (50 ng/ml), FLT3L (10 ng/ml), and TPO (10 ng/ml). 10 μl of concentrated retrovirus was added in a final volume of 0.5 ml. Then the cells and virus were centrifuged at 1000 × g at room temperature for 2 h, after which the plates were incubated at 37 °C in a 5% CO2 incubator. A second aliquot of concentrated virus was added after 12 h, followed by the second round of centrifugation as described above. Cells were collected 2 days after the last infection, and GFP+ (infected) cells were FACS-sorted for further studies.
Proliferation and Colony Forming Assays
Triplicate cultures of TF-1 cells were grown in GM-CSF-free medium for 24 h before being switched to fresh culture medium containing the indicated concentration(s) of the specified cytokine. After 3 days of culture, ATP levels were quantified as a measure of increase in viable cell number using the CellTiterGlo ATP bioluminescence reagent, according to supplier recommendations (Promega). For HSPCs, GFP-positive cells were directly FACS-sorted at 50 cells/well into triplicate wells of 96-well U-bottom plates pre-filled with HSPC medium containing specific cytokines at the indicated concentrations. After 3–5 days of growth, proliferation was scored either by CellTiterGlo ATP bioluminescence or by direct counting of cells (pictures were taken for each well and number of cells counted using ImageJ). For the colony forming assay, GFP-positive (retrovirus-infected) HSPCs were FACS-sorted, and 10,000 cells were plated in triplicate in 35-mm dishes in MethoCult M3234 medium (StemCell Technologies) containing the indicated cytokines. After 7–14 days of growth, colonies were scored by visual counting in images obtained using an inverted microscope.
Immunoprecipitation/Western Blotting
TF-1 cells were GM-CSF-deprived for 24 h and then stimulated with 100 ng/ml recombinant human SCF for the indicated times. At each time point, the plates were placed in an ice water bath, and cells were rinsed twice with cold PBS. Cells were then lysed in Triton X-100 lysis buffer (1% Triton X-100, 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 1 mm NaVO3, 10 mm NaF) containing a mixture of protease inhibitors (Sigma), and lysate protein was quantified using the BCA reagent (Thermo Fisher Scientific). 100-μg aliquots of lysate protein were resolved on 8–9% polyacrylamide gels, transferred to PVDF membranes (Millipore) overnight, blocked for at least 2 h in 4% BSA in 50 mm Tris, pH 7.5, 100 mm NaCl, 0.1% Tween 20 (Bio-Rad) (TBS-T), and immunoblotted (primary antibodies at 1:1000 and secondary antibodies at 1:20,000), as described (66, 72). For immunoprecipitation, 1–2 mg of lysate protein in 500 μl of Triton X-100 lysis buffer were incubated overnight with 3 μg of anti-HA antibody, and immune complexes were captured using protein G-Sepharose beads followed by immunoblotting as above.
Phosphokinase Antibody Array Analysis
The ProteomeProfiler antibody array kit, which can provide quantitative assessment of site-specific phosphorylation of tyrosine kinases, were purchased from R&D Systems. TF-1 cells expressing the empty vector, CBL-Y371H, or CBL-Y371H-ΔPRR were grown in GM-CSF-free medium for 24 h and stimulated with SCF for 15 min. Cell lysates were prepared in the antibody array sample buffer supplied by the manufacturer and analyzed following the manufacturer's protocol.
GST Fusion Protein Pulldown Analyses
The GST fusion proteins that include the CBL TKB domain (GST-CBL-N; amino acids 1–357 of human CBL) or the CBL PRR (GST-CBL-C; amino acids 358–906) were prepared as described previously (63, 77). The indicated amounts of cell lysate protein and GST fusion proteins non-covalently immobilized on glutathione-Sepharose CL4B beads (GE Healthcare) (amounts indicated in figure legends) were incubated overnight at 4 °C. After washing the beads, the bound proteins were resolved on 9% polyacrylamide gels followed by immunoblotting with anti-Tyr(P) antibody (4G10) and re-probed with an anti-phospho-c-KIT antibody. To carry out phosphokinase antibody array analysis of CBL-TKB- or CBL-PRR-binding proteins, pulldowns were carried out from 5-mg aliquots of lysate protein aliquots with 5 mg of GST fusion proteins (GST-CBL-N and GST-CBL-C), and bound proteins were eluted by re-suspending packed beads (after the last wash) with an equal volume of TBS, 1% SDS and incubation at 95 °C for 5 min. The mixture was cooled to room temperature and diluted with 10 volumes of phosphokinase antibody array sample buffer (from R&D Systems). After a brief centrifugation, the supernatants were collected and used for antibody array analysis as above.
Statistics
Unpaired Student's t test on Prism was used to calculate the p values. Values <0.05 were considered significant.
Author Contributions
H. B. and V. B. conceived the study. H. B., S. A. N., W. A., and B. C. M. designed the experiments. S. A. N., W. A., B. C. M., I. M., T. A. B., H. L., N. Z., G. A., and M. D. S. performed the experiments. S. A. N., W. A., B. C. M., M. D. S., and H. B. analyzed the data. S. A. N., W. A., B. C. M., and H. B. wrote and S. O. and V. B. edited the manuscript. M. S. and S. O. provided key reagents for the study.
Acknowledgments
We thank Dr. Inder Verma (Salk Institute) for the gift of pCL-Eco plasmid and Dr. David Root (Broad Institute) for plK plasmid. We thank the Band laboratory members for discussion and the staff of the University of Nebraska Medical Center Flow Cytometry Research Facility and the Center for Comparative Medicine for assistance. The University of Nebraska Medical Center Confocal, Flow Cytometry, and other Core facilities were supported by National Institutes of Health NCI Cancer Center Support Grant P30CA036727 (to the Fred and Pamela Buffett Cancer Center and the Nebraska Research Initiative).
This work was supported in part by National Institutes of Health Grants CA87986, CA105489, CA99163, and CA116552 (to H. B.) and CA96844 and CA144027 (to V. B.), Department of Defense Grants W81XWH-07-1-0351 and W81XWH-11-1-0171 (to V. B.), Nebraska Department of Health and Human Services LB-506 Grant 2014-01 and LB606 18123-Y3 (to H. B.), National Institutes of Health Institutional Development Award P30 GM106397 (IDeA) from the NIGMS. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PTK
- protein-tyrosine kinase
- HSPC
- hematopoietic stem/progenitor cell
- TKB
- tyrosine kinase-binding
- HSPC
- hematopoietic stem/progenitor
- PRR
- proline-rich region
- SCF
- stem cell factor
- RF
- RING finger
- LHR
- linker helical region
- TPO
- thrombopoietin
- RTK
- receptor tyrosine kinase
- JMML
- juvenile myelomonocytic leukemia
- SH
- Src homology
- UBA
- ubiquitin-associated
- RTK
- receptor tyrosine kinase
- IRES
- internal ribosome entry site
- EGFP
- enhanced GFP
- DKO
- double knock-out
- PLC
- phospholipase C
- SFK
- Src family kinase.
References
- 1. Mohapatra B., Ahmad G., Nadeau S., Zutshi N., An W., Scheffe S., Dong L., Feng D., Goetz B., Arya P., Bailey T. A., Palermo N., Borgstahl G. E., Natarajan A., Raja S. M., et al. (2013) Protein-tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 1833, 122–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meisner H., Conway B. R., Hartley D., and Czech M. P. (1995) Interactions of Cbl with Grb2 and phosphatidylinositol 3′-kinase in activated Jurkat cells. Mol. Cell. Biol. 15, 3571–3578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rivero-Lezcano O. M., Sameshima J. H., Marcilla A., and Robbins K. C. (1994) Physical association between Src homology 3 elements and the protein product of the c-cbl proto-oncogene. J. Biol. Chem. 269, 17363–17366 [PubMed] [Google Scholar]
- 4. Tezuka T., Umemori H., Fusaki N., Yagi T., Takata M., Kurosaki T., and Yamamoto T. (1996) Physical and functional association of the cbl proto-oncogene product with an src-family protein-tyrosine kinase, p53/56lyn, in the B cell antigen receptor-mediated signaling. J. Exp. Med. 183, 675–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Soubeyran P., Kowanetz K., Szymkiewicz I., Langdon W. Y., and Dikic I. (2002) Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 416, 183–187 [DOI] [PubMed] [Google Scholar]
- 6. Fukazawa T., Reedquist K. A., Trub T., Soltoff S., Panchamoorthy G., Druker B., Cantley L., Shoelson S. E., and Band H. (1995) The SH3 domain-binding T cell tyrosyl phosphoprotein p120. Demonstration of its identity with the c-cbl protooncogene product and in vivo complexes with Fyn, Grb2, and phosphatidylinositol 3-kinase. J. Biol. Chem. 270, 19141–19150 [DOI] [PubMed] [Google Scholar]
- 7. Reedquist K. A., Fukazawa T., Panchamoorthy G., Langdon W. Y., Shoelson S. E., Druker B. J., and Band H. (1996) Stimulation through the T cell receptor induces Cbl association with Crk proteins and the guanine nucleotide exchange protein C3G. J. Biol. Chem. 271, 8435–8442 [DOI] [PubMed] [Google Scholar]
- 8. Sawasdikosol S., Ravichandran K. S., Lee K. K., Chang J. H., and Burakoff S. J. (1995) Crk interacts with tyrosine-phosphorylated p116 upon T cell activation. J. Biol. Chem. 270, 2893–2896 [DOI] [PubMed] [Google Scholar]
- 9. Marengère L. E., Mirtsos C., Kozieradzki I., Veillette A., Mak T. W., and Penninger J. M. (1997) Proto-oncoprotein Vav interacts with c-Cbl in activated thymocytes and peripheral T cells. J. Immunol. 159, 70–76 [PubMed] [Google Scholar]
- 10. Bartkiewicz M., Houghton A., and Baron R. (1999) Leucine zipper-mediated homodimerization of the adaptor protein c-Cbl. A role in c-Cbl's tyrosine phosphorylation and its association with epidermal growth factor receptor. J. Biol. Chem. 274, 30887–30895 [DOI] [PubMed] [Google Scholar]
- 11. Davies G. C., Ettenberg S. A., Coats A. O., Mussante M., Ravichandran S., Collins J., Nau M. M., and Lipkowitz S. (2004) Cbl-b interacts with ubiquitinated proteins; differential functions of the UBA domains of c-Cbl and Cbl-b. Oncogene 23, 7104–7115 [DOI] [PubMed] [Google Scholar]
- 12. Thien C. B., and Langdon W. Y. (2001) Cbl: many adaptations to regulate protein-tyrosine kinases. Nat. Rev. Mol. Cell. Biol. 2, 294–307 [DOI] [PubMed] [Google Scholar]
- 13. Kales S. C., Ryan P. E., and Lipkowitz S. (2012) Cbl exposes its RING finger. Nat. Struct. Mol. Biol. 19, 131–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bonita D. P., Miyake S., Lupher M. L. Jr, Langdon W. Y., and Band H. (1997) Phosphotyrosine binding domain-dependent upregulation of the platelet-derived growth factor receptor α signaling cascade by transforming mutants of Cbl: implications for Cbl's function and oncogenicity. Mol. Cell. Biol. 17, 4597–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanada M., Suzuki T., Shih L. Y., Otsu M., Kato M., Yamazaki S., Tamura A., Honda H., Sakata-Yanagimoto M., Kumano K., Oda H., Yamagata T., Takita J., Gotoh N., Nakazaki K., et al. (2009) Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 460, 904–908 [DOI] [PubMed] [Google Scholar]
- 16. Rathinam C., Thien C. B., Flavell R. A., and Langdon W. Y. (2010) Myeloid leukemia development in c-Cbl RING finger mutant mice is dependent on FLT3 signaling. Cancer Cell 18, 341–352 [DOI] [PubMed] [Google Scholar]
- 17. Fang D., and Liu Y. C. (2001) Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat. Immunol. 2, 870–875 [DOI] [PubMed] [Google Scholar]
- 18. Wang H. Y., Altman Y., Fang D., Elly C., Dai Y., Shao Y., and Liu Y. C. (2001) Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of Zap-70. J. Biol. Chem. 276, 26004–26011 [DOI] [PubMed] [Google Scholar]
- 19. Schmidt M. H., and Dikic I. (2005) The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol. 6, 907–918 [DOI] [PubMed] [Google Scholar]
- 20. Murphy M. A., Schnall R. G., Venter D. J., Barnett L., Bertoncello I., Thien C. B., Langdon W. Y., and Bowtell D. D. (1998) Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol. Cell. Biol. 18, 4872–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bachmaier K., Krawczyk C., Kozieradzki I., Kong Y. Y., Sasaki T., Oliveira-dos-Santos A., Mariathasan S., Bouchard D., Wakeham A., Itie A., Le J., Ohashi P. S., Sarosi I., Nishina H., Lipkowitz S., and Penninger J. M. (2000) Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 403, 211–216 [DOI] [PubMed] [Google Scholar]
- 22. Naramura M., Kole H. K., Hu R. J., and Gu H. (1998) Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 15547–15552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Naramura M., Jang I. K., Kole H., Huang F., Haines D., and Gu H. (2002) c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat. Immunol. 3, 1192–1199 [DOI] [PubMed] [Google Scholar]
- 24. Chiang Y. J., Kole H. K., Brown K., Naramura M., Fukuhara S., Hu R. J., Jang I. K., Gutkind J. S., Shevach E., and Gu H. (2000) Cbl-b regulates the CD28 dependence of T-cell activation. Nature 403, 216–220 [DOI] [PubMed] [Google Scholar]
- 25. Caligiuri M. A., Briesewitz R., Yu J., Wang L., Wei M., Arnoczky K. J., Marburger T. B., Wen J., Perrotti D., Bloomfield C. D., and Whitman S. P. (2007) Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood 110, 1022–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dunbar A. J., Gondek L. P., O'Keefe C. L., Makishima H., Rataul M. S., Szpurka H., Sekeres M. A., Wang X. F., McDevitt M. A., and Maciejewski J. P. (2008) 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 68, 10349–10357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niemeyer C. M., Kang M. W., Shin D. H., Furlan I., Erlacher M., Bunin N. J., Bunda S., Finklestein J. Z., Sakamoto K. M., Gorr T. A., Mehta P., Schmid I., Kropshofer G., Corbacioglu S., Lang P. J., et al. (2010) Germ line CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet. 42, 794–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loh M. L., Sakai D. S., Flotho C., Kang M., Fliegauf M., Archambeault S., Mullighan C. G., Chen L., Bergstraesser E., Bueso-Ramos C. E., Emanuel P. D., Hasle H., Issa J. P., van den Heuvel-Eibrink M. M., Locatelli F., et al. (2009) Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114, 1859–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muramatsu H., Makishima H., Jankowska A. M., Cazzolli H., O'Keefe C., Yoshida N., Xu Y., Nishio N., Hama A., Yagasaki H., Takahashi Y., Kato K., Manabe A., Kojima S., and Maciejewski J. P. (2010) Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 115, 1969–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grand F. H., Hidalgo-Curtis C. E., Ernst T., Zoi K., Zoi C., McGuire C., Kreil S., Jones A., Score J., Metzgeroth G., Oscier D., Hall A., Brandts C., Serve H., Reiter A., et al. (2009) Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 113, 6182–6192 [DOI] [PubMed] [Google Scholar]
- 31. Bacher U., Haferlach C., Schnittger S., Kohlmann A., Kern W., and Haferlach T. (2010) Mutations of the TET2 and CBL genes: novel molecular markers in myeloid malignancies. Ann. Hematol. 89, 643–652 [DOI] [PubMed] [Google Scholar]
- 32. Javadi M., Richmond T. D., Huang K., and Barber D. L. (2013) CBL linker region and RING finger mutations lead to enhanced granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling via elevated levels of JAK2 and LYN. J. Biol. Chem. 288, 19459–19470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sargin B., Choudhary C., Crosetto N., Schmidt M. H., Grundler R., Rensinghoff M., Thiessen C., Tickenbrock L., Schwäble J., Brandts C., August B., Koschmieder S., Bandi S. R., Duyster J., Berdel W. E., et al. (2007) Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 110, 1004–1012 [DOI] [PubMed] [Google Scholar]
- 34. Nadeau S., An W., Palermo N., Feng D., Ahmad G., Dong L., Borgstahl G. E., Natarajan A., Naramura M., Band V., and Band H. (2012) Oncogenic signaling by leukemia-associated mutant Cbl proteins. Biochem. Anal. Biochem. 6, 7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Griffiths E. K., Sanchez O., Mill P., Krawczyk C., Hojilla C. V., Rubin E., Nau M. M., Khokha R., Lipkowitz S., Hui C. C., and Penninger J. M. (2003) Cbl-3-deficient mice exhibit normal epithelial development. Mol. Cell. Biol. 23, 7708–7718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Naramura M., Nandwani N., Gu H., Band V., and Band H. (2010) Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 107, 16274–16279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. An W., Nadeau S. A., Mohapatra B. C., Feng D., Zutshi N., Storck M. D., Arya P., Talmadge J. E., Meza J. L., Band V., and Band H. (2015) Loss of Cbl and Cbl-b ubiquitin ligases abrogates hematopoietic stem cell quiescence and sensitizes leukemic disease to chemotherapy. Oncotarget 6, 10498–10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rathinam C., Thien C. B., Langdon W. Y., Gu H., and Flavell R. A. (2008) The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 22, 992–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bunda S., Qin K., Kommaraju K., Heir P., and Ohh M. (2015) Juvenile myelomonocytic leukaemia-associated mutation in Cbl promotes resistance to apoptosis via the Lyn-PI3K/AKT pathway. Oncogene 34, 789–797 [DOI] [PubMed] [Google Scholar]
- 40. Bunda S., Kang M. W., Sybingco S. S., Weng J., Favre H., Shin D. H., Irwin M. S., Loh M. L., and Ohh M. (2013) Inhibition of SRC corrects GM-CSF hypersensitivity that underlies juvenile myelomonocytic leukemia. Cancer Res. 73, 2540–2550 [DOI] [PubMed] [Google Scholar]
- 41. Cancer Genome Atlas Research Network. (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Duyvestyn J. M., Taylor S. J., Dagger S. A., Orandle M., Morse H. C. 3rd., Thien C. B., and Langdon W. Y. (2014) Dasatinib targets B-lineage cells but does not provide an effective therapy for myeloproliferative disease in c-Cbl RING finger mutant mice. PLoS ONE 9, e94717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fernandes M. S., Reddy M. M., Croteau N. J., Walz C., Weisbach H., Podar K., Band H., Carroll M., Reiter A., Larson R. A., Salgia R., Griffin J. D., and Sattler M. (2010) Novel oncogenic mutations of CBL in human acute myeloid leukemia that activate growth and survival pathways depend on increased metabolism. J. Biol. Chem. 285, 32596–32605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Makishima H., Sugimoto Y., Szpurka H., Clemente M. J., Ng K. P., Muramatsu H., O'Keefe C., Saunthararajah Y., and Maciejewski J. P. (2012) CBL mutation-related patterns of phosphorylation and sensitivity to tyrosine kinase inhibitors. Leukemia 26, 1547–1554 [DOI] [PubMed] [Google Scholar]
- 45. Yee N. S., Hsiau C. W., Serve H., Vosseller K., and Besmer P. (1994) Mechanism of down-regulation of c-kit receptor. Roles of receptor tyrosine kinase, phosphatidylinositol 3′-kinase, and protein kinase C. J. Biol. Chem. 269, 31991–31998 [PubMed] [Google Scholar]
- 46. Zeng S., Xu Z., Lipkowitz S., and Longley J. B. (2005) Regulation of stem cell factor receptor signaling by Cbl family proteins (Cbl-b/c-Cbl). Blood 105, 226–232 [DOI] [PubMed] [Google Scholar]
- 47. Masson K., Heiss E., Band H., and Rönnstrand L. (2006) Direct binding of Cbl to Tyr568 and Tyr936 of the stem cell factor receptor/c-Kit is required for ligand-induced ubiquitination, internalization and degradation. Biochem. J. 399, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andoniou C. E., Thien C. B., and Langdon W. Y. (1994) Tumour induction by activated abl involves tyrosine phosphorylation of the product of the cbl oncogene. EMBO J. 13, 4515–4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghosh A. K., Reddi A. L., Rao N. L., Duan L., Band V., and Band H. (2004) Biochemical basis for the requirement of kinase activity for Cbl-dependent ubiquitinylation and degradation of a target tyrosine kinase. J. Biol. Chem. 279, 36132–36141 [DOI] [PubMed] [Google Scholar]
- 50. Ota S., Hazeki K., Rao N., Lupher M. L. Jr, Andoniou C. E., Druker B., and Band H. (2000) The RING finger domain of Cbl is essential for negative regulation of the Syk tyrosine kinase. J. Biol. Chem. 275, 414–422 [DOI] [PubMed] [Google Scholar]
- 51. Lill N. L., Douillard P., Awwad R. A., Ota S., Lupher M. L. Jr., Miyake S., Meissner-Lula N., Hsu V. W., and Band H. (2000) The evolutionarily conserved N-terminal region of Cbl is sufficient to enhance down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 275, 367–377 [DOI] [PubMed] [Google Scholar]
- 52. Rao N., Lupher M. L. Jr, Ota S., Reedquist K. A., Druker B. J., and Band H. (2000) The linker phosphorylation site Tyr292 mediates the negative regulatory effect of Cbl on ZAP-70 in T cells. J. Immunol. 164, 4616–4626 [DOI] [PubMed] [Google Scholar]
- 53. Bao J., Gur G., and Yarden Y. (2003) Src promotes destruction of c-Cbl: implications for oncogenic synergy between Src and growth factor receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 2438–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Magnifico A., Ettenberg S., Yang C., Mariano J., Tiwari S., Fang S., Lipkowitz S., and Weissman A. M. (2003) WW domain HECT E3s target Cbl RING finger E3s for proteasomal degradation. J. Biol. Chem. 278, 43169–43177 [DOI] [PubMed] [Google Scholar]
- 55. Adapala N. S., Barbe M. F., Langdon W. Y., Nakamura M. C., Tsygankov A. Y., and Sanjay A. (2010) The loss of Cbl-phosphatidylinositol 3-kinase interaction perturbs RANKL-mediated signaling, inhibiting bone resorption and promoting osteoclast survival. J. Biol. Chem. 285, 36745–36758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miura-Shimura Y., Duan L., Rao N. L., Reddi A. L., Shimura H., Rottapel R., Druker B. J., Tsygankov A., Band V., and Band H. (2003) Cbl-mediated ubiquitinylation and negative regulation of Vav. J. Biol. Chem. 278, 38495–38504 [DOI] [PubMed] [Google Scholar]
- 57. Donovan J. A., Wange R. L., Langdon W. Y., and Samelson L. E. (1994) The protein product of the c-cbl protooncogene is the 120-kDa tyrosine-phosphorylated protein in Jurkat cells activated via the T cell antigen receptor. J. Biol. Chem. 269, 22921–22924 [PubMed] [Google Scholar]
- 58. Reedquist K. A., Fukazawa T., Druker B., Panchamoorthy G., Shoelson S. E., and Band H. (1994) Rapid T-cell receptor-mediated tyrosine phosphorylation of p120, an Fyn/Lck Src homology 3 domain-binding protein. Proc. Natl. Acad. Sci. U.S.A. 91, 4135–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Panchamoorthy G., Fukazawa T., Miyake S., Soltoff S., Reedquist K., Druker B., Shoelson S., Cantley L., and Band H. (1996) p120cbl is a major substrate of tyrosine phosphorylation upon B cell antigen receptor stimulation and interacts in vivo with Fyn and Syk tyrosine kinases, Grb2 and Shc adaptors, and the p85 subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 271, 3187–3194 [DOI] [PubMed] [Google Scholar]
- 60. Bard F., Patel U., Levy J. B., Jurdic P., Horne W. C., and Baron R. (2002) Molecular complexes that contain both c-Cbl and c-Src associate with Golgi membranes. Eur. J. Cell Biol. 81, 26–35 [DOI] [PubMed] [Google Scholar]
- 61. Graham L. J., Stoica B. A., Shapiro M., DeBell K. E., Rellahan B., Laborda J., and Bonvini E. (1998) Sequences surrounding the Src-homology 3 domain of phospholipase Cγ-1 increase the domain's association with Cbl. Biochem. Biophys. Res. Commun. 249, 537–541 [DOI] [PubMed] [Google Scholar]
- 62. Aranaz P., Hurtado C., Erquiaga I., Miguéliz I., Ormazábal C., Cristobal I., García-Delgado M., Novo F. J., and Vizmanos J. L. (2012) CBL mutations in myeloproliferative neoplasms are also found in the gene's proline-rich domain and in patients with the V617FJAK2. Haematologica 97, 1234–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lupher M. L. Jr., Reedquist K. A., Miyake S., Langdon W. Y., and Band H. (1996) A novel phosphotyrosine-binding domain in the N-terminal transforming region of Cbl interacts directly and selectively with ZAP-70 in T cells. J. Biol. Chem. 271, 24063–24068 [DOI] [PubMed] [Google Scholar]
- 64. Miyake S., Lupher M. L. Jr., Druker B., and Band H. (1998) The tyrosine kinase regulator Cbl enhances the ubiquitination and degradation of the platelet-derived growth factor receptor alpha. Proc. Natl. Acad. Sci. U.S.A. 95, 7927–7932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reddi A. L., Ying G., Duan L., Chen G., Dimri M., Douillard P., Druker B. J., Naramura M., Band V., and Band H. (2007) Binding of Cbl to a phospholipase Cγ1-docking site on platelet-derived growth factor receptor β provides a dual mechanism of negative regulation. J. Biol. Chem. 282, 29336–29347 [DOI] [PubMed] [Google Scholar]
- 66. Duan L., Raja S. M., Chen G., Virmani S., Williams S. H., Clubb R. J., Mukhopadhyay C., Rainey M. A., Ying G., Dimri M., Chen J., Reddi A. L., Naramura M., Band V., and Band H. (2011) Negative regulation of EGFR-Vav2 signaling axis by Cbl ubiquitin ligase controls EGF receptor-mediated epithelial cell adherens junction dynamics and cell migration. J. Biol. Chem. 286, 620–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sanjay A., Miyazaki T., Itzstein C., Purev E., Horne W. C., and Baron R. (2006) Identification and functional characterization of an Src homology domain 3 domain-binding site on Cbl. FEBS J. 273, 5442–5456 [DOI] [PubMed] [Google Scholar]
- 68. Ribon V., Herrera R., Kay B. K., and Saltiel A. R. (1998) A role for CAP, a novel, multifunctional Src homology 3 domain-containing protein in formation of actin stress fibers and focal adhesions. J. Biol. Chem. 273, 4073–4080 [DOI] [PubMed] [Google Scholar]
- 69. Waterman H., Levkowitz G., Alroy I., and Yarden Y. (1999) The RING finger of c-Cbl mediates desensitization of the epidermal growth factor receptor. J. Biol. Chem. 274, 22151–22154 [DOI] [PubMed] [Google Scholar]
- 70. Andoniou C. E., Lill N. L., Thien C. B., Lupher M. L. Jr, Ota S., Bowtell D. D., Scaife R. M., Langdon W. Y., and Band H. (2000) The Cbl proto-oncogene product negatively regulates the Src-family tyrosine kinase Fyn by enhancing its degradation. Mol. Cell. Biol. 20, 851–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rao N., Ghosh A. K., Ota S., Zhou P., Reddi A. L., Hakezi K., Druker B. K., Wu J., and Band H. (2001) The non-receptor tyrosine kinase Syk is a target of Cbl-mediated ubiquitylation upon B-cell receptor stimulation. EMBO J. 20, 7085–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rao N., Miyake S., Reddi A. L., Douillard P., Ghosh A. K., Dodge I. L., Zhou P., Fernandes N. D., and Band H. (2002) Negative regulation of Lck by Cbl ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 99, 3794–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kassenbrock C. K., and Anderson S. M. (2004) Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations. J. Biol. Chem. 279, 28017–28027 [DOI] [PubMed] [Google Scholar]
- 74. Thien C. B., Walker F., and Langdon W. Y. (2001) RING finger mutations that abolish c-Cbl-directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol. Cell 7, 355–365 [DOI] [PubMed] [Google Scholar]
- 75. Langdon W. Y., Hartley J. W., Klinken S. P., Ruscetti S. K., and Morse H. C. 3rd (1989) v-cbl, an oncogene from a dual-recombinant murine retrovirus that induces early B-lineage lymphomas. Proc. Natl. Acad. Sci. U.S.A. 86, 1168–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang L., Rudert W. A., Loutaev I., Roginskaya V., and Corey S. J. (2002) Repression of c-Cbl leads to enhanced G-CSF Jak-STAT signaling without increased cell proliferation. Oncogene 21, 5346–5355 [DOI] [PubMed] [Google Scholar]
- 77. Lupher M. L. Jr., Rao N., Lill N. L., Andoniou C. E., Miyake S., Clark E. A., Druker B., and Band H. (1998) Cbl-mediated negative regulation of the Syk tyrosine kinase. A critical role for Cbl phosphotyrosine-binding domain binding to Syk phosphotyrosine 323. J. Biol. Chem. 273, 35273–35281 [DOI] [PubMed] [Google Scholar]