

Abstract
Cool-associated tyrosine-phosphorylated protein 1 (Cat-1) is a signaling scaffold as well as an ADP-ribosylation factor-GTPase-activating protein. Although best known for its role in cell migration, we recently showed that the ability of Cat-1 to bind paxillin, a major constituent of focal complexes, is also essential for the anchorage-independent growth of HeLa cervical carcinoma cells. Here we set out to learn more about the underlying mechanism by which Cat-paxillin interactions mediate this effect. We show that knocking down paxillin expression in HeLa cells promotes their ability to form colonies in soft agar, whereas ectopically expressing paxillin in these cells inhibits this transformed growth phenotype. Although knocking down Cat-1 prevents HeLa cells from forming colonies in soft agar, when paxillin is knocked down together with Cat-1, the cells are again able to undergo anchorage-independent growth. These results suggest that the requirement of Cat-1 for this hallmark of cellular transformation is coupled to its ability to bind paxillin and abrogate its actions as a negative regulator of anchorage-independent growth. We further show that knocking down Cat-1 expression in HeLa cells leads to a reduction in Akt activation, which can be reversed by knocking down paxillin. Moreover, expression of constitutively active forms of Akt1 and Akt2 restores the anchorage-independent growth capability of HeLa cells depleted of Cat-1 expression. Together, these findings highlight a novel mechanism whereby interactions between Cat-1 and its binding partner paxillin are necessary to ensure sufficient Akt activation so that cancer cells are able to grow under anchorage-independent conditions.
Keywords: ADP ribosylation factor (ARF), Akt PKB, CDC42, mammalian target of rapamycin (mTOR), transformation, Cat-1/Git-1, p70S6 kinase, paxillin
Introduction
Cool-associated tyrosine-phosphorylated protein 1 (Cat-1),2 also referred to as G protein-coupled receptor kinase interactor 1 (GIT-1), is thought to serve as a signaling scaffold/adaptor protein that binds a number of different protein partners to mediate actin cytoskeletal changes and cell migration (1–3). It also functions as a GAP for Arf1 and Arf6, two members of the Arf family of small GTPases that have been implicated in the regulation of trafficking events in cells (4–6). Recently, we found that Cat-1 is overexpressed in a large number of human cervical carcinomas and that knocking down its expression in HeLa cells using siRNAs blocked the ability of these cells to exhibit anchorage-independent growth (6) (i.e. as assayed by colony formation in soft agar), a hallmark of cancer and transformed cells (7). We then went on to show that introducing an siRNA-insensitive form of wild-type Cat-1 into HeLa cells, where endogenous Cat-1 expression was knocked down, restored their ability to form colonies in soft agar. On the other hand, introducing an siRNA-insensitive mutant form of Cat-1, defective in binding paxillin, was unable to restore this transformed phenotype. These findings indicated that the interaction between Cat-1 and paxillin was critical for the Cat-1-mediated anchorage-independent growth of HeLa cells.
Paxillin is another signaling scaffold/adapter protein that has been shown to play important roles in regulating focal adhesion dynamics and integrin-mediated signaling events (8). As one of the first proteins to be identified as a constituent of focal complexes (9), paxillin was shown to accumulate at nascent focal complexes in migrating cells (10). It was also demonstrated through mutagenesis studies that disrupting the phosphorylation of paxillin by tyrosine kinases such as the focal adhesion kinase or blocking the ability of paxillin to interact with proteins like Cat or tubulin, alters focal complex dynamics, resulting in irregular cell spreading and defects in cell migration (11, 12).
In addition to being important for the adhesion and migration of a wide variety of cell types, various reports have also implicated paxillin in the growth and survival of certain forms of human cancer. Indeed, the transcript and protein levels of paxillin are frequently up-regulated in several types of cancer, including oral, bone, and colorectal tumors (13–17). In colorectal tumors, survival analyses performed on patients revealed a correlation between the extent of paxillin expression and clinical outcome; the prognosis of patients showing a relatively high expression of paxillin was poorer compared with those with low paxillin expression (16). In such cases, the potential roles of paxillin in cell migration and invasiveness are likely to come into play in the development of these aggressive cancers. It has also been reported that paxillin can contribute to the promotion of anchorage-independent growth of certain colon cancer cell lines, DLD1 and HCT116, as well as fibroblasts stably expressing the constitutively active H-Ras G12V mutant (16, 18). However, there has also been a report where paxillin expression was negatively correlated with metastasis (19), and, as described below, how paxillin contributes to the ability of cancer cells to exhibit anchorage-independent growth appears to be context-dependent.
In this study, we set out to understand the underlying mechanism by which the paxillin binding partner, Cat-1, promoted the anchorage-independent growth of human cervical carcinoma cells (6). Given our previous findings highlighting an essential role played by Cat-1 in HeLa cell transformation (6), together with the suggestions that paxillin contributes to cancer progression (13–18), we initially suspected that the two proteins might work together in a signaling complex to send a stimulatory signal that would promote anchorage-independent growth. However, we found that paxillin exerts a negative regulatory effect on this transformed growth phenotype, whereas Cat-1, by binding to paxillin, is able to repress its negative regulatory activity and thereby promote anchorage-independent growth. Thus, the inhibition of anchorage-independent growth caused by knocking down Cat-1 expression in HeLa cells can be overcome by knockdown of paxillin expression. Moreover, these effects on anchorage-independent growth and transformation appear to be driven by changes in Akt activity. Specifically, knockdown of Cat-1 resulted in lower levels of Akt activation, whereas knocking down paxillin enhanced Akt activity. We then found that expressing activated forms of Akt1 and Akt2 was able to restore anchorage-independent growth in cells where Cat-1 expression had been knocked down. Collectively, these results point to new and unexpected roles for Cat-1 and paxillin in the regulation of anchorage-independent growth in human cervical carcinoma cells, whereby Cat-1 promotes this transformed phenotype by mitigating the negative regulatory actions of paxillin.
Results
Knocking Down Paxillin Expression Enhances Anchorage-independent Growth
The Cat-1-mediated anchorage-independent growth of HeLa cervical carcinoma cells is dependent on its ability to bind paxillin, a major scaffold/adaptor protein found in focal complexes (6). Here we wanted to build upon these findings by determining the mechanism by which Cat-1-paxillin interactions promote this transformed phenotype. To begin, HeLa cells that had been either mock-transfected or transfected with a construct encoding a GFP-tagged form of wild-type paxillin (GFP-Pxn WT) to label focal complexes were stained with GFP and Cat-1 antibodies. Fig. 1A shows that little GFP signal was detected in mock-transfected cells (Mock Transfected, GFP), whereas Cat-1 was expressed in structures that resembled focal complexes (Mock Transfected, Cat-1). When paxillin was ectopically expressed in the cells, it localized with Cat-1 (Fig. 1A, Transfected with GFP-Pxn WT, GFP and Cat-1), confirming that both of these proteins do indeed reside in focal complexes.
FIGURE 1.
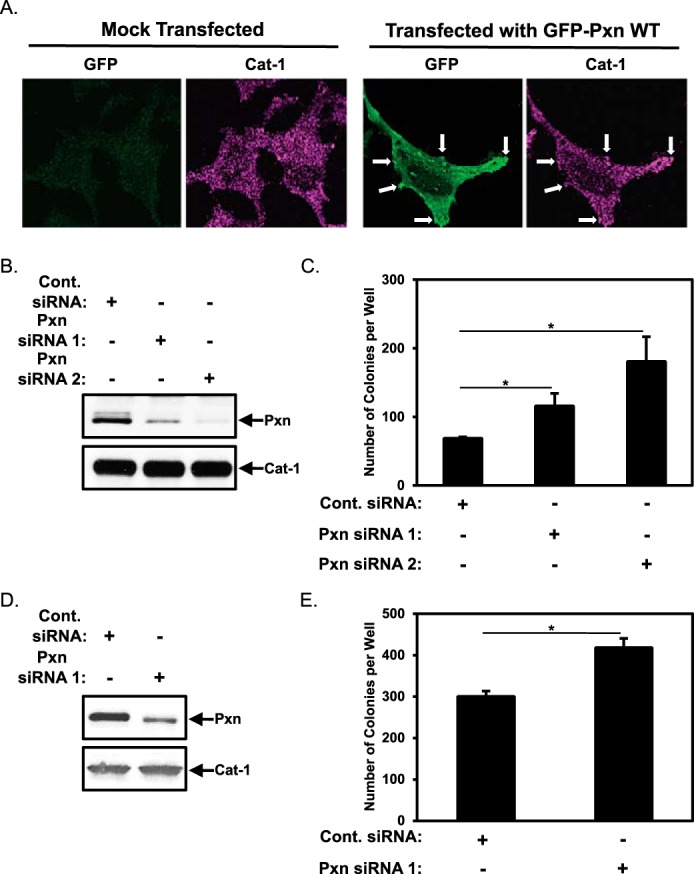
Paxillin negatively regulates the anchorage-independent growth of HeLa cells. A, images of HeLa cells that had been either mock-transfected or transfected with GFP-Pxn WT, as indicated, and then stained with GFP and Cat-1 antibodies. Arrows indicate some of the focal complexes where both GFP-tagged Pxn WT and Cat-1 were detected. B and C, two sets of HeLa cells were transfected with either Cont. siRNA or Pxn siRNA 1 or Pxn siRNA 2. One set (B) was lysed and then immunoblotted with Pxn and Cat-1 antibodies. Soft agar assays were performed (C) on the other set of cells. The number of colonies that formed for each condition was determined. D and E, two sets of NIH3T3 cells stably expressing an activated form of Cdc42, Cdc42 F28L, were transfected with either Cont. siRNA or Pxn siRNA 1 as indicated. One set (D) was lysed and then immunoblotted with Pxn and Cat-1 antibodies. Soft agar assays were performed (E) on the other set of cells. The number of colonies that formed for each condition was determined. All of the experiments shown were performed three separate times, each yielding similar results. The data shown in C and E represent mean ± S.D. Student's t tests were performed. *, p < 0.05.
We introduced a control siRNA or two different siRNAs targeting paxillin into HeLa cells and then assayed the ability of these cells to form colonies in soft agar (i.e. anchorage-independent growth). Fig. 1B, top panel, shows that both paxillin siRNAs (Pxn siRNA 1 and Pxn siRNA 2) knocked down endogenous paxillin expression compared with the control siRNA (Cont. siRNA). Pxn siRNA 1 reduced paxillin levels by ∼70%, whereas Pxn siRNA 2 consistently reduced paxillin expression by ∼95%. Given the findings from several studies linking paxillin to cell growth and survival, we initially suspected that knocking down paxillin in HeLa cells would compromise their ability to grow in soft agar. However, Fig. 1C shows that this was not the case. In fact, knocking down paxillin expression led to the formation of more colonies in soft agar compared with cancer cells expressing control siRNA (Fig. 1C, compare the Cont. siRNA column with the Pxn siRNA 1 and 2 columns), with the largest increase in colony formation being achieved when using the paxillin siRNA that most effectively reduced paxillin expression (i.e. Pxn siRNA 2, Fig. 1C).
An analogous set of experiments was carried out using NIH3T3 mouse fibroblasts that were transformed by the stable expression of a constitutively active Cdc42 mutant (Cdc42(F28L)), as we had found previously that these cells exhibited anchorage-independent growth in a manner dependent on Cat-1, similar to human cervical carcinoma cell lines (6). Control siRNA or Pxn siRNA 1 was introduced into fibroblasts expressing the activated Cdc42(F28L) mutant (Fig. 1D, top panel), prior to the cells being placed in soft agar. Fig. 1E shows that knocking down paxillin expression by 30%-50% resulted in a corresponding increase in the amount of colonies formed by NIH3T3 cells expressing Cdc42(F28L).
Because decreasing paxillin levels in HeLa cancer cells using siRNAs promoted their ability to grow under anchorage-independent conditions, we then examined whether overexpressing paxillin in these cells yielded the opposite effect. Compared with cells expressing vector alone, the amount of colonies generated by HeLa cells ectopically expressing GFP-tagged wild-type paxillin (Fig. 2A, top panel, Pxn WT) was reduced by ∼45% (Fig. 2B, compare the Vector and Pxn WT columns), which was similar to the reduction in colonies observed when HeLa cells were transfected with a V5-tagged dominant-negative form of Cat-1 that is unable to function as an Arf GAP or bind paxillin (6), referred to as Cat-1 KK-RA (Fig. 2, A, center panel, Cat-1 KK-RA, and B, compare the Cat-1 KK-RA and Pxn WT columns).
FIGURE 2.
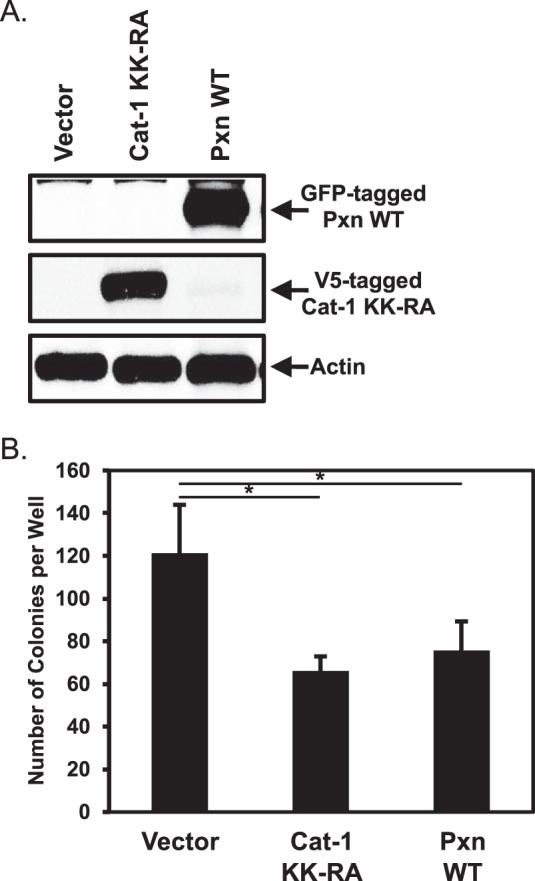
The ectopic expression of paxillin in HeLa cells causes them to lose their transformed phenotype. A and B, two sets of HeLa cells were transfected with vector alone, GFP-tagged wild-type paxillin (Pxn WT), or V5-tagged Cat-1 KK-RA. One set (A) was lysed and then immunoblotted with GFP, V5, and actin antibodies. Soft agar assays were performed (B) on the other set of cells. The number of colonies that formed for each condition was determined. All of the experiments shown were performed three separate times, each yielding similar results. The data shown in B represent mean ± S.D. Student's t tests were performed. *, p < 0.05.
The Binding of Cat-1 to Paxillin Reverses Its Negative Regulatory Effects on Anchorage-independent Growth
The findings described in the preceding section suggest that paxillin might function in certain types of cells (i.e. cervical carcinoma cells and transformed fibroblasts) as a negative regulator of anchorage-independent growth. When considered together with our previous findings that Cat-1 needs to interact with paxillin to promote the growth of HeLa cells in soft agar (6), these results raised the intriguing possibility that Cat-1, by binding to paxillin, represses a negative regulatory effect exerted by paxillin on anchorage-independent cell growth. This further suggested that the inability of HeLa cells to exhibit a transformed growth phenotype under conditions where Cat-1 has been knocked down should be reversed by the simultaneous knockdown of paxillin. To directly investigate this possibility, we knocked down the expression of Cat-1 alone versus knocking down both Cat-1 and paxillin expression (Fig. 3A, compare the Cont. siRNA lane with the Cat-1 siRNA and Cat-1 siRNA and Pxn siRNA lanes) and then performed anchorage-independent growth assays on the cells. As shown previously (6), knocking down Cat-1 expression in HeLa cells markedly reduced their ability to form colonies in soft agar (Fig. 3B, compare the Cont. siRNA and Cat-1 siRNA columns). However, under conditions where both Cat-1 and paxillin expression was knocked down, HeLa cells again formed colonies nearly as well as cells expressing the control siRNA (Fig. 3B, Cat-1 siRNA and Pxn siRNA). These findings are consistent with the idea that Cat-1 binds paxillin and represses its ability to inhibit anchorage-independent growth.
FIGURE 3.
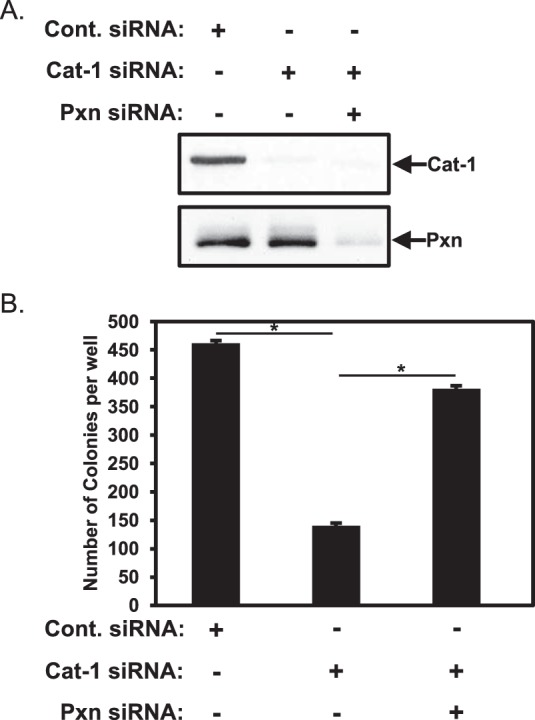
The inability of HeLa cells depleted of Cat-1 to grow under anchorage-independent conditions is restored by knocking down paxillin expression. A and B, two sets of HeLa cells were transfected with the indicated combinations of Cont. siRNA, Cat-1 siRNA, and Pxn siRNA. One set (A) was lysed and then immunoblotted with Cat-1 and paxillin antibodies. Soft agar assays were performed (B) on the other set of cells. The number of colonies that formed for each condition was determined. All of the experiments shown were performed three separate times, each yielding similar results. The data shown in B represent mean ± S.D. Student's t tests were performed. *, p < 0.05.
Cat-1 Maintains Akt Activation
We next set out to better understand how the formation of Cat-1·paxillin complexes positively supported anchorage-independent cell growth. Various intracellular signaling events have been implicated in the growth and survival responses required for cells to grow under anchorage-independent conditions, with one of the more common examples being the activation of the mTOR complex 1 and 2 (mTORC1 and mTORC2)-Akt pathway (20). To determine whether the binding of Cat-1 to paxillin could alter the activation of this pathway, HeLa cells were grown in suspension, and the resulting spheres that formed were stained with various combinations of antibodies that recognize paxillin, Cat-1, and Akt when it is phosphorylated at Ser-473. As a negative control, spheres were also stained with only the fluorescently conjugated secondary antibodies. Fig. 4 shows that very low levels of signal were detected in spheres stained with the secondary antibodies (Green or Red Secondary Ab), whereas paxillin appeared as small puncta throughout the cytosol of many of the HeLa cells that comprised the spheres (Pxn). Importantly, both Cat-1 (Fig. 4, Cat-1) and phosphorylated Akt (Fig. 4, P-Akt (Ser473)) localized with paxillin, suggesting that there might be a functional connection between these proteins.
FIGURE 4.
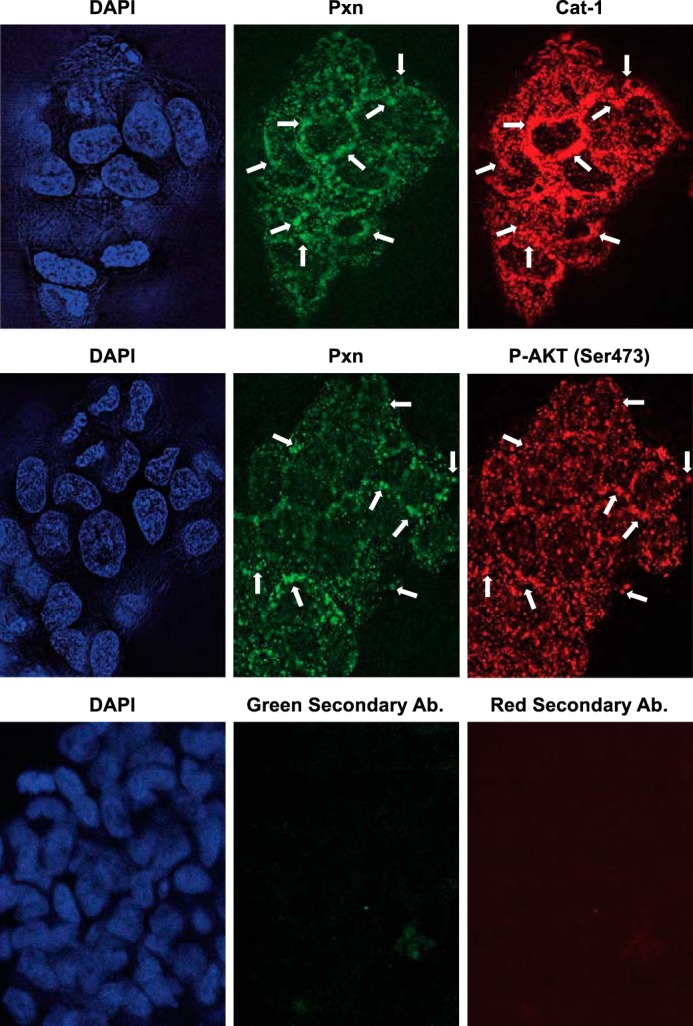
Paxillin, Cat-1, and activated AKT localize together in HeLa cells grown under anchorage-independent conditions as spheres. HeLa cells were cultured under anchorage-independent conditions by plating them on bacteria-grade dishes and allowing them to form spheres in suspension. The spheres were then collected and stained with the indicated combinations of Pxn, Cat-1, and phospho-Akt (Ser-473) antibodies. As a negative control, spheres of HeLa cells were also stained with only the fluorescently conjugated secondary antibodies (Green Secondary Ab and Red Secondary Ab). DAPI was used to label nuclei. Arrows indicate some of the areas where paxillin localizes with either Cat-1 or phospho-Akt (Ser-473). The experiments shown were performed three separate times, each yielding similar results.
We next introduced control siRNA or a Cat-1-targeted siRNA into HeLa cells and then analyzed the lysates generated from these cells by Western blotting analysis using antibodies that detect the phosphorylated (activated) forms of Akt and ERK, another protein that is important for mediating the anchorage-independent growth of some cancer cell types (20). Although we were unable to consistently detect changes in ERK activation following knockdown of Cat-1 in these cells (Fig. 5A, third panel, compare the Cont. siRNA and Cat-1 siRNA lanes), the phosphorylation levels of Akt at Ser-473 were significantly reduced (Fig. 5A, first panel, compare the Cont. siRNA and Cat-1 siRNA lanes). Akt phosphorylation was then restored by knocking down paxillin expression in cells that were already depleted of Cat-1 (Fig. 5A, first panel, compare the Cat-1 siRNA and Cat-1 and Pxn siRNA lanes). Interestingly, HeLa cells in which only paxillin was knocked down showed a much greater degree of phosphorylated Akt than their counterparts expressing control siRNA (Fig. 5B, top panel). These findings, combined with the fact that Akt phosphorylation is reduced in HeLa cells ectopically expressing wild-type paxillin (Fig. 5C, top panel), suggests that paxillin in some way imparts a negative regulation of Akt activation that can be abrogated upon binding of Cat-1 to paxillin.
FIGURE 5.
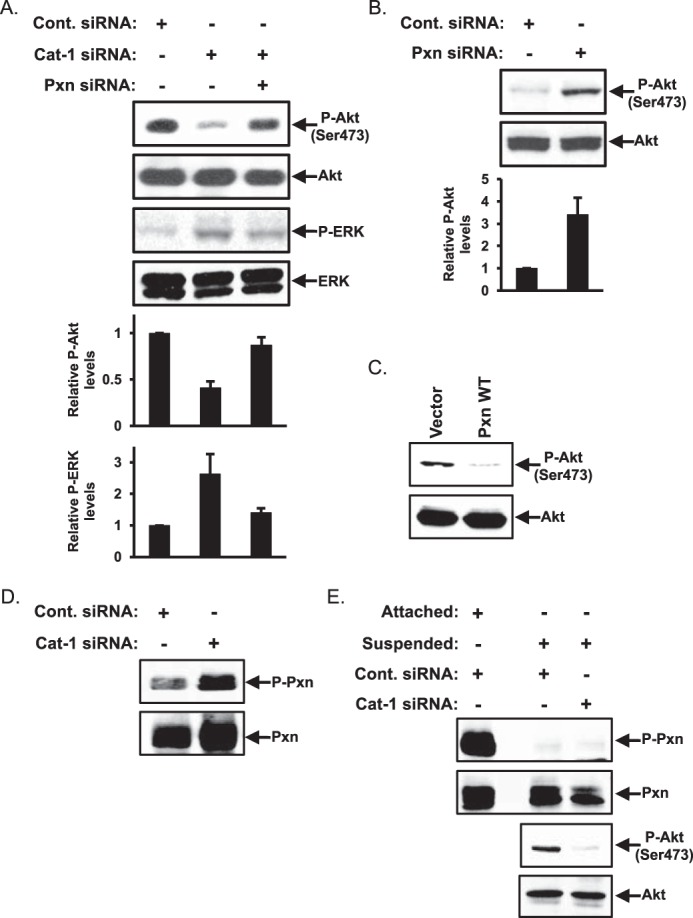
The levels of Akt phosphorylation are dependent on Cat-1 and paxillin expression. A, HeLa cells transfected with the indicated combinations of Cont. siRNA, Cat-1 siRNA, and Pxn siRNA were lysed and then immunoblotted with phospho-Akt (Ser-473), Akt, phospho-ERK, and ERK antibodies. B, HeLa cells transfected with either control siRNA or paxillin siRNA were lysed and then immunoblotted with phospho-Akt (Ser-473) and Akt antibodies. C, HeLa cells transfected with either the vector-alone (Vector) or GFP-tagged wild-type paxillin (Pxn WT) were lysed and then immunoblotted with phospho-AKT (Ser-473) and Akt antibodies. D, HeLa cells transfected with either Cont. siRNA or Cat-1 siRNA were lysed and then immunoblotted with phospho-Pxn (Tyr-118) and Pxn antibodies. E, HeLa cells transfected with either Cont. siRNA or Cat-1 siRNA were grown in a monolayer (Attached) or placed in suspension (Suspended) before being lysed and immunoblotted with phospho-Pxn (Tyr-118), Pxn, phospho-Akt (Ser-473), and Akt antibodies. All of the experiments shown were performed three separate times, each yielding similar results. The relative changes in ERK and AKT phosphorylation for each condition in A and B was determined.
Because the phosphorylation state of paxillin has been shown to influence its ability to regulate some cellular processes (i.e. promote focal complex turnover) (8–12), we determined whether paxillin phosphorylation might be important for inhibiting Akt activity. Lysates of HeLa cells expressing control siRNA or a Cat-1 siRNA were immunoblotted with an antibody that detects paxillin when it is phosphorylated at Tyr-118, a major regulatory site (8, 11, 12). However, only a slight increase in the levels of phosphorylated paxillin could be detected in cells in which Cat-1 expression was knocked down (Fig. 5D, top panel). The same experiment was also carried out on HeLa cells maintained in suspension, a condition that more closely reflects cells grown under anchorage-independent (soft agar) conditions. As expected, the levels of paxillin phosphorylation at Tyr-118 dramatically decreased in suspended cells expressing control siRNA compared with the same cells grown attached to a culture plate (Fig. 5E, first panel, compare the Attached cells expressing Cont. siRNA lane with the Suspended cells expressing Cont. siRNA lane). The phosphorylation of paxillin in suspended HeLa cells depleted of Cat-1 expression did not change (Fig. 5E, first panel, compare the Suspended cells expressing Cont. siRNA lane with the Suspended cells expressing Cat-1 siRNA lane) even though Akt phosphorylation decreased under these same conditions (Fig. 5E, third panel). Thus, changes in the phosphorylation state of paxillin do not appear to be directly responsible for its ability to negatively regulate the activation of Akt in HeLa cells.
We then determined whether the effect of knocking down Cat-1 expression on the phosphorylation of Akt was specific for a particular Akt isoform. The Akt family of proteins is made up of three highly related members, Akt1, Akt2, and Akt3. Akt1 and Akt2 are ubiquitously expressed and have been linked to promoting cell growth and survival responses, whereas Akt3 is expressed primarily in the brain (21), making it unlikely that this isoform would be playing a role in cervical carcinoma cells. Thus, we focused on Akt1 and Akt2. The lysates collected from HeLa cells expressing either control siRNA or Cat-1-targeted siRNA (Fig. 6A) were immunoblotted with Akt1- and Akt2-specific antibodies. Under conditions where Cat-1 expression was markedly reduced (Fig. 6A, top panel, Cat-1 siRNA), the overall levels of Akt1 and Akt2 were unchanged (Fig. 6A, center and bottom panels, compare the Cont. siRNA and Cat-1 siRNA lanes). To determine the extent to which each Akt isoform was activated, the same cell lysates were then probed with antibodies that can distinguish between Akt1 and Akt2 when they are phosphorylated at Ser-473. Fig. 6B shows that, under conditions where Cat-1 expression was knocked down in HeLa cells, the phosphorylation levels of Akt1 (left panel) and Akt2 (right panel) were decreased, suggesting that Cat-1 regulates the activity of both of these Akt isoforms.
FIGURE 6.
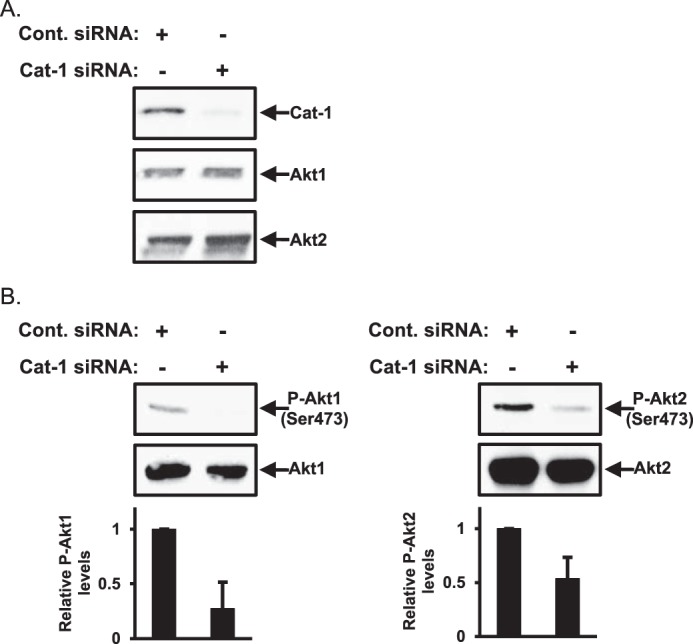
Cat-1 regulates the phosphorylation of both Akt1 and Akt2. A, HeLa cells transfected with either Cont. siRNA or Cat-1 siRNA were lysed and then immunoblotted with Cat-1, Akt1, and Akt2 antibodies. B, the same cell lysates generated in A were further immunoblotted with antibodies that specifically detect phospho-Akt1 (Ser-473) and Akt1 (left panel) and phospho-Akt2 (Ser-473) and Akt2 (right panel). All of the experiments shown were performed three separate times, each yielding similar results. The relative changes in Akt1 and Akt2 phosphorylation for each condition in B was determined.
Akt Activation Is Necessary for Anchorage-independent Growth
We next examined whether Akt activity was important for the Cat-1-mediated anchorage-independent growth of HeLa cancer cells by knocking down either Akt1 or Akt2, individually or in combination (Fig. 7A), and assaying their ability to form colonies in soft agar. HeLa cells expressing control siRNA (as a positive control) or Cat-1 siRNA (as a negative control) were also assayed. Knocking down the expression of either Akt isoform reduced the amount of colonies formed in soft agar, although knockdown of Akt2 consistently had a greater effect (Fig. 7B, compare the Cont. siRNA column with the Akt1 siRNA and Akt2 siRNA columns). Interestingly, simultaneous knockdown of both Akt1 and Akt2 or treatment of HeLa cells with an Akt inhibitor (Akt inhibitor VIII), a cell-permeable inhibitor that blocks both Akt isoforms, decreased colony formation by HeLa cells to a similar extent as knocking down Cat-1 (Fig. 7B, compare the Cont. siRNA column with the Akt1 plus Akt2 siRNA and Akt inhibitor VIII columns). These results suggest that the role of Cat-1 in helping maintain the activation of Akt is necessary for the anchorage-independent growth of HeLa cells.
FIGURE 7.
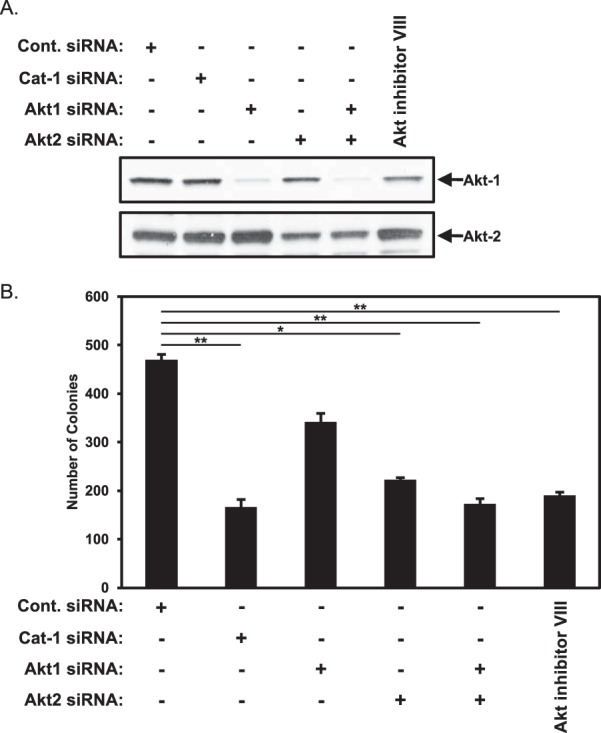
Akt1 and Akt2 contribute to the anchorage-independent growth of HeLa cells. A and B, two sets of HeLa cells were transfected with the indicated combinations of Cat-1 siRNA, Akt1 siRNA, and Akt2 siRNA or treated with Akt inhibitor VIII (2 μm). One set (A) was lysed and then immunoblotted with Akt1 and Akt2 antibodies. Soft agar assays were performed (B) on the other set of cells. The number of colonies that formed for each condition was determined. All of the experiments were performed three separate times, each yielding similar results. The data shown in B represent mean ± S.D. Student's t tests were performed. *, p < 0.05; **, p < 0.01.
We then went on to determine whether maintaining Akt activity in HeLa cells, under conditions where Cat-1 was knocked down, restored their ability to exhibit anchorage-independent growth. Specifically, we tested constitutively active forms of Akt1 and Akt2 in which aspartic acid residues replaced Thr-308 and Ser-473, i.e. the key residues that need to be phosphorylated for activation (20–23). The cells were then transfected with siRNAs targeting Cat-1 and assayed for colony formation in soft agar to see whether the constitutively active Akt mutants could reverse the inhibition of colony formation caused by knocking down Cat-1 expression. Fig. 8, A and B, shows that the expression of a phosphomimetic Akt1 or Akt2 mutant in cells where endogenous Cat expression had been knocked down, gave rise to a partial recovery of the ability of HeLa cells to grow in soft agar. However, co-expression of the phosphomimetic Akt1 and Akt2 mutants in HeLa cells in which Cat-1 expression was knocked down led to a complete rescue of anchorage-independent growth (Fig. 8B, compare the Cat-1 siRNA column with the Cat-1 siRNA plus Akt1 DD and Akt2 DD columns).
FIGURE 8.
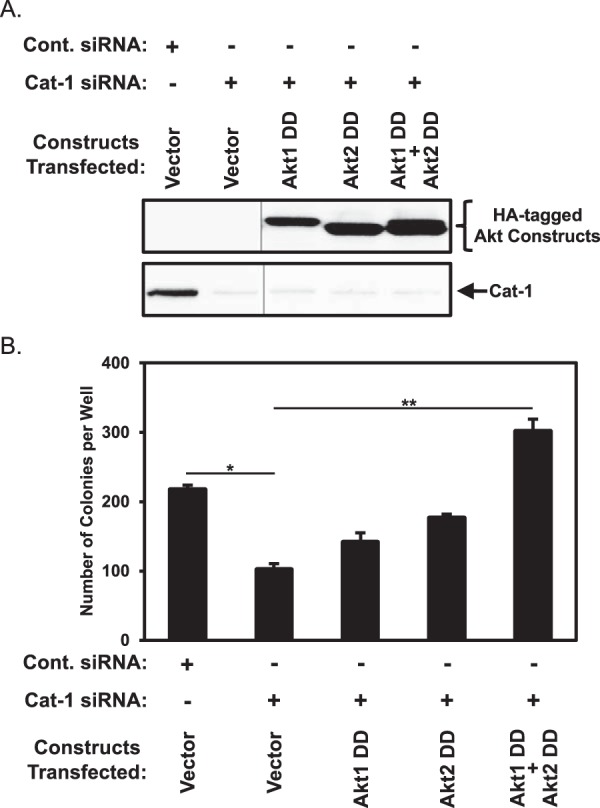
The block in HeLa cell transformation caused by the depletion of Cat-1 is rescued by the ectopic expression of activated forms of AKT. A and B, two sets of HeLa cells stably expressing the indicated combinations of the vector alone and constitutively active forms of Akt1 (Akt1 DD) and Akt2 (Akt2 DD) were transfected with either Cont. siRNA or Cat-1 siRNA. One set (A) was lysed and then immunoblotted with Cat-1 and HA antibodies. The vertical lines represent a portion of the blots that were cropped. Soft agar assays were performed (B) on the other set of cells. The number of colonies that formed for each condition was determined. All of the experiments shown were performed three separate times, each yielding similar results. The data shown in B represent mean ± S.D. Student's t tests were performed. *, p < 0.05; **, p < 0.01.
Cat-1 Mediates an Inverse Relationship between Akt and p70S6 Kinase Activation
These results indicate that Akt activation plays a key role in the ability of Cat-1 to promote anchorage-independent growth, although the molecular mechanism linking Cat-1 to Akt was unknown. However, we found that, in HeLa cells, there was an inverse relationship between the activation of Akt and p70S6 kinase so that when Akt activation was reduced as a result of knocking down Cat-1 expression, there was a concomitant increase in p70S6 kinase activation (Fig. 9A, first and third panels, Cat-1 siRNA). The opposite was true when paxillin expression was knocked down; Akt activity was increased, whereas the activating phosphorylation of p70S6 kinase was decreased (Fig. 9A, first and third panels, Pxn siRNA). These results are consistent with findings that show that excessive mTORC1-p70S6 kinase activity leads to the down-regulation of PI3 kinase and, in turn, mTORC2 and Akt (22, 23).
FIGURE 9.
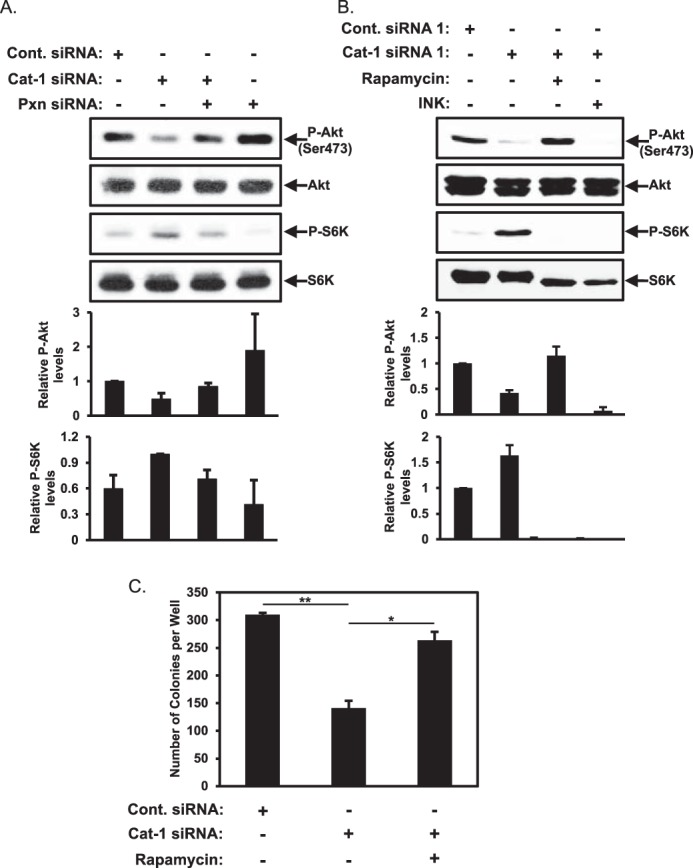
Paxillin inhibits Akt activity through an mTORC1-mediated negative feedback loop. A, HeLa cells transfected with the indicated combinations of Cont. siRNA, Cat-1 siRNA, and Pxn siRNA were lysed and then immunoblotted with phospho-Akt (Ser-473), Akt, phospho-p70S6 kinase (p-S6K), and p70S6 kinase (S6K) antibodies. B and C, two sets of HeLa cells were treated with the indicated combinations of Cont. siRNA, Cat-1 siRNA, rapamycin, and INK 128 (INK). One set (B) was lysed and then immunoblotted with phospho-Akt (Ser-473), Akt, phospho-p70S6 kinase, and p70S6 kinase antibodies. Soft agar assays were performed (C) on the other set of cells. The number of colonies that formed for each condition was determined. All of the experiments shown were performed three separate times, each yielding similar results. The relative changes in Akt and p70 S6 kinase phosphorylation for each condition in A and B was determined, whereas the data shown in C represent mean ± S.D. Student's t tests were performed. *, p < 0.05; **, p < 0.01.
We hypothesized that the down-regulation of Akt activity in HeLa cells where Cat-1 expression had been knocked down was caused by the up-regulation of p70S6 kinase activity. This led us to examine whether treating cells in which Cat-1 expression had been knocked down with rapamycin to reduce mTORC1 and p70S6 kinase activation would restore Akt activity. Indeed, rapamycin treatment restored Akt activation in cells where Cat-1 expression was knocked down (Fig. 9B, first panel, compare the Cat-1 siRNA lane with the Cat-1 siRNA plus Rapamycin lane). As further confirmation that the mTORC1-p70S6 kinase negative feedback loop is responsible for mediating this effect, we found that treatment of cells depleted of Cat-1 expression with INK 128 to block the activation of both mTORC1 and mTORC2 effectively ablated the phosphorylation of p70S6 kinase as well as that of Akt (Fig. 9B, first and third panels, Cat-1 siRNA plus INK). Next, we examined whether the restoration of Akt activity by rapamycin treatment is sufficient to reverse the blockage of anchorage-independent growth caused by knocking down Cat-1 expression. In fact, we found that treating HeLa cells where Cat-1 expression had been knocked down with rapamycin restored most of the anchorage-independent growth (Fig. 9C, compare the Cat-1 siRNA and Cat-1 siRNA plus Rapamycin columns). These results suggest that Cat-1 and potentially paxillin are regulating p70S6 kinase activity at the level of mTORC1.
Cat-1 Influences mTORC1/p70S6 Kinase through Its Regulatory Effects on the Arf1 GTPase
How is Cat-1 able to negatively influence mTORC1 activity and, in turn, increase the levels of Akt activity? Recent reports indicated that the Arf1 GTPase is capable of mediating the activation of mTORC1 (24, 25). Given that Cat-1 is a negative regulator of Arf1 GTPase activity by serving as a GAP (4, 5, 6), we hypothesized that Cat-1 might influence mTORC1 activity through its ability to negatively regulate Arf1. To test this idea, we monitored the levels of phosphorylated p70S6 kinase as a measure of mTORC1 activity in cells where Cat-1 expression had been knocked down and compared them with p70S6 kinase activity in cells where both Cat-1 and Arf1 had been knocked down. We found that knocking down both Cat-1 and Arf1 expression reduced the phosphorylation levels of p70S6 kinase so that they were comparable with those found in HeLa cells treated with just control siRNA (Fig. 10A, third panel, compare the Cat-1 siRNA and Cat-1 siRNA plus Arf1 siRNA lanes). This reduction in phospho-p70S6 kinase levels was accompanied by an increase in Akt activation (Fig. 10A, first panel, compare the Cat-1 siRNA and Cat-1 siRNA plus Arf1 siRNA lanes) as well as a restoration of anchorage-independent growth (Fig. 10B, compare the Cat-1 siRNA and Cat-1 siRNA plus Arf1 siRNA columns).
FIGURE 10.

The block in HeLa cell transformation caused by the depletion of Cat-1 is rescued by knocking down Arf1. A and B, two sets of HeLa cells were transfected with the indicated combinations of Cont. siRNA, Cat-1 siRNA, and Arf1 siRNA. One set (A) was lysed and then immunoblotted with phospho-Akt (Ser-473), Akt, phospho-p70S6 kinase, and p70S6 kinase antibodies. Soft agar assays were performed (B) on the other set of cells. The number of colonies that formed for each condition was determined. C, HeLa cells transfected with the indicated combinations of Cont. siRNA, Cat-1 siRNA, and Pxn siRNA were lysed and then used in pulldown assays using the Golgi-associated, γ adaptin ear-containing, Arf-binding protein 3 (GGA3)-GST fusion protein prebound to beads. The cell lysates (Input) as well as the cell lysate proteins that bound the GGA3 fusion protein (GGA3 pulldown) were immunoblotted with an Arf1 antibody. All of the experiments shown were performed three separate times, each yielding similar results. The relative changes in Akt phosphorylation for each condition in A and Arf1 activation in C were determined. The data shown in B represent mean ± S.D. Student's t tests were performed. *, p < 0.05; **, p < 0.01.
Thus, these findings suggest a mechanism by which Cat-1, through the negative regulation of Arf1 and a corresponding reduction in mTORC1 signaling to p70S6 kinase, resulted in the activation of Akt, thereby contributing a positive signal for anchorage-independent growth. We then asked how paxillin fit into this picture. Given that Cat-1 and paxillin undergo a direct interaction (26), we suspected that paxillin may be regulating the mTOR/Akt pathway by influencing the activation of Arf1. Thus, we monitored the cellular activation of Arf1 under different conditions where we knocked down Cat-1 and paxillin by using a pulldown assay that takes advantage of the ability of GTP-bound Arf1 to bind specifically to its signaling effector, GGA3. As expected, we found that knocking down Cat-1 expression led to a marked increase in GTP-bound (activated) Arf1 in HeLa cells (Fig. 10C, top panel, compare the Cont. siRNA and Cat-1 siRNA lanes). However, the majority of the increase in the levels of activated Arf1 that accompanied the knockdown of its negative regulator Cat-1 was lost when paxillin expression was knocked down together with Cat-1 (Fig. 10C, compare the Cat-1 siRNA and Cat-1 siRNA plus Pxn siRNA lanes). Concomitantly, there was a corresponding decrease in phospho-p70S6 kinase and an increase in phosphorylated Akt (Fig. 9A). We are currently further investigating how this might occur, although one intriguing possibility is a role for paxillin in promoting Arf1 activation through its ability to interact with the Arf guanine nucleotide exchange factor Arf nucleotide-binding site opener (ARNO) (27, 28).
Discussion
Cat-1 is a GAP that can inactivate members of the Arf GTPase family (i.e. Arf1 and Arf6). It also serves as a protein scaffold that has the potential to interact with a variety of binding partners, including Cool-1/β-Pix-1 and paxillin (1–3). Although Cat-1 is best known for its roles in regulating cell morphology and promoting cell migration (2, 29, 30), there has been accumulating evidence suggesting that Cat-1 can also contribute to several different aspects of cancer progression. For example, Cat-1 has been shown to be important for highly aggressive forms of breast (31), lung (32), and oral cancers (33) to metastasize to secondary sites as well as for the growth of liver and colon cancer cells (34). Moreover, we have recently shown that Cat-1 is overexpressed in a large majority of cervical carcinomas and is essential for the HeLa cervical cancer cell line to grow under anchorage-independent conditions. However, the mechanisms through which Cat-1 mediates its effects, especially as they pertain to the growth of cancer and transformed cells, are still not well understood.
Here we show that Cat-1 promotes the anchorage-independent growth of HeLa cells as well as fibroblasts that are transformed through the ectopic expression of an activated Cdc42 mutant as an outcome of its ability to interact with paxillin. Given the several connections linking paxillin to cancer progression (13–18), we initially assumed that Cat-1 and paxillin worked together to send a stimulatory signal that promoted this transformed phenotype. Instead, however, we discovered that Cat-1 interacts with paxillin to negatively regulate mTORC1/p70S6 kinase activation to an extent that ensures maximal activation of the cell growth/survival protein Akt (35). A working model that is consistent with the results we have obtained thus far is presented in Fig. 11. In this model, the role of Cat-1 is to antagonize the positive regulation of Arf1 by paxillin and thereby prevent it from signaling the activation of mTORC1 and p70S6 kinase, which, in turn, removes a negative regulation of mTORC2 and Akt normally mediated by activated p70S6 kinase. Thus, the positive actions of Cat-1 on Akt activation and, ultimately, anchorage-independent growth are an outcome of Cat-1 binding to paxillin and exerting its Arf-GAP activity, which would reverse any activation of Arf1 induced by paxillin alone. Knocking down Cat-1 has a negative effect on anchorage-independent growth through the loss of its antagonistic properties on the Arf1-mTORC1-p70S6 kinase pathway. However, knocking down paxillin removes a positive stimulatory input into this pathway (i.e. by eliminating the paxillin-mediated activation of Arf1) and thereby eliminates the deleterious effects caused by knocking down Cat-1. It is interesting to note that this newly described role for Cat-1 is similar to that of DEP domain-containing mTOR-interacting protein (DEPTOR), a protein that was identified as an inhibitor of mTORC1 activation (36). When overexpressed in multiple myeloma cells, DEPTOR promotes cell survival by suppressing mTORC1 activity and promoting Akt activity.
FIGURE 11.
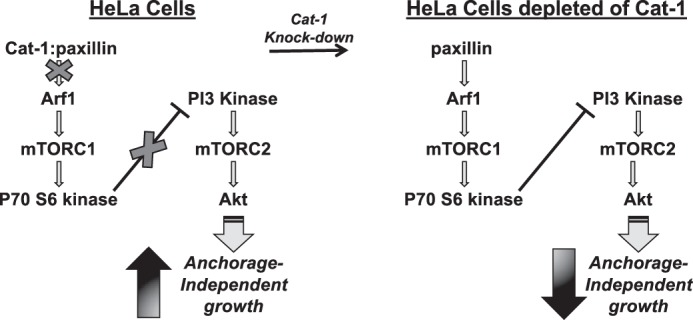
Diagram showing a plausible mechanism through which Cat-1 functions in HeLa cells to promote cellular transformation. Cat-1 binds to paxillin to abrogate its actions as a negative regulator of anchorage-independent growth (left). Specifically, the interaction between Cat-1 and paxillin perturbs the ability of paxillin to activate the Arf1-mTORC1-p70S6 kinase pathway, which ensures that the necessary level of AKT activation needed for supporting anchorage-independent growth (i.e. colony formation in soft agar) is met. However, under conditions where Cat-1 is knocked down (right), the Arf1-mTORC1-p70S6 kinase pathway is activated, leading to a p70S6 kinase-mediated negative regulation of Akt activity and loss of anchorage-independent growth.
It seems likely that the positive and negative inputs provided by paxillin and Cat-1 into the Arf1-mTORC1-p70S6 kinase pathway need to be very carefully balanced. Moreover, it is easy to imagine how, in some types of cancers, the stimulatory input provided by paxillin into the mTORC1/p70S6 kinase pathway may have a positive role in cancer cell growth and survival, as has sometimes been reported. Our future efforts will be directed toward understanding how the Cat-1-mediating signaling balance outlined in Fig. 11 is achieved in different contexts of malignant transformation as well as identifying additional proteins that play a role in this pathway. It will also be important to learn more about how Akt contributes stimulatory signals to the ability of cancer cells to exhibit anchorage-independent growth.
Experimental Procedures
Reagents
The Cat-1/Git-1 (catalog no. sc-13961), Arf1 (catalog no. sc-53168), and GFP (catalog no. sc-8334) antibodies were purchased from Santa Cruz Biotechnology and used at 1:500 dilutions. The phospho-p70S6 kinase (catalog no. 9205), p70S6 kinase (catalog no. 9202), phospho-Akt (Ser-473, catalog no. 4060), pan-Akt (catalog no. 9272), Akt1 (catalog no. 2938), Akt2 (catalog no. 2964), phospho-Akt1 (Ser-473, catalog no. 9018), phospho-Akt2 (Ser-474, catalog no. 8599), phospho-ERK (catalog no. 4370), ERK (catalog no. 9102), V5 (catalog no. 13202), HA (catalog no. 3724), actin (catalog no. 8456), and phospho-paxillin (Tyr-118, catalog no. 2541) primary antibodies as well as the mouse and rabbit IgG horseradish peroxidase-conjugated secondary antibodies (catalog nos. 7074 and 7076, respectively), were from Cell Signaling Technology and used at 1:2000 dilutions. The paxillin antibody (catalog no. 05-417) was from EMD Millipore and used at a 1:1000 dilution. AKT inhibitor VIII (catalog no. 124018) and rapamycin (catalog no. 553210) were also from EMD Millipore and used at 0.2 μm and 0.1 μm, respectively. INK 128 (catalog no. S2811) was from Selleckchem and used at 0.5 μm. The precast 4–20% Tris/glycine acrylamide gels were from Thermo Fisher Scientific. The PVDF transfer membrane and the Western Lighting Plus ECL reagent were from PerkinElmer Life Sciences. The protein G-agarose beads and all cell culture reagents, including RPMI 1640 medium, DMEM, calf serum, FBS, Lipofectamine, and puromycin, as well as the control siRNA and siRNAs targeting Cat-1, paxillin, Akt1, Akt2, and Arf1, were purchased from Invitrogen.
Plasmids
The V5-tagged pcDNA3 expression construct encoding a dominant negative form of rat Cat-1 (Cat-1 KK-RA), the pEGFP expression construct encoding chicken paxillin, and the HA-tagged lentiviral vector pCDH-CMV-MOS-EF1-Puro (System BioSciences) encoding human Akt1 and Akt2 were generated previously (6). The constitutively active mutant forms of Akt1, termed Akt1 DD (T308D/S473D), and Akt2, termed Akt2 DD (T309D/S474D), were generated using the QuikChangeTM site-directed mutagenesis kit (Stratagene).
Lentivirus Generation
The HA-tagged lentiviral vector pCDH-CMV-MOS-EF1-Puro encoding wild-type and mutant forms of Akt1 and Akt2 were transfected into HEK293T cells using Lipofectamine (Invitrogen). The mature lentiviruses shed into the culturing medium from the transfectants were collected and processed according to the instructions of the manufacturer (System BioSciences).
Cell Culture
HeLa cells were grown in RPMI 1640 medium containing 10% FBS, whereas NIH3T3 cells stably expressing an activated form of Cdc42 (Cdc42 F28L) were grown in DMEM containing 10% calf serum. The siRNAs and the expression constructs encoding various forms of Cat-1 and paxillin were transfected into cells using Lipofectamine, whereas the Akt constructs were introduced into cells via lentiviral infection. HeLa cells expressing HA-tagged forms of Akt were selected for and maintained by supplementing the growth medium with 0.175 μg/ml puromycin. As indicated, cells were treated with 2.0 μm AKT inhibitor VIII, 0.1 μm rapamycin, and 0.5 μm INK 128.
Immunofluorescence
HeLa cells grown on glass coverslips were either mock-transfected, or transfected with a construct encoding GFP-tagged wild-type paxillin. In certain cases, HeLa cells were also grown under anchorage-independent conditions by plating them on bacteria-grade dishes and allowing them to form spheres. The cells grown on coverslips or in suspension as spheres were fixed with 3.7% formaldehyde, permeabilized with PBS containing 0.1% Triton X-100, and then blocked with 10% bovine serum albumin diluted in PBS. The cells were incubated with GFP, phospho-Akt (Ser-473), and Cat-1 antibodies at 1:500 dilutions, whereas the paxillin clone 165 antibody (catalog no. 610620) from BD Biosciences was used at a 1:200 dilution. The primary antibodies were detected using Alexa Fluor 488-conjugated anti-mouse (catalog no. A-11034) and Rhodamine-conjugated anti-rabbit (catalog no. R-6394) secondary antibodies from Molecular Probes at 1:800 dilutions. DAPI was used to label nuclei. Digital images of the cells were captured using a Zeiss fluorescence microscope outfitted with a Sensicam qe camera (Cooke Co.) and processed using IPLABS software (BD Biosciences).
Immunoblot Analysis
Cells were lysed with cell lysis buffer (25 mm Tris (pH 7.4), 100 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1 mm DTT, 1 mm NaVO4, 1 mm β-glycerol phosphate, 1 mm aprotinin, 1 mm leupeptin, and 1 mm PMSF). The lysates were resolved by SDS-PAGE, and then the proteins were transferred to PVDF membranes. The membranes were incubated with the indicated primary antibodies diluted in 20 mm Tris (pH 7.4), 135 mm NaCl, and 0.02% Tween 20. The primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies followed by exposure to ECL reagent.
Immunoprecipitations
Cell extracts (typically 500 μg of each) were incubated with primary antibodies for 2 h at 4 °C with rotation. Protein G-agarose beads were then added to the lysates for an additional hour, at which point the beads were washed extensively with cell lysis buffer before being resuspended in Laemmli sample buffer and boiled.
Soft-agar Assays
HeLa cells and NIH3T3 cells stably expressing Cdc42 F28L, transfected with various siRNAs or expression plasmids or infected with a lentivirus as indicated, were plated at a density of 5 × 103 cells/ml in medium containing 0.3% agarose onto underlays composed of growth medium containing 0.6% agarose in 6-well dishes. The cultures were fed once a week, and, after 14 days, the colonies were counted.
Arf GTPase Activity Assays
A GST fusion protein containing the N-terminal portion of Golgi-associated, γ adaptin ear-containing, Arf-binding protein 3 bound to a glutathione resin was purchased from Pierce. The activity assays were performed according to the instructions of the manufacturer.
Author Contributions
S. M. Y., A. L., M. A. A., and R. A. C. designed the study, performed the experiments, and wrote the manuscript.
Acknowledgments
We thank Cindy Westmiller for expert assistance with preparing the manuscript.
This work was supported by National Institutes of Health Grants GM040654, GM047458, and CA201402 (to R. A. C.) The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Cat-1
- Cool-associated tyrosine phosphorylated protein 1
- ARF
- ADP-ribosylation factor
- GAP
- GTPase-activating protein
- Pxn
- paxillin
- Cont.
- control
- mTOR
- mammalian target of rapamycin.
References
- 1. Bagrodia S., Bailey D., Lenard Z., Hart M., Guan J. L., Premont R. T., Taylor S. J., and Cerione R. A. (1999) A tyrosine-phosphorylated protein that binds to an important regulatory region on the cool family of p21-activated kinase-binding proteins. J. Biol. Chem. 274, 22393–22400 [DOI] [PubMed] [Google Scholar]
- 2. Hoefen R. J., and Berk B. C. (2006) The multifunctional GIT family of proteins. J. Cell Sci. 119, 1469–1475 [DOI] [PubMed] [Google Scholar]
- 3. Inoue H., and Randazzo P. A. (2007) Arf GAPs and their interacting proteins. Traffic 8, 1465–1475 [DOI] [PubMed] [Google Scholar]
- 4. Mazaki Y., Hashimoto S., Okawa K., Tsubouchi A., Nakamura K., Yagi R., Yano H., Kondo A., Iwamatsu A., Mizoguchi A., and Sabe H. (2001) An ADP-ribosylation factor GTPase-activating protein Git2-short/KIAA0148 is involved in subcellular localization of paxillin and actin cytoskeletal organization. Mol. Biol. Cell 12, 645–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vitale N., Patton W. A., Moss J., Vaughan M., Lefkowitz R. J., and Premont R. T. (2000) GIT proteins, a novel family of phosphatidylinositol 3,4,5-trisphosphate-stimulated GTPase-activating proteins for ARF6. J. Biol. Chem. 275, 13901–13906 [DOI] [PubMed] [Google Scholar]
- 6. Yoo S. M., Antonyak M. A., and Cerione R. A. (2012) The adaptor protein and Arf GTPase-activating protein Cat-1/Git-1 is required for cellular transformation. J. Biol. Chem. 287, 31462–31470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hamburger A. W., and Salmon S. E. (1977) Primary bioassay of human tumor stem cells. Science 197, 461–463 [DOI] [PubMed] [Google Scholar]
- 8. Deakin N. O., and Turner C. E. (2008) Paxillin comes of age. J. Cell Sci. 121, 2435–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Turner C. E., Glenney J. R. Jr, and Burridge K. (1990) Paxillin: a new vinculin-binding protein present in focal adhesions. J. Cell Biol. 111, 1059–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Digman M. A., Brown C. M., Horwitz A. R., Mantulin W. W., and Gratton E. (2008) Paxillin dynamics measured during adhesion assembly and disassembly by correlation spectroscopy. Biophys. J. 94, 2819–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. West K. A., Zhang H., Brown M. C., Nikolopoulos S. N., Riedy M. C., Horwitz A. F., and Turner C. E. (2001) The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J. Cell Biol. 154, 161–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Efimov A., Schiefermeier N., Grigoriev I., Ohi R., Brown M. C., Turner C. E., Small J. V., and Kaverina I. (2008) Paxillin-dependent stimulation of microtubule catastrophes at focal adhesion sites. J. Cell Sci. 121, 196–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Azuma K., Tanaka M., Uekita T., Inoue S., Yokota J., Ouchi Y., and Sakai R. (2005) Tyrosine phosphorylation of paxillin affects the metastatic potential of human osteosarcoma. Oncogene 24, 4754–4764 [DOI] [PubMed] [Google Scholar]
- 14. Nagata M., Fujita H., Ida H., Hoshina H., Inoue T., Seki Y., Ohnishi M., Ohyama T., Shingaki S., Kaji M., Saku T., and Takagi R. (2003) Identification of potential biomarkers of lymph node metastasis in oral squamous cell carcinoma by cDNA microarray analysis. Int. J. Cancer 106, 683–689 [DOI] [PubMed] [Google Scholar]
- 15. Zhao Y., Zhang X., Guda K., Lawrence E., Sun Q., Watanabe T., Iwakura Y., Asano M., Wei L., Yang Z., Zheng W., Dawson D., Willis J., Markowitz S. D., Satake M., and Wang Z. (2010) Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc. Natl. Acad. Sci. U.S.A. 107, 2592–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao C. J., Du S. K., Dang X. B., and Gong M. (2015) Expression of paxillin is correlated with clinical prognosis in colorectal cancer patients. Med. Sci. Monit. 21, 1989–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deakin N. O., Pignatelli J., and Turner C. E. (2012) Diverse roles for the paxillin family of proteins in cancer. Genes Cancer 3, 362–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wade R., Brimer N., Lyons C., and Vande Pol S. (2011) Paxillin enables attachment-independent tyrosine phosphorylation of focal adhesion kinase and transformation by RAS. J. Biol. Chem. 286, 37932–37944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salgia R., Li J. L., Ewaniuk D. S., Wang Y. B., Sattler M., Chen W. C., Richards W., Pisick E., Shapiro G. I., Rollins B. J., Chen L. B., Griffin J. D., and Sugarbaker D. J. (1999) Expression of the focal adhesion protein paxillin in lung cancer and its relation to cell motility. Oncogene 18, 67–77 [DOI] [PubMed] [Google Scholar]
- 20. Guadamillas M. C., Cerezo A., and Del Pozo M. A. (2011) Overcoming anoikis: pathways to anchorage-independent growth in cancer. J. Cell Sci. 124, 3189–3197 [DOI] [PubMed] [Google Scholar]
- 21. Gonzalez E., and McGraw T. E. (2009) The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 8, 2502–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Efeyan A., and Sabatini D. M. (2010) mTOR and cancer: many loops in one pathway. Curr. Opin. Cell Biol. 22, 169176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li L., Kim E., Yuan H., Inoki K., Goraksha-Hicks P., Schiesher R. L., Neufeld T. P., and Guan K. L. (2010) Regulation of mTORC1 by the Rab and Arf GTPases. J. Biol. Chem. 285, 19705–19709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jewell J. L., Kim Y. C., Russell R. C., Yu F. X., Park H. W., Plouffe S. W., Tagliabracci V. S., and Guan K. L. (2015) Metabolism: differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng Q., Baird D., Yoo S., Antonyak M., and Cerione R. A. (2010) Phosphorylation of the cool-1/β-Pix protein serves as a regulatory signal for the migration and invasive activity of Src-transformed cells. J. Biol. Chem. 285, 18806–18816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Torii T., Miyamoto Y., Sanbe A., Nishimura K., Yamauchi J., and Tanoue A. (2010) Cytohesin-2/ARNO, through its interaction with focal adhesion adaptor protein paxillin, regulates preadipocyte migration via the downstream activation of Arf6. J. Biol. Chem. 285, 24270–24281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen L. A., Honda A., Varnai P., Brown F. D., Balla T., and Donaldson J. G. (2007) Active Arf6 recruits ARNO/cytohesin GEFs to the PM by binding their PH domains. Mol. Biol. Cell 18, 2244–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu J. A., Deakin N. O., and Turner C. E. (2010) Emerging role of paxillin-PKL in regulation of cell adhesion, polarity and migration. Cell Adh. Migr. 4, 342–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frank S. R., and Hansen S. H. (2008) The PIX-GIT complex: a G protein signaling cassette in control of cell shape. Semin. Cell Dev. Biol. 19, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chan S. H., Huang W. C., Chang J. W., Chang K. J., Kuo W. H., Wang M. Y., Lin K. Y., Uen Y. H., Hou M. F., Lin C. M., Jang T. H., Tu C. W., Lee Y. R., Lee Y. H., Tien M. T., and Wang L. H. (2014) MicroRNA-149 targets GIT1 to suppress integrin signaling and breast cancer metastasis. Oncogene 33, 4496–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang J. S., Su C. Y., Yu W. H., Lee W. J., Liu Y. P., Lai T. C., Jan Y. H., Yang Y. F., Shen C. N., Shew J. Y., Lu J., Yang C. J., Huang M. S., Lu P. J., Lin Y. F., et al. (2015) GIT1 promotes lung cancer cell metastasis through modulating Rac1/Cdc42 activity and is associated with poor prognosis. Oncotarget 6, 36278–36291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang W. C., Chan S. H., Jang T. H., Chang J. W., Ko Y. C., Yen T. C., Chiang S. L., Chiang W. F., Shieh T. Y., Liao C. T., Juang J. L., Wang H. C., Cheng A. J., Lu Y. C., and Wang L. H. (2014) miRNA-491–5p and GIT1 serve as modulators and biomarkers for oral squamous cell carcinoma invasion and metastasis. Cancer Res. 7, 751–764 [DOI] [PubMed] [Google Scholar]
- 34. Peng H., Dara L., Li T. W., Zheng Y., Yang H., Tomasi M. L., Tomasi I., Giordano P., Mato J. M., and Lu S. C. (2013) MAT2B-GIT1 interplay activates MEK1/ERK 1 and 2 to induce growth in human liver and colon cancer. Hepatology 57, 2299–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vivanco I., and Sawyers C. L. (2002) The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 [DOI] [PubMed] [Google Scholar]
- 36. Peterson T. R., Laplante M., Thoreen C. C., Sancak Y., Kang S. A., Kuehl W. M., Gray N. S., and Sabatini D. M. (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886 [DOI] [PMC free article] [PubMed] [Google Scholar]