

Abstract
Cullin-RING ligase 4 (CRL4), a complex of Cul4 and DDB1, regulates the cell cycle, DNA damage repair, and chromatin replication by targeting a variety of substrates for ubiquitination. CRL4 is also hijacked by viral proteins or thalidomide-derived compounds to degrade host restriction factors. Here we report that the c-Abl non-receptor kinase phosphorylates DDB1 at residue Tyr-316 to recruit a small regulatory protein, DDA1, leading to increased substrate ubiquitination. Pharmacological inhibition or genetic ablation of the Abl-DDB1-DDA1 axis decreases the ubiquitination of CRL4 substrates, including IKZF1 and IKZF3, in lenalidomide-treated multiple myeloma cells. Importantly, panobinostat, a recently approved anti-myeloma drug, and dexamethasone enhance lenalidomide-induced substrate degradation and cytotoxicity by activating c-Abl, therefore providing a mechanism underlying their combination with lenalidomide to treat multiple myeloma.
Keywords: cancer biology, cancer therapy, molecular cell biology, ubiquitin ligase, ubiquitylation (ubiquitination), CRL4, DDA1, c-Abl, lenalidomide
Introduction
The cullin-RING ubiquitin ligases (CRLs),2 comprising the scaffold cullins, the E2-interacting RING finger protein ROC1/2, the adaptor proteins specific for each cullin family member, and adaptor-interacting substrate receptors, are the largest family of E3 ligases in eukaryotes, regulating diverse cellular pathways (1). CRL4 uses the damaged DNA binding protein 1 (DDB1) as the adaptor protein to assemble with a subset of WD40-containing proteins called DDB1- and Cullin4 (Cul4)-associated factors to target diverse substrates to regulate the cell cycle, DNA damage repair, and chromatin functions (1–3). CRL4 ubiquitinates cellular proteins such as CDT1 (4), P21 (5, 6), and DDB2 (7) for proteasomal degradation and ten-eleven translocation (TET) proteins (8) and histones H2A (9), H3 (10, 11), and H4 (10) to regulate their chromatin-mediated functions. Several viral proteins can hijack CRL4 to turn over host restriction factors to promote virus replication, including STAT1 targeted by SV5 virus V protein (12–14), SAMHD1 by simian HIV Vpr protein (15), and Smc5/6 by hepatitis B virus X protein (16).
Additionally, immunomodulatory drugs (IMiDs), including thalidomide and its derivatives lenalidomide and pomalidomide, can repurpose CRL4 to target and destroy IKZF1 and IKZF3 (17, 18), two lymphoid transcription factors essential for multiple myeloma (MM) cell survival. Similarly, lenalidomide induces ubiquitination and degradation of casein kinase 1A1 (CK1α), which accounts for the clinical efficacy of lenalidomide in myelodysplastic syndrome (MDS) with deletion of chromosome 5q (del(5q)) (19). Crystal structure studies reveal that the glutarimide moiety of lenalidomide directly inserts into a hydrophobic pocket of cereblon (CRBN), a CRL4 substrate receptor, and that the exposed chemical moiety, together with CRBN, creates a new surface for receiving substrates (20–22). Based on these results, novel chemicals conjugating glutarimide to other protein-interacting chemical structures are designed to control protein stability and thus enable targeting of previously intractable proteins (23). CRBN by itself mediates the ubiquitination of several putative substrates, including the large conductance, Ca2+- and voltage-activated K+ (BK) channels (24), MEIS2 (20) and glutamine synthetase (25), suggesting that the CRL4CRBN ubiquitin ligase activity is diverted by IMiDs or its derived chemicals from its physiological functions.
Human DDA1 (DDB1- and DET1-associated protein 1) is a small (12-kDa) DDB1-interacting protein that co-purifies with the Cul4-DDB1 complex (26, 27). DDA1 associates with some DDB1- and Cul4-associated factors and is assembled into CRL4 complexes (28), but how this process is regulated remains elusive. Here we present evidence that the c-Abl non-receptor kinase phosphorylates DDB1 at residue Tyr-316 to recruit DDA1, leading to increased substrate ubiquitination. Furthermore, the newly approved anti-myeloma drug LBH589 (panobinostat) (29), a histone deacetylase (HDAC) inhibitor, stimulates c-Abl kinase activity to promote lenalidomide-dependent ubiquitination and degradation of IKZF transcription factors in MM cells, justifying their combination in treating MM patients for better clinical benefits.
Results
Inhibition of c-Abl Kinase Desensitizes MM Cells to Lenalidomide
IMiDs target the CRL4CRBN ubiquitin ligase to induce the ubiquitination and degradation of IKZF1 and IKZF3 to block MM cell proliferation (17, 18). c-Abl was reported to bind and phosphorylate DDB1 (30), an adaptor protein of the CRL4 complex, but the exact role of c-Abl in regulating CRL4 is not clear. We first tested whether c-Abl kinase affects the cytotoxicity of lenalidomide in MM cells. In our initial experiments, imatinib (Gleevec), an inhibitor of Abl family kinases, ameliorated the anti-proliferative activity of lenalidomide in several MM cell lines, including MM1S, MM1R, and U266 (Fig. 1, A–C). Deletion of Abl1, which encodes c-Abl, by CRISPR-Cas9 genome editing via lentiviral vectors similarly desensitized the cells to lenalidomide-induced cell death (Fig. 1, D and E). These results show that the activity of c-Abl kinase is required for optimal lenalidomide-induced inhibition of MM cell proliferation.
FIGURE 1.

c-Abl kinase is required for lenalidomide-dependent cytotoxicity of MM cells. A–C, viability assays of MM1S cells (A), MM1R cells (B), or U266 cells (C) treated with the indicated concentrations of lenalidomide (Len) in combination with 10 μm imatinib for 5 days. D, validation of Abl deletion in MM1S-derived cells by immunoblot analysis. CRISPR-Cas9 lentiviral vectors carrying two single guide RNAs targeting Abl (sgAbl) or two non-targeting sequences (sgCtrl) were constructed to infect MM1S cells, which were harvested as pools for analysis after selection with 0.5 μg/ml puromycin for 3 days. E, viability assays of MM1S cells expressing wild-type or knockout c-Abl after treatment with the indicated concentrations of Len for 5 days. F, immunoblot analysis of MM1S cells stably expressing c-Abl and treated with the indicated concentrations of LBH589 (LBH) in the presence or absence of 10 μm imatinib for 24 h. G, proliferation of MM1S cells treated with 0.05 μm Len, 0.5 nm LBH, 10 μm imatinib, or their combination for the indicated time. For all cell viability assays, error bars represent mean ± S.E. (n = 4).
Activation of c-Abl Enhances Lenalidomide-induced Cytotoxicity
c-Abl can be activated by multiple stress signals, such as cell adhesion (31) and DNA damage (32). Trichostatin A, a pan-histone deacetylase inhibitor, was reported to activate c-Abl by inducing chromatin distortion (33). LBH589 (panobinostat), a pan-histone deacetylase inhibitor, has recently been approved to treat relapsed MM patients in combination with bortezomib and dexamethasone (29). We first generated an MM1S cell line stably expressing c-Abl by lentiviral vectors and treated these cells with increasing concentrations of LBH589. Using c-Abl self-phosphorylation (Tyr(P)-245) as a readout, we confirmed that LBH589 activated c-Abl kinase in a dose-dependent manner in these MM cells (Fig. 1F). To test whether LBH589 would affect lenalidomide cytotoxicity in MM cells by activating c-Abl, we treated MM1S cells with lenalidomide and LBH589 in the presence or absence of imatinib. Proliferation of MM1S cells was better inhibited by combination treatment with LBH589 and lenalidomide, but the enhanced inhibition could be abolished by imatinib treatment (Fig. 1G).
Dexamethasone is a glucocorticoid approved to treat MM in combination with IMiDs and proteasome inhibitors (34, 35). Little is known about the mechanism underlying how dexamethasone enhances the anti-myeloma activity of these drugs. We found that imatinib similarly reversed the dexamethasone-dependent increase of lenalidomide-induced MM cell proliferation arrest (Fig. 3A). We conclude that LBH589 and dexamethasone increase the cytotoxic effect of lenalidomide on MM cells by activating the c-Abl kinase.
FIGURE 3.

Dexamethasone-potentiated lenalidomide activity requires c-Abl kinase activity. A, proliferation of MM1S cells treated with 0.1 μm Len, 2.5 nm dexamethasone (Dexa), 10 μm imatinib, or their combinations for the indicated time. B, CHX chase analysis for IKZF3 stability in MM1S cells treated with 25 nm dexamethasone for 12 h, 10 μm imatinib for 12 h, 2 μm Len for 1 h, or the indicated combination before addition of 100 μg/ml CHX for the indicated time. C, immunoblot for IKZF1/IKZF3 in OPM2 cells treated with 25 nm dexamethasone for 24 h, 0.5 μm Len for 12 h, or both. SE, short exposure of detection; LE, long exposure. D, proliferation of OPM2 cells treated with 4 nm dexamethasone, 1.8 μm Len, or their combination for the indicated time. For the cell viability assay, error bars represent mean ± S.E. (n = 4).
Activation of c-Abl Promotes Lenalidomide-dependent Substrate Degradation
We next sought to determine whether c-Abl enhanced lenalidomide cytotoxicity by stimulating the turnover of the lenalidomide substrates IKZF1 and IKZF3. LBH589 increased lenalidomide-induced degradation of IKZF1/IKZF3 and loss of viability in lenalidomide-sensitive MM cell lines, including MM1S cells (Fig. 2A), OPM2 cells (Fig. 2B), and U266 cells (Fig. 2C), with no significant effect on the abundance of CRBN protein (Fig. 2, A–C). LBH589 did not alter IKZF1/IKZF3 levels or cause significant cytotoxicity in these sensitive cell lines as a single agent (Fig. 2, A–C, second lanes). We generated pomalidomide-resistant MM1S-P5000 cells by continuous culture of MM1S cells in medium containing gradually increasing concentrations of pomalidomide. The final pomalidomide concentration in medium was 5 μm to maintain the growth of these cells. MM1S-P5000 cells are also lenalidomide-resistant, likely because of the loss of CRBN expression (Fig. 2A). We found that LBH589 did not alter IKZF1/IKZF3 levels or cause obvious cytotoxicity in MM1S-P5000 cells as a single agent or even in combination with lenalidomide (Fig. 2A), suggesting that LBH589-induced cytotoxicity is dependent on lenalidomide. Deletion of c-Abl mitigated the decrease of IKZF1/IKZF3 protein levels and the arrest of cell proliferation in cells treated with LBH589 and lenalidomide (Fig. 2D). Interestingly, treatment with MLN4924, a neddylation inhibitor that inactivates all CRLs, including CRL4CRBN (36), completely rescued the decrease in IKZF1/IKZF3 protein abundance after treatment with either lenalidomide alone or a combination of LBH589 and lenalidomide in these three MM cell lines (Fig. 2E). As LBH589 activated c-Abl, we then examined whether c-Abl regulates the lenalidomide-dependent ubiquitination of IKZF1/IKZF3. IKZF1/IKZF3 ubiquitination was detectable in HEK 293T cells only when the cells were treated with lenalidomide, and expression of exogenous wild-type c-Abl, but not the kinase-defective c-Abl (Abl-KR), enhanced the levels of IKZF3 and IKZF1 ubiquitination (Fig. 2, F and G). These data suggest that LBH589 stimulates the ubiquitination and proteasomal degradation of IKZF1/IKZF3 in the presence of lenalidomide.
FIGURE 2.

LBH589 enhances lenalidomide-dependent degradation of IKZF1 and IKZF3 and cytotoxicity of MM cells. A, immunoblot for IKZF1/IKZF3 in MM1S or pomalidomide-resistant MM1S-P5000 cells that were treated with 6 nm LBH for 24 h, 0.1 μm Len for 12 h, or both (top panel). Also shown are viability assays of these cells treated with 0.5 nm LBH, 0.05 μm Len, or both on day 5 (bottom panel). B, immunoblot for IKZF1/IKZF3 in OPM2 cells that were treated with 12 nm LBH for 24 h, 0.5 μm Len for 12 h, or both (top panel). Also shown are viability assays of OPM2 cells treated with 1.5 nm LBH, 1.6 μm Len, or both on day 5 (bottom panel). C, immunoblot for IKZF1/IKZF3 in U266 cells that were treated with 12 nm LBH for 24 h, 0.5 μm Len for 12 h, or both (top panel). The asterisk indicates a nonspecific band. Also shown are viability assays of U266 cells treated with 1.5 nm LBH, 3 μm Len, or both on day 5 (bottom panel). D, immunoblot for IKZF1/IKZF3 in control or c-Abl knockout MM1S cells that were treated with 6 nm LBH for 24 h, 0.1 μm Len for 12 h, or both (top panel). Also shown are viability assays of MM1S cells treated with 0.5 nm LBH, 0.05 μm Len, or both on day 5 (bottom panel). Each band of the WB for IKZF3 was quantified with Gel-Pro Analyzer 4.0 software (Media Cybernetics) and normalized to the corresponding actin band (A–D). E, immunoblot for IKZF1/IKZF3 in MM1S, OPM2, and U266 cells treated with LBH or Len under the same conditions as in A–C, respectively, or their combination with DMSO or 1 μm MLN4924 (MLN) for 12 h as indicated. The asterisk indicates a nonspecific band. F and G, ubiquitination of IKZF3 (F) or IKZF1 (G) by immunoprecipitation (IP) in HEK 293T cells expressing c-Abl or Abl-KR and treated with 10 μm MG132 for 12 h and 5 μm Len for 12 h as indicated. HC, heavy chain. For all cell viability assays, error bars represent mean ± S.E. (n = 4). The p values were determined by Student's t test. ***, p < 0.001.
Like LBH589, dexamethasone further destabilized IKZF3 protein (Fig. 3B) and reduced IKZF1/IKZF3 abundance (Fig. 3C) in lenalidomide-treated MM cells, leading to enhanced inhibition of MM1S (Fig. 3A) and OPM2 cell (Fig. 3D) proliferation. Co-treatment with imatinib abolished the dexamethasone-stimulated destabilization of IKZF3 (Fig. 3B) and inhibition of cell proliferation in lenalidomide-treated MM1S cells (Fig. 3A). Put together, these results suggest that LBH589 and dexamethasone promote lenalidomide-induced IKZF1/IKZF3 ubiquitination and degradation by activating c-Abl kinase.
c-Abl Phosphorylates Multiple Tyrosines of DDB1 Protein
c-Abl was reported to bind and phosphorylate DDB1 (30) and promote the ubiquitination and proteasomal degradation of DDB2 in cells exposed to UV damage (37). We confirmed that c-Abl kinase phosphorylated DDB1 in a kinase-dependent manner (Fig. 4A). However, which sites of DDB1 are phosphorylated is not clear. There are three potential c-Abl target sites in the primary sequence of DDB1 protein with the consensus YXXP sequence (Fig. 4B): Tyr-182 and Tyr-718 on the exposed surface and Tyr-678 inside the DDB1-Cul4 binding scaffold (27). Mutation at Tyr-182 or Tyr-718, but not Tyr-678, reduced the phosphorylation of DDB1 (Fig. 4C). These results suggest that c-Abl phosphorylates diverse tyrosines of DDB1 protein.
FIGURE 4.

c-Abl phosphorylates multiple tyrosines of DDB1 protein. A, phosphorylation of DDB1 by IP in HEK 293T cells expressing FLAG-Abl or FLAG-Abl-KR or treated with 2 μm imatinib for 24 h as indicated. B, c-Abl substrate motif (YXXP) and the Tyr-316 site on DDB1. C, phosphorylation of WT or mutant DDB1 by IP in HEK 293T cells expressing FLAG-Abl or FLAG-Abl-KR.
c-Abl Phosphorylates DDB1 to Recruit DDA1
Interestingly, we found that c-Abl kinase promoted the interaction of DDB1 with DDA1 (Fig. 5A), a small protein (12 kDa) with no obvious motif and likely a positive regulator of the CRL4 ligase (28). The kinase activity of c-Abl is essential for the increased DDB1-DDA1 interaction because treating cells with imatinib or expressing kinase-defective Abl-KR abolished this effect (Fig. 5A). A conserved cluster of residues (Tyr-316/Asp-318/Asn-319) on one of the three seven-bladed WD40 β propellers (BPA, BPB, and BPC) of DDB1 (27) is required for DDA1 interaction (26). Conversion of Tyr-316 of DDB1 to Phe completely abrogated the interaction of DDB1 and DDA1, which also failed to be enhanced by c-Abl expression (Fig. 5B). However, the total tyrosine phosphorylation of DDB1 was only marginally reduced by this mutation (Fig. 5B), consistent with DDB1 harboring additional sites (Tyr-182 and Tyr-718) phosphorylated by c-Abl. Besides, c-Abl-induced recruitment of DDA1 was independent of phosphorylation at these two sites (Fig. 5C). Triple mutations (TMs) in Tyr-316/Tyr-182/Tyr-718 of DDB1 exhibited a more severe phosphorylation defect than the double mutations (DMs, Tyr-182/Tyr-718), indicating that all three tyrosine residues are phosphorylated (Fig. 5C). c-Abl phosphorylated DDB1 mostly in the cytoplasm, enriched DDA1 in the nucleus, and promoted the assembly of the DDB1-DDA1 complex in both the cytoplasm and nucleus (Fig. 5D). These data demonstrate that DDB1 is phosphorylated by c-Abl at Tyr-316 to recruit DDA1.
FIGURE 5.

c-Abl phosphorylates DDB1 at Tyr-316 to recruit DDA1. A, DDB1 phosphorylation and its interaction with DDA1 by co-IP in HEK 293T cells expressing FLAG-Abl/Abl-KR or treated with 2 μm imatinib for 24 h as indicated. B, interaction of WT or mutant DDB1 (Y316F) with DDA1 by co-IP in HEK 293T cells expressing FLAG-Abl. SE, short exposure of detection; LE, long exposure. C, phosphorylation of DDB1 mutants and their interaction with DDA1 by co-IP in HEK 293T cells expressing FLAG-Abl. DM, double mutant DDB1 (Y182F/Y718F); TM, triple mutant DDB1 (Y316F/Y182F/Y718F). D, phosphorylation of DDB1 and its interaction with DDA1 in cytoplasmic and nuclear fractions of HeLa cells.
Lenalidomide-dependent IKZF3 Degradation Requires DDB1 to Recruit DDA1
To address the role of DDA1 in lenalidomide-induced substrate turnover, we examined the ubiquitination and stability of IKZF3 protein overexpressed in HEK 293T cells with knockdown of DDA1 expression. Expression of c-Abl increased the levels of ubiquitinated IKZF3, whereas knocking down DDA1 reduced both the basal and Abl-induced IKZF3 ubiquitination levels (Fig. 6, A and B). Similarly, lenalidomide-dependent destabilization of IKZF3 was abrogated by knockdown of DDA1 expression but enhanced by overexpression of c-Abl with intact kinase activity (Fig. 6, C and D). Expression of c-Abl without lenalidomide treatment did not alter IKZF3 stability (Fig. 6C). Put together, these results suggest that DDA1 is recruited to DDB1 at phosphorylated Tyr-316 to promote lenalidomide-induced ubiquitination and degradation of IKZF3.
FIGURE 6.

Lenalidomide-dependent IKZF3 degradation requires DDB1 to recruit DDA1. A, ubiquitination of IKZF3 by IP in HEK 293T cells expressing c-Abl, siDDA1, or both and treated with 5 μm Len for 12 h and 10 μm MG132 for 12 h as indicated. B, RT-PCR assay to determine the efficiency of DDA1 knockdown in HEK 293T cells. Data are mean ± S.D.; n = 3; *, p < 0.05. C, CHX chase analysis for IKZF3 stability in HEK 293T cells expressing IKZF3-HA, FLAG-CRBN, c-Abl, Abl-KR, siDDA1, or combination of c-Abl and siDDA1. Cells were treated with 2.5 μm imatinib for 12 h, 2 μm Len for 12 h, or the indicated combination before addition of 100 μg/ml CHX for the indicated time. D, time course of IKZF3 protein decay in cells treated with lenalidomide from C. Each band of the WB for IKZF3 was quantified with Gel-Pro Analyzer 4.0 software (Media Cybernetics) and normalized to the corresponding actin band. Half-life (t½) was estimated as the time for degradation of 50% of the protein.
Discussion
In summary, we have uncovered a mechanism underlying clinically approved combination treatment options with lenalidomide for MM (Fig. 7). Other than their inherent cytotoxicity, small-molecule drugs such as LBH589 and dexamethasone can activate the c-Abl kinase, which phosphorylates DDB1 at Tyr-316 to recruit DDA1, leading to increased substrate ubiquitination.
FIGURE 7.
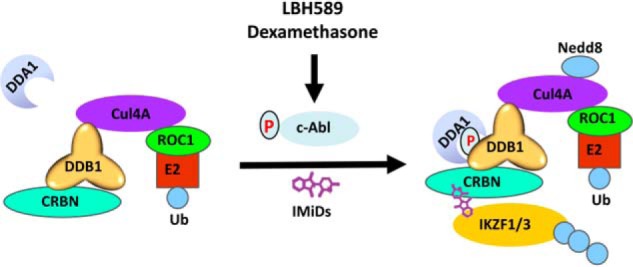
Schematic depicting how the Abl-DDB1-DDA1 cascade regulates CRL4 substrate ubiquitination and potentiates IMiD cytotoxicity.
CRL4 assembles with a subset of WD40 proteins to target a variety of substrates to regulate the cell cycle, chromatin functions, and DNA damage repair (1–3). c-Abl-induced activation of CRL4 would presumably affect the ubiquitination of all CRL4 substrates. Therefore we checked the ubiquitination of histone H3 (10, 11) and CK1α (19), substrates of CRL4, and found that c-Abl increased ubiquitination of the two proteins (Fig. 8, A and B). These data suggest that c-Abl kinase can increase the ubiquitination of diverse substrates of CRL4 E3 ligase. In this way, it would be interesting to explore the functional significance of c-Abl activation in regulating the turnover of the various CRL4 substrates in physiological or pathological processes.
FIGURE 8.

c-Abl stimulates ubiquitination of CRL4 substrates. A, ubiquitination of chromatin-bound histone H3 by IP in HEK 293T cells expressing c-Abl or Abl-KR. LC, light chain. B, ubiquitination of CK1α by IP in HEK 293T cells expressing c-Abl or Abl-KR and treated with 10 μm MG132 for 12 h and 5 μm Len for 12 h. HC, heavy chain.
DDA1 is a small protein without an obvious motif that co-purifies with the Cul4-DDB1 complex (26, 27) and has been suggested to be a positive regulator of the CRL4 ligase (28), but how DDA1 is assembled into CRL4 is not clear. Here we show evidence that c-Abl kinase phosphorylates DDB1 at Tyr-316 to recruit DDA1. Next, we would like to explore how DDA1 recruitment accelerates the ubiquitination of various substrates.
Lenalidomide is empowered by the activated CRL4CRBN ubiquitin ligase to target its substrates more effectively. Therefore, any intervention that activates Abl kinases would, in principle, potentiate the activities of lenalidomide or other IMiDs that engage CRL4CRBN to treat MM (17, 18) or del(5q) MDS (19). For example, DNA damage induced by ionizing radiation or cisplatin can activate c-Abl (32), begging a closer examination of these agents in DDA1-CRL4 complex assembly, leading to CRL4 activation and justifying clinical investigation into the combination of IMiDs with conventional radiotherapy or chemotherapy in treating MM and del(5q) MDS patients. Additionally, the development of new drugs to hijack the CRL4CRBN ubiquitin ligase to degrade previously intractable proteins should take into account the critical role of Abl kinase activation in achieving better targeting efficacy (23).
Experimental Procedures
Cell Lines
HEK 293T, 293FT, HeLa, MM1S, MM1R, OPM2, and U266 cell lines were obtained from the ATCC. Cells were cultured in media recommended by the ATCC and maintained at 37 °C in 5% CO2. Pomalidomide-resistant MM1S-P5000 cells were generated by culturing MM1S cells in medium containing gradually increasing concentrations of pomalidomide. The final pomalidomide concentration in medium was 5 μm to maintain the growth of these cells.
cDNA and Plasmids
pMT21 expression vectors carrying a C-terminal Myc-tagged c-Abl and Abl-KR were gifts from Stephen Goff (Columbia University, New York, NY). The FLAG-ub expression vector was a kind gift from Zongping Xia (Zhejiang University). The full-length cDNAs (Human ORFeome Collection) of DDA1, CRBN, Abl1, and Abl1-KR were amplified and subcloned into the pXF6H expression vector (3×FLAG) from Xinhua Feng (Zhejiang University) between the EcoRI and BamHI restriction sites and named FLAG-DDA1, FLAG-CRBN, FLAG-Abl, and FLAG-Abl-KR plasmids. The full-length human DDB1 was amplified and subcloned into the pXF4H expression vector (2×HA) from Xinhua Feng (Zhejiang University) between the XbaI and HindIII restriction sites and named HA-DDB1 plasmid. Site-directed mutagenesis to generate HA-DDB1 (Y316F), HA-DDB1 (Y182F), HA-DDB1 (Y678F), HA-DDB1 (Y718F), HA-DDB1-DM (Y182F/Y718F), and HA-DDB1-TM (Y182F/Y718F/Y316F) mutant expression vectors were carried out utilizing the QuikChange method (Agilent Stratagene). Human IKZF3 and IKZF1 cDNA clones were amplified with an HA tag in the C terminus and then cloned into the pXF4H expression vector between the ClaI and EcoRI sites and named IKZF3-HA and IKZF1-HA plasmids. The human CRBN cDNA clone was amplified with a Myc tag in the N terminus and then cloned into the pXF4H expression vector between the ClaI and EcoRI sites and named Myc-CRBN plasmid.
Compounds and Antibodies
MG132 (S2619), MLN4924 (S7109), LBH589 (S1030), Dexamethasone (S1322), imatinib (S2475), and lenalidomide (S1029) were purchased from Selleck Chemicals. All compounds were dissolved in DMSO. Anti-FLAG (F3165, Sigma-Aldrich), anti-HA (H6908 and H3663, Sigma-Aldrich), anti-Myc (2272, Cell Signaling Technology), anti-Cul4A (A300-739A, Bethyl), anti-Tyr(P) (9411S, Cell Signaling Technology), anti-c-Abl (sc-131, Santa Cruz Biotechnology), anti-p-c-Abl(Tyr-245) (2861S, Cell Signaling Technology), anti-DDB1 (37-6200, Invitrogen), anti-Actin (1844-1, Epitomics), anti-IKZF3 (NBP2-24495, Novus Biologicals), anti-IKZF1 (AP10222B, Abgent), anti-DDA1 (AP14094C, Abgent), anti-β-tubulin (1559509A, Life Technologies), anti-Lamin A/C (sc-20681, Santa Cruz Biotechnology), anti-HA affinity gel (E6779, Sigma-Aldrich), and anti-CRBN (SAB045910, Sigma-Aldrich) were purchased and used according to the recommendations of the manufacturer. Secondary antibodies were goat anti-mouse IgG-HRP (sc-2005, Santa Cruz Biotechnology) and goat anti-rabbit IgG-HRP (sc-2004, Santa Cruz Biotechnology).
Immunoblot Analysis
All Protein lysates were resolved by standard SDS-PAGE and transferred to PVDF membranes. Blots were blocked in 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) and incubated with primary antibodies in 5% BSA at 4 °C overnight. Blots were washed three times with 1× TBS-T the next day and then incubated with secondary antibodies for 1 h at room temperature. Detection was performed with ECL Western blotting detection reagent (Thermo Scientific).
Co-immunoprecipitation Assays
Cells were transfected with the indicated plasmids and lysed in NETN lysis buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1% Nonidet P-40, and 1 mm Na3VO4 supplemented with complete protease inhibitors (Roche)). After centrifugation at 13,000 rpm for 15 min, supernatants were subjected to immunoprecipitation by incubation with anti-HA affinity gel for either 4 h or overnight at 4 °C. Samples were washed three times with lysis buffer before being separated by SDS-PAGE and analyzed by immunoblot.
In Vivo Ubiquitination Assay
Plasmids were transfected into HEK 293T cells as indicated. Cell lysates were prepared for ubiquitination assays as described previously (7). For ubiquitination of histone H3, histones were extracted and purified as described elsewhere (38) and then subjected to the ubiquitination assay.
Preparation of Nuclear and Cytoplasmic Extracts
HeLa cells were transfected as indicated, and the fractions were prepared as described previously (30).
Protein Degradation Analysis and Protein Quantification
Cells were treated with cycloheximide (CHX, 100 μg/ml), and cells were harvested for lysate preparation using NETN lysis buffer at the indicated time points and subjected to immunoblot analysis. For quantification, each band of the WB for IKZF3 was quantified with Gel-Pro Analyzer 4.0 software (Media Cybernetics) and normalized to the corresponding actin band. The relative IKZF3 protein level at the starting time (0 h) in each group was set to 1. The relative IKZF3 protein levels at other time points were compared with that at the starting time. Data points were adjusted to a one-exponential decay curve using GraphPad Prism software (GraphPad), and the protein degradation rate (t½) was expressed as the time for degradation of 50% of the protein.
siRNA Sequences and Transfection
The siRNA sequences for DDA1 were CCUCAUAGGAGCCGAUGUAdTdT and GCGAGUACCCGUCUGAACAdTdT. Cells were transfected using Lipofectamine RNAiMax (Invitrogen). Lipofectamine 2000 (Invitrogen) was used for transfection with vectors and siRNAs simultaneously.
Real-time RT-PCR
Total RNA was extracted using TRIzol reagent (Life Technologies) and reverse-transcribed into cDNA using the PrimeScript RT Master Mix Kit (RR036A, TaKaRa) according to the protocol of the manufacturer. Real-time PCR was performed in triplicate using Power SYBR Green PCR Master Mix (4367659, Life Technologies) and an Applied Biosystems 7500 real-time PCR system. PCR primers were as follows: DDA1-F, GTACCCGTCTGAACAGATCATCG; DDA1-R, GGCAGCGTTCTTTTTGTCCC; GAPDH-F, CGACCACTTTGTCAAGCTCA; and GAPDH-R, TTACTCCTTGGAGGCCATGT.
Lentiviral Vectors and Virus Infection
Single guide RNA sequences targeting Abl (#1, GCAGCCCTTACCCCAGACGT; #2, GGTTCATCATCATTCAACGG) and sgControl (#1, GCCATATCGGGGCGAGACATG; #2, GTGAGGATCATGTCGAGCGCC) sequences were annealed and subcloned into lentiCRISPRv2 vectors (39, 40). The c-Abl coding sequences were amplified and cloned into the PLVX-IRES-Neo vector. The vectors were transfected into HEK 293FT cells together with pVSVg and psPAX2 vectors using Lipofectamine 2000. The medium was replaced after 24 h. The viral supernatant was collected 72 h post-transfection and ultracentrifuged at 25,000 rpm at 4 °C for 2 h. For viral infection, MM1S cells were seeded in 12-well plates and supplemented with 6 μg/ml Polybrene (Sigma). Cells were centrifuged at 2000 rpm for 2 h at 37 °C. Following the spin, the medium was aspirated, and fresh medium without Polybrene was added.
Viability Assay
MM cells were seeded in 96-well plates and treated with compounds at the indicated concentration and time. Proliferation rates were analyzed using Cell Counting Kit 8 (Dojindo Laboratories, Kumamoto, Japan) according to the protocol of the manufacturer.
Statistical Analysis
The data presented were acquired from a minimum of two independent experiments unless otherwise indicated. Statistical significance in assays with identical cell lines was assessed with Student's t test (two-tailed). GraphPad Prism 5 (GraphPad) was used for all statistical analyses.
Author Contributions
S. G. and C. G. performed the experiments. T. S., J. L., and X. L. provided technical assistance. S. G., Z. C., and Y. C. analyzed the data. S. G. and Y. C. designed the study and wrote the manuscript.
Acknowledgments
We thank X. H. Feng for expression vectors; the staff of the core facilities at the Life Sciences Institute and School of Medicine of Zhejiang University for technical assistance; Z. G. Peng, G. F. Li, and T. Ji for discussions; and W. J. Wang for administrative assistance.
This work was supported in part by National 973 Plan for Basic Research Grant 2015CB553803, National Natural Science Foundation Grant 91429302, and Fundamental Research Funds for the Central Universities and Key Construction Program of the National 985 Project. The authors declare that they have no conflicts of interest with the contents of this article.
- CRL
- cullin-RING ubiquitin ligase
- IMiD
- immunomodulatory drug
- MM
- multiple myeloma
- MDS
- myelodysplastic syndrome
- CRBN
- cereblon
- TM
- triple mutant
- DM
- double mutant
- CHX
- cycloheximide
- WB
- Western blot
- Len
- lenalidomide
- LBH
- LBH589
- IP
- immunoprecipitation
- ub
- ubiquitin
- IKZF
- Ikaros family zinc finger proteins
- CRISPR-Cas9
- clustered regularly interspaced short palindromic repeats-associated nuclease Cas9.
References
- 1. Jackson S., and Xiong Y. (2009) CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 34, 562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Havens C. G., and Walter J. C. (2011) Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 25, 1568–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee J., and Zhou P. (2007) DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 26, 775–780 [DOI] [PubMed] [Google Scholar]
- 4. Hu J., McCall C. M., Ohta T., and Xiong Y. (2004) Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 6, 1003–1009 [DOI] [PubMed] [Google Scholar]
- 5. Kim Y., Starostina N. G., and Kipreos E. T. (2008) The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 22, 2507–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., and Dutta A. (2008) PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 22, 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen X., Zhang Y., Douglas L., and Zhou P. (2001) UV-damaged DNA-binding proteins are targets of CUL-4A-mediated ubiquitination and degradation. J. Biol. Chem. 276, 48175–48182 [DOI] [PubMed] [Google Scholar]
- 8. Nakagawa T., Lv L., Nakagawa M., Yu Y., Yu C., D'Alessio A. C., Nakayama K., Fan H. Y., Chen X., and Xiong Y. (2015) CRL4(VprBP) E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Mol. Cell 57, 247–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kapetanaki M. G., Guerrero-Santoro J., Bisi D. C., Hsieh C. L., Rapić-Otrin V., and Levine A. S. (2006) The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc. Natl. Acad. Sci. U.S.A. 103, 2588–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang H., Zhai L., Xu J., Joo H. Y., Jackson S., Erdjument-Bromage H., Tempst P., Xiong Y., and Zhang Y. (2006) Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 22, 383–394 [DOI] [PubMed] [Google Scholar]
- 11. Han J., Zhang H., Zhang H., Wang Z., Zhou H., and Zhang Z. (2013) A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 155, 817–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Didcock L., Young D. F., Goodbourn S., and Randall R. E. (1999) The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 73, 9928–9933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parisien J. P., Lau J. F., Rodriguez J. J., Sullivan B. M., Moscona A., Parks G. D., Lamb R. A., and Horvath C. M. (2001) The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology 283, 230–239 [DOI] [PubMed] [Google Scholar]
- 14. Ulane C. M., and Horvath C. M. (2002) Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 304, 160–166 [DOI] [PubMed] [Google Scholar]
- 15. Hrecka K., Hao C., Gierszewska M., Swanson S. K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M. P., and Skowronski J. (2011) Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Decorsière A., Mueller H., van Breugel P. C., Abdul F., Gerossier L., Beran R. K., Livingston C. M., Niu C., Fletcher S. P., Hantz O., and Strubin M. (2016) Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531, 386–389 [DOI] [PubMed] [Google Scholar]
- 17. Krönke J., Udeshi N. D., Narla A., Grauman P., Hurst S. N., McConkey M., Svinkina T., Heckl D., Comer E., Li X., Ciarlo C., Hartman E., Munshi N., Schenone M., Schreiber S. L., et al. (2013) Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu G., Middleton R. E., Sun H., Naniong M., Ott C. J., Mitsiades C. S., Wong K. K., Bradner J. E., and Kaelin W. G. (2014) The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krönke J., Fink E. C., Hollenbach P. W., MacBeth K. J., Hurst S. N., Udeshi N. D., Chamberlain P. P., Mani D. R., Man H. W., Gandhi A. K., Svinkina T., Schneider R. K., McConkey M., Järås M., Griffiths E., et al. (2015) Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischer E. S., Böhm K., Lydeard J. R., Yang H., Stadler M. B., Cavadini S., Nagel J., Serluca F., Acker V., Lingaraju G. M., Tichkule R. B., Schebesta M., Forrester W. C., Schirle M., Hassiepen U., et al. (2014) Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chamberlain P. P., Lopez-Girona A., Miller K., Carmel G., Pagarigan B., Chie-Leon B., Rychak E., Corral L. G., Ren Y. J., Wang M., Riley M., Delker S. L., Ito T., Ando H., Mori T., et al. (2014) Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 21, 803–809 [DOI] [PubMed] [Google Scholar]
- 22. Petzold G., Fischer E. S., and Thomä N. H. (2016) Structural basis of lenalidomide-induced CK1α degradation by the CRL4 ubiquitin ligase. Nature 532, 127–130 [DOI] [PubMed] [Google Scholar]
- 23. Winter G. E., Buckley D. L., Paulk J., Roberts J. M., Souza A., Dhe-Paganon S., and Bradner J. E. (2015) Drug Development: phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu J., Ye J., Zou X., Xu Z., Feng Y., Zou X., Chen Z., Li Y., and Cang Y. (2014) CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat. Commun. 5, 3924. [DOI] [PubMed] [Google Scholar]
- 25. Nguyen T. V., Lee J. E., Sweredoski M. J., Yang S. J., Jeon S. J., Harrison J. S., Yim J. H., Lee S. G., Handa H., Kuhlman B., Jeong J. S., Reitsma J. M., Park C. S., Hess S., and Deshaies R. J. (2016) Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol. Cell 61, 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin J., Arias E. E., Chen J., Harper J. W., and Walter J. C. (2006) A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 [DOI] [PubMed] [Google Scholar]
- 27. Angers S., Li T., Yi X., MacCoss M. J., Moon R. T., and Zheng N. (2006) Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 [DOI] [PubMed] [Google Scholar]
- 28. Olma M. H., Roy M., Le Bihan T., Sumara I., Maerki S., Larsen B., Quadroni M., Peter M., Tyers M., and Pintard L. (2009) An interaction network of the mammalian COP9 signalosome identifies Dda1 as a core subunit of multiple Cul4-based E3 ligases. J. Cell Sci. 122, 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laubach J. P., Moreau P., San-Miguel J. F., and Richardson P. G. (2015) Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 21, 4767–4773 [DOI] [PubMed] [Google Scholar]
- 30. Cong F., Tang J., Hwang B. J., Vuong B. Q., Chu G., and Goff S. P. (2002) Interaction between UV-damaged DNA binding activity proteins and the c-Abl tyrosine kinase. J. Biol. Chem. 277, 34870–34878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lewis J. M., Baskaran R., Taagepera S., Schwartz M. A., and Wang J. Y. (1996) Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc. Natl. Acad. Sci. U.S.A. 93, 15174–15179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kharbanda S., Ren R., Pandey P., Shafman T. D., Feller S. M., Weichselbaum R. R., and Kufe D. W. (1995) Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 376, 785–788 [DOI] [PubMed] [Google Scholar]
- 33. Kaidi A., and Jackson S. P. (2013) KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 498, 70–74 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Meijer E., and Sonneveld P. (2009) Hematology: lenalidomide plus dexamethasone is effective in multiple myeloma. Nat. Rev. Clin. Oncol. 6, 247–248 [DOI] [PubMed] [Google Scholar]
- 35. Avigan D., and Rosenblatt J. (2014) Current treatment for multiple myeloma. N. Engl. J. Med. 371, 961–962 [DOI] [PubMed] [Google Scholar]
- 36. Soucy T. A., Smith P. G., Milhollen M. A., Berger A. J., Gavin J. M., Adhikari S., Brownell J. E., Burke K. E., Cardin D. P., Critchley S., Cullis C. A., Doucette A., Garnsey J. J., Gaulin J. L., Gershman R. E., et al. (2009) An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 [DOI] [PubMed] [Google Scholar]
- 37. Chen X., Zhang J., Lee J., Lin P. S., Ford J. M., Zheng N., and Zhou P. (2006) A kinase-independent function of c-Abl in promoting proteolytic destruction of damaged DNA binding proteins. Mol. Cell 22, 489–499 [DOI] [PubMed] [Google Scholar]
- 38. Shechter D., Dormann H. L., Allis C. D., and Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 39. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelsen T. S., Heckl D., Ebert B. L., Root D. E., Doench J. G., and Zhang F. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]