

Abstract
The antibody Fc region regulates antibody cytotoxic activities and serum half-life. In a therapeutic context, however, the cytotoxic effector function of an antibody is often not desirable and can create safety liabilities by activating native host immune defenses against cells expressing the receptor antigens. Several amino acid changes in the Fc region have been reported to silence or reduce the effector function of antibodies. These earlier studies focused primarily on the interaction of human antibodies with human Fc-γ receptors, and it remains largely unknown how such changes to Fc might translate to the context of a murine antibody. We demonstrate that the commonly used N297G (NG) and D265A, N297G (DANG) variants that are efficacious in attenuating effector function in primates retain potent complement activation capacity in mice, leading to safety liabilities in murine studies. In contrast, we found an L234A, L235A, P329G (LALA-PG) variant that eliminates complement binding and fixation as well as Fc-γ-dependent, antibody-dependent, cell-mediated cytotoxity in both murine IgG2a and human IgG1. These LALA-PG substitutions allow a more accurate translation of results generated with an “effectorless” antibody between mice and primates. Further, we show that both human and murine antibodies containing the LALA-PG variant have typical pharmacokinetics in rodents and retain thermostability, enabling efficient knobs-into-holes bispecific antibody production and a robust path to generating highly effector-attenuated bispecific antibodies for preclinical studies.
Keywords: antibody engineering; complement; Fc receptor; Fc-γ receptor; protein stability; recombinant protein expression; aglycosylation; antibody-dependent, cell-mediated cytotoxicity (ADCC); bispecific antibody; complement-dependent cytotoxicity (CDC)
Introduction
Antibodies have a large and growing role as protein therapeutics, with over 50 approved molecules at the end of 2014 (1, 2). As the range of therapeutic applications for antibodies expands, the complexity of antibody engineering approaches has increased beyond traditional monospecific antibodies. This is reflected in the rapid expansion of clinical trials with bispecific antibodies in recent years (3, 4). Additionally, many therapeutic approaches require modifications to the Fc region to silence antibody effector function or to extend antibody serum half-life. As these novel antibody formats and altered Fc properties are often combined, there is an increasing need to identify compatible amino acid changes in the antibody constant region. Furthermore, these changes in Fc need to translate between preclinical animal models and humans (5, 6).
The challenges in translating antibody properties from animals to humans have long been recognized and partially result from the protein sequence differences between human and murine antibodies (7). The most common human IgG isotype used in therapeutic development is IgG1 (1), which is highly abundant in human serum. IgG1 antibodies have a high level of native cytotoxic function and a long half-life and can be efficiently produced with recombinant technologies. In preclinical animal studies, particularly those conducted in mice with intact immune systems, species-specific antibodies are often used to avoid immunogenicity, and a murine isotype with Fc properties corresponding to the human antibody is selected. Murine IgG2a has high functional similarity to human IgG1 in terms of pharmacokinetics (PK)5 and Fc-mediated effector function and is therefore often chosen as a surrogate antibody for murine studies. In addition, the murine IgG2a isotype is convenient for PK assays, as this isotype is not expressed in the common laboratory mouse strain c57BL/6J in the absence of specific antigen stimulation (8). The cytotoxic potential of an antibody is primarily mediated through the Fc region of the antibody, either through the complement-dependent cytotoxicity (CDC) or antibody-dependent, cell-mediated cytotoxicity (ADCC) pathways.
The CDC response involves activation of a biochemical cascade through which protein components of serum directly attack a pathogen (9). The classical CDC pathway is initiated by binding of the C1q complex to the antibody-antigen complex, which ultimately leads to C3 fixation and membrane attachment. Several components of the complement pathway can also directly recruit macrophages to opsonize cells in a process known as antibody-dependent cellular phagocytosis (ADCP) (10).
Activation and recruitment of lymphocytes during ADCC is mediated by binding of the antibody Fc region to Fc-γ receptors. The human Fc-γ receptor family consists of Fc-γ receptors I (CD64), IIa/b/c (CD32a/b/c), and RIIIa/b (CD16a/b). They are expressed on numerous cell types such as myeloid lineage cells, including macrophages. Fc-γ receptor I, IIa/c, and IIIa are activating receptors, and Fc-γ receptor IIb is an inhibitory receptor. Fc-γ receptor IIIb is unique among the human Fc-γ receptors because it does not have a transmembrane domain and is expressed on the surface of neutrophils by a glycophosphatidylinositol anchor. The mouse Fc-γ receptor family consists of Fc-γ receptors I, IIb, III, and IV. Only Fc-γ receptor IIb is an inhibitory receptor. Fc-γRIV selectively binds the IgG2a/b antibodies (11–13). The most prevalent Fc-γ receptors on natural killer cells are Fc-γRIIIa in humans, although a subset of human natural killer cells express Fc-γRIIc (14), or RIII in mice (13, 15). With the exception of Fc-γRI, binding to all Fc-γ receptors is strongly enhanced by avidity effects following immune complex formation.
A number of strategies have been developed to reduce or eliminate antibody cytotoxicity (16). One effective strategy in human antibodies is the elimination of the N-linked glycosylation at residue Asn-297, achieved by substituting Asn-297 with an alanine, glycine, or aspartic acid (17, 18), or modification of the serine/threonine residue at position 299 (19). The elimination of glycosylation results in reduced antibody binding to C1q and Fc-γ receptors via allosteric changes in the antibody CH2 domain. Although this strategy is effective at reducing both CDC and ADCC activity of human antibodies, the reduction in effector function is much less complete in mice. Aglycosylation and the associated allosteric changes also have the considerable disadvantage of reducing antibody stability. To further reduce the effector function of aglycosylated murine antibodies, changes that impair glycosylation at Asn-297 are often combined with the D265A mutation (20). The two most commonly used pairs of amino acid changes at these positions are designated D265A, N297A or D265A, N297G (DANG). Although the DANG substitutions are intended to eliminate effector functions of human and murine antibodies in mice, it has been shown recently that anti-transferrin receptor (TfR) bispecific antibodies containing DANG or NG substitutions can still lead to a potentially toxic reduction of TfR-expressing reticulocytes in mice via ADCP or CDC activity (21).
Alternative strategies have also been employed to mitigate antibody effector function, including substitutions of residues in the antibody lower hinge such as L234A and L235A (LALA) (22). These residues form part of the Fc-γ receptor binding site on the CH2 domain (23), and the exchange of these residues between antibody isotypes with greater or lesser effector function identified their importance in ADCC. Although alanine substitutions at these sites are effective in reducing ADCC in both human and murine antibodies (24–26), these substitutions are less effective at reducing CDC activity. Another single variant, P329A, identified by a random mutagenesis approach to map the C1q binding site of the Fc, was shown to be highly effective at reducing CDC activity while retaining ADCC activity (27). The location of these amino acid changes on the Fc lower hinge, together with the interaction site of Fc-γRIII, is shown in Fig. 1 (23).
FIGURE 1.

Illustration of the location of the effector reduction substitutions on the antibody Fc lower hinge region. A, structure of the antibody Fc-γRIII receptor complex (PDB code 1E4K). The two chains of the antibody Fc are shown in teal and green. The Fc-γ receptor is shown in pink, and residues on the Fc within 5 Å are colored gray in the surface rendering. The location of the Asp-265 and Asn-297 variants (DANG) that eliminate glycosylation and reduce effector functions are indicated in red. The locations of the Leu-234, Leu-235, and Pro-329 positions are highlighted in yellow. B, alignment of relevant regions of the hinge (gray fill) and CH2 domain (cyan fill) of murine IgG2a and human IgG. The naturally effector-attenuated human IgG4 sequence is also included for reference. The positions of the LALA-PG and DANG variants are indicated by yellow and red, respectively. The numbering of the CH2 residues for human IgG1 is according to Eu convention.
Bispecific antibodies have increasing importance in research and clinical development, as they provide the ability to target two specific antigens using a single antibody molecule. These bispecific antibodies also provide the opportunity to expand the function of a monospecific antibody; e.g. one binding moiety can be leveraged to deliver the antibody to a specific tissue or organ, whereas the second binding moiety is directed at the target. One such example is the recently described anti-TfR/BACE1 bispecific antibody, designed to efficiently deliver an anti-β secretase (BACE1) antibody to the brain via TfR-mediated transcytosis (21, 28, 29). This particular application requires a bispecific molecule that is devoid of effector function because of toxicities associated with effector function-competent antibodies binding to TfR expressed on red blood cell progenitors (21). Several antibody engineering solutions were therefore explored to create bispecific antibodies that might address the safety liabilities mechanistically associated with TfR binding-related, Fc-γ-mediated ADCC and complement-mediated ADCP activities that were detectable even at low doses of antibody (3). Evaluations of effector-attenuation strategies in this study were based on this highly sensitive in vivo readout related to the fate of precursor red blood cells following anti-TfR antibody administration (21) within the context of the knobs-into-holes bispecific technology (30, 31). In addition, this work highlights a specific need for new methods that can significantly attenuate antibody effector function for murine bispecific antibodies without introducing undesirable production liabilities.
To identify effector silencing substitutions that could both improve the production of murine IgG2a-bispecific antibodies and effectively reduce effector function, we screened several antibody variants, including the recently identified combination of L234A, L235A, and P329A (LALA-PG) substitutions that have been shown to effectively silence the effector function of human IgG1 antibodies (32). In contrast to the allosteric effects of aglycosylation, the LALA-PG substitutions directly block the interaction of the Fc with the Fc-γ receptors and C1q. In addition to being more effective at reducing effector function than the allosteric mechanism of aglycosylation, they maintain antibody stability. In this work, we show that introduction of the LALA-PG substitutions into murine IgG2a antibodies vastly improves the expression and assembly of the knobs-into-holes half-antibodies, and the LALA-PG variant retains the thermal stability of the respective wild-type murine IgG2a and human IgG1 antibodies in contrast to the aglycosylated DANG and NG variants. In vitro assays demonstrate that the LALA-PG substitution in a murine IgG2a antibody highly attenuates Fc-γ receptor binding as well as complement fixation relative to both aglycosylated NG and DANG antibodies. Furthermore, our murine studies demonstrate that LALA-PG effector silencing translates to an in vivo setting and improves the safety profile of a TfR-targeting bispecific antibody. With the use of the murine IgG2a LALA-PG antibody variant, we have established a murine surrogate system to mimic the effector function of the LALA, NG, and LALA-PG versions of human IgG1 antibodies in the clinic.
Results
The LALA-PG Variant Retains a Similar Expression Level and Stability as Wild-type Antibodies
The production of knob-and-hole half-antibodies is the first step in the production of a bispecific antibody (21, 29, 33). After purification, the half-antibodies are assembled into the intact bispecific antibody. For an efficient production process, a high expression level of each half-antibody and efficient assembly of the two half-antibodies is necessary. Typical recovery of knob or hole murine IgG2a half-antibodies is 25–50 mg/liter in CHO. Unexpectedly, half-antibody expression is greatly attenuated in context with the DANG variant (Fig. 2A). We hypothesize that the impact on expression level could be a consequence of protein instability resulting from aglycosylation. To determine whether effectorless variants that retain the N-linked glycosylation at Asn-297 show improved expression over the DANG antibodies, we tested several previously identified variants that silence effector function. The expression of murine IgG2a half-antibody with LALA substitutions as well as in combination with either the D265A substitution (LALA-DA) or LALA-PG were tested. All variants express at levels at least comparable with the WT half-antibodies, with the exception of the LALA-DA, Hole half-antibody variant (Fig. 2A).
FIGURE 2.
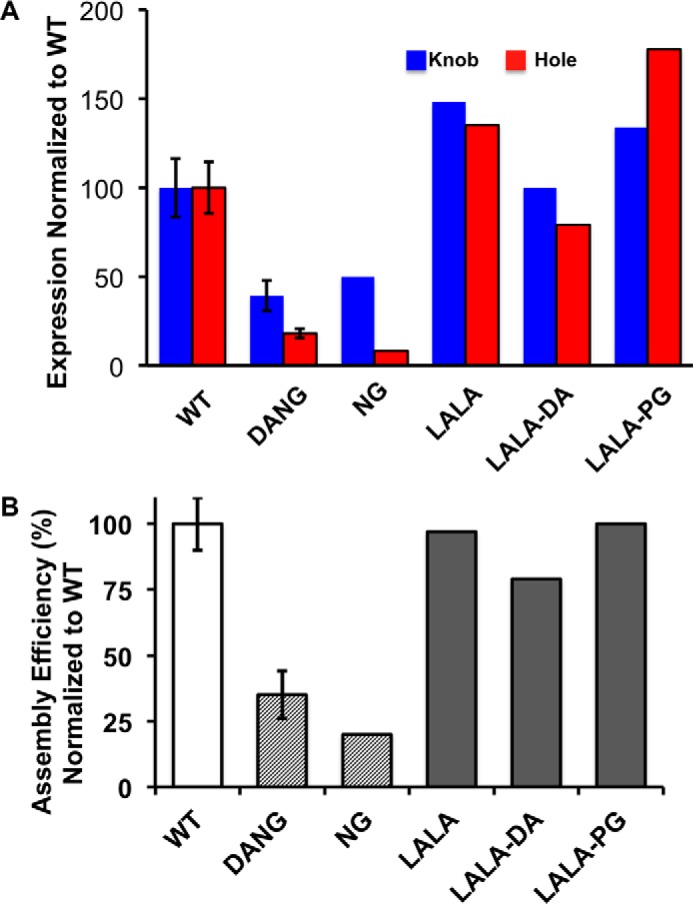
LALA variants improve half-antibody expression and bispecific antibody assembly compared with NG variants. A, knob (blue) and hole (red) murine IgG2a half-antibodies were expressed in CHO, followed by MabSelect SuRe affinity purification. Recoveries were normalized to wild-type knob and hole half-antibodies. A significant impact on expression is observed for the aglycosylated NG and DANG variants. All glycosylated variants, including LALA and LALA-PG, show expression profiles similar to the wild-type antibody. B) Assembly efficiency of murine IgG2a bispecific antibodies from knob and hole half-antibodies with substitution in the CH2 domain. Representative S.E. for three or more experimental replicates is shown for wild-type and DANG antibody variants.
LALA-PG Variants Retain Assembly Properties of the Wild-type Half-antibodies
Murine IgG2a half-antibodies containing the effector function-reducing substitutions were evaluated for efficiency of assembly into intact bispecific antibody. Analogous to our observation with the expression of half-antibodies, the assembly into bispecific antibody from aglycosylated DANG and NG half-antibodies is dramatically impacted. The assembly efficiencies are reduced by 65%-80% compared with the WT half-antibodies (Fig. 2B). In contrast, all of the glycosylated variants, LALA, LALA-DA and LALA-PG, retain an assembly efficiency that is similar to the wild-type antibody (Fig. 2B).
The LALA-PG Variant Retains a Thermal Stability Comparable with the Wild-type Antibody
We had speculated that the impact on expression and assembly could be related to reduced thermal stability of the aglycosylated antibodies. Differential scanning calorimetry (DSC) was performed with the murine WT and aglycosylated (NG) mIgG2a antibodies to confirm the impact of aglycosylation on stability (Fig. 3, A and B). The NG antibody shows a reduction in stability typically assigned to destabilization of the aglycsoylated CH2 domain. To study whether murine and human LALA-PG antibodies retain the thermal stability of their respective wild-type antibodies, we repeated the DSC experiments with the annealed bispecific antibodies. Both the murine IgG2a-LALA-PG bispecific antibody (Fig. 3C) and the human IgG1-LALA-PG bispecific antibody (Fig. 3D) lack the characteristic reduction in stability associated with aglycosylated antibodies and demonstrate thermal stabilites consistent with previous reports of wild-type glycosylated human IgG1 and murine IgG2a antibodies (34–36). The calculated inflection points for the deconvoluted isotherms are shown in Fig. 3E.
FIGURE 3.

LALA-PG antibodies retain the stability of wild-type antibodies. A–D, differential scanning calorimetry isotherms of wild-type murine IgG2a antibody (A; experimental data are shown in blue, and calculated isotherms are shown in orange), aglycosylated (NG) murine IgG2a (B; experimental data are shown in black, and calculated isotherms are shown in orange), murine IgG2a monospecific WT (blue) bispecific LALA-PG (red) antibodies (C), and human IgG1 and bispecific WT (blue) bispecific LALA-PG (red) antibodies (D). E, summary of the first and second thermal melt transition for murine (m) and human (h) Fc fragments and full-length antibodies.
The LALA-PG Variant Reduces the Effector Function of Murine IgG2a More Effectively Than LALA Alone
In murine serum, the LALA-PG variant displays reduced C1q binding (Fig. 4A) and C3 fixation (Fig. 4B) relative to both wild-type Fc and the DANG variants. The reduction in C1q binding with the LALA-PG antibody is even more pronounced with isolated murine C1q complex because of the absence of competitive binding by other proteins in serum. Experiments with human IgG1 did not show a reduction in serum C3 fixation (supplemental Fig. S1B) and very little difference between LALA-PG and NG variants in complement C1q binding in either the serum (supplemental Fig. S1A) or with isolated human C1q (supplemental Fig. S1C).
FIGURE 4.
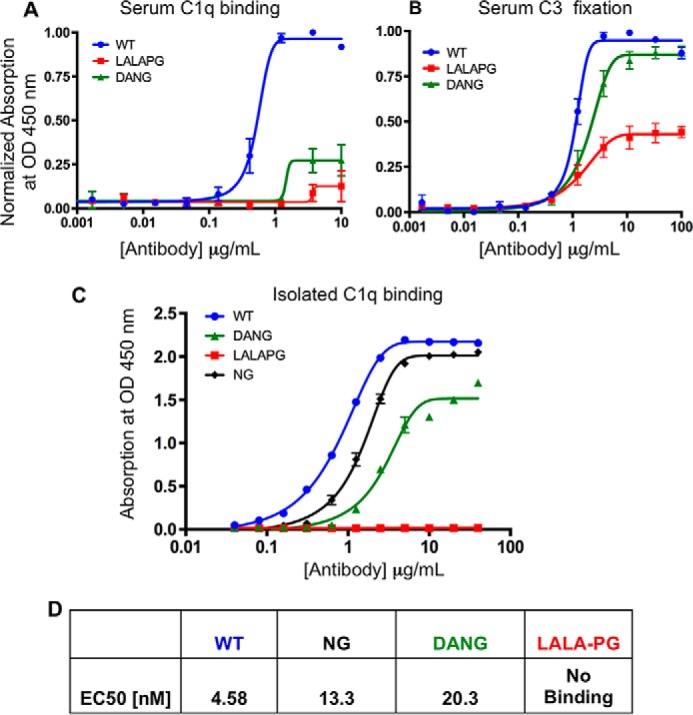
Murine C1q binding and C3 fixation are significantly reduced by LALA-PG substitutions. A–D, ELISAs of serum murine C1q binding (A), serum murine C3 fixation (B), isolated murine C1q binding (C), and tabulated EC50 values for antibody complement fixation and binding (D). WT, blue; NG, black; DANG, green; LALA-PG, red. LALA-PG substitutions show a significant reduction in complement binding and fixation relative to NG and DANG substitutions. The data shown are representative of multiple experimental replicates. Error bars represent standard error. The data include two or more experimental replicates in A and B. All experiments include two or more technical replicates.
We further characterized the binding of effector-reduced antibodies to murine Fc-γ receptors by ELISA and observed residual binding of the murine IgG2a-NG antibodies to all Fc-γ receptors (Fig. 5). Addition of the D265A variant in conjunction with the NG variant further reduces binding to the Fc-γRI receptor and eliminates residual binding to all other Fc-γ receptors in the assay. In contrast, the LALA-PG variant shows residual Fc-γ binding only to the Fc-γRIII receptor. However, this binding is attenuated by more than 50-fold from the wild-type murine IgG2a antibody (Fig. 5). In human IgG1 antibodies, both the LALA-PG and the NG variants are equally effective at eliminating Fc-γ receptor binding (supplemental Fig. S2) and ADCC activity (supplemental Fig. S3). The LALA-PG and NG variants in human IgG1 were also assessed for macrophage-mediated ADCP activity in a highly avid assay format where aglycosylated antibodies have previously demonstrated activity comparable with IgG1 (37). Consistent with the previous report, the human IgG1 and its NG variant demonstrated comparable ADCP activity, whereas the LALA-PG variant had low to undetectable ADCP activity (supplemental Fig. S4).
FIGURE 5.
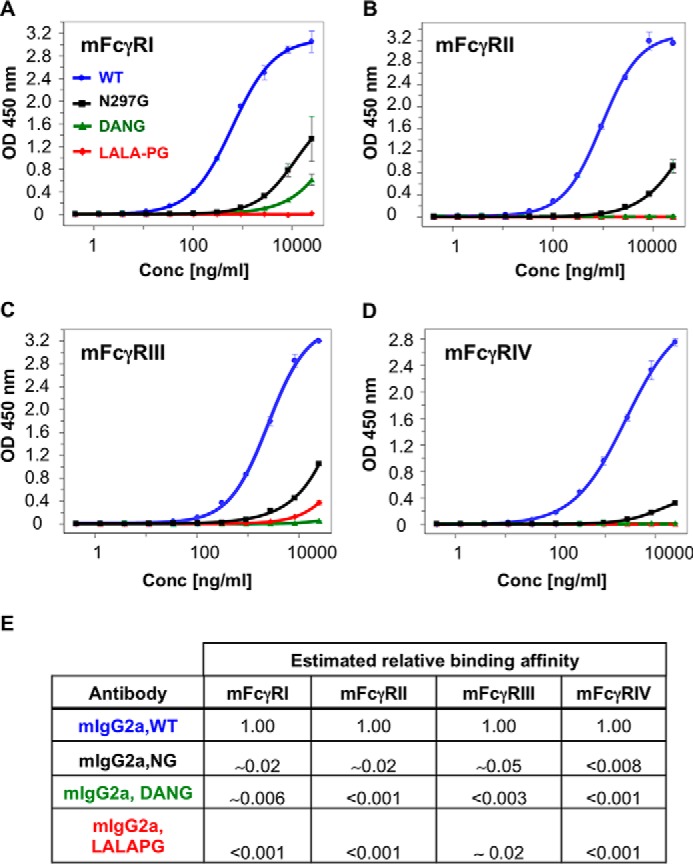
Murine Fc-γ binding is significantly reduced by LALA-PG substitutions. A–D, ELISAs of representative mIgG2a antibody binding to Fc-γRI (A), Fc-γRII (B), Fc-γRIII (C), and Fc-γRIV (D). Tabulated binding was estimated by dividing the equivalent nanograms per milliliter wild type concentration at 25,000 ng/ml variant by 25000 ng/ml. The tabulated binding ratio was calculated as WT/variant at 25,000 ng/ml. The color code is as in Fig. 4. LALA-PG substitutions show a significant reduction in Fc-γ binding. In all cases, the LALA-PG substitutions demonstrate greater reduction in Fc-γ binding than N297G alone and equivalent or greater reduction in Fc-γ binding than DANG, except Fc-γRIII. Error bars represent standard deviation for two technical replicates.
The LALA-PG Variant Demonstrates an Improved Safety Profile in Vivo
Bispecific antibodies targeting the murine TfR have been shown recently to evoke complement-dependent reticulocyte depletion even in the presence of the N297G effector-reducing substitution (21). These earlier studies utilized a rat variable domain targeting a murine TfR (21, 29). In this work, we have grafted this anti-TfR domain (anti-TfRD) onto bispecific frameworks for human IgG1 and murine IgG2a. An additional murine IgG2a-bispecific antibody containing murinized complement-determining regions from the anti-TfRD variable domain was also generated (noted as anti-TfRm). Based on the in vitro data, the LALA-PG variant should be as effective as the DANG and NG variants in reduction of Fc-γ-mediated effector function and superior to DANG in reduction of CDC and ADCP effector functions in vivo. As expected, the bispecific anti-TfRD/control and the anti-TfRm/control bispecific antibodies with the LALA-PG variants eliminated the residual reticulocyte reduction observed with the DANG variant (Fig. 6A). This effect was not a result of any change in clearance of the anti-TfRD/control LALA-PG bispecific antibody, as confirmed by similar serum levels of all antibodies evaluated (Fig. 6B). Finally, as would be predicted based on the locations of the LALA-PG variants, the PK profile is similar to values for historic control antibodies (Fig. 6C).
FIGURE 6.

Anti-TfR LALA-PG bispecific antibodies do not deplete reticulocytes. A, no statistically significant reticulocyte depletion was observed with mIgG2a LALA-PG at 24 h post-dose. B, antibody levels are within the expected historical range. Some increase in antibody clearance is observed with anti-TfRm, a murinized version of anti-TfRD. C, pharmacokinetics analysis of non-targeting murine IgG2a LALA-PG (red) antibodies is similar to historic values (gray) for other non-targeting antibodies with wild-type effector regions.
Discussion
Using both in vitro and in vivo experiments, we have demonstrated that the LALA-PG variant is highly effective in eliminating both Fc-γ- and complement-mediated antibody effector functions for both human IgG1 and murine IgG2a antibodies. In particular, we show a notable reduction in ADCP and/or CDC activity for murine IgG2a antibodies in a highly sensitive in vivo context using anti-TfR antibodies with a known safety liability (21) (e.g. reduction in reticulocytes via multiple effector-mediated mechanisms). This assay can reveal even small levels of effector function masked in less sensitive assays. Furthermore, we have shown that murine IgG2a antibodies with the LALA-PG substitutions retain favorable biophysical and manufacturing properties. Additionally, the LALA-PG variant is compatible with the knobs-into-holes technology for generation of bispecific antibodies, a property not shared by either the NG or the DANG variant, both of which destabilize the antibody Fc. Thus, we expect that LALA-PG will also be compatible with other bispecific formats or antibody engineering technologies.
In vitro, there are several differences in Fc-γ receptor binding of murine IgG2a antibodies containing either the LALA-PG or the DANG variants. The LALA-PG variant is superior to the historic NG or the DANG variants in reducing binding to two high-affinity murine Fc-γ receptors, RI and RIV. The DANG variant, however, is superior to LALA-PG in reducing binding of the murine IgG2a to the murine Fc-γRIII receptor. Although this is the most widely expressed murine Fc-γ receptor and has a role in ADCC activity, the weak residual binding of the LALA-PG variant to this receptor is insufficient to trigger detectable ADCC activity in vivo, based on our data with the anti-TfR bispecific antibody (Fig. 4) or in vitro using the anti-human TfR antibody described in Ref. 28 (supplemental Fig. S3). In human IgG1 antibodies, the LALA-PG variant is equivalent to the NG variant at eliminating binding to Fc-γ receptors. Our data further demonstrate the superiority of using the LALA-PG variant over the LALA variant to silence effector function, as previous work has shown that the LALA variant in murine IgG2a retains attenuated binding to the murine Fc-γ receptors RIII and RIV (26). In contrast to previous studies (26), we demonstrate residual binding of the murine IgG2a-DANG to Fc-γRI. This may be explained by an avidity component of the ELISA, enabling us to identify low-affinity interactions. We note that others have reported the binding of aglycosylated human IgG1 molecules to Fc-γRI-expressing cells following cross-linking (37).
The reduction in complement fixation by the LALA-PG variant is unexpected and significant in the case of the murine IgG2a antibodies. Although the murine IgG2a-DANG variant increases the effective antibody concentration for some complement reactions, the maximum activity remains almost the same as the wild-type antibody for both the serum C3 fixation assay and isolated C1q binding (Fig. 4). In contrast, the LALA-PG variant in murine IgG2a antibodies has the dramatic effect of reducing C1q binding and C3 fixation in serum to just above background levels and completely eliminating antibody binding by isolated C1q.
It has been shown previously that anti-TfR/BACE1 bispecific antibodies containing the effector function-attenuating NG variant deplete reticulocytes in mice, and this reticulocyte depletion is eliminated in C3 knockout mice (21). The reduced in vitro complement activation of the murine IgG2a-LALA-PG variant was confirmed by our in vivo experiments and resulted in an improved safety profile for the anti-TfR LALA-PG bispecific antibody relative to the DANG bispecific antibody. Further work will be necessary to understand why the reduction in complement activity by the LALA-PG variant is more pronounced for the murine IgG2a antibodies than for human IgG1 antibodies, and it is possible that the effect is a function of the species-specific variation in the strength of complement response, as has been noted previously (38). However, it is clear from the data in supplemental Fig. S4 that ADCP activity in the human IgG1 is completely eliminated by the LALA-PG variant, something not accomplished by the antibody N297G aglycosylation variant.
In this study, we characterize the LALA-PG variant in murine IgG2a and human IgG1 antibodies. Additional work will be necessary to determine whether the results translate to other antibody isotypes and species. However, as glycosylation has a structural role in the antibody CH2, the LALA-PG substitutions will be always preferred over aglycosylation for antibody stability. The impact of aglycosylation on expression and assembly of murine IgG2a half-antibodies is independent of the antibody variable domains over all antibodies evaluated.
We have not evaluated the effect of the LALA-PG substitutions on the posttranslational modification of antibodies. It was shown recently that expression of glycosylated half-antibodies results in production of heterogeneous glycosylation patterns (39). This altered glycosylation could potentially complicate analytical characterization, although this heterogeneity did not present any obstacles to this study.
In summary, we have identified the LALA-PG variant as a solution to efficiently attenuate both Fc-γ-dependent ADCC and C1q/C3B-dependent CDC activity in murine antibodies. The amino acid changes do not impact the thermal stability of the molecule, making them attractive for use in a regular monoclonal antibody as well as engineered antibody formats that suffer from reduced stability. The lack of detectable interaction of murine IgG2a LALA-PG with murine Fc-γ receptors and C1q now enables a safe path for murine in vivo studies with antibodies to targets that do not tolerate any cytotoxic activity, thus improving the translation of preclinical animal models to the clinic.
Experimental Procedures
Research Ethics
All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (Publication 8523, revised 1985). The Institutional Animal Care and Use Committee at Genentech reviewed and approved all animal protocols.
Plasmid Construction and Antibody Expression
Antibodies and antibody Fc regions were cloned by standard molecular biology techniques into mammalian expression vectors (40) as described previously. IgGs were expressed as 1 liter of transient transfection cultures of CHO (41). All antibody positions are according to the Eu numbering convention.
Half-antibody and Bispecific Antibody Production
The bispecific antibodies, including the anti-TfR/control (29), were generated using the knobs-into-holes technology described previously (21). The half-antibodies were expressed in CHO cells and affinity-purified over a GE mAb Select SuRe column (GE Healthcare). The purified half-antibodies were assembled under reducing conditions with reduced glutathione and were allowed to assemble over 3 days at room temperature at pH 8.5 for human IgG1 and pH 9.5 for murine IgG2a. The bispecific antibodies were purified with a hydrophobic interaction column (ProPac HIC-10, Thermo Scientific, Sunnyvale, CA). Assembly efficiency was calculated based on the theoretic maximum yield using the input amounts of the half-antibodies and then normalized to the recovery from wild-type bispecific antibody.
Melting Temperature and Enthalpy Calculations
The thermostability of the murine IgG2a variants was determined by measuring the melting temperatures (Tm) and enthalpies (ΔHm) of the antibodies via differential scanning calorimetry (MicroCal VP-DSC, Northampton, MA). Raw melt data were fit to a non-two-state model to calculate Tm and ΔHm.
Serum C1q Binding and C3 Fixation
ELISA for C1q binding and C3 fixation were adapted from Hessell et al. (25). Briefly, purified bispecific antibodies were serially diluted in 1× PBS and coated onto microtiter plates overnight at 4 °C. The plates were blocked with 5% gelatin/PBST (0.5× PBS and 1% (v/v) Tween 20), followed by incubation with murine or human serum (10% (v/v) in PBST). C1q binding was detected by HRP-conjugated rabbit anti-C1q (Bioss Inc.) at a 1:500 dilution in PBST. To test C3 fixation, rabbit anti-C3 (Abcam) was added at a 1:1000 dilution, followed by HRP-conjugated chicken anti-rabbit IgG (Abcam) at a 1:2000 dilution. The plates were developed with TMB (KPL, Gaithersburg, MD). The reaction was stopped with 1 m H3PO4 and read at 450 nm using 620 nm for background reading on a Tecan M1000 fluorescence reader (Tecan, Mannedort, Switzerland). EC50 values were calculated by fitting the data to a log (agonist) versus response variable slope (four-parameter) model using GraphPad Prism (Sunnyvale, CA).
Isolated C1q Binding ELISA
MaxiSorp 384-well plates (Thermo Scientific, Nunc, Roskilde, Denmark) were coated with serially diluted antibodies (0.039–40 μg/ml mouse IgG2a or 0.049–50 μg/ml human IgG1) in 50 mm carbonate buffer (pH 9.6) (coat buffer) at 4 °C overnight. Plates were washed with PBS containing 0.05% polysorbate 20 (pH 7.4) and blocked with PBS containing 0.5% BSA, 0.05% polysorbate 20, 15 ppm Proclin, and 10% blocker casein (Thermo Scientific) (pH 7.4). After 1-h incubation at room temperature, plates were washed. Mouse C1q (0.5 μg/ml, Genentech) or human C1q (1 μg/ml, Quidel, San Diego, CA) in the same buffer was added and incubated for 1.5 h. Bound C1q was detected by adding biotinylated mouse anti-mouse C1q (20 ng/ml, Hycult Biotech, Plymouth Meeting, PA; cross-reacting with human C1q) for 1.5 h, followed by HRP-conjugated streptavidin (GE Healthcare Life Sciences) for 1 h. To check for coating efficiency, some coated wells received buffer only for the first two incubation steps and received goat anti-mouse F(ab′)2-HRP (for murine IgG2a-coated wells) or anti-human F(ab′)2-HRP (for human IgG1-coated wells) when the wells used for measuring C1q binding received streptavidin-HRP. Plates were washed after each incubation step. Peroxidase activity was detected with the substrate 3,3′,5,5′-tetramethyl benzidine (TMB) (Kirkegaard and Perry Laboratories). The reaction was stopped with 1 m H3PO4, and absorbance was measured at 450 nm using a Multiskan Ascent reader (Thermo Scientific, Hudson, NH). Dose-response binding curves were fitted with a four-parameter model, and EC50 values were calculated using KaleidaGraph (Synerg Software, Reading, PA).
Isolation of Murine C1q
Murine C1q was purified directly from mouse serum similar to methods described previously (42). Briefly, 10 mm EDTA and 1 mm PMSF, final concentrations, were added to raw mouse serum and 0.2-μm-filtered. The serum was then applied to immobilized mouse and/or rabbit IgG-Sepharose (Sigma-Aldrich), and C1q was eluted in PBS with an additional 1 m NaCl and 10 mm EDTA. Fractions containing C1q, as determined by immunoblot against C1q-A, were pooled and dialyzed to 100 mm NaCl for cation exchange chromatography (Mono S, GE Healthcare), washed with 50 mm HEPES (pH 7.2), 100 mm NaCl, 0.1% Triton X-114, and 0.02% Triton X-100, followed by 50 mm HEPES (pH 7.2), 100 mm NaCl to remove detergent. C1q was eluted with a 25 column volumes gradient to 500 mm NaCl. Peak fractions containing C1q were pooled from the eluate. To remove any IgG contaminants, the eluate from the cation exchange step was applied directly onto MabSelect SuRe (GE Healthcare), and the IgG-free C1q was collected from the flow-through. Finally, the material was applied to a gel filtration column (Superdex 200, GE Healthcare) in PBS supplemented with 1 m NaCl. C1q peak fractions were pooled and dialyzed into 50 mm HEPES (pH 7.2), 150 mm NaCl, and 20% glycerol, and then 0.2-μm sterile-filtered. The identity of the purified material was confirmed by immunoblotting as well as mass spectrometric analysis. The native molecular weight was verified by size exclusion chromatography-multi-angle laser light scatter (MALLS).
Soluble Fc-γ Receptor Binding ELISAs
For mouse FcγR binding, soluble mouse Fc-γRI, Fc-γRIIb, and Fc-γRIII were expressed as recombinant fusion proteins with Gly-His6-GST at the C terminus of the extracellular domain of the receptor α chains (Fc-γR His-GST) (Genentech). Soluble mouse Fc-γRIV was expressed as Fc-γR-His. MaxiSorp 384-well plates were coated with mouse Fc-γRs (1 μg/ml) in coat buffer. Plates were washed and blocked with PBS containing 0.5% BSA and 15 ppm Proclin (pH 7.4). After a 1-h incubation, plates were washed, and mouse IgG2a (0.42–25,000 ng/ml in 3-fold serial dilution in duplicate) in PBS containing 0.5% BSA, 0.05% polysorbate 20, and 15 ppm Proclin (pH 7.4) was added to the plates and incubated for 2 h. Bound IgG2a was detected with goat F(ab′)2 anti-mouse F(ab′)2-HRP (Jackson ImmunoResearch Laboratories) using TMB as a substrate. The reaction was stopped, and the plate was read as described above. The dose-dependent binding curve of the wild-type antibody was fitted with a four-parameter curve-fitting program (KaleidaGraph, Synergy Software). The relative affinity of the variant versus the wild type was estimated by dividing the equivalent nanograms per milliliter wild type concentration at 25,000 ng/ml variant by 25,000 ng/ml.
Human Fc-γR binding ELISAs were carried out similarly using plates coated with human Fc-γR-His-GST molecules (43). For measuring binding to human Fc-γRII and Fc-γRIIII, human IgG1 was preincubated with anti-human κ antibody to form a complex to increase binding avidity. Serially diluted IgG1 (0.0085–500 ng/ml for Fc-γRI and 0.42–25,000 for Fc-γRII and Fc-γRIII) was added to the plate. Bound IgG1 was detected using goat F(ab′)2 anti-human F(ab′)2-HRP as described previously (44). The relative affinity of the variant versus the wild type was estimated as described above, except that, for Fc-γRI binding, the equivalent nanograms per milliliter wild type concentration at 500 ng/ml variant was divided by 500 ng/ml.
Antibody Affinity Measurements
The anti-TfR competition ELISA was performed in MaxiSorp plates (Nunc) coated with purified muTfR-His (2.5 μg/ml) in PBS at 4 °C overnight. Plates were washed with PBS/0.05% Tween 20 and blocked with SuperBlock blocking buffer in PBS (Thermo Scientific). A 1:3 serially titrated IgG was combined with1 nm biotinylated anti-TfRA and added to the plate for 1 h at room temperature. Plates were washed with PBS/0.05% Tween 20, and HRP-streptavidin (SouthernBiotech) was added to the plate and incubated for 1 h at room temperature. Plates were washed with PBS/0.05% Tween 20, and biotinylated anti-TfRA bound to the plate was detected with TMB substrate (BioFX Laboratories).
Hematology and Chemistry Analysis
Reticulocyte counts were determined on K-EDTA blood with a Sysmex XT-2000iV. The Sysmex detects and classifies reticulocytes by flow cytometry with a fluorescent polymethine dye to bind cellular RNA and cell light scatter characteristics.
Measuring Antibody Concentrations in Mouse Plasma
Total antibody concentrations in mouse plasma were measured with a generic human Fc ELISA. Nunc 384-well MaxiSorp immunoplates were coated with an F(ab′)2 fragment of donkey anti-human IgG and Fc fragment-specific polyclonal antibody (Jackson ImmunoResearch Laboratories) overnight at 4 °C. Plates were blocked with PBS and 0.5% BSA for 1 h at 25 °C. Each antibody (control IgG and anti-TfR/control-bispecific) was used as a standard to quantify respective antibody concentrations. Plates were washed with PBS and 0.05% Tween 20 using a microplate washer (Bio-Tek Instruments Inc.), and standards and samples were diluted in PBS containing 0.5% BSA, 0.35 m NaCl, 0.25% CHAPS, 5 mm EDTA, and 0.05% Tween 20, and 15 ppm was added for 2 h at 25 °C. Bound antibody was detected with HRP-conjugated F(ab′)2 goat anti-human IgG and Fc-specific polyclonal antibody (Jackson ImmunoResearch Laboratories) and developed with TMB (KPL Inc.), and absorbance was measured at 450 nm on a Multiskan Ascent reader (Thermo Scientific). Concentrations were determined from the standard curve with a four-parameter nonlinear regression program. Wild-type female C57B/6 mice aged 6–8 weeks were used for all studies.
Author Contributions
J. A. E. and C. S. conceived the project. M. L., H. S. K., R. K. T., T. W. B., J. M. V., Y. G. M., Y. Z., Y. J. Y. Z., R. J. B., C. S., and J. A. E. designed and performed the assays. M. L., R. K. T., T. W. B., and J. A. E. contributed to the purification of recombinant proteins. Y. J. Y. Z. performed the mouse experiments. Y. J. Y. Z., J. A. C., R. J. W., and J. L. designed and analyzed the murine reticulocyte and pharmacokinetic analyses. M. L., H. S. K., R. K. T., T. W. B., J. M. V., Y. G. M., Y. Z., Y. J. Y. Z., R. J. W., M. S. D., Y. L. L., S. C., R. J. B., C. S., and J. A. E. analyzed the data. M. L., H. S. K., R. T., T. W. B., Y. G. M., C. S., and J. A. E. drafted the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank our Genentech colleagues in Protein Chemistry, Antibody Engineering, Neuroscience, and Safety Assessment, the Biomolecular Engineering Group, and Laboratory Animal Resources for technical assistance.
This work was funded by Genentech, Inc. All authors are present or former paid employees of Genentech Inc.

This article contains supplemental Figs. S1–S4 and Methods.
- PK
- pharmacokinetics
- CDC
- complement-dependent cytotoxicity
- ADCC
- antibody-dependent, cell-mediated cytotoxicity
- ADCP
- antibody-dependent cellular phagocytosis
- DANG
- D265A, N297G
- TfR
- transferrin receptor
- NG
- N297G
- LALA
- L234A, L235A
- PG
- P329G
- DA
- D265A
- DSC
- differential scanning calorimetry
- TMB
- 3,3′,5,5′-tetramethyl benzidine.
References
- 1. Reichert J. M. (2012) Marketed therapeutic antibodies compendium. MAbs 4, 413–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walsh G. (2014) Biopharmaceutical benchmarks 2014. Nat. Biotechnol. 32, 992–1000 [DOI] [PubMed] [Google Scholar]
- 3. Spiess C., Zhai Q., and Carter P. J. (2015) Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 67, 95–106 [DOI] [PubMed] [Google Scholar]
- 4. Sheridan C. (2015) Amgen's bispecific antibody puffs across finish line. Nat. Biotechnol. 33, 219–221 [DOI] [PubMed] [Google Scholar]
- 5. Chan A. C., and Carter P. J. (2010) Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 10, 301–316 [DOI] [PubMed] [Google Scholar]
- 6. Weiner G. J. (2015) Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mestas J., and Hughes C. C. (2004) Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 [DOI] [PubMed] [Google Scholar]
- 8. Epstein S. L., and Gottlieb P. D. (1977) Quantitative measurement of mouse IgG subclasses with the use of heteroantisera: the importance of allotype considerations. J. Immunol. 118, 935–942 [PubMed] [Google Scholar]
- 9. Gelderman K. A., Tomlinson S., Ross G. D., and Gorter A. (2004) Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 25, 158–164 [DOI] [PubMed] [Google Scholar]
- 10. Bakema J. E., and van Egmond M. (2014) Fc receptor-dependent mechanisms of monoclonal antibody therapy of cancer. Curr. Top Microbiol. Immunol. 382, 373–392 [DOI] [PubMed] [Google Scholar]
- 11. Bruhns P. (2012) Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119, 5640–5649 [DOI] [PubMed] [Google Scholar]
- 12. Nimmerjahn F., Bruhns P., Horiuchi K., and Ravetch J. V. (2005) FcγRIV: a novel FcR with distinct IgG subclass specificity. Immunity 23, 41–51 [DOI] [PubMed] [Google Scholar]
- 13. Lazar G. A., and Desjarlais J. R. (2009) in Therapeutic Monoclonal Antibodies: From Bench to Clinic (An Z., ed.), pp. 350–363, John Wiley & Sons, Hoboken, NJ [Google Scholar]
- 14. Ernst L. K., Metes D., Herberman R. B., and Morel P. A. (2002) Allelic polymorphisms in the FcγRIIC gene can influence its function on normal human natural killer cells. J. Mol. Med. 80, 248–257 [DOI] [PubMed] [Google Scholar]
- 15. Nimmerjahn F., and Ravetch J. V. (2008) Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 [DOI] [PubMed] [Google Scholar]
- 16. Leabman M. K., Meng Y. G., Kelley R. F., DeForge L. E., Cowan K. J., and Iyer S. (2013) Effects of altered FcγR binding on antibody pharmacokinetics in cynomolgus monkeys. MAbs 5, 896–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tao M. H., and Morrison S. L. (1989) Studies of aglycosylated chimeric mouse-human IgG: role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 143, 2595–2601 [PubMed] [Google Scholar]
- 18. Hristodorov D., Fischer R., and Linden L. (2013) With or without sugar? (A)glycosylation of therapeutic antibodies. Mol. Biotechnol. 54, 1056–1068 [DOI] [PubMed] [Google Scholar]
- 19. Sazinsky S. L., Ott R. G., Silver N. W., Tidor B., Ravetch J. V., and Wittrup K. D. (2008) Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 20167–20172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lund J., Takahashi N., Pound J. D., Goodall M., and Jefferis R. (1996) Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc γ receptor I and influence the synthesis of its oligosaccharide chains. J. Immunol. 157, 4963–4969 [PubMed] [Google Scholar]
- 21. Couch J. A., Yu Y. J., Zhang Y., Tarrant J. M., Fuji R. N., Meilandt W. J., Solanoy H., Tong R. K., Hoyte K., Luk W., Lu Y., Gadkar K., Prabhu S., Ordonia B. A., Nguyen Q., et al. (2013) Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci. Transl. Med. 5, 183ra157, 1–12 [DOI] [PubMed] [Google Scholar]
- 22. Chappel M. S., Isenman D. E., Everett M., Xu Y. Y., Dorrington K. J., and Klein M. H. (1991) Identification of the Fc gamma receptor class I binding site in human IgG through the use of recombinant IgG1/IgG2 hybrid and point-mutated antibodies. Proc. Natl. Acad. Sci. U.S.A. 88, 9036–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sondermann P., Huber R., Oosthuizen V., and Jacob U. (2000) The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc γRIII complex. Nature 406, 267–273 [DOI] [PubMed] [Google Scholar]
- 24. Hezareh M., Hessell A. J., Jensen R. C., van de Winkel J. G., and Parren P. W. (2001) Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 75, 12161–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hessell A. J., Hangartner L., Hunter M., Havenith C. E., Beurskens F. J., Bakker J. M., Lanigan C. M., Landucci G., Forthal D. N., Parren P. W., Marx P. A., and Burton D. R. (2007) Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104 [DOI] [PubMed] [Google Scholar]
- 26. Arduin E., Arora S., Bamert P. R., Kuiper T., Popp S., Geisse S., Grau R., Calzascia T., Zenke G., and Kovarik J. (2015) Highly reduced binding to high and low affinity mouse Fc γ receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol. Immunol. 63, 456–463 [DOI] [PubMed] [Google Scholar]
- 27. Idusogie E. E., Presta L. G., Gazzano-Santoro H., Totpal K., Wong P. Y., Ultsch M., Meng Y. G., and Mulkerrin M. G. (2000) Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J. Immunol. 164, 4178–4184 [DOI] [PubMed] [Google Scholar]
- 28. Yu Y. J., Atwal J. K., Zhang Y., Tong R. K., Wildsmith K. R., Tan C., Bien-Ly N., Hersom M., Maloney J. A., Meilandt W. J., Bumbaca D., Gadkar K., Hoyte K., Luk W., Lu Y., et al. (2014) Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 6, 261ra154. [DOI] [PubMed] [Google Scholar]
- 29. Yu Y. J., Zhang Y., Kenrick M., Hoyte K., Luk W., Lu Y., Atwal J., Elliott J. M., Prabhu S., Watts R. J., and Dennis M. S. (2011) Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 3, 84ra44. [DOI] [PubMed] [Google Scholar]
- 30. Ridgway J. B., Presta L. G., and Carter P. (1996) “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 9, 617–621 [DOI] [PubMed] [Google Scholar]
- 31. Atwell S., Ridgway J. B., Wells J. A., and Carter P. (1997) Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 270, 26–35 [DOI] [PubMed] [Google Scholar]
- 32. Schlothauer T., Herter S., Koller C. F., Grau-Richards S., Steinhart V., Spick C., Kubbies M., Klein C., Umaña P., and Mössner E. (2016) Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 29, 457–466 [DOI] [PubMed] [Google Scholar]
- 33. Spiess C., Merchant M., Huang A., Zheng Z., Yang N. Y., Peng J., Ellerman D., Shatz W., Reilly D., Yansura D. G., and Scheer J. M. (2013) Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat. Biotechnol. 31, 753–758 [DOI] [PubMed] [Google Scholar]
- 34. Mimura Y., Sondermann P., Ghirlando R., Lund J., Young S. P., Goodall M., and Jefferis R. (2001) Role of oligosaccharide residues of IgG1-Fc in Fc γ RIIb binding. J. Biol. Chem. 276, 45539–45547 [DOI] [PubMed] [Google Scholar]
- 35. Welfle K., Misselwitz R., Hausdorf G., Höhne W., and Welfle H. (1999) Conformation, pH-induced conformational changes, and thermal unfolding of anti-p24 (HIV-1) monoclonal antibody CB4–1 and its Fab and Fc fragments. Biochim. Biophys. Acta 1431, 120–131 [DOI] [PubMed] [Google Scholar]
- 36. Vermeer A. W., and Norde W. (2000) The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys. J. 78, 394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nesspor T. C., Raju T. S., Chin C. N., Vafa O., and Brezski R. J. (2012) Avidity confers FcγR binding and immune effector function to aglycosylated immunoglobulin G1. J. Mol. Recognit. 25, 147–154 [DOI] [PubMed] [Google Scholar]
- 38. Dangl J. L., Wensel T. G., Morrison S. L., Stryer L., Herzenberg L. A., and Oi V. T. (1988) Segmental flexibility and complement fixation of genetically engineered chimeric human, rabbit and mouse antibodies. EMBO J. 7, 1989–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shatz W., Chung S., Li B., Marshall B., Tejada M., Phung W., Sandoval W., Kelley R. F., and Scheer J. M. (2013) Knobs-into-holes antibody production in mammalian cell lines reveals that asymmetric afucosylation is sufficient for full antibody-dependent cellular cytotoxicity. MAbs 5, 872–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eaton D. L., Wood W. I., Eaton D., Hass P. E., Hollingshead P., Wion K., Mather J., Lawn R. M., Vehar G. A., and Gorman C. (1986) Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochemistry 25, 8343–8347 [DOI] [PubMed] [Google Scholar]
- 41. Wong A. W., Baginski T. K., and Reilly D. E. (2010) Enhancement of DNA uptake in FUT8-deleted CHO cells for transient production of afucosylated antibodies. Biotechnol. Bioeng. 106, 751–763 [DOI] [PubMed] [Google Scholar]
- 42. McKay E. J. (1981) A simple two-step procedure for the purification of plasma C1q from different animal species. Immunol. Lett. 3, 303–308 [DOI] [PubMed] [Google Scholar]
- 43. Shields R. L., Namenuk A. K., Hong K., Meng Y. G., Rae J., Briggs J., Xie D., Lai J., Stadlen A., Li B., Fox J. A., and Presta L. G. (2001) High resolution mapping of the binding site on human IgG1 for Fc γ RI, Fc γ RII, Fc γ RIII, and FcRn and design of IgG1 variants with improved binding to the Fc γ R. J. Biol. Chem. 276, 6591–6604 [DOI] [PubMed] [Google Scholar]
- 44. Lu Y., Vernes J. M., Chiang N., Ou Q., Ding J., Adams C., Hong K., Truong B. T., Ng D., Shen A., Nakamura G., Gong Q., Presta L. G., Beresini M., Kelley B., et al. (2011) Identification of IgG(1) variants with increased affinity to FcγRIIIa and unaltered affinity to FcγRI and FcRn: comparison of soluble receptor-based and cell-based binding assays. J. Immunol. Methods 365, 132–141 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.