

Abstract
A chronic high fat diet results in hepatic mitochondrial dysfunction and induction of peroxisomal fatty acid oxidation (FAO); whether specific inhibition of peroxisomal FAO benefits mitochondrial FAO and reactive oxygen species (ROS) metabolism remains unclear. In this study a specific inhibitor for the rate-limiting enzyme involved in peroxisomal FAO, acyl-CoA oxidase-1 (ACOX1) was developed and used for the investigation of peroxisomal FAO inhibition upon mitochondrial FAO and ROS metabolism. Specific inhibition of ACOX1 by 10,12-tricosadiynoic acid increased hepatic mitochondrial FAO via activation of the SIRT1-AMPK (adenosine 5′-monophosphate-activated protein kinase) pathway and proliferator activator receptor α and reduced hydrogen peroxide accumulation in high fat diet-fed rats, which significantly decreased hepatic lipid and ROS contents, reduced body weight gain, and decreased serum triglyceride and insulin levels. Inhibition of ACOX1 is a novel and effective approach for the treatment of high fat diet- or obesity-induced metabolic diseases by improving mitochondrial lipid and ROS metabolism.
Keywords: acetyl coenzyme A (acetyl-CoA), fatty acid, fatty acid metabolism, fatty acid oxidation, inhibitor, metabolism
Introduction
Hepatic lipid and reactive oxygen species (ROS)2 accumulation due to hyperlipidemia and obesity are critical factors associated with the prevalence of metabolic and cardiovascular diseases (1, 2). Decreased mitochondrial fatty acid oxidation (FAO) and increased lipogenesis are major contributors to lipid accumulation in liver and other tissues (3). Excessive hepatic ROS generation and decreased cellular antioxidative activity lead to oxidative stress and hepatic oxidative injury (2, 4). Agents that are able to promote mitochondrial fatty acid oxidation or have antilipogenic and antioxidative effects will improve lipid and ROS metabolism and attenuate hepatic steatosis and oxidative injury.
Peroxisomes are subcellular respiratory organelles that are critical for the metabolism of long chain and branched-chain fatty acids (5). Peroxisomes are sensitive to external signals and are easy to proliferate under conditions of high fat diet (HFD) (6, 7), diabetes (8), or hypolipidemic drug treatment (9); peroxisomal FAO is induced, oxidation capacity is increased by 2–10-fold, and peroxisomal FAO is regulated by the peroxisome proliferator activator receptor α isoform (PPARα) (5).
Acyl-CoA oxidase-1 (ACOX1, EC 1.3.3.6) is a flavoenzyme that catalyzes the initial and rate-determining reaction of the classical peroxisomal FAO using straight-chain fatty acyl-CoAs as the substrates, which donates electrons to molecular oxygen generating hydrogen peroxide; this byproduct is further decomposed by catalase, as shown in Fig. 1. The classical peroxisomal FAO has been known for nearly 40 years (9), but it is still not clear about the exact role of peroxisomal FAO in cellular fatty acid metabolism and especially its effect on mitochondrial fatty acid oxidation. Several lines of evidence have shown that chronic induction of peroxisomal FAO may cause oxidative stress and is potentially detrimental to mitochondrial fatty acid metabolism.
FIGURE 1.
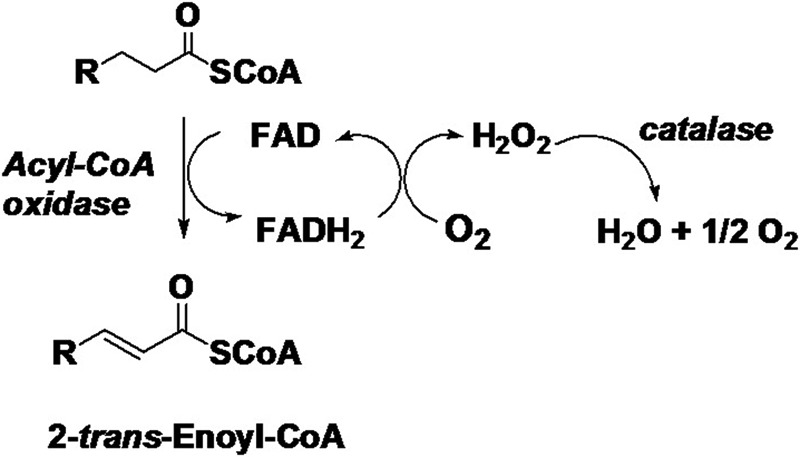
Reaction catalyzed by acyl-CoA oxidase. In the reductive half-reaction, the substrate acyl-CoA is α,β-dehydrogenated into the corresponding 2-trans-enoyl-CoA, with electrons transferred to FAD, which becomes reduced, whereas in the oxidative half-reaction reduced FAD is reoxidized by molecular oxygen, generating hydrogen peroxide.
A number of reports indicated that the hydrogen peroxide-generating enzyme ACOX1 was inducible, whereas the hydrogen peroxide-scavenging enzyme catalase was not induced under conditions of HFD or exposure to PPARα ligands, which resulted in a net increase of hydrogen peroxide in peroxisomes and accumulation of cellular ROS (9–12) and further led to induction of the NF-κB signal pathway and caused cellular oxidative injury (12).
The acetyl-CoA derived from peroxisomal FAO may also be used for malonyl-CoA synthesis and negatively regulates mitochondrial FAO via inhibition of CPT-1A (13, 14). Under conditions of high fat diet or obesity, induction of peroxisomal FAO generates more acetyl-CoA for the synthesis of malonyl-CoA in the cytosol, thereby inhibiting CPT-1A, the rate-limiting enzyme in mitochondrial FAO.
Peroxisomal FAO consumes NAD+ and generates NADH, which will then be transported to the cytosol via redox shuttles, and increases redox potential in the cytosol because, unlike mitochondria, a NAD+ regenerating system is absent in peroxisomes (15–17). Enhanced peroxisomal FAO results in increased NADH generation in peroxisomes, which further increases the cytosolic NADH/NAD+ ratio, thereby inhibiting the activity of SIRT1 and its downstream targets and negatively regulating mitochondrial FAO and cellular ROS metabolism.
To address the above issues, a specific ACOX1 inhibitor will be critical to illustrate the effects of ACOX1 on mitochondrial FAO and cellular ROS metabolism. Acetylenic acids were proposed to have the potential to inhibit ACOX1 according to previous reports (18, 19). We screened from a series of acetylenic acids and obtained a highly specific ACOX1 inhibitor, 10,12-tricosadiynoic acid (TDYA) (Fig. 2); this acetylenic acid is a suicide substrate of ACOX1 with high affinity to the target and high selectivity in vivo.
FIGURE 2.
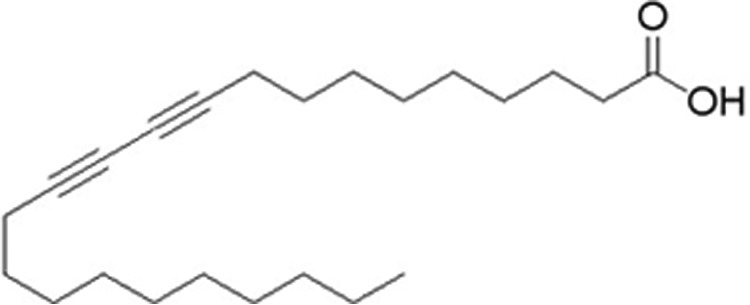
Structure of 10,12-tricosadiynoic acid.
The purpose of this study was to investigate the effects of TDYA on liver lipid and ROS metabolism in rats fed HFD. The influence of this inhibitor upon hepatic SIRT1-AMPK signal pathways as well as mitochondrial FAO was also studied.
Results
Specific Inhibition of ACOX1 by TDYA
First we tested the inhibitory activity using recombinant purified ACOX1. TDYA-CoA rapidly inhibited ACOX1 activity in a time- and concentration-dependent manner as shown in Fig. 3A. The activity of ACOX1 decreased by nearly 95% after 5 min of incubation with 10 eq of TDYA-CoA. ACOX1 activity was inhibited only if free TDYA was activated as the CoA thioester, the substrate form. Inhibition of ACOX1 by TDYA-CoA is irreversible, as we observed the activity of ACOX1 could not be restored after overnight dialysis of the ACOX1-TDYA-CoA mixture (data not shown). KI and kinact were thus used to characterize inhibition kinetics, and the kinetics parameters KI and kinact were calculated to be 680 nm and 3.18 min−1, respectively, as shown in Fig. 3B. TDYA-CoA is a suicide substrate of ACOX1 with a low KI value; it is possible that TDYA-CoA forms a covalent bond with a key residue in the catalytic center of ACOX1 and irreversibly inhibits the enzyme.
FIGURE 3.

A, analysis of inhibition of purified ACOX activity by TDYA-CoA. ACOX1 was dissolved in 20 mm PBS buffer (pH 7.4) at a concentration of 80 nm, then 80 nm (▾), 240 nm (▵), 480 nm (■), 960 nm (□) TDYA-CoA, and 800 nm (○) TDYA free acid were added into the enzyme solution, an equal volume of distilled water was added as the control group (●), and residual ACOX enzyme activity was determined after incubation at 25 °C for various periods. The specific activity that corresponded to 100% activity was 1.43 μmol/min/mg of protein. B, determination of the inactivation kinetic parameter for ACOX1. C, effect of TDYA-CoA on the activity of ACOX1 from rat liver peroxisomes. 200 μg peroxisome (in protein) was suspended in 20 mm PBS buffer (pH 7.4), 5 μm TDYA-CoA (inhibitor group) was added to the solution, and a corresponding volume of water was added to the control group; after incubation at 25 °C for 0 min, 2 min, 5 min, 10 min, and 20 min, ACOX activity for each group was determined. The specific activity that corresponded to 100% activity was 1.16 nmol/min/mg protein. D, inhibition of the activity of ACOX1 in the livers of Wistar rats. n = 6 for each group. *, p < 0.05, **, p < 0.01, ***, p < 0.001.
The inhibition of ACOX1 by TDYA-CoA was also investigated using isolated rat liver peroxisomes. TDYA-CoA rapidly inhibited the activity of ACOX1 from rat liver peroxisomes in a time-dependent manner (Fig. 3C).
TDYA is the precursor of TDYA-CoA and was transformed into TDYA-CoA by peroxisomal very long chain acyl-CoA synthetase (VLACS) after entering into cells, and it inhibited ACOX1 in vivo. In this study TDYA-CoA was isolated and identified by LC/MS analysis in the livers of the rats treated with TDYA (data not shown).
We further tested the in vivo inhibitory activity of TDYA in rats. The results are shown in Fig. 3D; liver ACOX1 activity decreased significantly compared with control group after treatment with TDYA. TDYA inhibited rat liver ACOX1 in a dose-dependent manner in the range of 0–200 μg/kg, and the activity decreased by 70% after treatment with TDYA at an oral dose of 160 μg/kg.
Finally, the inhibitory activity of TDYA-CoA upon key enzymes involved in peroxisomal FAO besides ACOX1 were also investigated, as shown in Table 1. TDYA-CoA showed no inhibition activity toward ACOX-2 (branched-chain ACOX), L-bifunctional protein, thiolase, enoyl-CoA isomerase, dienoyl-CoA reductase, and VLACS. Furthermore, TDYA-CoA is a thioester of very long chain fatty acid, which could not enter into mitochondria because very long chain acyl-CoA synthetase is absent in mitochondria, and both CPT1 and CPT2 show no obvious activity toward very long chain fatty acyl-CoAs (5); therefore TDYA-CoA did not affect mitochondrial fatty acid metabolism directly.
TABLE 1.
Inhibitory activities of TDYA-CoA upon the enzymes involved in peroxisomal FAO
Purified enzymes were dissolved in 20 mm PBS buffer (pH 7.4) at a concentration of 80 nm, then 800 nm TDYA-CoA was added into the enzyme solution, the controls were added with an equal volume of distilled water. After incubation at 25 °C for 30 min, residual activities for all the tested enzymes were determined. Specific activities were obtained from the mean of three repeated tests and expressed as μmol/min/mg. −, with addition of distilled water; +, with addition of TDYA-CoA.
| Enzymes | TDYA-CoA |
|
|---|---|---|
| − | + | |
| ACOX1 | 1.39 | 0.12 |
| ACOX2 | 0.74 | 0.71 |
| L-bifunctional protein | 3.76 | 3.43 |
| 3-Ketoacyl-CoA thiolase | 18.76 | 19.03 |
| 3,2-Enoyl-CoA isomerase | 21.75 | 20.82 |
| 2,4-Dienoyl-CoA reductase | 6.57 | 6.95 |
| VLACS | 0.13 | 0.12 |
Effect of TDYA on Body Weight, Fasting Glucose, and Insulin Levels
HFD slightly increased food intake compared with normal diet group. TDYA treatment did not affect food intake compared with HFD group. Body weight gain was reduced by 20% in the HFD-fed rats treated with TDYA for 8 weeks compared with the HFD control (Table 2, p < 0.05), suggesting body weight loss effect of TDYA.
TABLE 2.
Body weight gain, food intake, plasma glucose, plasma insulin, serum triglyceride, serum total cholesterol (TC), and liver index in rats fed a ND, HFD, or HFD+TDYA
BW, body weight.
| Metabolic parameters | ND | HFD | HFD+TDYA |
|---|---|---|---|
| Initial body weight (g) | 224.6 ± 2.7 | 226.7 ± 3.3 | 225.9 ± 3.1 |
| Final body weight (g) | 183.7 ± 4.5 | 248.4 ± 7.9a | 197.8 ± 5.4b |
| Daily food intake (g/d/rat) | 23.4 ± 0.2 | 25.1 ± 0.5 | 24.6 ± 0.3 |
| Plasma glucose (mmol/liter) | 6.5 ± 0.2 | 7.4 ± 0.3 | 6.6 ± 0.1 |
| Serum insulin (ng/ml) | 1.28 ± 0.13 | 12.73 ± 0.31c | 4.65 ± 0.19d |
| Serum triglyceride (mmol) | 1.02 ± 0.08 | 1.96 ± 0.13a | 1.34 ± 0.10b |
| Serum TC (mmol/liter) | 1.24 ± 0.08 | 1.59 ± 0.13 | 1.37 ± 0.10 |
| Liver index (g liver/100 g BW) | 3.12 ± 0.06 | 4.33 ± 0.09c | 3.52 ± 0.08d |
a ND vs. HFD, p < 0.05. n = 8 for each group.
b HFD vs. HF+TDYA, p < 0.05. n = 8 for each group.
c ND vs. HFD, p < 0.01. n = 8 for each group.
d HFD vs. HF+TDYA, p < 0.01. n = 8 for each group.
After 8 weeks of TDYA treatment, fasting blood glucose was slightly decreased in HFD-fed rats, whereas circulating insulin levels were significantly reduced in the TDYA-treated group compared with the HFD group (Table 2, p < 0.05).
Effect of TDYA on Plasma and Liver Lipid Levels
TDYA treatment significantly decreased serum triglyceride in the HFD-fed rats (by 32%, p < 0.05); serum cholesterol levels were not significantly affected after TDYA treatment, as shown in Table 2.
HFD significantly increased both liver index and triglyceride level in rats (p < 0.01); TDYA treatment significantly decreased liver weight and triglyceride in HFD-fed rats (by 19 and 40%, respectively, p < 0.01), as shown in Table 2 and Fig. 4A. Liver sections were observed by optical microscope with analytic software, and the density of lipid droplets was calculated to be 25.7 ± 6.3/1000 μm2 and 14.6 ± 1.9/1000 μm2,respectively (p < 0.05, n = 4 for each group), which indicated that the quantity of fat droplets significantly decreased in the HFD+TDYA group compared with HFD group. Liver lipid droplets in the HFD group were mainly macrovesicular, whereas lipid deposition in the livers of HFD+TDYA group was mainly composed of microvesicular fat droplets, as shown in Fig. 4B. The diameters of the hepatic lipid droplets in the HFD-fed rats and TDYA-treated rats were measured to be 5.28 ± 1.62 μm and 1.71 ± 0.43 μm, respectively (p < 0.01, n = 4 for each group).
FIGURE 4.

A, effect of ACOX inhibitor TDYA treatment on hepatic triglyceride level in high fat diet fed rats. B, effects of TDYA upon hepatic steatosis was determined by HE staining in rats treated with a normal diet (Normal), a HFD, or HFD with TDYA (HFD+TDYA) for 8 weeks. Magnification: 200×. Scale bar = 20 μm. ##, p < 0.01, **, p < 0.01.
Effect of TDYA on Liver Hydrogen Peroxide and Malondialdehyde Level
HFD increased hepatic generation of hydrogen peroxide as well as increased hepatic ROS levels compared with the normal diet group (Fig. 5, A and B, p < 0.05). Compared with the HFD control group, TDYA intake significantly lowered hepatic hydrogen peroxide (by 30%, Fig. 5A, p < 0.05) and malondialdehyde (MDA) levels (by 29%, Fig. 5B, p < 0.01), indicating that ACOX1 inhibitor has the potential to alleviate HFD-induced ROS formation and accumulation.
FIGURE 5.

A, effect of TDYA treatment on hepatic H2O2 level in rats fed a high fat diet for 8 weeks. B, effect of TDYA treatment on liver MDA level in rats fed a high fat diet for 8 weeks. n = 8 for each group. ##, p < 0.01, **, p < 0.01.
Effect of TDYA on Hepatic ACOX1 and Catalase Activity
HFD significantly increased hepatic ACOX1 activity by 35% due to induction of peroxisomal FAO; as expected, catalase activity was not changed in the HFD-fed rats compared with normal rats (Table 3). TDYA treatment significantly decreased ACOX1 activity and increased catalase activity in the rats fed a HFD (ACOX1 by 30%; catalase by 29%, p < 0.01). Furthermore, the catalase/ACOX1 ratio in the TDYA-treated group increased significantly compare with the HFD control group (Table 3, p < 0.01).
TABLE 3.
Hepatic ACOX1 and catalase activities in rats fed a ND, HFD, or HFD+TDYA for 8 weeks
| Enzymes | ND | HFD | HFD+TDYA |
|---|---|---|---|
| ACOX1 (nmol/min/mg protein) | 0.78 ± 0.18 | 1.24 ± 0.21a | 0.83 ± 0.16b |
| Catalase (μmol/min/mg protein) | 1.28 ± 0.13 | 1.39 ± 0.22 | 1.76 ± 0.29c |
| Catalase/ACOX1 (×103) | 1.66 ± 0.21 | 1.12 ± 0.15d | 2.16 ± 0.33b |
a ND vs. HFD, p < 0.01. n = 8 for each group.
b HFD vs. HF+TDYA, p < 0.01. n = 8 for each group.
c HFD vs. HF+TDYA, p < 0.05, n = 8 for each group.
d ND vs. HFD, p < 0.05, n = 8 for each group.
Effect of TDYA on NAD+/NADH Levels, the Expression Level of SIRT1, PGC-1α, and Phosphorylation Levels of AMPK, ACC, and p70S6K
TDYA treatment increased the liver NAD+/NADH ratio by 36% in the rats fed HFD compared with HFD control (Fig. 6A, p < 0.01)). Western blot analysis indicated that the expression of SIRT1 was increased in the TDYA-treated group compared with the HFD control group (p < 0.01), as shown in Fig. 6B. The phosphorylation levels of AMPK (Thr-172) and ACC (Ser-79) in the livers of TDYA-treated rats were also significantly higher than that in HFD-fed rats (by 82 and 105%, respectively; Fig. 6, C and D, p < 0.01). These results suggested the SIRT1-AMPK pathway could be activated by inhibiting ACOX1 activity through mediating cellular redox potential. p70S6K is a downstream target of AMPK through phosphorylation by mTOR, which will mediate the insulin signal pathway by phosphorylation and inhibition of IRS-1. Our Western blot analysis further showed that the phosphorylation level of p70S6K (Thr-389) in the livers of TDYA-treated rats decreased by 49% compared with the HFD group (Fig. 6E, p < 0.01). As expected, a high fat diet decreased liver PGC-1α expression compared with normal group (by 28%, p < 0.05); after treatment with TDYA, liver PGC-1α expression was enhanced significantly (by 83%, p < 0.01), as shown in Fig. 6F.
FIGURE 6.

A, effect of TDYA treatment on liver NAD+/NADH level in rats fed a high fat-diet. B, hepatic expression of SIRT1 in rats treated with normal diet, high fat diet, or HFD with TDYA for 8 weeks. C, effect of TDYA on the phosphorylation level of AMPK (Thr-172) in livers of the high fat diet-fed rats. D, effect of TDYA on the phosphorylation level of ACC (Ser-79) in livers of high fat diet-fed rats. E, effect of TDYA on the phosphorylation level of p70S6K (Thr-389) in the livers of the high fat diet-fed rats. F, effect of TDYA treatment on liver mRNA expression level of PGC-1α. n = 6–8 for each group. AU, arbitrary units. #, p < 0.05, ##, p < 0.01, **, p < 0.01.
Effect of TDYA on Liver Malonyl-CoA Levels, Gene Expression of CPT-1A, MCAD, L-bifunctional protein (L-BP), and 3,2-enoyl-CoA isomerase (ECI), Mitochondrial FAO, and Cytochrome c Oxidase (COX) Activity
HFD significantly increased liver malonyl-CoA content compared with normal diet group (by 225%, p < 0.01), TDYA treatment markedly reduced malonyl-CoA content in livers of the HFD-fed rats (by 60%, p < 0.01), as shown in Fig. 7A. CPT-1A, medium-chain acyl-CoA dehydrogenase (MCAD), L-BP, and mitochondrial ECI are key enzymes involved in mitochondrial and peroxisomal FAO that are marker genes under control of PPARα. TDYA treatment significantly increased the mRNA expression levels of CPT-1A, MCAD, L-BP, and ECI in the livers of the HFD-fed rats (by 63% for CPT-1A, 91% for MCAD, 136% for L-BP, and 123% for ECI, respectively, p < 0.01), as shown in Fig. 7, B–E, which suggested that PPARα was activated and its target genes were up-regulated by inhibition of ACOX1. Hepatic mitochondrial fatty acid oxidation was markedly increased in HFD-fed rats after treatment with TDYA (by 62%, p < 0.01, Fig. 7F). Cytochrome c oxidase is the rate-limiting enzyme involved in mitochondrial oxidative phosphorylation, which was also increased by 34% in rats fed HFD and TDYA (Fig. 7G, p < 0.01).
FIGURE 7.

A, effect of TDYA treatment on liver malonyl-CoA level in rats fed a high fat diet for 8 weeks. B, effect of TDYA treatment on liver mRNA expression level of CPT-1A. C, effect of TDYA treatment on liver mRNA expression level of MCAD. D, effect of TDYA treatment on liver mRNA expression level of L-BP. E, effect of TDYA treatment on liver mRNA expression level of mitochondrial ECI. F, effect of TDYA treatment on liver mitochondrial fatty acid oxidation. G, effect of TDYA treatment on liver cytochrome c oxidase activity. n = 6–8 for each group. AU, arbitrary units. #, p < 0.05, ##, p < 0.01, **, p < 0.01.
Discussion
The high fat diet caused repression of mitochondrial FAO and induction of peroxisomal FAO, which led to lipid accumulation in liver. We found that treatment of TDYA markedly attenuated high fat diet-induced hyperlipidemia and hepatic steatosis. Our histological data indicated that TDYA treatment reduced fat deposits in hepatocytes. Western blot data revealed that TDYA treatment effectively activated SIRT1-AMPK pathway via decreasing cellular redox potential as reflected by an increase in NAD+/NADH ratio. Furthermore, TDYA treatment alleviated HFD-induced hepatic oxidative stress. These findings supported that specific inhibition of ACOX1 could protect the liver by increasing mitochondrial FAO and reducing ROS levels to improve high fat diet-caused fatty liver and oxidative injury.
A study from the ACOX1−/− mice model indicated that the ob/ob mice lacking ACOX1 showed sustained activation of PPARα in liver and resistance to obesity with improved glucose tolerance and insulin sensitivity (20, 21). However, these ACOX−/− ob/ob mice developed hepatocellular carcinomas due to hepatic superimposed activation of PPARα and endoplasmic reticulum (ER) stress. Our animals with measured inhibition of ACOX1 are different from the knock-out model. First, inhibition of ACOX1 enhanced mitochondrial FAO by activation of the SIRT1-AMPK pathway. PPARα was also activated in our study, possibly via the SIRT1-PGC-1α pathway, but the extent of the activation was much more gentle than that in ACOX1−/− mice due to efficient metabolism of the endogenous ligands of PPARα, which increased liver FAO in a relatively mild way (∼2-fold induction in the target mRNA level). In ACOX−/− mice, PPARα is robust and progressively activated, and its downstream targets including ω-oxidation system genes are strongly induced (5–10-fold induction), which resulted in excess fatty acid oxidation, ROS formation, and cell proliferation (20, 21). Second, ACOX−/− mice showed strong ER stress, whereas in our study, ER stress is not induced, and cellular ROS level was reduced. In fact, pharmacological inhibition of ACOX1 has a potential anti-oxidative activity. Third, VLCFAs were accumulated in the liver and blood of the ACOX−/− mice due to the block of straight-chain and very long chain fatty acids metabolism, which may cause X-linked adrenoleukodystrophy and degenerative diseases, whereas in our study there were no significant changes of VLCFA levels in the liver and blood in the rats fed TDYA (data not shown), and the VLCFA can be effectively metabolized in peroxisomes because PPARα is activated by inhibition of ACOX1.
In another study mice deficiency of very long chain acyl-CoA dehydrogenase (VLCAD) were protected from HFD-induced obesity and insulin resistance due to chronic activation of AMPK, which resulted in increased fatty acid oxidation and decreased hepatic lipid content (22). Our results showed that specific inhibition of ACOX1 had a similar physiological phenotype to that of the VLCAD−/− mice. In both cases, hepatic FAO was increased and insulin resistance was improved via activation of the AMPK pathway, and PPARα was mildly activated. It should be noted that both VLCAD and ACOX1 shared similar substrate preference and catalyzed the same reaction.
Excessive fatty acid uptake due to HFD or obesity results in overload of mitochondrial fatty acid oxidation and induction of peroxisomal fatty acid oxidation. Several lines of evidence suggested that enhanced peroxisomal turnover of acyl-CoAs was potentially detrimental to mitochondrial FAO and caused oxidative stress; the proposed mechanism is shown in Fig. 8.
FIGURE 8.
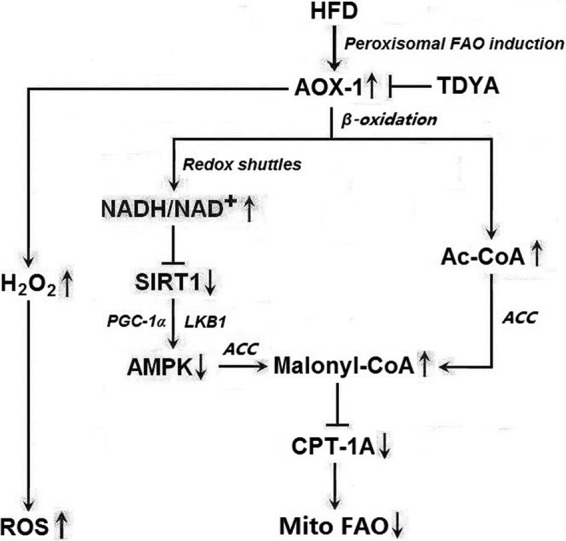
Proposed mechanism by which induction of ACOX1 causes repression of mitochondrial fatty acid oxidation and ROS accumulation.
Enhanced peroxisomal FAO leads to increased acetyl-CoA production in peroxisomes followed by increased transport of the acetyl group into the cytosol via redox shuttles, which will increase malonyl-CoA synthesis in the cytosol, thereby decreasing CPT1 activity and mitochondrial fatty acid oxidation.
Inhibition of ACOX reduces NADH generation by peroxisomal FAO, thereby decreasing the transport of the reducing equivalents from peroxisomes to the cytosol and decreasing the cytosolic NADH/NAD+ ratio. The decreased NADH/NAD+ ratio will increase the activity of SIRT1, a NAD+-dependent deacetylase, resulting in activation of AMPK and inhibition of ACC via increasing the deacetylation level to LKB1 and PGC-1α, thereby increasing mitochondrial FAO (23, 24). SIRT1 and AMPK are known as fuel-sensing molecules, and activation of the SIRT1/AMPK pathway plays a central role in regulating hepatic fatty acid metabolism. Decreased SIRT1 activity may also reduce the deacetylation of LKB1 and inhibit this kinase, which in turn inhibits AMPK and its downstream targets (24, 25). The activated form of AMPK is responsible for metabolic changes via phosphorylation of various downstream substrates; it promotes mitochondrial FAO through phosphorylation of ACC and mitochondrial biogenesis via increasing the transcription activity of PGC-1α and expression of the proteins vital for mitochondrial function such as citrate synthase and cytochrome c oxidase.
A number of physiological processes have been shown to be able to activate AMPK, including conditions that lead to increases of the intracellular AMP/ATP ratio (e.g. hypoxia, glucose deprivation) and calcium concentration as well as the action of various hormones, cytokines (26, 27). Our results suggested that the SIRT1-AMPK pathway could be activated by inhibition of ACOX1, the first enzyme in peroxisomal FAO, via increasing cytosolic NAD+ levels and the expression of SIRT1.
Elevated substrate flux through ACOX1 due to HFD results in increased hydrogen peroxide generation in peroxisomes; however, hydrogen peroxide turnover is not elevated because the activity of catalase is not elevated accordingly, which leads to excessive hydrogen peroxide accumulation in peroxisomes and increased diffusion of hydrogen peroxide from peroxisomes to the cytosol and mitochondria, which will disturb the mitochondrial tricarboxylic acid cycle and impair respiratory chain in liver and possible heart, leading to enhanced oxidative stress and consequently activated MAPK and NF-κB pathways, causing hepatic oxidative damage.
It was reported that ACOX1 overexpression in Cos-1 cells activated NF-κB-regulated reporter gene in the presence of ACOX1 substrates. The antioxidants vitamin E and catalase inhibited this activation (28). Conversely, specific inhibition of catalase resulted in peroxisome-derived oxidative imbalance, which rapidly impaired mitochondrial function and caused mitochondrial damage (29). In another study, liver-specific catalase expression in transgenic mice inhibits NF-κB activation induced by the peroxisome proliferator ciprofibrate; active oxygen plays a critical role in the induction of cell proliferation by the peroxisome proliferator and, therefore, may be important in the carcinogenicity of these agent (30).
Our study revealed that a high fat diet led to induction of peroxisomal FAO (ACOX1) and decreased catalase activity, resulting in increased hepatic ROS levels. Inhibition of ACOX1 by TDYA significantly increased the catalase/ACOX1 ratio, thereby attenuating lipid-induced ROS accumulation in the livers of the HFD-fed rats, which may be benefit to liver against mitochondrial dysfunction and oxidative damage.
It is widely accepted that lipid and ROS are two critical factors that cause insulin resistance via activation of a series of protein kinase and inhibition of the insulin signal pathway (31, 32). After 8 weeks of HFD feeding, liver insulin resistance was fully developed in rats due to hepatic lipid and ROS accumulation, as shown in Figs. 4 and 5, and serum insulin levels will be elevated as a compensatory mechanism; our data indicated that TDYA treatment decreased fasting blood glucose and alleviated hyperinsulinemia in HFD rats. The increased hepatic fatty acid oxidation and reduced ROS formation were proposed to be responsible for the increased insulin sensitivity in these TDYA-treated HFD rats.
Polyacetylenic acids were reported to have a broad range of biological activities, including inhibition of prostaglandin biosynthesis (33), antitumor (34), anti-HIV reverse transcriptase (35), antifungal and antibacterial (34), and pesticidal (36). In this study a conjugated acetylenic acid TDYA reduced liver lipids and ROS accumulation and improved insulin resistance in HFD-fed rats via specific inhibition to ACOX1. Therefore, conjugated acetylenic acids, whether in natural or synthetic sources with ACOX1 inhibition potential, may be promising compounds for the treatment of metabolic diseases such as fatty liver and hypertriglyceridemia and possibly diabetes.
In conclusion, specific inhibition of ACOX1 by 10,12-tricosadiynoic acid increased hepatic mitochondrial FAO via activation of the SIRT1-AMPK pathway and PPARα and reduced hepatic hydrogen peroxide formation in HFD-fed rats, which significantly decreased hepatic lipid and ROS contents, reduced body weight gain, and decreased serum triglyceride and insulin levels. Therefore, inhibition of ACOX1 will be a novel and effective approach for the treatment of HFD or obesity-induced metabolic diseases by improving mitochondrial lipid and ROS metabolism and increasing hepatic insulin sensitivity.
Experimental Procedures
Materials
10,12-Tricosadiynoic acid and palmitoyl-CoA were purchased from Sigma, coenzyme A sodium salt was purchased from USB (Cleveland, OH). Standard rodent diet and high fat diet containing 58% of calories as fat were purchased from Shanghai Slac Laboratory Animal Co. Ltd (Shanghai, China). All the chemical reagents used were of analytical grade or better.
Animals
Male Wistar rats weighing 200–220 g were obtained from Shanghai Slac Laboratory Animal Co. Ltd. Use of the rats was approved by the Hunan University of Science and Technology Animal Care Committee.
Preparation of TDYA-CoA and 3-Indolepropionyl-CoA
Preparation of TDYA-CoA and 3-indolepropionyl-CoA was performed according to the methods as described previously (37). Crude TDYA-CoA and 3-indolepropionyl-CoA after the coupling reactions were further isolated by reversed phase high performance liquid chromatography (RP-HPLC) followed by freeze-drying to remove solvent to obtain the powder of TDYA-CoA and 3-indolepropionyl-CoA; the purity of the purified TDYA-CoA and 3-indolepropionyl-CoA was >90%.
Isolation of Mitochondria and Peroxisomes Fraction from Liver Homogenate
Isolation of liver mitochondria and peroxisome fractions by differential centrifugation in 0.25 m sucrose was performed as described by de Duve et al. (38).
In Vitro Inhibition
Recombinant rat liver ACOX1 was expressed in Escherichia coli and purified by affinity chromatography with purity >95% according to Zeng and Li (39). Purified ACOX1 was dissolved in 20 mm PBS buffer (pH 7.4) at a concentration of 80 nm, then varied concentrations of TDYA-CoA were added into the enzyme solution, and equal volumes of distilled water were added as the control group. After incubation at 25 °C for different times, residual activities for all the groups were determined. ACOX1 activity was assayed by measuring absorbance change at 263 nm with a Shimadzu UV-1800 UV-visible spectrophotometer (40). The reaction mixture contained 50 mm PBS buffer (pH 7.4), 30 μm palmitoyl-CoA, and 80 nm ACOX1 with a total volume of 500 μl. The inactivation kinetic parameters KI and kinact were used to characterize the inhibition kinetics according to previous report (37).
In Vivo Inhibition
After fasting for 12 h, male Wistar rats weighing 220–250 g were divided into a control group and inhibitor groups, 6 rats for each group. For the control group, olive oil was treated by intragastric administration at 0.2 ml/rat, and for the inhibitor groups, TDYA was dissolved in olive oil and then administered at a dose of 20–160 μg/kg. Five hours later, all the rats were sacrificed, and livers were removed rapidly and stored in liquid nitrogen immediately. Then liver ACOX1 activity for each sample was assayed.
Tissue ACOX1 Activity Assay
The ACOX1 activity in tissue extract was measured directly by following the increase in absorbance at 367 nm due to the formation of reaction products trans-3-indoleacryloyl-CoA (ϵ367 nm = 26.5 mm−1 cm−1) from the substrate 3-indolepropionyl-CoA (40). The enzyme was routinely assayed in a 500-μl reaction volume on a Shimadzu UV-1800 UV-visible spectrophotometer containing 50 mm phosphate buffer (pH 7.4), 200 μg of isolated peroxisomes, 30 μmol/liter FAD (Sigma), and 0.02% Triton X-100. The reaction was started by adding 50 μm 3-indolepropionyl-CoA into the reaction mixture.
Animal Experiment
24 male Wistar rats with body weights of 210–230 g were divided into 3 groups, namely normal a control group, a HFD control group, and a HFD+TDYA group, 8 rats for each group. A normal diet group (ND) was fed a regular diet (12% fat by calories), and the HFD groups were fed a high fat diet (58% fat by calories). For ND and HFD control groups, the rats were treated with 0.2 ml/rat olive oil by gavage; for the HFD+TDYA group, the rats were administered TDYA at a dose of 100 μg/kg after dissolving it in appropriate volume of olive oil. The rats were treated once per day and continued treating for 8 weeks. Body weights were measured every 2 weeks. On day 57, fasting blood glucose was measured via a glucometer from tail vein after 6 h of fasting (6 a.m.-12 p.m.). Then all the rats were bled from the eyes for preparation of serum, and then stored in a −80 °C freezer. All the rats were then killed, and livers were removed and weighed, then stored in a −80 °C freezer.
Hepatic TG Analysis
0.5 g of liver sample was homogenate in 0.9% NaCl, then 1 ml of homogenate was mixed with 3 ml of chloroform/methanol (2:1, v/v). The chloroform layer was collected and concentrated. After mixing with 10% Triton X-100 in isopropyl alcohol, the sample was assayed by using Wako Triglyceride E-Test kit (Wako Pure Chemical).
Histological Analysis
Liver tissues of the killed rats were cut quickly from a definite part of right lobe of the livers, and immediately fixed with 4% paraformaldehyde for 24 h. Then the paraffin sections were prepared and cut into 5–7-μm-thick sections and stained by hematoxylin-eosin (HE), hepatic steatosis was observed by optical microscope, and 4 samples were used for each group to observe the lipid droplets in liver tissues.
Quantitative Real Time PCR
Cells were harvested, and total RNA was extracted with TRIzol reagent (Invitrogen). Quantitative real-time PCR was performed in a 7500 Fast Real-time PCR System (Applied Biosystems), and SYBR Green was used as the fluorescent dye. Real time PCR was performed in triplicate with 10-fold diluted first-strand cDNAs in a final volume of 20 μl containing 2×SYBR Green Supermix (Applied Biosystems) and 200 nm or 400 nm of both primers. Results were normalized to the housekeeping gene 18S rRNA to determine the target gene expression. The following primers were used: PGC-1α, 5′-CAGCAAAAGCCACAAAGACG-3′ (forward (F)) and 5′-AGTTCCAGAGAGTTCCACAC-3′ (reverse (R)); CPT-1A, 5′-GAGAAGGGAGGACAGAGACT-3′ (F) and 5′-GCTCAGACAATACCTCCTTC-3′ (R); MCAD, 5′-AAACCATCTCTCAAGCAGGA-3′ (F) and 5′-ATTTTTATACTTTTCAATGTGCTC-3′(R); L-BP, 5′-AAATACAGAGATACCAGAAGCCG-3′ (F) and 5′-AAGAATCCCCAGTGTGACTTC-3′(R); ECI, 5′-GACTACAGGATAATGGCGGAC-3′ (F) and 5′-AAGCGTCCCCAGTTGAAG-3′(R); 18S rRNA, 5′-GTTATGGTCTTTGGTCGC-3′ (F) and 5′-CGTCTGCCCTATCAACTTTC-3′ (R). PCR reaction mixture was denatured at 95 °C for 10 min before the first PCR cycle. The Thermal Cycling program was 1) denaturation for 15 s at 95 °C, 2) annealing for 30 s at 60 °C, and 3) extension for 30 s at 72 °C. A total of 40 PCR cycles was used. Data were collected at the DNA extension step. Relative expression of mRNA levels was calculated using the comparative ΔCT method.
Western Blot Analysis
Liver tissue was homogenized in radioimmune precipitation assay buffer containing the protease inhibitor mixture and phosphatase inhibitor (PhosSTOP) (Roche Applied Science). The homogenate was further mixed with buffer containing 50 mm Tris-HCl, 2% SDS, and 2% β-mercaptoethanol and boiled for 5 min. 50 μg of sample was applied to 10% SDS-polyacrylamide gel electrophoresis and then transferred to a PVDF membrane (Millipore, Bedford, MA). After blocking with 5% BSA at room temperature for 2 h, the membrane was incubated with anti-SIRT1, anti-AMPKα, anti-phosphorylated AMPKα (Thr-172), anti-phosphorylated ACC (Ser-79), anti-p70S6K, anti-phosphorylated p70S6K (Thr-389) (Cell Signaling, Danvers, MA), and β-actin (Sigma) monoclonal antibodies at 4 °C overnight followed by reactions with horseradish peroxidase-conjugated antibody for 3 h at room temperature. The detected bands were quantified by an image analyzer (Bio-Rad), and β-actin was used as the internal control. The blots were quantified by densitometric analysis, and the results were normalized to β-actin and given as arbitrary units.
NAD+/NADH Assay
Liver NAD+/NADH ratio was measured using a NAD+/NADH quantitation kit (Sigma) according to the manufacturer's instruction.
Biochemical Analysis
Protein concentration was determined with Bio-Rad DC protein assay kit. Serum triglyceride and total cholesterol were measured using commercial assay kits (Wako Pure Chemical). Serum non-esterified fatty acid (NEFA) was measured by a NEFA assay kit (Nanjing Jiancheng Biotech., Nanjing, China). Serum insulin was measured by Insulin Elisa kit (Millipore). Hepatic hydrogen peroxide levels were measured by a hydrogen peroxide testing kit (Beyotime Biotech., Haimen, China). Hepatic lipoperoxide level was tested using a MDA testing kit (Nanjing Jiancheng Biotech.). Hepatic catalase was assayed by a colormetric method using a catalase testing kit (Nanjing Jiancheng Biotech.). Mitochondrial FAO rate was determined in intact isolated mitochondria from the 14C-labeled acid-soluble metabolites in a sealed system using [1-14C]palmitate (GE Healthcare) as the substrate according to the method described previously (22).
Statistical Analysis
Data are presented as the means ± S.E. The significance of the differences in mean values was evaluated using Student's test by SPSS 18.0. p < 0.05 was considered statistically significant.
Author Contributions
J. Z. conceived the idea for the project, conducted most of the experiments, analyzed the results, and wrote most of the paper. S. D. conducted assays of enzyme activity and wrote the paper with J. Z. Y. W. and P. L. conducted the animal experiments. L. T. and Y. P. conducted real time PCR.
This work was supported by the National Natural Science Fund of China (30900024) and the Doctoral Program of Ministry of Education of China (200805331034). The authors declare that they have no conflicts of interest with the contents of this article.
- ROS
- reactive oxygen species
- AMPK
- adenosine 5′-monophosphate-activated protein kinase
- ACOX1
- acyl-CoA oxidase-1
- ECI
- 3,2-enoyl-CoA isomerase
- FAO
- fatty acid oxidation
- HFD
- high fat diet
- L-BP
- L-bifunctional protein
- MCAD
- medium-chain acyl-CoA dehydrogenase
- MDA
- malondialdehyde
- PPARα
- peroxisome proliferator activator receptor α isoform
- TDYA
- 10,12-tricosadiynoic acid
- VLACS
- very long chain acyl-CoA synthetase
- ER
- endoplasmic reticulum
- VLCAD
- very long chain acyl-CoA dehydrogenase
- ND
- normal diet
- ACC
- acetyl-CoA carboxylase.
References
- 1. Perry R. J., Samuel V. T., Petersen K. F., and Shulman G. I. (2014) The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510, 84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rani V., Deep G., Singh R. K., Palle K., and Yadav U. C. (2016) Oxidative stress and metabolic disorders: pathogenesis and therapeutic strategies. Life Sci. 148, 183–193 [DOI] [PubMed] [Google Scholar]
- 3. Nsiah-Sefaa A., and McKenzie M. (2016) Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 36, e00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marí M., Colell A., Morales A., von Montfort C., Garcia-Ruiz C., and Fernández-Checa J. C. (2010) Redox control of liver function in health and disease. Antioxid. Redox. Signal. 12, 1295–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reddy J. K., and Hashimoto T. (2001) Peroxisomal β-oxidation and peroxisome proliferator-activated receptor α: an adaptive metabolic system. Annu. Rev. Nutr. 21, 193–230 [DOI] [PubMed] [Google Scholar]
- 6. Neat C. E., Thomassen M. S., and Osmundsen H. (1980) Induction of peroxisomal β-oxidation in rat liver by high-fat diets. Biochem. J. 186, 369–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomassen M. S., Christiansen E. N., and Norum K. R. (1982) Characterization of the stimulatory effect of high-fat diets on peroxisomal β-oxidation in rat liver. Biochem. J. 206, 195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas H., Schladt L., Knehr M., and Oesch F. (1989) Effect of diabetes and starvation on the activity of rat liver epoxide hydrolases, glutathione S-transferases, and peroxisomal β-oxidation. Biochem. Pharmacol. 38, 4291–4297 [DOI] [PubMed] [Google Scholar]
- 9. Lazarow P. B., and De Duve C. (1976) A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. U.S.A. 73, 2043–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turecký L., Kupčová V., Uhlíková E., and Mojto V. (2014) Peroxisomal enzymes in the liver of rats with experimental diabetes mellitus type 2. Physiol. Res. 63, S585–S591 [DOI] [PubMed] [Google Scholar]
- 11. Goel S. K., Lalwani N. D., and Reddy J. K. (1986) Peroxisome proliferation and lipid peroxidation in rat liver. Cancer Res. 46, 1324–1330 [PubMed] [Google Scholar]
- 12. Fransen M., Nordgren M., Wang B., and Apanasets O. (2012) Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim. Biophys. Acta 1822, 1363–1373 [DOI] [PubMed] [Google Scholar]
- 13. Reszko A. E., Kasumov T., David F., Jobbins K. A., Thomas K. R., Hoppel C. L., Brunengraber H., and Des Rosiers C. (2004) Peroxisomal fatty acid oxidation is a substantial source of the acetyl moiety of malonyl-CoA in rat heart. J. Biol. Chem. 279, 19574–19579 [DOI] [PubMed] [Google Scholar]
- 14. Kasumov T., Adams J. E., Bian F., David F., Thomas K. R., Jobbins K. A., Minkler P. E., Hoppel C. L., and Brunengraber H. (2005) Probing peroxisomal β-oxidation and the labelling of acetyl-CoA proxies with [1-13C]octanoate and [3-13C]octanoate in the perfused rat liver. Biochem. J. 389, 397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rognstad R. (1991) Estimation of peroxisomal and mitochondrial fatty acid oxidation in rat hepatocytes using tritiated substrates. Biochem. J. 279, 147–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pardridge W. M., Casanello-Ertl D., and Duducgian-Vartavarian L. (1980) Branched chain amino acid oxidation in cultured rat skeletal muscle cells: selective inhibition by clofibric acid. J. Clin. Invest. 66, 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shin M., Iwamoto N., Yamashita M., Sano K., and Umezawa C (1998) Pyridine nucleotide levels in liver of rats fed clofibrate or pyrazinamide-containing diets. Biochem. Pharmacol. 55, 367–371 [DOI] [PubMed] [Google Scholar]
- 18. Zeng J., Wu L., Zhang X., Liu Y., Deng G., and Li D. (2008) Oct-2-en-4-ynoyl-CoA as a specific inhibitor of acyl-CoA oxidase. Org. Lett. 10, 4287–4290 [DOI] [PubMed] [Google Scholar]
- 19. Zeng J., Deng G., and Li D. (2006) Intrinsicenoyl-CoA isomerase activity of rat acyl-CoA oxidase I. Biochim. Biophys. Acta 1760, 78–85 [DOI] [PubMed] [Google Scholar]
- 20. Huang J., Jia Y., Fu T., Viswakarma N., Bai L., Rao M. S., Zhu Y., Borensztajn J., and Reddy J. K. (2012) Sustained activation of PPARα by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J. 26, 628–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang J., Viswakarma N., Yu S, Jia Y., Bai L., Vluggens A., Cherkaoui-Malki M., Khan M., Singh I., Yang G., Rao M. S., Borensztajn J., Reddy J. K. (2011) Progressive endoplasmic reticulum stress contributes to hepatocarcinogenesis in fatty acyl-CoA oxidase 1-deficient mice. Am. J. Pathol. 179, 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang D., Christianson J., Liu Z. X., Tian L., Choi C. S., Neschen S., Dong J., Wood P. A., and Shulman G. I. (2010) Resistance to high-fat diet-induced obesity and insulin resistance in mice with very long-chain acyl-CoA dehydrogenase deficiency. Cell Metab. 11, 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hou X., Xu S., Maitland-Toolan K. A., Sato K., Jiang B., Ido Y., Lan F., Walsh K., Wierzbicki M., Verbeuren T. J., Cohen R. A., and Zang M. (2008) SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 283, 20015–20026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lan F., Cacicedo J. M., Ruderman N., and Ido Y. (2008) SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1 possible role in AMP-activated protein kinase activation. J. Biol. Chem. 283, 27628–27635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee Y. H., Chen H. Y., Su L. J., and Chueh P. J. (2015) Sirtuin 1 (SIRT1) deacetylase activity and NAD+/NADH ratio are imperative for capsaicin-mediated programmed cell death. J. Agric. Food Chem. 63, 7361–7370 [DOI] [PubMed] [Google Scholar]
- 26. Steinberg G. R., and Kemp B. E. (2009) AMPK in health and disease. Physiol. Rev. 89, 1025–1078 [DOI] [PubMed] [Google Scholar]
- 27. Zhang B. B., Zhou G., and Li C. (2009) AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 9, 407–416 [DOI] [PubMed] [Google Scholar]
- 28. Li Y., Tharappel J. C., Cooper S., Glenn M., Glauert H. P., and Spear B. T. (2000) Expression of the hydrogen peroxide-generating enzyme fatty acyl CoA oxidase activates NF-κB. DNA Cell Biol. 19, 113–120 [DOI] [PubMed] [Google Scholar]
- 29. Nilakantan V., Spear B. T., and Glauert H. P. (1998) liver-specific catalase expression in transgenic mice inhibits NF-κB activation and DNA synthesis induced by the peroxisome proliferator ciprofibrate. Carcinogenesis 19, 631–637 [DOI] [PubMed] [Google Scholar]
- 30. Walton P. A., and Pizzitelli M. (2012) Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 3, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Samuel V. T., Petersen K. F., and Shulman G. I. (2010) Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsuda M., and Shimomura I. (2013) Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 7, e330-e341 [DOI] [PubMed] [Google Scholar]
- 33. Nugteren D. H., and Christ-Hazelhof E. (1987) Naturally occurring conjugated octadecatrienoic acids are strong inhibitors of prostaglandin biosynthesis. Prostaglandins 33, 403–417 [DOI] [PubMed] [Google Scholar]
- 34. Kuklev D. V., Domb A. J., and Dembitsky V. M. (2013), Bioactive acetylenic metabolites. Phytomedicine 20, 1145–1159 [DOI] [PubMed] [Google Scholar]
- 35. Ishiyama H., Ishibashi M., Ogawa A., Yoshida S., et al. (1997) Taurospongin A. A novel acetylenic fatty acid derivative inhibiting DNA polymerase B and HIV reverse transcriptase from Sponge Hippospongia sp. J. Org. Chem. 62, 3831–3836 [Google Scholar]
- 36. Fatope M. O., Adoum O. A., and Takeda Y. (2000) C18 acetylenic fatty acids of Ximenia americana with potential pesticidal activity. J. Agric. Food Chem. 48, 1872–1874 [DOI] [PubMed] [Google Scholar]
- 37. Li D., Agnihotri G., Dakoji S., Oh E., Lantz M., and Liu H. W. (2001) The toxicity of methylenecyclopropylglycine: studies of the inhibitory effects of (methylenecyclopropyl) formyl-CoA on enzymes involved in fatty acid metabolism and the molecular basis of its inactivation of enoyl-CoA hydratases. J. Am. Chem. Soc. 121, 9034–9042 [Google Scholar]
- 38. De Duve C., Pressman B., Gianetto R., Wattiaux R., and Appelmans F. (1955) Tissue fractionation studies 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zeng J., and Li D. (2004) Expression and purification of his-tagged rat peroxisomal acyl-CoA oxidase I wild-type and E421 mutant proteins. Protein Expr. Purif. 38, 153–160 [DOI] [PubMed] [Google Scholar]
- 40. Luo Y. S., Wang H. J., Gopalan K. V., Srivastava D. K., Nicaud J. M., and Chardot T. (2000) Purification and characterization of the recombinant form of Acyl CoA oxidase 3 from the yeast Yarrowia lipolytica. Arch. Biochem. Biophys. 384, 1–8 [DOI] [PubMed] [Google Scholar]