

Abstract
Nonalcoholic fatty liver disease (NAFLD), characterized by excessive fat accumulation in liver, is prevalent in obesity. Genetic factors that link obesity to NAFLD remain obscure. Apolipoprotein C3 (APOC3) is a lipid-binding protein with a pivotal role in triglyceride metabolism. Humans with APOC3 gain-of-function mutations and mice with APOC3 overproduction are associated with hypertriglyceridemia. Nonetheless, it remains controversial whether APOC3 is culpable for diet-induced NAFLD. To address this fundamental issue, we fed APOC3-transgenic and wild-type littermates a high fructose diet or high fat diet, followed by determination of the effect of APOC3 on hepatic lipid metabolism and inflammation and the progression of NAFLD. To gain mechanistic insight into NAFLD, we determined the impact of APOC3 on hepatic triglyceride synthesis and secretion versus fatty acid oxidation. APOC3-transgenic mice were hypertriglyceridemic, culminating in marked elevation of triglycerides, cholesterols, and non-esterified fatty acids in plasma. Despite the prevailing hypertriglyceridemia, APOC3-transgenic mice, relative to wild-type littermates, had similar weight gain and hepatic lipid content without alterations in hepatic expression of key genes involved in triglyceride synthesis and secretion and fatty acid oxidation. APOC3-transgenic and wild-type mice had similar Kupffer cell content without alterations in hepatic expression of pro- and anti-inflammatory cytokines. APOC3 neither exacerbated diet-induced adiposity nor aggravated the degree of steatosis in high fructose or high fat-fed APOC3-transgenic mice. These effects ensued independently of weight gain even after 10-month high fat feeding. We concluded that APOC3, whose dysregulation is liable for hypertriglyceridemia, is not a predisposing factor for linking overnutrition to NAFLD in obesity.
Keywords: apolipoprotein, lipid metabolism, liver, obesity, triglyceride, Apolipoprotein C3, Hypertriglyceridemia, Nonalcoholic fatty liver disease, Steatosis, Triglyceride metabolism
Introduction
Nonalcoholic fatty liver disease (NAFLD)4 is characterized by excessive lipid deposition in the liver of subjects with little alcohol consumption. Patients with NAFLD are at heightened risk of developing steatohepatitis with potential complications of fibrosis and cirrhosis (1–4). Currently, NAFLD affects about 30% of the general population, and its prevalence is increasing along with the rising epidemic of obesity in both adults and children (1, 5–10). Although it is known that NAFLD results from increased lipogenesis and decreased fatty acid oxidation in the liver, accompanied often by hepatic overproduction of TG-rich very low density lipoprotein (VLDL-TG), genetic factors that tip the balance of these three intertwining pathways in hepatic lipid metabolism at the expense of NAFLD are incompletely characterized.
Apolipoprotein C3 (APOC3) (79 amino acids in length) is one of the most abundant apolipoproteins in the blood, where it is present as an exchangeable moiety between high density lipoproteins (HDLs) and triglyceride (TG)-rich particles, such as VLDL and chylomicrons (11). Secreted mainly from the liver and to a lesser extent from the intestine, APOC3 impacts TG metabolism via multiple mechanisms. APOC3 functions as an inhibitor of lipoprotein lipase (LPL) and hepatic lipase (HL), key enzymes responsible for the hydrolysis of TG in VLDL and chylomicrons in the post-absorption phase (12–14). Elevated APOC3 levels also perturb hepatic uptake and clearance of TG-rich lipoprotein remnants (15, 16). This action is mediated by the low density lipoprotein (LDL) family receptors independently of LPL (17). Apart from its extracellular functions, APOC3 plays an intracellular role in promoting VLDL-TG secretion from the liver (18–21). As a result, elevated plasma APOC3 levels are associated with increased hepatic VLDL-TG production and decreased systemic clearance of TG-rich lipoproteins, contributing to the development of hypertriglyceridemia. Indeed, APOC3 transgenic mice manifest chronic hypertriglyceridemia after birth (22). Conversely, APOC3 deficiency is associated with enhanced TG hydrolysis and clearance, contributing to hypotriglyceridemia in APOC3 knock-out mice (23–25). Likewise, human subjects with APOC3 loss-of-function mutations are associated with decreased plasma TG levels and reduced risk of coronary artery disease (26–28). Antisense oligonucleotide-mediated reduction of plasma APOC3 levels results in significantly lower plasma TG levels in nonhuman primates as well as patients with familial chylomicronemia (29, 30). Taken together, these data have revealed a causal relationship between APOC3 overproduction and hypertriglyceridemia, validating APOC3 as a potential target for the development of anti-hypertriglyceridemia therapy for lowering the cardiovascular risk (31, 32).
Nevertheless, it remains controversial as to whether APOC3 is a predisposing factor for NAFLD. Petersen et al. (33) report that human subjects with genetic variants (C-482T or T-455C) within the APOC3 promoter region are at increased risk of developing NAFLD. Lee et al. (34) demonstrate that APOC3-transgenic mice develop mild steatosis in response to high fat feeding. In contrast, Kozlitina et al. (35) report that neither of these two APOC3 variants (C-482T and T-455C) is associated with NAFLD in humans, although both alleles are associated with hypertriglyceridemia. This latter observation was supported by three independent clinical studies showing that carriers of either or both of the two APOC3 alleles (C-482T and T-455C) are not associated with NAFLD (36–38). Added to this controversy is the study by Duivenvoorden et al. (39), who show that APOC3 knock-out mice are susceptible to developing diet-induced obesity with a concomitant induction of NAFLD. However, this observation was contradicted by clinical studies, in which humans with APOC3 loss-of-function alleles are associated with decreased plasma TG levels but not with increased susceptibility to developing steatosis (26–28). Thus, it remains controversial whether APOC3 is an independent risk factor for NAFLD. To address this controversy, we fed APOC3 transgenic (APOC3-tg) mice and age/sex-matched wild-type (WT) littermates for 4 months a high fructose diet and separately a high fat diet, two feeding conditions that are both deleterious to hepatic lipid metabolism. High fructose consumption induces steatosis without inducing excessive weight gain, whereas high fat feeding elicits steatosis with the induction of obesity. To consolidate our findings, we prolonged high fat feeding of APOC3-tg and WT littermates for up to 10 months. Our goal is to answer the longstanding question as to whether APOC3 is a predisposing factor for exacerbating the development of NAFLD in diet-induced obesity.
Results
Effect of APOC3 on the Pathogenesis of NAFLD in APOC3-tg Mice
To determine the effect of APOC3 on hepatic lipid metabolism, we monitored APOC3-tg and age/sex-matched WT littermates (n = 8/group) on regular chow for up to 28 weeks. APOC3-tg mice, as opposed to WT littermates, exhibited significantly elevated plasma levels of TG, cholesterol, and non-esterified fatty acid (NEFA), characteristic of hypertriglyceridemia (supplemental Table 1). This pro-atherogenic lipid profile was reproduced in both male and female APOC3-tg mice, independently of body weight. To determine the contribution of APOC3 to NAFLD, we euthanized APOC3-tg and WT littermates at 28 weeks of age, followed by the determination of hepatic TG and cholesterol contents. APOC3-tg and WT littermates had similar hepatic levels of TG and cholesterols, regardless of sex (supplemental Table 1). Because both male and female APOC3-tg mice had similar phenotypes in terms of plasma and hepatic TG metabolism, we chose male APOC3-tg mice for further characterization.
Does High Fructose Feeding Exacerbate NAFLD in APOC3-tg Mice?
Excessive fructose consumption, which is deleterious to hepatic lipid metabolism, is considered an independent risk factor of NAFLD in animal models and human subjects (6, 40–44). We hypothesized that APOC3 transgenic production would compound the effect of high fructose consumption on steatosis. To address this hypothesis, we fed APOC3-tg and WT littermates (male, n = 8/group) a high fructose diet for 16 weeks. When compared with WT littermates, APOC3-tg mice maintained similar weight gain but exhibited markedly elevated plasma levels of TG, cholesterols, and NEFA (Table 1). APOC3-tg and WT mice were euthanized after 16 weeks of high fructose feeding, followed by the quantification of hepatic lipid content. APOC3-tg and WT mice had similar hepatic TG and cholesterol contents (Table 1).
TABLE 1.
Plasma lipid profiles in WT and APOC3-tg mice on high fructose diet
APOC3-tg and WT littermates (male, 8 weeks old) were fed a high fructose diet for 16 weeks, followed by the determination of plasma TG, cholesterol, and NEFA levels after 16 h of fasting. Mice were euthanized after 16 weeks of high fructose feeding under non-fasting conditions for the determination of hepatic TG and cholesterol content.
| Genotype | WT | APOC3-tg |
|---|---|---|
| Body weight (g) | 25.6 ± 1.2 | 25.2 ± 1.7 |
| Plasma TG (mg/dl) | 87 ± 6 | 580 ± 60a |
| Plasma cholesterol (mg/dl) | 149 ± 13 | 236 ± 49b |
| Plasma NEFA (mmol/liter) | 1.2 ± 0.07 | 2.6 ± 0.27b |
| Hepatic TG (mg/g protein) | 510 ± 61 | 615 ± 194 |
| Hepatic cholesterol (mg/g protein) | 66 ± 7 | 76 ± 8 |
a p < 0.001 versus WT control.
b p < 0.05 versus WT control.
Impact of APOC3 on Hepatic Lipid Metabolism in APOC3-tg Mice on High Fat Diet
To address the hypothesis that APOC3 transgenic production would aggravate high fat-elicited steatosis, we fed APOC3-tg and WT littermates (male, 8 weeks old, n = 8/group) a high fat diet for 16 weeks. When compared with WT littermates, APOC3-tg mice maintained similar weight gain (Fig. 1A) but exhibited markedly elevated plasma levels of TG (Fig. 1B), cholesterols (Fig. 1C), and NEFA (Fig. 1D). APOC3-tg mice, as opposed to WT controls, had milk-like serum (Fig. 1E). To determine postprandial TG clearance, we performed a fat tolerance test. APOC3-tg mice were associated with impaired fat tolerance, as evidenced by markedly elevated plasma TG profiles in response to an oral bolus of olive oil (Fig. 1F). To determine the effect of APOC3 on hepatic VLDL-TG production, we determined hepatic VLDL-TG production in APOC3-tg versus WT mice, using tyloxapol to inhibit systemic TG clearance, as described (45, 46). APOC3-tg mice had significantly higher plasma TG levels at all time points after tyloxapol administration (Fig. 1G). This effect correlated with a 2-fold induction in the rate of hepatic VLDL-TG secretion in APOC3-tg versus WT littermates (Fig. 1H). After 16 weeks of high fat feeding, we euthanized APOC3-tg and WT mice. Liver tissues were procured for the quantification of hepatic lipid content. APOC3-tg and WT mice had similar hepatic TG (Fig. 1I) and cholesterol contents (Fig. 1J).
FIGURE 1.

TG metabolism in APOC3-tg mice after 4 months of high fat feeding. APOC3-tg and WT littermates (male, 8 weeks old, n = 8/group) were fed a high fat diet for 4 months, followed by the determination of TG metabolism. A, body weight; B, plasma TG levels; C, plasma cholesterol levels; D, plasma NEFA levels. Plasma TG, cholesterol, and NEFA levels were determined in mice after 16 h of fasting. E, sera of APOC3-tg and WT littermates. Sera in capillary tubes were visualized under light. F, fat tolerance test. G, hepatic VLDL-TG production. H, rates of hepatic VLDL-TG production. Hepatic VLDL-TG production rates, defined as mg of TG/g of body weight/h, were calculated from the data in G. I, hepatic TG content. J, hepatic cholesterol content. *, p < 0.05; **, p < 0.001 versus WT. Error bars, S.E.
Does High Fat Feeding Aggravate NAFLD in APOC3-tg Mice?
We presumed that the lack of association between APOC3 and steatosis in APOC3-tg mice might be due to the relatively short duration of high fat feeding (16 weeks). To address this possibility, we fed APOC3-tg and WT littermates (male, 6 weeks old, n = 17/group) a high fat diet for 10 months. APOC3-tg and WT littermates had an equivalent weight gain over time and become morbidly obese after 10 months of high fat feeding (Fig. 2A). APOC3-tg and WT littermates had a similar fat versus lean mass distribution (Fig. 2B). When compared with WT littermates, APOC3-tg mice had significantly elevated plasma TG (Fig. 2C), cholesterol (Fig. 2D), and NEFA levels (Fig. 2E). To determine the effect of APOC3 on lipoprotein metabolism in fat-induced obesity, we subjected sera from obese APOC3-tg versus WT littermates to gel filtration column chromatography for the fractionation of lipoproteins. APOC3-tg mice, as opposed to WT controls, had markedly higher levels of VLDL-TG particles (Fig. 2F), consistent with APOC3-mediated induction of hepatic VLDL-TG production and retardation in systemic VLDL-TG clearance. Furthermore, APOC3-tg mice had higher VLDL-cholesterol and LDL-cholesterol levels, accompanied by lower HDL-cholesterol levels (Fig. 2G), characteristic of pro-atherogenic lipoprotein profiles. To assess the effect of APOC3 on lipid excretion, we determined fecal TG and cholesterol levels. No significant differences in fecal TG and cholesterol contents during a 24-h period were detected between APOC3-tg and WT mice (Fig. 2, H and I).
FIGURE 2.

TG and lipoprotein metabolism in APOC3-tg mice after 10 months of high fat feeding. APOC3-tg and WT littermates (male, 6 weeks old, n = 17/group) were fed a high fat diet for 10 months, followed by the determination of TG and lipoprotein metabolism. A, growth curves; B, fat mass and lean mass; C, plasma TG levels; D, plasma cholesterol levels; E, plasma NEFA levels. Plasma TG, cholesterol, and NEFA levels were determined in mice after 16 h of fasting. F, plasma TG distribution among lipoproteins. G, plasma cholesterol distribution among lipoproteins. Aliquots (500 μl) of plasma pooled from APOC3-tg and WT littermates were fractionated by gel filtration in a FPLC system. Fractions (500 μl) were eluted for the determination of TG levels (F) and cholesterol levels (G). H, fecal TG content. I, fecal cholesterol content. Feces were collected from individual mice under fed conditions during a 24-h period. Fecal lipid content was expressed as mg of TG or cholesterol/g of feces. *, p < 0.05; **, p < 0.001 versus WT. Error bars, S.E.
To determine APOC3 contribution to steatosis, we euthanized APOC3-tg and WT littermates after 10 months of high fat feeding. Liver tissues were procured for histological examination after staining with Oil Red O and H&E (Fig. 3, A–D). As expected, chronic high fat feeding resulted in significant fat accumulation in the liver. Nonetheless, APOC3-tg and WT littermates had similar degrees of fat infiltration into the liver. To corroborate these findings, we quantified hepatic lipid content, demonstrating that APOC3-tg and WT littermates had similar excessive fat deposition (Fig. 3E), accompanied by similar hepatic cholesterol content in the liver (Fig. 3F).
FIGURE 3.

Liver histology and hepatic gene expression profiles in high fat-fed APOC3 mice. WT control (A and C) and APOC3-tg mice (B and D) (male, 11 months old, n = 7–9/group) were euthanized under non-fasting conditions after 10 months of high fat feeding. Liver tissues were procured for histological examination after staining with H&E (A and B) and Oil Red O (C and D). Aliquots of liver tissues were used for the determination of hepatic TG (E) and cholesterol (F) contents. Total liver RNA was prepared and was subjected to a real-time qRT-PCR assay for profiling hepatic expression of genes involved in hepatic lipid metabolism (G). Bar, 200 μm. Error bars, S.E.
To gain mechanistic insight into the lack of association between APOC3 and steatosis, we profiled hepatic expression of key functions involved in VLDL-TG production, lipogenesis, and fatty acid oxidation, three independent pathways in hepatic lipid metabolism (Fig. 3G). We did not detect significant differences between APOC3-tg and WT littermates in hepatic mRNA levels of apob (apolipoprotein B) and mttp (microsomal triglyceride transfer protein), two genes involved in hepatic VLDL-TG production. Likewise, APOC3-tg and WT littermates had similar hepatic mRNA levels of lipogenic genes srebp-1c (sterol regulatory element-binding protein 1c), fas (fatty acid synthase), acc (acetyl-CoA carboxylase), pgc-1β (peroxisome proliferator-activated receptor γ coactivator 1-β), and ppar-γ (peroxisome proliferator-activated receptor γ) and of genes responsible for fatty acid oxidation, including ppar-α, cpt1 (carnitine palmitoyltransferase 1), and acox1 (acyl-coenzyme A oxidase 1).
To address whether human APOC3 transgenic production would suppress endogenous APOC3 production in the liver, we subjected total hepatic RNA from APOC3-tg and WT mice to a real-time qRT-PCR assay. This assay did not reveal differences in mouse APOC3 mRNA levels between APOC3-tg and WT mice (supplemental Fig. 1). To corroborate these findings, we subjected aliquots of plasma from APOC3-tg and WT mice to a mouse-specific ApoC3 ELISA assay, demonstrating that APOC3-tg and WT mice had similar mouse APOC3 protein levels in plasma. As a control, we detected human APOC3 mRNA expression in the liver using human APOC3-specific primers, in accordance with transgenic production of human APOC3 proteins in APOC3-tg mice, as determined by human-specific APOC3 ELISA assay (supplemental Fig. 1). In contrast, WT littermates had non-detectable human APOC3 mRNAs in liver and human APOC3 proteins in plasma. These results precluded the possibility that human APOC3-tg production inhibits endogenous APOC3 expression in the liver of APOC3-tg mice.
Impact of APOC3 on Glucose Metabolism in Chronic Fat-fed APOC3-tg Mice
To determine the effect of APOC3 transgenic production on glucose metabolism, we determined blood glucose profiles, demonstrating that APOC3-tg and WT mice had similar blood glucose levels under both fed and fasting conditions (Fig. 4A). To determine postprandial glucose disposal rates, we performed intraperitoneal glucose tolerance test. APOC3-tg and WT mice had similar blood glucose profiles during the glucose tolerance test. This result was reproduced in APOC3-tg and WT mice at different months of high fat feeding (Fig. 4, B and C). To rule out the possibility that the lack of changes in glucose tolerance in APOC3-tg mice, relative to that in WT littermates, was due to the intraperitoneal route of glucose injection, we performed an oral glucose tolerance test. Both APOC3-tg and WT groups had similar profiles of postprandial blood glucose excursion in response to an oral bolus of glucose (Fig. 4D). These results were unexpected, given that APOC3-tg mice were obese and hypertriglyceridemic after 10 months of high fat feeding. We postulated that the prevailing hypertriglyceridemia provoked a compensatory increase in insulin secretion, which in turn suppressed the development of hyperglycemia and glucose intolerance in obese APOC3-tg mice. To address this hypothesis, we determined plasma insulin levels under fasting conditions and at 15 and 30 min after intraperitoneal glucose injection. We detected significant increases in basal and glucose-stimulated insulin secretion (Fig. 4E), in accordance with relatively higher HOMA-IR (homeostasis model for insulin resistance) levels in APOC3-tg versus WT mice (Fig. 4F). In contrast, no significant differences in plasma glucagon levels were seen in APOC3-tg versus WT mice (Fig. 4G). As a control, we isolated islets from high fat-fed APOC3-tg and WT mice (n = 6/group). Aliquots of islets in equivalent sizes (n = 50 hand-picked islets/mouse) were examined for ex vivo insulin secretion at low (2.8 mm) and high (20 mm) glucose concentrations in culture medium. APOC3-tg islets had relatively higher levels of glucose-stimulated insulin release (Fig. 4H), although the amplitude of ex vivo glucose-stimulated insulin secretion from APOC3-tg islets did not reach a significant level when compared with control islets from WT mice.
FIGURE 4.

Glucose metabolism in high fat-fed APOC3 mice. APOC3-tg and WT littermates (male, 6 weeks old, n = 7–9/group) were fed a high fat diet for 10 months, followed by the determination of glucose metabolism. A, blood glucose levels. Blood glucose levels were determined in APOC3-tg and WT littermates under fed conditions and after 16 h of fasting. B, intraperitoneal glucose tolerance test at 9 months of age. C, intraperitoneal glucose tolerance test at 11 months of age. D, oral glucose tolerance test at 11 months of age. Mice were fasted for 16 h, followed by either intraperitoneal injection or oral gavage of 2 g/kg glucose. E, glucose-stimulated insulin secretion. During the intraperitoneal glucose tolerance test, aliquots of blood (25 μl) were sampled at fasting and at 15 and 30 min after glucose injection for determining basal and glucose-stimulated insulin secretion. F, HOMA-IR. HOMA-IR was calculated from fasting blood glucose and plasma insulin levels. G, plasma glucagon levels. Plasma glucagon levels were determined after 16 h of fasting in APOC3-tg and WT mice at the end of a 10-month high fat feeding. H, ex vivo glucose-stimulated insulin secretion. The amplitude of glucose-stimulated insulin secretion was defined as the amount of insulin secreted within a 30-min time window from islets normalized to total islet cell proteins in the presence of 2.8 and 20 mm glucose. *, p < 0.05; **, p < 0.001 versus WT control. Error bars, S.E.
Impact of APOC3 on β-Cell Mass in Chronic Fat-fed APOC3-tg Mice
To address whether the significant induction in basal and glucose-stimulated insulin secretion was attributable to increased islet mass, we performed anti-insulin and anti-glucagon immunohistochemistry on pancreas tissues retrieved from euthanized APOC3-tg and WT mice after 10 months of high fat feeding (Fig. 5, A and B). APOC3-tg mice had significantly increased β-cell mass (Fig. 5C), consistent with increased plasma insulin levels under basal and glucose-stimulated conditions (Fig. 4E). In contrast, no significant differences in α-cell mass were detectable (Fig. 5D), coinciding with the lack of changes in plasma glucagon levels in APOC3-tg versus WT mice (Fig. 4G). Likewise, APOC3-tg and WT mice had similar pancreas weight (Fig. 5E). Furthermore, we showed that APOC3-tg mice had significantly increased large sized islets (diameter ≥100 mm) than WT mice (Fig. 5F).
FIGURE 5.

Impact of APOC3 on β-cell and α-cell masses. APOC3-tg and WT mice (male, 6 weeks old, n = 5–7/group) were fed a high fat diet for 10 months. Mice were euthanized at fasting conditions, and the pancreas tissues were subjected to dual immunohistochemistry using anti-insulin and anti-glucagon antibodies. Representative images of pancreatic islets of WT (A) and APOC3-tg (B) mice are shown. β-Cells were immunostained green, and α-cells were immunostained red. C, β-cell mass. D, α-cell mass. E, pancreas weight. The pancreas weight (mg) was normalized to body weight (g). F, islet distribution in size. Islets were scored per pancreas section and were assigned to four subgroups according to islet size, ranging in diameter from <50 to 50–100, 100–200, and >200 μm. 3–5 nonconsecutive sections were scored per pancreas for determining the number of islets in different subgroups according to islet size. The percentage of islets in each subgroup out of total islets was calculated and compared between APOC3-tg and WT mice. G, islet expression of insulin genes. H, islet mRNA levels. Islet mRNA levels were determined by a real-time qRT-PCR assay, using 18S RNA as control. *, p < 0.05 versus WT control. Bar, 50 μm. Error bars, S.E.
To gain mechanistic insight into the increases in glucose-stimulated insulin secretion in APOC3-tg mice, we profiled islet expression of genes whose functions are critical for insulin synthesis and secretion (Ins1 (insulin 1), Ins2, Pdx1 (pancreatic and duodenal homeobox 1), NeuroD (neurogenic differentiation factor), and FoxA2 (forkhead box A2)), insulin signaling (Irs1 (insulin receptor substrate 1) and Irs2), and β-cell glucose sensing (Gck (glucokinase) and Glut2 (glucose transporter 2)). We detected a significant induction in both Ins1 and Ins2 expression in APOC3-tg islets (Fig. 5G). These effects corrected with the significant induction in islet expression of Pdx1, the key transcription factor that is responsible for promoting insulin gene expression (Fig. 5H). In addition, APOC3-tg islets had increased expression of Irs2 (Fig. 5H), whose function is critical for β-cell function and mass regulation (47, 48). In contrast, no significant differences were seen in Glut2 and Gck expression in islets of high fat-fed APOC3-tg mice.
Effect of APOC3 on Hepatic Inflammation in Fat-induced Obese APOC3-tg Mice
To determine the effect of APOC3 on hepatic inflammation, we performed anti-F4/80 immunohistochemistry to determine hepatic infiltration by Kupffer cells, known as resident macrophages in the liver (49). Hepatic inflammation, accompanied by increased Kupffer cell infiltration into the liver, is a prominent pathological feature of NAFLD (50). Anti-F4/80 immunohistochemistry in combination with morphometric analysis revealed that high fat-induced obese APOC3-tg and WT littermates had similar Kupffer cell content (Fig. 6, A–C). We then profiled hepatic expression of inflammatory cytokines, demonstrating that hepatic mRNA levels of pro-inflammatory proteins TNFα, IL-6, and IL-1β and anti-inflammatory gene IL-10 remained unchanged in APOC3-tg versus WT mice (Fig. 6D). Likewise, no significant differences in hepatic F4/80 mRNA abundance were detected, consistent with the lack of changes in Kupffer cell content in the liver of APOC3-tg versus WT mice.
FIGURE 6.

Effect of APOC3 on Kupffer cell content in the liver of high fat-fed APOC3 mice. Liver tissues were obtained from high fat-fed WT control (A) and APOC3-tg mice (B) as described in the legend to Fig. 1. Frozen sections of liver tissues were subjected to anti-F4/80 immunohistochemistry. F4/80-positive cells (stained red) were counted and normalized to the total number of cells (nuclei stained blue by DAPI) in liver sections (C). Aliquots of liver tissues (20 mg) were used for the preparation of total RNA, which was subjected to real-time qRT-PCR for determining F4/80, TNFα, IL-1β, IL-6, and IL-10 mRNA levels (D). n = 7–9/group. Bar, 200 μm. Error bars, S.E.
Effect of APOC3 on Hepatic Fibrosis in Fat-induced Obese APOC3-tg Mice
To determine the impact of APOC3 on hepatic fibrosis in high fat-fed APOC3 versus WT mice, we subjected liver sections to Sirius Red staining. Both APOC3-tg and WT mice exhibited relatively low levels of periportal fibrosis, as reflected in the deposition of collagen fibers in the liver. Morphometric analysis did not reveal significant differences in the degree of fibrosis between high fat-fed APOC3-tg and WT mice (Fig. 7, A–C). To support this finding, we determined hepatic collagen levels, demonstrating that APOC3-tg and WT mice had similar total collagen content in the liver (Fig. 7D).
FIGURE 7.
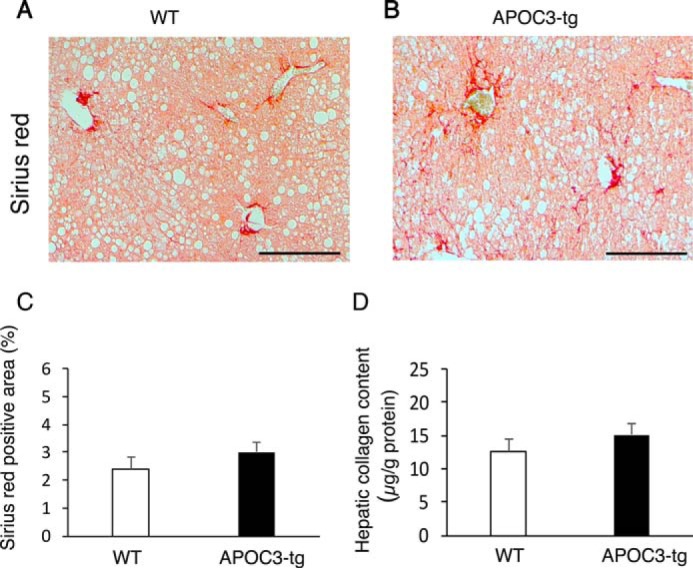
Effect of APOC3 on hepatic fibrosis in high fat-fed APOC3 mice. Liver tissues were derived from high fat-fed WT control (A) and APOC3-tg mice (B) as described in the legend to Fig. 1. Paraffin sections of liver tissues were subjected to Sirius Red staining for collagen. The Sirius Red-positive area in non-consecutive sections was determined by morphometric analysis (C). Aliquots of liver tissues were subject to a collagen assay for determining hepatic collagen content (D), expressed as μg of collagen/g of total liver protein. n = 7–9/group. Bar, 200 μm. Error bars, S.E.
Discussion
APOC3 is a lipid-binding protein that is circulating mainly in TG-rich lipoproteins. APOC3 modulates TG metabolism via at least three distinct mechanisms: by inhibiting LPL- and HL-mediated TG hydrolysis, by promoting hepatic VLDL-TG production, and by retarding hepatic uptake and clearance of TG-rich lipoprotein remnants. As a result, APOC3 dysregulation is associated with abnormal TG metabolism (31, 33, 51–56). Notwithstanding its importance in the pathogenesis of hypertriglyceridemia, its functional contribution to NAFLD remains obscure. To unveil the direct effect of APOC3 on NAFLD, we determined hepatic lipid metabolism in APOC3-tg mice versus WT littermates. We showed that APOC3-tg mice, despite prevailing hypertriglyceridemia, were not predisposed to developing NAFLD. We recapitulated this finding in APOC3-tg mice on regular chow for 7 months, high fructose diet for 4 months, and high fat diet for up to 10 months. Although high fructose feeding and separately high fat feeding resulted in excessive fat accumulation in the liver, the degree of steatosis was indistinguishable between APOC3-tg and WT mice, as determined by liver histology in combination with quantification of hepatic fat content. This effect correlated with the lack of changes in hepatic expression of genes involved in de novo lipogenesis and fatty acid oxidation. Our data argue against the notion that APOC3 is an independent risk factor for NAFLD.
Consistent with our data are the clinical observations that two common APOC3 variants (C-482T and T-455C), which are associated with APOC3 gain of function and are linked to the inheritance of hypertriglyceridemia, are not associated with NAFLD in humans. This finding was reproducible in humans harboring either or both of the C-482T and T-455C alleles in different ethnic groups, including African Americans, European Americans, Hispanics, Southern Europeans, Italians, and Chinese (35, 36, 38, 57, 58). Further physiological underpinning of the dissociation between APOC3 and NAFLD is derived from the clinical study by Zampino et al. (59), who evaluated the genetic influence of APOC3 polymorphism on steatosis in an Italian patient population with chronic hepatitis C infection. Whereas chronic hepatitis C infection is an independent risk factor for NAFLD, this clinical study showed that neither of the two APOC3 variants, namely C-482T and T-455C, is a compounding factor for hepatitis C-elicited steatosis (59).
APOC3 has emerged as an important regulator of TG metabolism. APOC3 is initially characterized as an inhibitor of LPL and HL, two lipases that are responsible for systemic TG clearance (12–14). Subsequent studies unveiled that APOC3 also plays an intracellular role in regulating hepatic VLDL-TG secretion. Yao and colleagues (18, 60, 61) showed that APOC3 is responsible for the formation of luminal lipid droplets as well as for the bulk incorporation of TG-enriched luminal lipid droplets into nascent VLDL, contributing to the expansion of TG-rich VLDL1 particles. This effect is attributable to the lipid-binding domain within the carboxyl region encompassing amino acid resides 41–79 of APOC3 protein. Genetic mutations, which perturb the carboxyl lipid-binding domain, result in loss-of-function of APOC3 in promoting the formation of TG-enriched LLD and VLDL1 secretion (18, 60, 61). This effect ensues without changes in hepatic MTP activity, suggesting that APOC3 promotes the formation of LLD and secretion of TG-enriched large VLDL1 particles independently of MTP (18). In keeping with these findings, we showed that APOC3-tg mice had significantly increased hepatic VLDL-TG secretion, without alterations in MTP expression in the liver. Likewise, we did not detect significant changes in hepatic APOB mRNA levels in APOC3-tg versus WT mice, consistent with the fact that APOB mRNA is constitutively expressed and APOB protein lipidation is tightly regulated by NEFA substrate availability at the co-translational level (62). In the absence of substrates, APOB proteins undergo proteolytic degradation. In this context, we showed that APOC3-tg mice had markedly elevated NEFA levels, and this effect was exacerbated in response to high fat feeding. Our interpretation is that increased NEFA influx into the liver fueled the production of TG via fatty acid re-esterification in the liver. This effect along with APOC3-mediated induction of TG-rich LLD formation promoted hepatic VLDL-TG secretion and contributed to the removal of lipids from the liver, congruent with the lack of exacerbating effects of APOC3 on steatosis in APOC3-tg mice.
Despite hypertriglyceridemia, APOC3-tg mice had blood glucose levels similar to that in WT controls under both fed and fasting conditions. Furthermore, APOC3-tg and WT mice maintained similar postprandial blood glucose excursion in response to both intraperitoneal and oral glucose tolerance. These effects ensued despite high fat-induced steatosis in APOC3-tg and WT mice. Of particular significance, we showed that high fat-induced obese APOC3-tg mice had increased basal and glucose-stimulated insulin secretion, which were attributable to a compensatory increase in β-cell mass in APOC3-tg mice. As a result, hypertriglyceridemic APOC3-tg mice, despite fat-elicited steatosis and insulin resistance, were able to maintain normal blood glucose metabolism. Consistent with our data are clinical observations that human subjects with genetic APOC3 variants exhibit hypertriglyceridemia without displaying altered glucose metabolism (35, 38, 57).
β-Cell compensation is an adaptive mechanism by which β-cells increase insulin secretion to overcome insulin resistance in peripheral tissues for maintaining euglycemia in the body. β-Cell compensation culminates in β-cell mass expansion, increased insulin synthesis and secretion, or enhanced glucose sensing in islets. To gain mechanistic insight into β-cell compensation in APOC3-tg mice, we showed that APOC3-tg islets had increased expression of key genes involved in β-cell insulin synthesis and secretion, including the insulin genes Ins1 and Ins2 and Pdx1. APOC3-tg islets also had significantly increased expression of Irs2, whose product is required for promoting β-cell compensation for insulin resistance in obesity (48, 63). At present, we do not know the mechanism underlying the induction of β-cell compensation in APOC3-transgenic mice. However, we do know that such a compensatory induction in β-cell mass and function did not take place in APOC3-tg mice under regular chow (64) but ensued in response to overnutrition in APOC3-transgenic mice. These results suggest that hypertriglyceridemia does not directly stimulate β-cell mass expansion in the absence of insulin resistance but acts as an accessory factor for amplifying the effect of overnutrition on β-cell compensation for insulin resistance in APOC3-tg mice. Further investigation is required for dissecting the molecular basis that connects hypertriglyceridemia to β-cell compensation in diet-induced obese APOC3-tg mice.
It is noteworthy that our data deviate partly from Lee et al. (34), who examined the effect of APOC3 on steatosis in human APOC3-tg mice. In their study, APOC3-tg mice were fed a high fat diet with 55% calories from fat obtained from Harlan Teklad. In our study, we fed APOC3-tg mice a high fat diet with 60% calories from fat purchased from Research Diet Inc. Overall, their and our results were in agreement with respect to the lack of changes in weight gain, fat mass, plasma lipid profiles, and blood glucose levels, with a discrepancy in hepatic TG content in APOC3-tg versus WT mice. They detected about 30% higher mean TG content in the liver of APOC3-tg mice following 12 weeks of high fat feeding. In contrast, we showed that although both groups develop fat-induced steatosis, no significant differences were detectable in hepatic TG and cholesterol contents between APOC3-tg and WT littermates after 4- and 10-month high fat feeding, respectively. We recapitulated this finding in APOC3-tg and WT littermates on a high fructose diet, a feeding condition that is conducive to the development of steatosis. APOC3 failed to aggravate high fructose-induced steatosis. Aside from the difference in diets from different vendors, one variable for the inconsistency is the duration of high fat feeding. It is possible that a 30% increase in hepatic fat content is due to the acute effect of APOC3 overproduction on hepatic lipid metabolism in APOC3-tg mice following 12-week high fat feeding in their studies (34). Another variable that could account for the apparent discrepancy in the score of steatosis in APOC3-tg versus WT mice between their and our studies is the experimental conditions. Although their and our APOC3-tg mice were derived originally from the same transgenic line (22), those APOC3-tg mice were maintained as separate colonies in different animal facilities, a confounding factor that might contribute to the discrepancy. Indeed, they showed that high fat-fed APOC3-tg mice were associated with chronic inflammation, as evidenced by marked induction of pro-inflammatory cytokine TNF-α production (34). In contrast, we demonstrated that high fat-fed APOC3-tg and WT mice had similar hepatic expression profiles of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α and anti-inflammatory cytokine IL-10, in accordance with the lack of changes in Kupffer cell content in the liver of APOC3-tg versus WT mice.
In conclusion, we have provided compelling evidence that APOC3, which is culpable for hypertriglyceridemia, is not a compounding factor for the pathogenesis of NAFLD in high fat-fed APOC3-tg mice. Although APOC3 is being targeted for the development of anti-hypertriglyceridemia therapy for reducing the cardiovascular risk (31, 32), cautions must be exercised for applying such therapy in patients with established NAFLD. Given its intracellular role in facilitating VLDL-TG assembly and secretion (18–21), APOC3 is probably an ill target for therapeutic intervention of NAFLD. Selective APOC3 inhibition may impair hepatic VLDL-TG secretion, resulting in excessive fat accumulation in the liver. Indeed, APOC3 deficiency is associated with diet-induced steatosis, obesity, and insulin resistance in APOC3 knock-out mice (39). Further research is warranted to determine whether chronic APOC3 inhibition for lowering plasma TG levels is inadvertently coupled with an increased risk of developing NAFLD.
Experimental Procedures
Animal Studies
Transgenic mice expressing human APOC3 (APOC3-tg) from its endogenous promoter were originally described previously (22, 65, 66). We obtained the APOC3 transgenic line G1 with relatively higher levels of APOC3 transgenic production. We maintained this APOC3 transgenic line in C57BL/6J genetic background in the animal facility of Children's Hospital of Pittsburgh of UPMC (64, 66). We crossed male APOC3-tg mice with female wild-type mice. The resulting APOC-tg progenies, which are uniformly heterozygous in terms of the human APOC3 transgene, were used in this study. Mice were fed standard rodent chow and water ad libitum in sterile cages with a 12-h light/dark cycle in a pathogen-free barrier facility. For fructose feeding, mice were fed a high fructose diet containing 60% fructose (w/w) (Dyets, Bethlehem, PA), as described (40). To induce obesity, APOC3-tg mice and WT littermates were fed a high fat diet (fat content, 60 kcal%, Research Diets Inc., New Brunswick, NJ). For blood chemistry, mice were fasted for 16 h, and tail vein blood was sampled. Blood glucose levels were measured, using Glucometer Elite (Bayer, IN). Plasma insulin levels were determined, using the ultrasensitive mouse insulin enzyme-linked immunosorbent assay (ALPCO, Windham, NH). The homeostasis model for insulin resistance (HOMA-IR) was determined by multiplying fasting blood glucose (mmol/liter) and fasting plasma insulin (μIU/ml) levels, divided by 22.5. Plasma levels of TG and cholesterol were determined using Thermo Infinity TG and cholesterol reagents (ThermoFisher Scientific, Middletown, VA). Plasma NEFA levels were determined using the Wako NEFA assay kit (Wako Chemical USA, Richmond, VA). Mice were euthanized under non-fasting conditions to collect liver tissues for analysis. All procedures were approved by the institutional animal care and use committee of the University of Pittsburgh.
VLDL-TG Production Assay
Mice were fasted for 16 h, followed by intraperitoneal injection of tyloxapol (1 g/kg; Sigma-Aldrich) to inhibit systemic TG clearance. Aliquots of tail vein blood were taken at different times for determining plasma TG levels. VLDL-TG production rates were defined as the amount of hepatic TG produced per unit time, as described (46).
Fat Tolerance Test
Mice were fasted for 16 h, followed by an oral bolus of olive oil (10 μl/g). Aliquots of blood (25 μl) were taken from the tail vein at different times for determining plasma TG, as described (51).
Glucose Tolerance Test
Mice were fasted for 16 h, followed by intraperitoneal injection of glucose (2 g/kg) or oral gavage of glucose (2 g/kg). Blood glucose levels were determined before and at different times after glucose administration.
Fat Mass Determination
Fat and lean masses of mice were determined using the EchoMRI-100 system (Echo Medical Systems, Houston, TX).
Islet Isolation
Mice were euthanized, followed by pancreatic intraductal infusion of 3 ml of cold Hanks' buffer containing 1.95 mg/ml collagenase-V (Sigma). The pancreas was procured for islet isolation, as described (47).
Ex Vivo Glucose-stimulated Insulin Secretion
Aliquots of islets (n = 50) equivalent in size between APOC3-tg and WT mice (n = 6/group) were cultured in RPMI 1640 medium overnight, followed by incubation in Kreb buffer containing 2.8 or 20 mm glucose for 30 min. Aliquots (50 μl) of conditioned medium were collected for determining insulin concentrations. Medium insulin levels were normalized to total cellular proteins of islets used in the assay.
RNA Isolation and Real-time qRT-PCR
Total RNA was prepared from the liver or islets using TRIzol reagent (Invitrogen). Real-time qRT-PCR was used for quantifying mRNA concentrations using the Roche LightCycler-RNA amplification kit (Roche Diagnostics), as described (46). As tabulated in supplemental Table 2, all primers were obtained commercially from Integrated DNA Technologies (Coralville, IA).
Liver Histology
Liver tissues were embedded in the Histoprep tissue-embedding medium and snap-frozen. Frozen sections (6 μm in thickness) were cut and stained with hematoxylin and eosin. For visualizing lipid in the liver, frozen sections were stained with Oil Red O, followed by counterstaining with hematoxylin. For detecting fibrosis in the liver, paraffin sections were stained with Sirius Red using the Picro Sirius Red Stain Kit (Abcam, Cambridge, MA).
FPLC Fractionation of Lipoproteins
Aliquots (500 μl) of plasma pooled from APOC3-tg mice and control littermates were applied to two head-to-tail linked Tricorn high performance Superose S-6 10/300GL columns using an FPLC system (GE Healthcare), followed by elution with PBS at a constant flow rate of 0.25 ml/min. Fractions (500 μl) were eluted for determining TG and cholesterol levels, as described (46, 51).
Hepatic TG Content
Liver tissues (20 mg) were homogenized in 400 μl of HPLC grade acetone. After incubation with agitation at room temperature overnight, aliquots (50 μl) of acetone-extracted lipid suspension were used for determining TG concentration, using the Infinity triglyceride reagent (ThermoFisher Scientific). Hepatic TG content was defined as mg of TG/g of total liver proteins.
Hepatic Cholesterol Content
Liver tissues (20 mg) were homogenized in 400 μl of hexane/isopropyl alcohol (3:2 in volume). After incubation with agitation at room temperature overnight, aliquots (50 μl) of hexane/isopropyl alcohol-extracted cholesterol suspension were used for determining cholesterol concentration, using the Infinity cholesterol reagent (ThermoFisher Scientific). Hepatic cholesterol content was defined as mg of cholesterol/g of total liver proteins.
Hepatic Collagen Content
Liver tissues (10 mg) were homogenized in 100 μl of H2O. The homogenates were mixed with 100 μl of 12 m HCl, followed by incubation at 120 °C for 3 h for acid hydrolysis. Aliquots of the hydrolyzed samples (20 μl) were subjected to a collagen assay, using the colorimetric Collagen Assay Kit (BioVision, Milpitas, CA).
APOC3 Assay
Plasma APOC3 levels were determined, using APOC3 ELISA kits that are species-specific for human APOC3 protein (catalog no. ab154131, Abcam) and for mouse APOC3 protein (catalog no. MBS944394, MyBioSource), respectively.
Fecal Lipid Content
Feces were collected from individual mice during a 24-h period under fed conditions. Aliquots of feces (150 mg) were homogenized in 500 μl of HPLC grade acetone, followed by incubation with agitation at room temperature overnight. Fecal TG content, defined as mg of TG/g of feces, was determined in acetone-extract lipid suspension, using the Infinity triglyceride reagent (ThermoFisher Scientific). Likewise, aliquots of feces (150 mg) were homogenized in 500 μl of hexane/isopropyl alcohol (3:2 in volume). After incubation with agitation at room temperature overnight, aliquots (50 μl) of hexane/isopropyl alcohol-extracted fecal suspension were used for determining fecal cholesterol content, defined as mg of cholesterol/g of feces.
Immunohistochemistry
Liver tissues were fixed in 4% paraformaldehyde and cryopreserved in 30% sucrose. Frozen sections (6 μm) were subjected to immunohistochemistry using rat anti-F4/80 antibody (Invitrogen) and Cy3-conjugated donkey anti-rat IgG (Jackson ImmunoResearch Laboratories). The nuclei of cells were stained with DAPI (Sigma-Aldrich) before fluorescent microscopy.
β-Cell and α-Cell Masses
Mice were euthanized, and the pancreas was subjected to immunohistochemistry, using guinea pig anti-insulin (DAKO; 1:300) and mouse anti-glucagon (Sigma, catalog no. G2654; 1:500). The second antibodies were Cy2-conjugated donkey anti-guinea pig IgG and Cy3-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories). The nuclei of cells were stained with DAPI (Sigma) before immunofluorescent microscopy. Digital images of pancreas sections immunostained by anti-insulin or anti-glucagon antibody were subjected to morphometric analysis, using MetaMorph image analysis software (Molecular Devices, Downingtown, PA). β-cell area/total pancreas area/section was determined in 3–5 non-consecutive sections for determining β-cell mass, as described (46). Likewise, α-cell mass was determined in APOC3-tg and WT groups.
Immunoblot Analysis
Aliquots of plasma at a fixed concentration of 15 μg of proteins/lane were applied to 4–20% SDS-polyacrylamide gels. After electrophoresis, the gels were subjected to an immunoblot assay, using polyclonal anti-APOC3 antibody (catalog no. ab55984; Abcam). This anti-APOC3 antibody cross-reacted with both human and mouse APOC3 proteins, allowing us to determine total plasma APOC3 proteins in APOC3-tg mice. As control, we used polyclonal goat anti-albumin antibody (catalog no. ab19194; Abcam).
Statistics
Statistics of data were analyzed by Student's t test and were further validated by an analysis of variance post hoc test, using JMP statistics software (Cary, NC). Data are expressed as means ± S.E. p values of <0.05 were considered statistically significant.
Author Contributions
X. C., J. Y., S. Q., and H. H. D. conceived the idea and coordinated the in vivo and ex vivo studies. X. C. and J. Y. conducted the animal studies. X. C. and S. L. performed liver histology studies. T. Z. performed the real-time qRT-PCR assay. Z. G. and R. M. were responsible for blood chemistry and Western blotting analysis. X. C. and H. H. D. analyzed the data and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This study was supported by National Institutes of Health Grant R01 DK098437. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. 1 and Tables 1 and 2.
- NAFLD
- nonalcoholic fatty liver disease
- TG
- triglyceride
- NEFA
- nonesterified fatty acids
- LPL
- lipoprotein lipase
- HL
- hepatic lipase
- VLDL-TG
- TG-rich VLDL
- APOC3-tg
- APOC3 transgenic
- qRT-PCR
- quantitative RT-PCR.
References
- 1. Vuppalanchi R., and Chalasani N. (2009) Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: selected practical issues in their evaluation and management. Hepatology 49, 306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brunt E. M. (2010) Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 7, 195–203 [DOI] [PubMed] [Google Scholar]
- 3. Nugent C., and Younossi Z. M. (2007) Evaluation and management of obesity-related nonalcoholic fatty liver disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 4, 432–441 [DOI] [PubMed] [Google Scholar]
- 4. Torres D. M., and Harrison S. A. (2008) Diagnosis and therapy of nonalcoholic steatohepatitis. Gastroenterology 134, 1682–1698 [DOI] [PubMed] [Google Scholar]
- 5. Sanyal A. J., Chalasani N., Kowdley K. V., McCullough A., Diehl A. M., Bass N. M., Neuschwander-Tetri B. A., Lavine J. E., Tonascia J., Unalp A., Van Natta M., Clark J., Brunt E. M., Kleiner D. E., Hoofnagle J. H., et al. (2010) Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 362, 1675–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lim J. S., Mietus-Snyder M., Valente A., Schwarz J. M., and Lustig R. H. (2010) The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 7, 251–264 [DOI] [PubMed] [Google Scholar]
- 7. Burgert T. S., Taksali S. E., Dziura J., Goodman T. R., Yeckel C. W., Papademetris X., Constable R. T., Weiss R., Tamborlane W. V., Savoye M., Seyal A. A., and Caprio S. (2006) Alanine aminotransferase levels and fatty liver in childhood obesity: associations with insulin resistance, adiponectin, and visceral fat. J. Clin. Endocrinol. Metab. 91, 4287–4294 [DOI] [PubMed] [Google Scholar]
- 8. Lerret S. M., and Skelton J. A. (2008) Pediatric nonalcoholic fatty liver disease. Gastroenterol. Nurs. 31, 115–119 [DOI] [PubMed] [Google Scholar]
- 9. Dunn W., and Schwimmer J. B. (2008) The obesity epidemic and nonalcoholic fatty liver disease in children. Curr. Gastroenterol. Rep. 10, 67–72 [DOI] [PubMed] [Google Scholar]
- 10. Nobili V., Alisi A., and Raponi M. (2009) Pediatric non-alcoholic fatty liver disease: preventive and therapeutic value of lifestyle intervention. World J. Gastroenterol. 15, 6017–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jong M. C., Hofker M. H., and Havekes L. M. (1999) Role of apoCs in lipoprotein metabolism: functional differences between apoC1, apoC2 and apoC3. Arterioscler. Thromb. Vasc. Biol. 19, 472–484 [DOI] [PubMed] [Google Scholar]
- 12. Wang C. S., McConathy W. J., Kloer H. U., and Alaupovic P. (1985) Modulation of lipoprotein lipase activity by apolipoproteins: effect of apolipoprotein C-III. J. Clin. Invest. 75, 384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McConathy W. J., Gesquiere J. C., Bass H., Tartar A., Fruchart J. C., and Wang C. S. (1992) Inhibition of lipoprotein lipase activity by synthetic peptides of apolipoprotein C-III. J. Lipid Res. 33, 995–1003 [PubMed] [Google Scholar]
- 14. Kinnunen P. K. J., and Ehnolm C. (1976) Effect of serum and C apolipoproteins from very low density lipoproteins on human post-heparin plasma hepatic lipase. FEBS Lett. 65, 354–357 [DOI] [PubMed] [Google Scholar]
- 15. Quarfordt S. H., Michalopoulos G., and Schirmer B. (1982) The effect of human C apolipoproteins on the in vitro hepatic metabolism of triglyceride emulsions in the rat. J. Biol. Chem. 257, 14642–14647 [PubMed] [Google Scholar]
- 16. Mann C. J., Troussard A. A., Yen F. T., Hannouche N., Najib J., Fruchart J. C., Lotteau V., André P., and Bihain B. E. (1997) Inhibitory effects of specific apolipoprotein C-III isoforms on the binding of triglyceride-rich lipoproteins to the lipolysis-stimulated receptor. J. Biol. Chem. 272, 31348–31354 [DOI] [PubMed] [Google Scholar]
- 17. Gordts P. L., Nock R., Son N. H., Ramms B., Lew I., Gonzales J. C., Thacker B. E., Basu D., Lee R. G., Mullick A. E., Graham M. J., Goldberg I. J., Crooke R. M., Witztum J. L., and Esko J. D. (2016) ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J. Clin. Invest. 126, 2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qin W., Sundaram M., Wang Y., Zhou H., Zhong S., Chang C. C., Manhas S., Yao E. F., Parks R. J., McFie P. J., Stone S. J., Jiang Z. G., Wang C., Figeys D., Jia W., and Yao Z. (2011) Missense mutation in APOC3 within the C-terminal lipid binding domain of human ApoC-III results in impaired assembly and secretion of triacylglycerol-rich very low density lipoproteins: evidence that ApoC-III plays a major role in the formation of lipid precursors within the microsomal lumen. J. Biol. Chem. 286, 27769–27780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan D. C., Watts G. F., Nguyen M. N., and Barrett P. H. (2006) Apolipoproteins C-III and A-V as predictors of very-low-density lipoprotein triglyceride and apolipoprotein B-100 kinetics. Arterioscler. Thromb. Vasc. Biol. 26, 590–596 [DOI] [PubMed] [Google Scholar]
- 20. Taskinen M. R., Adiels M., Westerbacka J., Söderlund S., Kahri J., Lundbom N., Lundbom J., Hakkarainen A., Olofsson S. O., Orho-Melander M., and Borén J. (2011) Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler. Thromb. Vasc. Biol. 31, 2144–2150 [DOI] [PubMed] [Google Scholar]
- 21. Cohn J. S., Patterson B. W., Uffelman K. D., Davignon J., and Steiner G. (2004) Rate of production of plasma and very-low-density lipoprotein (VLDL) apolipoprotein C-III is strongly related to the concentration and level of production of VLDL triglyceride in male subjects with different body weights and levels of insulin sensitivity. J. Clin. Endocrinol. Metab. 89, 3949–3955 [DOI] [PubMed] [Google Scholar]
- 22. Ito Y., Azrolan N., O'Connell A., Walsh A., and Breslow J. L. (1990) Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science 249, 790–793 [DOI] [PubMed] [Google Scholar]
- 23. Maeda N., Li H., Lee D., Oliver P., Quarfordt S. H., and Osada J. (1994) Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem. 269, 23610–23616 [PubMed] [Google Scholar]
- 24. Gerritsen G., Rensen P. C., Kypreos K. E., Zannis V. I., Havekes L. M., and Willems van Dijk K. (2005) ApoC-III deficiency prevents hyperlipidemia induced by apoE overexpression. J. Lipid Res. 46, 1466–1473 [DOI] [PubMed] [Google Scholar]
- 25. Jong M. C., Rensen P. C. N., Dahlmans V. E., van der Boom H., van Berkel T. J. C., and Havekes L. M. (2001) Apolipoprotein C-III deficiency accelerates triglyceride hydrolysis by lipoprotein lipase in wild-type and apoE knockout mice. J. Lipid Res. 42, 1578–1585 [PubMed] [Google Scholar]
- 26. Pollin T. I., Damcott C. M., Shen H., Ott S. H., Shelton J., Horenstein R. B., Post W., McLenithan J. C., Bielak L. F., Peyser P. A., Mitchell B. D., Miller M., O'Connell J. R., and Shuldiner A. R. (2008) A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 322, 1702–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jørgensen A. B., Frikke-Schmidt R., Nordestgaard B. G., and Tybjaerg-Hansen A. (2014) Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N. Engl. J. Med. 371, 32–41 [DOI] [PubMed] [Google Scholar]
- 28. TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, Crosby J., Peloso G. M., Auer P. L., Crosslin D. R., Stitziel N. O., Lange L. A., Lu Y., Tang Z. Z., Zhang H., Hindy G., Masca N., Stirrups K., Kanoni S., Do R., et al. (2014) Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 371, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graham M. J., Lee R. G., Bell T. A. 3rd, Fu W., Mullick A. E., Alexander V. J., Singleton W., Viney N., Geary R., Su J., Baker B. F., Burkey J., Crooke S. T., and Crooke R. M. (2013) Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ. Res. 112, 1479–1490 [DOI] [PubMed] [Google Scholar]
- 30. Gaudet D., Brisson D., Tremblay K., Alexander V. J., Singleton W., Hughes S. G., Geary R. S., Baker B. F., Graham M. J., Crooke R. M., and Witztum J. L. (2014) Targeting APOC3 in the familial chylomicronemia syndrome. N. Engl. J. Med. 371, 2200–2206 [DOI] [PubMed] [Google Scholar]
- 31. Norata G. D., Tsimikas S., Pirillo A., and Catapano A. L. (2015) Apolipoprotein C-III: from pathophysiology to pharmacology. Trends Pharmacol. Sci. 36, 675–687 [DOI] [PubMed] [Google Scholar]
- 32. Khetarpal S. A., Qamar A., Millar J. S., and Rader D. J. (2016) Targeting ApoC-III to reduce coronary disease risk. Curr. Atheroscler. Rep. 18, 54. [DOI] [PubMed] [Google Scholar]
- 33. Petersen K. F., Dufour S., Hariri A., Nelson-Williams C., Foo J. N., Zhang X. M., Dziura J., Lifton R. P., and Shulman G. I. (2010) Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N. Engl. J. Med. 362, 1082–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee H. Y., Birkenfeld A. L., Jornayvaz F. R., Jurczak M. J., Kanda S., Popov V., Frederick D. W., Zhang D., Guigni B., Bharadwaj K. G., Choi C. S., Goldberg I. J., Park J. H., Petersen K. F., Samuel V. T., and Shulman G. I. (2011) Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 54, 1650–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozlitina J., Boerwinkle E., Cohen J. C., and Hobbs H. H. (2011) Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance. Hepatology 53, 467–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valenti L., Nobili V., Al-Serri A., Rametta R., Leathart J. B., Zappa M. A., Dongiovanni P., Fracanzani A. L., Alterio A., Roviaro G., Daly A. K., Fargion S., and Day C. P. (2011) The APOC3 T-455C and C-482T promoter region polymorphisms are not associated with the severity of liver damage independently of PNPLA3 I148M genotype in patients with nonalcoholic fatty liver. J. Hepatol. 55, 1409–1414 [DOI] [PubMed] [Google Scholar]
- 37. Verrijken A., Beckers S., Francque S., Hilden H., Caron S., Zegers D., Ruppert M., Hubens G., Van Marck E., Michielsen P., Staels B., Taskinen M. R., Van Hul W., and Van Gaal L. (2013) A gene variant of PNPLA3, but not of APOC3, is associated with histological parameters of NAFLD in an obese population. Obesity 21, 2138–2145 [DOI] [PubMed] [Google Scholar]
- 38. Sentinelli F., Romeo S., Maglio C., Incani M., Burza M. A., Scano F., Coccia F., Cossu E., Leonetti F., and Baroni M. G. (2011) Lack of effect of apolipoprotein C3 polymorphisms on indices of liver steatosis, lipid profile and insulin resistance in obese Southern Europeans. Lipids Health Dis. 10, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Duivenvoorden I., Teusink B., Rensen P. C., Romijn J. A., Havekes L. M., and Voshol P. J. (2005) Apolipoprotein C3 deficiency results in diet-induced obesity and aggravated insulin resistance in mice. Diabetes 54, 664–671 [DOI] [PubMed] [Google Scholar]
- 40. Qu S., Su D., Altomonte J., Kamagate A., He J., Perdomo G., Tse T., Jiang Y., and Dong H. H. (2007) PPARα mediates the hypolipidemic action of fibrates by antagonizing FoxO1. Am. J. Physiol. Endocrinol. Metab. 292, E421–E434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang L., Perdomo G., Kim D. H., Qu S., Ringquist S., Trucco M., and Dong H. H. (2008) Proteomic analysis of fructose-induced fatty liver in hamsters. Metabolism 57, 1115–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lê K. A., Ith M., Kreis R., Faeh D., Bortolotti M., Tran C., Boesch C., and Tappy L. (2009) Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am. J. Clin. Nutr. 89, 1760–1765 [DOI] [PubMed] [Google Scholar]
- 43. Stanhope K. L., Schwarz J. M., Keim N. L., Griffen S. C., Bremer A. A., Graham J. L., Hatcher B., Cox C. L., Dyachenko A., Zhang W., McGahan J. P., Seibert A., Krauss R. M., Chiu S., Schaefer E. J., et al. (2009) Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Invest. 119, 1322–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Samuel V. T. (2011) Fructose induced lipogenesis: from sugar to fat to insulin resistance. Trends Endocrinol. Metab. 22, 60–65 [DOI] [PubMed] [Google Scholar]
- 45. Fan Y., Menon R. K., Cohen P., Hwang D., Clemens T., DiGirolamo D. J., Kopchick J. J., Le Roith D., Trucco M., and Sperling M. A. (2009) Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J. Biol. Chem. 284, 19937–19944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kamagate A., Qu S., Perdomo G., Su D., Kim D. H., Slusher S., Meseck M., and Dong H. H. (2008) FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J. Clin. Invest. 118, 2347–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang T., Kim D. H., Xiao X., Lee S., Gong Z., Muzumdar R., Calabuig-Navarro V., Yamauchi J., Harashima H., Wang R., Bottino R., Alvarez-Perez J. C., Garcia-Ocaña A., Gittes G., and Dong H. H. (2016) FoxO1 plays an important role in regulating beta-cell compensation for insulin resistance in male mice. Endocrinology 157, 1055–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Withers D. J., Gutierrez J. S., Towery H., Burks D. J., Ren J. M., Previs S., Zhang Y., Bernal D., Pons S., Shulman G. I., Bonner-Weir S., and White M. F. (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391, 900–904 [DOI] [PubMed] [Google Scholar]
- 49. Baffy G. (2009) Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J. Hepatol. 51, 212–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Olefsky J. M., and Glass C. K. (2010) Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246 [DOI] [PubMed] [Google Scholar]
- 51. Altomonte J., Cong L., Harbaran S., Richter A., Xu J., Meseck M., and Dong H. H. (2004) Foxo1 mediates insulin action on ApoC-III and triglyceride metabolism. J. Clin. Invest. 114, 1493–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Waterworth D. M., Hubacek J. A., Pitha J., Kovar J., Poledne R., Humphries S. E., and Talmud P. J. (2000) Plasma levels of remnant particles are determined in part by variation in the APOC3 gene insulin response element and the APOCI-APOE cluster. J. Lipid Res. 41, 1103–1109 [PubMed] [Google Scholar]
- 53. Hegele R. A., Connelly P. W., Hanley A. J. G., Sun F., Harris S. B., and Zinman B. (1997) Common genetic variation in the APOC3 promoter associated with variation in plasma lipoproteins. Arterioscler. Thromb. Vasc. Biol. 17, 2753–2758 [DOI] [PubMed] [Google Scholar]
- 54. Chen M., Breslow J. L., Li W., and Leff T. (1994) Transcriptional regulation of the apoC-III gene by insulin in diabetic mice: correction with changes in plasma triglyceride levels. J. Lipid Res. 35, 1918–1924 [PubMed] [Google Scholar]
- 55. Ebara T., Ramakrishnan R., Steiner G., and Shachter N. S. (1997) Chylomicronemia due to apolipoprotein CIII overexpression in apolipoprotein E-null mice: apolipoprotein CIII-induced hypertriglyceridemia is not mediated by effects on apolipoprotein E. J. Clin. Invest. 99, 2672–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Talmud P. J., and Humphries S. E. (1997) Apolipoprotein C-III gene variation and dyslipidemia. Curr. Opin. Lipidol. 8, 154–158 [DOI] [PubMed] [Google Scholar]
- 57. Niu T. H., Jiang M., Xin Y. N., Jiang X. J., Lin Z. H., and Xuan S. Y. (2014) Lack of association between apolipoprotein C3 gene polymorphisms and risk of nonalcoholic fatty liver disease in a Chinese Han population. World J. Gastroenterol. 20, 3655–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang H., Chen L., Xin Y., Lou Y., Liu Y., and Xuan S. (2014) Apolipoprotein c3 gene polymorphisms are not a risk factor for developing non-alcoholic fatty liver disease: a meta-analysis. Hepat. Mon. 14, e23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zampino R., Coppola N., Cirillo G., Boemio A., Pisaturo M., Marrone A., Macera M., Sagnelli E., Perrone L., Adinolfi L. E., and Miraglia del Giudice E. (2013) Abdominal fat interacts with PNPLA3 I148M, but not with the APOC3 variant in the pathogenesis of liver steatosis in chronic hepatitis C. J. Viral Hepat. 20, 517–523 [DOI] [PubMed] [Google Scholar]
- 60. Yao Z. (2012) Human apolipoprotein C-III: a new intrahepatic protein factor promoting assembly and secretion of very low density lipoproteins. Cardiovasc. Hematol. Disord. Drug Targets 12, 133–140 [DOI] [PubMed] [Google Scholar]
- 61. Sundaram M., Zhong S., Bou Khalil M., Zhou H., Jiang Z. G., Zhao Y., Iqbal J., Hussain M. M., Figeys D., Wang Y., and Yao Z. (2010) Functional analysis of the missense APOC3 mutation Ala23Thr associated with human hypotriglyceridemia. J. Lipid Res. 51, 1524–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ginsberg H. N., and Fisher E. A. (2009) The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J. Lipid Res. 50, S162–S166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kubota N., Tobe K., Terauchi Y., Eto K., Yamauchi T., Suzuki R., Tsubamoto Y., Komeda K., Nakano R., Miki H., Satoh S., Sekihara H., Sciacchitano S., Lesniak M., Aizawa S., et al. (2000) Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes 49, 1880–1889 [DOI] [PubMed] [Google Scholar]
- 64. Liu Y. Z., Cheng X., Zhang T., Lee S., Yamauchi J., Xiao X., Gittes G., Qu S., Jiang C. L., and Dong H. H. (2016) Effect of hypertriglyceridemia on beta cell mass and function in ApoC3 transgenic mice. J. Biol. Chem. 291, 14695–14705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aalto-Setälä K., Fisher E. A., Chen X., Chajek-Shaul T., Hayek T., Zechner R., Walsh A., Ramakrishnan R., Ginsberg H. N., and Breslow J. L. (1992) Mechanism of hypertriglyceridemia in human apolipoprotein (apo) CIII transgenic mice: diminished very low density lipoprotein fractional catabolic rate associated with increased apo CIII and reduced apo E on the particles. J. Clin. Invest. 90, 1889–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Qu S., Perdomo G., Su D., D'Souza F. M., Shachter N. S., and Dong H. H. (2007) Effects of apoA-V on HDL and VLDL metabolism in APOC3 transgenic mice. J. Lipid Res. 48, 1476–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.