

Abstract
The nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) is essential for placental development. Here, we show that the mucin gene Muc1 is a PPARγ target, whose expression is lost in PPARγ null placentas. During differentiation of trophoblast stem cells, PPARγ is strongly induced, and Muc1 expression is upregulated by the PPARγ agonist rosiglitazone. Muc1 promoter is activated strongly and specifically by liganded PPARγ but not PPARα or PPARδ. A PPAR binding site (DR1) in the proximal Muc1 promoter acts as a basal silencer in the absence of PPARγ, and its cooperation with a composite upstream enhancer element is both necessary and sufficient for PPARγ-dependent induction of Muc1. In the placenta, MUC1 protein is localized exclusively to the apical surface of the labyrinthine trophoblast around maternal blood sinuses, resembling its luminal localization on secretory epithelia. Last, variably penetrant maternal blood sinus dilation in Muc1-deficient placentas suggests that Muc1 regulation by PPARγ contributes to normal placental development but also that the essential functions of PPARγ in the organ are mediated by other targets.
Peroxisome proliferator-activated receptor γ (PPARγ) is an orphan nuclear receptor with diverse biological activities of prime clinical importance (20). It heterodimerizes with RXR to regulate transcription of target genes through response elements (PPREs) comprised of direct repeats of a core motif spaced by 1 bp (underlined) (AGGTCA N AGGTCA; DR1) (15). PPARγ is the molecular target for the thiazolidinedione class of insulin sensitizers, which are widely prescribed for the treatment of type II diabetes (9). It is also a key regulator of adipocyte differentiation and regulates genes mediating lipid homeostasis pathways in adipocytes and macrophages (4, 6, 30). In addition, PPARγ has been implicated as a differentiation factor and a potential anti-oncogenic target in breast and colon cancer (27).
PPARγ deficiency results in death by the 10th day of gestation (E10.0) (3). At this developmental stage, PPARγ is expressed abundantly and exclusively in the placenta, and rescue of PPARγ null embryos to term by tetraploid chimeras shows that its essential functions are confined to the trophoblast (3). During placentation, structures transducing either maternal or fetal blood interdigitate to form a labyrinthine network of vessels. Histological studies reveal that PPARγ null placentas fail to form this vascular labyrinth (3). Fetal blood circulates in the placenta in endothelium-lined vessels that adhere intimately to the trophoblast. In PPARγ-deficient placentas, the tight interface between the trophoblast and the fetal endothelium is severely disrupted, and consequently, fetal vessels arrest at the chorionic plate. Once in the placenta, maternal blood leaves the arterial system and bathes the trophoblast through a series of small blood pools, or lacunae, that are lined immediately by the labyrinthine trophoblast (2). These blood pools are dilated and torn in PPARγ null placentas, forming an abnormal, continuous blood sinus on the maternal side of the labyrinth, with overt phagocytosis of maternal erythrocytes by junctional zone trophoblasts (3). Normal labyrinthine trophoblast differentiates into a barrier epithelium that separates the maternal and fetal circulations while performing the essential exchange of metabolites between the two (7). This differentiation is critical for vascular remodeling, as demonstrated in various mouse mutants (21). However, the labyrinthine trophoblast of PPARγ null placentas fails to undergo typical morphological and cellular changes, such as compaction, syncytium formation, and lipid droplet accumulation (3). Embryos deficient for RXRα, alone or in combination with RXRβ, exhibit defects that are similar to those seen in PPARγ null placentas, demonstrating the functional dependency of PPARγ on RXR (22, 31).
Although the list of defects in PPARγ null placentas is extensive, no specific target genes have been established for these phenotypes. Here, an effort to identify and characterize transcriptional targets of PPARγ revealed that the mucin gene Muc1 is a tightly regulated PPARγ target in the placenta and differentiated trophoblast stem cells. This regulation is mediated by the cooperative action of PPARγ-binding and nonbinding elements in the proximal part of the Muc1 promoter, whose protein product is confined to the trophoblast layer surrounding the maternal lacunae. This asymmetric distribution is analogous to the previously established localization of MUC1 protein on luminal surfaces of simple secretory epithelia (5) and implicates the maternal lacunae in the placenta as the anatomical analogues of secretory lumens. About half of Muc1 null placentas exhibit dilation of the maternal lacunae, suggesting that Muc1 may participate in this aspect of the PPARγ null phenotype. Our data provide new mechanistic insights into PPARγ action in trophoblasts, both by implicating it in shared biological regulation of epithelia and trophoblast and by revealing novel combinatorial interactions of PPARγ in target regulation.
MATERIALS AND METHODS
Preparation of placental RNA.
Individual placentas were isolated from E9.5 embryo progeny of either PPARγ+/− (3) or RXRα+/− (28) breeder pairs and kept frozen at −80°C. The corresponding genotypes were determined by PCR of yolk sac DNA, as described previously (3), at which stage placentas with similar genotypes were pooled in groups of four, and RNA was extracted with Tri-Reagent. RNA preparations were further purified by treatment with RNase-free DNase and reextraction.
RDA.
Total RNA (1 μg) from either wild-type or PPARγ−/− placentas was converted to double-stranded cDNA, using the SMART PCR cDNA synthesis kit (Clontech). This cDNA was amplified through several rounds of long-range PCR, using Advantage Taq polymerase mix (Clontech). The amplified full-length cDNA was digested with DpnII and used to carry out reciprocal representational difference analysis (RDA) essentially as described previously (11), except that amplification of subtracted products was performed by using Advantage Taq polymerase mix and for 13 to 17 amplification cycles only. An additional modification was the supplementation of the subtracted driver cDNA population with Sau3AI-digested PPARγ (added to null driver) or lacZ and neo (added to the wild-type [wt] driver) to circumvent differential recloning of these genes. At the end of three rounds of subtraction-amplification, individual bands could be discerned on agarose gels, from which they were isolated and subcloned into pBluescript. Ten plasmid clones from each band were sequenced to determine its predominant composition, and sequences iterated more than once were subjected to BLAST analysis with the National Center for Biotechnology Information database to determine identity as well as being reprobed against RNA from PPARγ+/+, PPARγ+/−, and PPARγ−/− placentas to confirm true differentials.
Trophoblast stem (TS) cell culture.
GFP-Trf mouse trophoblast stem cells (29) were cultured on a feeder layer of embryonic fibroblasts in RPMI 1640 medium containing 20% serum, fibroblast growth factor 4 (FGF4; 25 ng/ml; Sigma), and heparin (1 μg/ml), with medium change every other day. Cells were passaged once in the absence of feeder cells in a similar medium supplemented with 70% embryonic fibroblast conditioned medium and then split for the various experiments. Differentiation was accomplished by withdrawing conditioned medium, FGF4, and heparin from the medium. Where appropriate, cultures were supplemented with 1 μM rosiglitazone.
Northern blots, EMSA, transfections, and reporter assays.
Northern blots and an electrophoretic mobility shift assay (EMSA) were carried out as described previously (3, 10). Supershift was performed using concentrated polyclonal α-PPARγ (H-100) or α-RXRα (D-20) antibodies (SantaCruz Biotech). Transfections of CV1 cells and reporter assays were carried out with a 48-well format as described previously (9), with some modifications. In short, wells containing 50 to 70% confluent CV1 cells were lipofected with the indicated plasmid combinations, using DOTAP (Avanti Polar Lipids, Inc.). Receptors, reporters, and cytomegalovirus (CMV)-lacZ controls were transfected at 25, 62, and 125 ng/well, respectively. Lipofection medium was replaced 3 to 5 h after transfection with Dulbecco's modified Eagle's medium containing 2% fetal calf serum and the indicated ligand combinations. Cells were extracted 24 to 36 h later and assayed for luciferase and β-galactosidase activities. Data shown reflect averages and standard deviations for normalized luciferase activity divided by β-galactosidase activity in triplicate wells from one representative experiment out of at least four repeats with qualitatively similar results.
Histology and immunofluorescence.
C57BL/6J Muc1+/− breeder pairs (26) were intercrossed, and pregnancies were timed by monitoring coital plugs. Embryos and placentas were retrieved from pregnant females at the indicated gestational day, and respective genotypes were determined by PCR of DNA from embryonic matter. For histology, placentas were fixed for 24 h in 10% formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. For immunofluorescence, placentas were fixed overnight in Bouin's solution, washed with running water for 6 h, embedded in paraffin, and sectioned at 5 μm. MUC1 was detected using a diluted hybridoma supernatant containing an Armenian hamster-derived monoclonal antibody against the short cytoplasmic tail (CT2) (S. J. Gendler, unpublished data) and secondary Cy3-conjugated goat anti-Armenian hamster antibody (10 μg/ml; Jackson Immunoresearch). Caveolin-1 was detected by using a polyclonal rabbit antiserum (5 μg/ml; Transduction Labs) and Alexa488-linked goat anti-rabbit antibody (5 μg/ml; Molecular Probes, Inc.). All incubations and washes were carried out in phosphate-buffered saline containing 0.05% Tween 20 and 5% normal goat serum.
RESULTS
Muc1 expression is lost in PPARγ null placentas.
To identify PPARγ target genes in trophoblasts, we screened for mRNA species enriched in wt versus PPARγ null placentas by using RDA (11). This screen identified the mucin-1 gene, Muc1 (25). Northern blot analysis confirmed that Muc1 is expressed in wt placentas at E9.5 (Fig. 1A, lanes 1 to 2, and 1B, lane 1) and is virtually absent from either PPARγ null or RXRα null placentas (Fig. 1A, lanes 5 and 6; Fig. 1B, lane 3, respectively). Moreover, placentas heterozygous for either PPARγ or RXRα express intermediate levels of Muc1 (Fig. 1A, lanes 3 and 4, and 1B, lane 2), suggesting that Muc1 expression is directly proportional to the amount of PPARγ-RXRα heterodimers in trophoblasts.
FIG. 1.
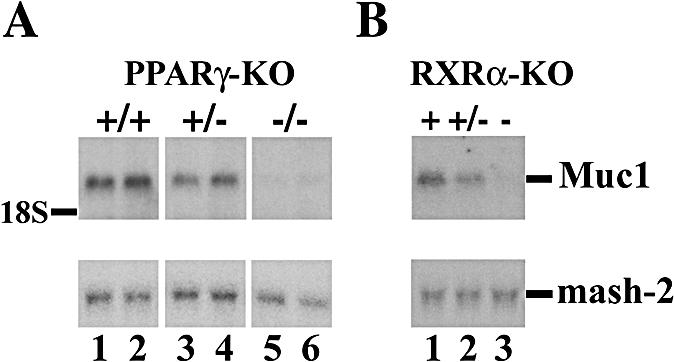
PPARγ and RXRα are obligatory for placental Muc1 expression. Northern blot analysis of RNA from pools of genotypically identical placentas at E9.5 reveals Muc1 expression in wt placentas and its loss in both PPARγ (A) and RXRα (B) null placentas. Placentas heterozygous for either nuclear receptor (+/−) express roughly half the wt level of Muc1. A Mash2 probe was used for quantitative normalization.
A PPARγ agonist stimulates Muc1 induction in murine trophoblast stem cells.
TS cells proliferate and retain stem cell status as long as they are supplemented with FGF4, heparin, and embryonic fibroblast conditioned medium (29). Once the additives are withdrawn, these cells undergo terminal differentiation, as manifested by the induction of the spongiotrophoblast-specific marker 4311 4 days later (Fig. 2A, lanes 5 and 9). While PPARγ is only minimally expressed in proliferating TS cells, it is dramatically induced with the onset of differentiation (Fig. 2B, lanes 1 to 5), recapitulating its association with trophoblast differentiation in the intact placenta (3). Interestingly, rosiglitazone treatment of differentiated TS cells attenuates PPARγ expression (Fig. 2B, compare lanes 6 through 9 to lanes 2 through 5), suggesting that PPARγ engages in negative autofeedback regulation.
FIG. 2.
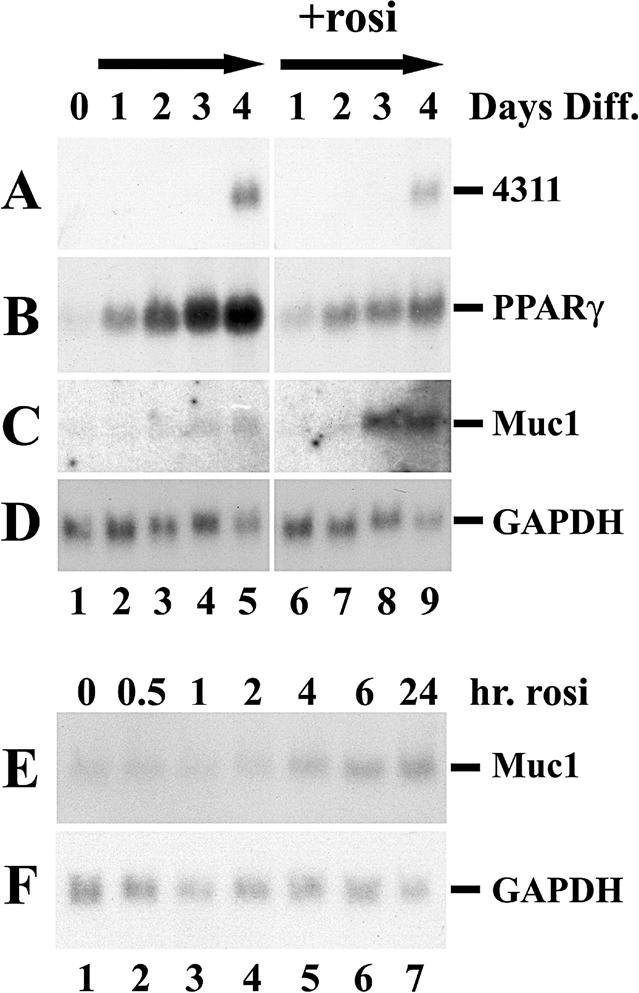
PPARγ and Muc1 induction during TS cell differentiation. (A-D) TS cells were grown continuously in the presence of FGF4 and embryonic fibroblast conditioned medium (lane 1) or induced to differentiate by factor withdrawal in the absence (lanes 2 to 5) or presence of 1 μM of the PPARγ agonist rosiglitazone (+rosi; lanes 6 to 9). RNA was extracted at daily intervals after the onset of differentiation (top), and expression of the spongiotrophoblast differentiation marker 4311 (A) and those of PPARγ (B), Muc1 (C), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (D) were analyzed by Northern blotting. (E-F) TS cells were induced to differentiate for 4 days and then treated with 1 μm rosiglitazone for the indicated durations (top). Expression of Muc1 (E) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (F) was analyzed.
Muc1 expression is detected in TS cells 3 days after the onset of differentiation (Fig. 2C, lane 4). When rosiglitazone is administered throughout differentiation, Muc1 is still induced only at the third day of differentiation but much more robustly (Fig. 2C, lanes 8 and 9), consistent with the notion of responsiveness to PPARγ. In contrast, if TS cells were allowed first to differentiate for 4 days, Muc1 can be induced as early as 4 h after rosiglitazone administration (Fig. 2E, lane 5); Muc1 transcripts continue to accumulate in predifferentiated TS cells for at least 48 h following ligand treatment (Fig. 2E, lanes 6 and 7; also data not shown). These observations suggest that Muc1 responds directly to liganded PPARγ in differentiated TS cells and that its failure to express prior to the third day of differentiation likely reflects delayed acquisition of transcriptional competence rather than a slow or indirect response to PPARγ.
Robust activation of the Muc1 promoter by PPARγ.
The Muc1 promoter was next characterized for response to PPARγ by reporter assays with CV1 cells. As shown previously (10), multimerized consensus PPAR response elements (3xDR1) respond readily to rosiglitazone in CV-1 cells in both the absence and presence of cotransfected RXRα and PPARγ (Fig. 3A). In contrast, the proximal Muc1 promoter (−715 to +33, with +1 denoting the 5′ end of Muc1 mRNA) required cotransfection of both PPARγ and RXRα to induce strong reporter activity (Fig. 3B). Unlike 3xDR1, Muc1 was induced weakly, albeit significantly, by PPARγ-RXRα even in the absence of an exogenous agonist (three- to fivefold in different experiments), and 1 μM rosiglitazone augmented its activity by an additional three- to fivefold (Fig. 3B). Similar response patterns and magnitudes were exhibited by fragments stretching from the +33 position to as far as −1836 or as near as the −535 position (data not shown). Thus, the proximal 535 bp of the Muc1 promoter contain sequence information that is necessary and sufficient for activation by PPARγ. In aggregate, the data presented thus far provide genetic, pharmacological, and transcriptional evidence that Muc1 is a primary PPARγ target gene.
FIG. 3.

Robust, PPARγ-specific activation of the Muc1 promoter by PPARγ/RXRα heterodimers. CV1 cells were transfected with luciferase reporters driven by either a trimerized consensus PPRE sequence (3xDR-1) (A, C) or a Muc1 promoter fragment stretching from −715 to +33 (Muc1 −715) (B, D). Luciferase activity was measured 24 to 48 h posttransfection and normalized to β-galactosidase activity of a cotransfected CMV-lacZ construct. (A, B) The indicated combinations of PPARs and RXRα were cotransfected with the reporters in the absence or presence of 1 μM rosiglitazone (rosi) as labeled. The canonical PPRE reporter responds strongly to endogenous PPARγ, while the Muc1 promoter requires transfection of both PPARγ and RXRα into the cells. (C, D) RXRα was cotransfected alone or with each of the PPARs in the absence or presence of ligands (1 μM LG268, 5 μM Wy-14,643, 1 μM rosiglitazone, and 1 μM cPGI for RXR alone, PPARα, PPARγ, and PPARδ, respectively). The canonical PPRE reporter responds to all three PPARs (see inset for enhanced PPARδ data), while the Muc1 promoter is entirely refractory to PPARα and PPARδ.
The Muc1 promoter is refractory to PPARα and PPARδ.
The distinct biological functions of PPARα, -γ, and -δ (14) imply that each must have at least some unique transcriptional targets. However, the promoters of PPAR targets identified so far, such as acyl-coenzyme A oxidase, aP2, lipoprotein lipase, CD36, LXRα, and ADRP, are at least in part responsive to more than one PPAR. This pan-specificity is exemplified by the 3xDR1 reporter, which is activated by any of the PPARs in the presence of their corresponding ligands, as shown previously and reiterated here (10) (Fig. 3C). In contrast, the Muc1 promoter is induced solely by PPARγ and is entirely refractory to either free or ligand-bound PPARα and PPARδ (Fig. 3D). The lack of compensatory activation of the Muc1 promoter by PPARα and PPARδ is unique so far among established PPAR targets but is consistent with the cessation of Muc1 expression in PPARγ null placentas despite ongoing expression of the two other PPARs (data not shown).
A DR1 element in the proximal Muc1 promoter is a low-affinity PPARγ-binding site.
To understand how Muc1 is regulated by PPARγ, we first scanned its promoter for potential PPAR response elements (PPREs). A reverse DR1 sequence (5′ AGGTGA C AGGTAA 3′; Fig. 4A) was found ∼65 bp upstream of the murine Muc1 transcription start site. The orthologous human Muc1 promoter sequence is highly similar (5′ AGGTGA C AGGTGA 3′) (17). A synthetic oligonucleotide duplex spanning the Muc1 DR1 was bound by a combination of in vitro-translated PPARγ and RXRα (Fig. 4C, lane 8) but not by RXRα or PPARγ alone (lane 7; also data not shown). However, combinations of RXRα with similar quantities of either PPARα or PPARδ (see Fig. 4B) exhibited only residual binding to the same sequence (Fig. 4C, lanes 9 to 12). Reverting the DR1 sequence to a consensus PPRE sequence (5′ AGGTCA C AGGTCA 3′) within the Muc1 promoter sequence context significantly improved PPARγ-RXRα binding (Fig. 4C, lane 2) and restored ligand-stimulated binding of PPARα-RXRα and PPARδ-RXRα (lanes 3 to 6). Template competition experiments demonstrated that a 10-fold excess of unlabeled consensus DR1 competed for binding to any of the PPAR-RXRα heterodimers as effectively as a 100-fold excess of native Muc1 DR1 (Fig. 4D, compare lanes 5 to 2, 10 to 7, and 15 to 12). Thus, the two-base variation of the Muc1 PPRE compromises its affinity towards all three PPARs equally; its poor interaction with PPARα and PPARδ simply reflects their lower inherent DR1 binding potential relative to that of PPARγ (Fig. 4C, compare lanes 4 and 6 to lane 2).
FIG. 4.

A PPARγ binding site in the proximal Muc1 promoter mediates basal repression. (A) Sequence alignment of mouse and human Muc1 promoter sequences, highlighting the reverse DR1 element at the −65 position (boxed). Sequences shown are identical to the plus strand of oligonucleotides used in panels C to E and H. The consensus PPRE sequence is aligned at the bottom. M1, M2, ΔDR-1 are the mutations introduced experimentally into the sequence in the context of either oligonucleotide probes or full promoter constructs. Equal quantities of in vitro-translated PPARs (B) were mixed with RXRα and used to shift oligonucleotides containing the Muc1 −78→−51 promoter sequence with either a consensus PPRE core or the native mouse Muc1 DR1 element (C). RXRα-PPARα or -δ heterodimers were used either without or with their cognate ligands (lanes 3 to 6 and 9 to 12), which reportedly improve DNA binding (10). (D) Complexes of RXRα-PPAR heterodimers with labeled consensus oligonucleotide were competed with 10× and 100× molar unlabeled excess of either the same oligonucleotide or the native Muc1 DR1 sequence. A tenfold excess of the consensus probe (lanes 2, 7, and 12) competes for any of the PPARs as effectively as a 100-fold excess of native Muc1 DR1 (lanes 5, 10, and 15), consistent with a ∼10-fold-lower affinity of Muc1 DR1 compared to that of consensus PPRE towards any of the PPARs. NS, nonspecific gel-shift activity. (E) Basal and PPARγ-induced activities of mouse Muc1 −715 reporter fragments containing the original DR1 sequences, deletion of DR1 (ΔDR-1), or two mutations (M1 and M2; see panel A). All transfections include CMV-lacZ, RXRα, and the respective reporter plasmid, with PPARγ and PPARγ plus rosiglitazone as variables. Fold induction, ratio of reporter activity to its activity with RXRα alone. (F) Complexes of RXRα-PPARγ with labeled native Muc1 oligonucleotide were competed with 10× and 100×molar unlabeled excess of either the same oligonucleotide, PPRE consensus, or M1 and M2 mutant oligonucleotides (see panel A). M1 and M2 fail to compete at up to 100× molar excess. (G) Activities of reporters with human Muc1 DR1 or a PPRE consensus. Conditions are as for panel E. (H, I) EMSA of consensus and Muc1 DR1 probes with extracts of TS cell differentiated for the indicated durations with or without rosiglitazone. IVT, in vitro-translated PPARγ/RXRα. Arrowheads, RXRα-PPARγ-probe complexes (H, lanes 1 to 6 and 12; I, lanes 1 and 4) that are shifted by antibodies to either PPARγ or RXRα (I, filled circles, lanes 2 and 5 and lanes 3 and 6, respectively). Asterisk, endogenous DNA-binding activity with preference for Muc1 DR1 over consensus PPRE sequence.
The proximal DR1 element mediates basal silencing of Muc1.
To examine the role of the proximal DR1 element in the response of Muc1 to PPARγ, we assayed a series of reporter constructs, where this element was inactivated by various mutations in the context of the −715→+33 promoter fragment (Fig. 4A). Surprisingly, basal Muc1 promoter activity was increased by either complete deletion of the DR1 or point mutations to either of its halves (Fig. 4E), both of which completely eliminated PPARγ binding (Fig. 4F). Although activities of DR1-deficient promoter mutants in the presence of PPARγ and rosiglitazone were moderately higher than that of the native Muc1 promoter, the overall effect of PPARγ was significantly blunted (Fig. 4E, bottom [fold induction]). These results demonstrate that Muc1 DR1 harbors a basal repressor, and Muc1 expression depends in part on derepression of this element by PPARγ. This dependence can be alleviated by eliminating basal repression at the outset.
The human DR1 ortholog (5′ AGGTGA C AGGTGA 3′) retained robust response to PPARγ despite a threefold increase in basal activity (Fig. 4G), demonstrating that Muc1 DR1 is functionally conserved during evolution. In contrast, altering Muc1 DR1 into a consensus PPRE increased basal promoter activity by ∼50-fold, and while maximal activity in the presence of liganded PPARγ was 12-fold higher than that of the native Muc1 promoter, the response differential was blunted from 32× to 7.2× over the basal level (Fig. 4G). Thus, the deviation of Muc1 PPRE from the consensus is critical for basal repression and in turn for tighter dependence of Muc1 on PPARγ-mediated derepression, albeit at the expense of maximal expression.
DR1 sites are established repression targets for members of the COUP-TF family of orphan nuclear receptors (32). However, while COUP-TFs bind to a consensus DR1, none bound to the Muc1 variant (data not shown). Moreover, cotransfection of COUP-TFI and -II did not further repress basal or PPARγ-dependent activity of the Muc1 promoter (data not shown), suggesting that repression of Muc1 DR1 is mediated by a different factor. To test whether trophoblasts contain activities that correlate with the basal DR1 repression activity from CV1 cells, electromobility shift assays were carried out with extracts of TS cells at various stages of differentiation. Two major DNA-binding activities were observed (Fig. 4H and I). The first activity interacted readily with consensus PPRE and substantially less with Muc1 DR1. It was identified as endogenous PPARγ-RXRα heterodimers by mobility that was identical to that of in vitro-translated PPARγ-RXRα, relative abundance which mirrored PPARγ expression during TS cell differentiation (see Fig. 2), and full attenuation by either anti-PPARγ or anti-RXRα antibodies (Fig. 4I, lanes 5 and 6). Most importantly, the extracts contained an additional DNA-binding activity, which migrated slower than PPARγ-RXRα heterodimers and was refractory to antibodies against either receptor (Fig. 4H, lanes 1 to 5 and 7 to 11; Fig. 4I, lanes 4 to 9, asterisks). This activity exhibited marked preference towards Muc1 DR1 over the consensus PPRE counterpart, similar to the basal repression pattern in CV1 cells. It peaked at the second day of differentiation, declining by the fourth; a repressor with such a temporal profile would potentially account for the delay in Muc1 expression until later in differentiation despite the earlier induction of PPARγ (see Fig. 2). These observations correlate Muc1 DR1-binding activity from TS cells to basal silencing activity in CV1 cells.
Induction of Muc1 by PPARγ requires both the DR1 motif and a composite enhancer element.
Although DR1 mutations blunted the response of the Muc1 promoter to PPARγ, a considerable response was nevertheless retained, implicating additional elements in coregulating Muc1 with PPARγ. This notion was confirmed by a series of successive 5′ truncations of the promoter. Fragments extending from as near as −76 or as far as −512 to +33, all containing an intact DR1 element, exhibited a markedly compromised response to PPARγ (Fig. 5A; also data not shown). Finer truncation experiments (data not shown) narrowed the critical element to a 56-bp sequence between positions −535 and −480 (56U) (Fig. 5B). Systematic point mutations along the entire length of this element (data not shown), as well as partial truncations (e.g., the −531→+33 fragment in Fig. 5A), caused only partial loss of the response to PPARγ, suggesting that 56U comprises several additive enhancer modules.
FIG. 5.

A 56-bp upstream element mediates the PPARγ response of the Muc1 promoter. (A) Truncation analysis of the Muc1 promoter. Numbers indicate 5′ termini of each fragment relative to the transcription start site. All fragments stretch to the +33 position of the Muc1 gene. (B) Nucleotide sequence of the murine 56-bp upstream element (56U) element, aligned with its human ortholog. Gray boxes mark mouse-human homologies. Note the apparent insertion within the human sequence. (C) Basal and PPARγ-dependent activities of “abbreviated” Muc1 promoter constructs. The 56U element was appended directly to either the basal −45 promoter fragment or the DR1-containing −108 one. Resulting reporter activities were compared to those of the corresponding −45 and −108 control fragments. Note the different scale compared to that for panel A.
When placed directly upstream of the basal Muc1 promoter, the 56U element drove robust luciferase expression (Fig. 5C) (56U/−45), indicating that it docks an endogenous transcriptional activator present constitutively in the cells. Although not harboring a recognizable PPRE, the 56U/−45 promoter construct still displayed modest three- and sixfold responses to free and ligand-stimulated PPARγ, respectively (Fig. 5C). However, placing the 56U element upstream of a 108-bp proximal Muc1 promoter fragment that includes the DR1 motif (56U/−108) restored basal silencing and in turn an ∼70-fold response to ligand-activated PPARγ. These analyses indicate that regulation of the Muc1 promoter by PPARγ is mediated cooperatively by two distinct elements, a low-affinity PPRE that serves as a basal silencer at ∼−65 and a composite enhancer at −500. Each of these elements is modestly responsive to PPARγ on its own, and together they control a robust and specific response to PPARγ.
Analogous localization of MUC1 in the placenta and luminal epithelia.
The MUC1 protein is localized to the apical-luminal surface of simple secretory epithelia, such as the milk ducts of the mammary gland (5, 19), as shown in Fig. 6A. To understand the biological significance of MUC1 expression in the placenta, we sought to determine whether it is localized in this organ to a comparable luminal structure.
FIG. 6.

MUC1 is localized to the trophoblast lining of maternal blood pools in the placenta. Paraffin sections of mammary glands (A, B) and E11.5 placentas (C to F) were probed with anti-MUC1 monoclonal antibody and a cy3-conjugated secondary antibody. In the pregnant mammary gland, MUC1 is restricted to the luminal surface of milk ducts (arrows in panels A and B). In the placental labyrinth (C, D), MUC1 is localized exclusively to the apical surface of layer I trophoblasts, surrounding the maternal blood pools (MV), and is not associated with fetal blood vessels (FV). Signal specificity is confirmed by its absence in placentas of Muc1−/− littermates (E, F). (A, C, and E) Fluorescence; (B, D, and F) corresponding Nomarski images. Scale bars, 50 μm. (G) Coimmunofluorescence with anti-MUC1 antibodies (red) and anti-caveolin-1 antibodies (green). Caveolin-1 is confined to the fetal endothelium and exhibits no signal overlap with MUC1, demonstrating the complete sequestration of MUC1 to the maternal side of the labyrinth. Scale bar, 50 μm.
Figures 6C and D demonstrate that the maternal lacunae in the labyrinth qualify as a lumen analog. Placental MUC1 is confined to the apical surface of layer I trophoblasts in the labyrinth, surrounding the lacunae, and is absent from the fetal endothelium, as well as layer II or III cells and the spongiotrophoblast. The immunofluorescent signal is absent from placentas of Muc1−/− embryos (Fig. 6E and F), confirming that the signal observed in wild-type placentas is specific to MUC1. Double immunofluorescence with anti-caveolin 1 antibodies, whose presence in the placenta is confined to the fetal endothelium, further demonstrates that MUC1 completely segregates to the maternal interface of the placenta (Fig. 6G). Thus, the maternal blood pools in the placenta share anatomical properties with secretory lumens, and by inference, PPARγ emerges as a potential common regulator of these analogous properties.
Low-penetrance maternal vascular defects in the Muc1 null labyrinth.
We next assessed the contribution of MUC1 to placental functions downstream of PPARγ by histological analysis of Muc1 null placentas. The normal Mendelian distribution and birth size, as well as the full viability of Muc1 null pups (26), provided no prior evidence of defects in Muc1 null placentas. Our analyses confirmed this notion, although detailed histological inspection revealed minor dilations and tears in the maternal lacunae of ∼50% of Muc1 null placentas between E12.5 and E18.5 (Compare Fig. 7B and D to Fig. 7A and C, respectively). Two out of nineteen Muc1 null placentas, but none of the wild-type placentas, exhibited expansive thrombi (see Fig. 7E), which could represent harsher manifestations of the same defect. Association of these defects with the maternal lacunae is consistent with the localization of MUC1 to layer I of the labyrinth, around these lacunae. Thus, MUC1 may cooperate with additional targets downstream of PPARγ to maintain the integrity of maternal blood pools, which are severely torn and hemorrhagic in PPARγ null placentas (3). All other PPARγ-dependent histopathies, including labyrinthine trophoblast differentiation, fetal vessel permeation, and lipid droplet accumulation, were normal in Muc1 null placentas (data not shown). These last functions are likely regulated by other PPARγ target genes.
FIG. 7.

Maternal blood pool defects in Muc1−/− placentas. (A to D) Hematoxylin and eosin-stained paraffin section of wt (A, C) and Muc1−/− labyrinths (B, D) at E12.5 (A, B) and E18.5 (C, D). Arrows point to dilated maternal lacunae within the mutant placentas. Note that the more dilated spaces in the E12.5 wt placenta (A) are mostly fetal blood vessels containing nucleated erythrocytes. Fetal vessel space is actually shrunken in the Muc1−/− placenta (B), perhaps due to excessive space allocation to the dilated maternal lacunae. Scale bars, 100 μm. (E) A hematoxylin- and eosin-stained E14.5 Muc1−/− placenta. Arrow points to a massive hemorrhage at the interface of the labyrinth and the spongiotrophoblast. Scale bar, 500 μm.
DISCUSSION
Here we use gene-targeted mice as a differential platform in order to understand the molecular functions of PPARγ in embryonic development. Our studies identify the Muc1 gene as a PPARγ target in trophoblasts, reveal a novel combinatorial mechanism of gene regulation by PPARγ, and implicate PPARγ in regulating epithelial functions of trophoblasts. Importantly, Muc1 is the only direct PPARγ target so far with no obvious ties to lipid or energy metabolism, demonstrating the versatile, nonredundant functions of this nuclear receptor in different biological systems.
Novel insights into gene regulation by PPARγ.
The dependence of Muc1 expression on PPARγ is manifested in every regulatory parameter tested. These include the complete shutdown of Muc1 expression in PPARγ null placentas, its strong upregulation by an agonist in TS cells, and the robust PPARγ-specific response of its promoter. Muc1 thus provides a new, biologically relevant template for mechanistic studies of PPARγ-regulated transcription.
Before discussing the details of Muc1 regulation by PPARγ, it is important to address the issue of cell type specificity. Because Muc1 is not expressed in all PPARγ-expressing tissues, most notoriously adipocytes or macrophages (data not shown), it is clear that its regulation by PPARγ in trophoblasts is tissue specific. Therefore, the activation of its promoter in kidney-derived CV1 cells is surprising. However, this activation is robust, specific for PPARγ over PPARα and PPARδ, and involves complex interactions between two cis regulatory elements, arguing that it is neither coincidental nor promiscuous. Most importantly, a DNA binding activity from TS cells (Fig. 4H and I), whose preference for Muc1 DR1 over consensus PPRE mirrors that of the basal silencing activity in CV1 cells, suggests that components of the Muc1 regulatory network are likely shared between trophoblasts and CV1 cells. In hindsight, the ability of CV1 cells to support the Muc1 response to PPARγ may not be as surprising, considering their reported renal epithelial origin (13) and the notion that emerges here of molecular and cellular similarities between trophoblasts and epithelia.
The response of Muc1 to PPARγ is an interplay between two cis elements: a direct repeat sequence (DR1) that comprises a variant PPRE and a composite 56-bp-wide enhancer (56U). The 56U element drives robust transcription independently of PPARγ, suggesting that it constitutively docks active transcription factors. However, in the basal state, such as in undifferentiated trophoblasts or nontransfected cells, PPARγ is absent, and the DR1 element dominantly silences the promoter.
PPARγ-RXRα heterodimers can bind the variant DR1, albeit with a 10-fold-lower affinity than consensus PPRE. The affinity of RXRα-PPARα and RXRα-PPARδ heterodimers is reduced similarly, suggesting that preferential activation of Muc1 by PPARγ is likely not due to deviation from the consensus. Mutating the DR1 motif not only increases basal Muc1 promoter activity but significantly attenuates the response differential, indicating that PPARγ regulates derepression of this element. Surprisingly, when canonical PPRE is restored in place of the original DR1, basal repression is lost and the degree of response to PPARγ is blunted. Thus, silencing and temporally controlled PPARγ-mediated derepression are critical for Muc1 regulation and require modification of the PPRE sequence. However, cell type specificity and expression intensity must be provided elsewhere. We hypothesize that this crucial biological context for Muc1 induction, as well as its remarkably specific response to PPARγ, are mediated by the 56U element, where an epithelium-specific enhancer has been previously characterized (1, 16, 23, 24).
One means of achieving transcriptional cooperativity between the 56U and DR1 elements is for a constitutive, tissue-specific 56U-bound transcription complex of factors and cofactors to tether PPARγ-RXR heterodimers to the Muc1 promoter. Ligands increase the interaction of PPARγ with various coactivators and could accordingly enhance cooperativity by recruiting PPARγ to an integral coactivator component of the 56U-bound complex. Such model would explain the puzzling ability of PPARγ and rosiglitazone to directly activate the 56U element (see Fig. 5C, 56U/45). Tethering through 56U should in turn greatly facilitate interaction of the PPARγ-RXR heterodimer with its low-affinity DR1 target, which would further cement an active transcription complex on the Muc1 promoter. This model envisions PPARγ agonists as mediators of cooperativity between promoter elements, and hence in the control of transcriptional context and specificity, beyond their known role as transcriptional pacemakers.
The combinatorial complexity of Muc1 regulation by PPARγ has not been documented previously with other PPARγ-regulated promoters. Future studies should reveal whether this form of regulation is unique to Muc1 or whether it has simply been overlooked heretofore.
PPARγ, MUC1, and the analogies between trophoblast and epithelia.
The identification of Muc1, a classical marker of luminal epithelia, as a PPARγ target in trophoblasts suggests an analogy between the placenta and prototypic luminal epithelia. In the placenta, MUC1 is confined exclusively to the apical surface of the labyrinth, surrounding the lacunae that conduct maternal blood. This pattern reiterates the luminal localization of MUC1 in prototypic glandular epithelia (5). Although glandular lumens contain gland secretions, and the placental “lumens” conduct blood, the analogous distribution of MUC1 in both highlights their architectural similarities and suggests that they share additional properties. The analogy extends to the induction of Muc1 expression upon differentiation of the mammary gland during pregnancy and lactation (19), which resembles its induction during trophoblast differentiation. PPARγ is expressed abundantly in the mammary epithelium and other luminal epithelia (12, 18), and it is therefore plausible that its role in these tissues may resemble its placental function.
Muc1 deficiency is not lethal, and does not cause classical manifestations of placental defects, such as intrauterine growth retardation. Approximately 50% of Muc1-deficient placentas exhibit mild structural anomalies in the maternal lacunae, which could reflect a partial role of Muc1 down-regulation in the overt dilation and breakage seen in the lacunae of PPARγ null placentas (3). It is equally possible that the major function of Muc1 downstream of PPARγ is nonstructural. For example, the MUC1 protein may primarily function to protect the placenta against other genetic or maternally borne insults, such as bacterial pathogens (8). The incomplete penetrance of the phenotype may reflect the variable extent or frequency of these putative insults. The full function of Muc1 has yet to be revealed, but regardless, its elaborate regulation by PPARγ suggests that it is a functionally important target rather than a coincidental one. At the same time, the mild Muc1 null phenotype implies that additional targets transduce the essential developmental signals of PPARγ in the placenta.
This study finds that the expression of PPARγ is tightly regulated in TS cells. In undifferentiated TS cells, PPARγ expression is minimal and is confined to rare cells that have differentiated spontaneously (immunofluorescence data [not shown]). Within hours of FGF4 and conditioned medium deprivation, PPARγ expression is induced dramatically in the vast majority of cells in the culture, suggesting that it is an early determinant in the differentiation of all trophoblast lineages. These observations align with the importance of PPARγ for trophoblast differentiation and placental development in the whole animal (3). The rapid induction of PPARγ in differentiating TS cells is reminiscent of its early induction during adipogenesis in vitro and in vivo (30). In contrast, Muc1 is expressed neither in nascent nor in mature adipocytes, whereas adipogenic PPARγ target genes, such as aP2, CD36, LXRα and lipoprotein lipase, are either absent from the placenta or impervious to the status of PPARγ in the organ (data not shown). These differences suggest that while PPARγ is intimately involved in early differentiation of both trophoblasts and adipocytes, its mechanisms of action and downstream targets are distinct in each cell type.
Acknowledgments
We thank Janet Rossant and Tilo Kunath for the TS cell line GFP-Trf and probes for 4311 and mash-2; Barbara Knowles and Karen Fancher for mammary gland sections; Greg Martin, Judy Ford, and members of the Jackson Laboratory Biological Imaging core for assistance with histology, immunofluorescence, and microscopy; Henry Juguilon for assistance with reporter assays; Timothy O'Brien and Susan Ackerman for insightful comments and discussions; Sarah Williamson for figure preparation; and Beth Whitney and Elaine Stevens for administrative assistance.
R.M.E. is an investigator of the Howard Hughes Medical Institute at The Salk Institute. This work was supported in part by NIH HD044103 to Y.B. and by CORE CA34196 to The Jackson Laboratory.
REFERENCES
- 1.Abe, M., and D. Kufe. 1993. Characterization of cis-acting elements regulating transcription of the human DF3 breast carcinoma-associated antigen (MUC1) gene. Proc. Natl. Acad. Sci. USA 90:282-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adamson, S. L., Y. Lu, K. J. Whiteley, D. Holmyard, M. Hemberger, C. Pfarrer, and J. C. Cross. 2002. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev. Biol. 250:358-373. [DOI] [PubMed] [Google Scholar]
- 3.Barak, Y., M. C. Nelson, E. S. Ong, Y. Z. Jones, P. Ruiz-Lozano, K. R. Chien, A. Koder, and R. M. Evans. 1999. PPARγ is required for placental, cardiac, and adipose tissue development. Mol. Cell 4:585-595. [DOI] [PubMed] [Google Scholar]
- 4.Berger, J., and D. E. Moller. 2002. The mechanisms of action of PPARs. Annu. Rev. Med. 53:409-435. [DOI] [PubMed] [Google Scholar]
- 5.Braga, V. M., L. F. Pemberton, T. Duhig, and S. J. Gendler. 1992. Spatial and temporal expression of an epithelial mucin, Muc-1, during mouse development. Development 115:427-437. [DOI] [PubMed] [Google Scholar]
- 6.Chawla, A., W. A. Boisvert, C. H. Lee, B. A. Laffitte, Y. Barak, S. B. Joseph, D. Liao, L. Nagy, P. A. Edwards, L. K. Curtiss, R. M. Evans, and P. Tontonoz. 2001. A PPARγ-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 7:161-171. [DOI] [PubMed] [Google Scholar]
- 7.Cross, J. C. 2000. Genetic insights into trophoblast differentiation and placental morphogenesis. Semin. Cell Dev. Biol. 11:105-113. [DOI] [PubMed] [Google Scholar]
- 8.DeSouza, M. M., G. A. Surveyor, R. E. Price, J. Julian, R. Kardon, X. Zhou, S. Gendler, J. Hilkens, and D. D. Carson. 1999. MUC1/episialin: a critical barrier in the female reproductive tract. J. Reprod. Immunol. 45:127-158. [DOI] [PubMed] [Google Scholar]
- 9.Forman, B. M., P. Tontonoz, J. Chen, R. P. Brun, B. M. Spiegelman, and R. M. Evans. 1995. 15-Deoxy-δ 12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 83:803-812. [DOI] [PubMed] [Google Scholar]
- 10.Forman, B. M., J. Chen, and R. M. Evans. 1997. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl. Acad. Sci. USA 94:4312-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubank, M., and D. G. Schatz. 1994. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucleic Acids Res. 22:5640-5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain, S., S. Pulikuri, Y. Zhu, C. Qi, Y. S. Kanwar, A. V. Yeldandi, M. S. Rao, and J. K. Reddy. 1998. Differential expression of the peroxisome proliferator-activated receptor γ (PPARγ) and its coactivators steroid receptor coactivator-1 and PPAR-binding protein PBP in the brown fat, urinary bladder, colon, and breast of the mouse. Am. J. Pathol. 153:349-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen, F. C., A. J. Girardi, R. V. Gilden, and H. Koprowski. 1964. Infection of human and simian tissue cultures with Rous sarcoma virus. Proc. Natl. Acad. Sci. USA 52:53-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kersten, S., B. Desvergne, and W. Wahli. 2000. Roles of PPARs in health and disease. Nature 405:421-424. [DOI] [PubMed] [Google Scholar]
- 15.Kliewer, S. A., K. Umesono, D. J. Noonan, R. A. Heyman, and R. M. Evans. 1992. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 358:771-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovarik, A., N. Peat, D. Wilson, S. J. Gendler, and J. Taylor-Papadimitriou. 1993. Analysis of the tissue-specific promoter of the MUC1 gene. J. Biol. Chem. 268:9917-9926. [PubMed] [Google Scholar]
- 17.Lancaster, C. A., N. Peat, T. Duhig, D. Wilson, J. Taylor-Papadimitriou, and S. J. Gendler. 1990. Structure and expression of the human polymorphic epithelial mucin gene: an expressed VNTR unit. Biochem. Biophys. Res. Commun. 173:1019-1029. [DOI] [PubMed] [Google Scholar]
- 18.Mueller, E., P. Sarraf, P. Tontonoz, R. M. Evans, K. J. Martin, M. Zhang, C. Fletcher, S. Singer, and B. M. Spiegelman. 1998. Terminal differentiation of human breast cancer through PPARγ. Mol. Cell 1:465-470. [DOI] [PubMed] [Google Scholar]
- 19.Parry, G., J. Li, J. Stubbs, M. J. Bissell, C. Schmidhauser, A. P. Spicer, and S. J. Gendler. 1992. Studies of Muc-1 mucin expression and polarity in the mouse mammary gland demonstrate developmental regulation of Muc-1 glycosylation and establish the hormonal basis for mRNA expression. J. Cell Sci. 101:191-199. [DOI] [PubMed] [Google Scholar]
- 20.Rosen, E. D., and B. M. Spiegelman. 2001. PPARγ: a nuclear regulator of metabolism, differentiation and cell growth. J. Biol. Chem. 276:37731-37734. [DOI] [PubMed] [Google Scholar]
- 21.Rossant, J., and J. C. Cross. 2001. Placental development: lessons from mouse mutants. Nat. Rev. Genet. 2:538-548. [DOI] [PubMed] [Google Scholar]
- 22.Sapin, V., P. Dolle, C. Hindelang, P. Kastner, and P. Chambon. 1997. Defects of the chorioallantoic placenta in mouse RXRα null fetuses. Dev. Biol. 191:29-41. [DOI] [PubMed] [Google Scholar]
- 23.Shirotani, K., J. Taylor-Papadimitriou, S. J. Gendler, and T. Irimura. 1994. Transcriptional regulation of the MUC1 mucin gene in colon carcinoma cells by a soluble factor. Identification of a regulatory element. J. Biol. Chem. 269:15030-15035. [PubMed] [Google Scholar]
- 24.Shirotani, K., and T. Irimura. 1998. Purification of nuclear proteins that potentially regulate transcription of the MUC1 mucin gene induced by a soluble factor. J. Biochem. 124:585-590. [DOI] [PubMed] [Google Scholar]
- 25.Spicer, A. P., G. Parry, S. Patton, and S. J. Gendler. 1991. Molecular cloning and analysis of the mouse homologue of the tumor-associated mucin, MUC1, reveals conservation of potential O-glycosylation sites, transmembrane, and cytoplasmic domains and a loss of minisatellite-like polymorphism J. Biol. Chem. 266:15099-15109. [PubMed] [Google Scholar]
- 26.Spicer, A. P., G. J. Rowse, T. K. Lidner, and S. J. Gendler. 1995. Delayed mammary tumor progression in Muc-1 null mice. J. Biol. Chem. 270:30093-30101. [DOI] [PubMed] [Google Scholar]
- 27.Sporn, M. B., N. Suh, and D. J. Mangelsdorf. 2001. Prospects for prevention and treatment of cancer with selective PPARγ modulators (SPARMs). Trends Mol. Med. 7:395-400. [DOI] [PubMed] [Google Scholar]
- 28.Sucov, H. M., E. Dyson, C. L. Gumeringer, J. Price, K. R. Chien, and R. M. Evans. 1994. RXR α mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 8:1007-1018. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka, S., T. Kunath, A. K. Hadjantonakis, A. Nagy, and J. Rossant. 1998. Promotion of trophoblast stem cell proliferation by FGF4. Science 282:2072-2075. [DOI] [PubMed] [Google Scholar]
- 30.Tontonoz, P., E. Hu, and B. M. Spiegelman. 1994. Stimulation of adipogenesis in fibroblasts by PPARγ 2, a lipid-activated transcription factor. Cell 79:1147-1156. [DOI] [PubMed] [Google Scholar]
- 31.Wendling, O., P. Chambon, and M. Mark. 1999. Retinoid X receptors are essential for early mouse development and placentogenesis. Proc. Natl. Acad. Sci. USA 96:547-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y., and M. L. Dufau. 2004. Gene silencing by nuclear orphan receptors. Vitam. Horm. 68:1-48. [DOI] [PubMed] [Google Scholar]