

Abstract
We identified Ras guanine-releasing protein 3 (RasGRP3) as a guanine exchange factor expressed in blood vessels via an embryonic stem (ES) cell-based gene trap screen to identify novel vascular genes. RasGRP3 is expressed in embryonic blood vessels, down-regulated in mature adult vessels, and reexpressed in newly formed vessels during pregnancy and tumorigenesis. This expression pattern is consistent with an angiogenic function for RasGRP3. Although a loss-of-function mutation in RasGRP3 did not affect viability, RasGRP3 was up-regulated in response to vascular endothelial growth factor (VEGF) stimulation of human umbilical vein endothelial cells, placing RasGRP3 regulation downstream of VEGF signaling. Phorbol esters mimic the second messenger diacylglycerol (DAG) in activating both protein kinase C (PKC) and non-PKC phorbol ester receptors such as RasGRP3. ES cell-derived wild-type blood vessels exposed to phorbol myristate acetate (PMA) underwent extensive aberrant morphogenesis that resulted in the formation of large endothelial sheets rather than properly branched vessels. This response to PMA was completely dependent on the presence of RasGRP3, as mutant vessels were refractory to the treatment. Taken together, these findings show that endothelial RasGRP3 is up-regulated in response to VEGF stimulation and that RasGRP3 functions as an endothelial cell phorbol ester receptor in a pathway whose stimulation perturbs normal angiogenesis. This suggests that RasGRP3 activity may exacerbate vascular complications in diseases characterized by excess DAG, such as diabetes.
Blood vessels provide cells with the nutrients and oxygen necessary for viability by forming a branched vascular network that links to larger vessels, a process called angiogenesis. For proper angiogenesis, endothelial cells must receive a number of extracellular signals and then assemble these cues into a coordinated biological response (for reviews, see references 16, 24, 30, 57, and 79). Vascular endothelial growth factor A (VEGF-A) and its two high-affinity receptors, flk-1 (VEGF receptor 2 [VEGFR-2]) and flt-1 (VEGFR-1), stimulate signal transduction pathways that regulate endothelial cell proliferation, migration, survival, and permeability (for reviews, see references 29, 64, and 72). VEGF-A is a secreted ligand expressed by nonendothelial cells developmentally and during physiological and pathological angiogenesis in the adult. The importance of VEGF-mediated signaling in vascular development has been clearly demonstrated genetically, since mutations in VEGF-A and both receptors lead to vascular perturbations and embryonic death (11, 28, 31, 63). Moreover, recent studies indicate that VEGF-A levels and perhaps spatial availability must also be regulated for blood vessels to form properly (15, 33, 50, 62).
Recently, the intracellular signaling pathways downstream of VEGF-A in endothelial cells have been elucidated (for a review, see reference 14). Signaling through Ras is one pathway downstream of VEGF stimulation of flk-1 (18, 38, 48). Ras is a key intracellular signaling hub that incorporates a number of upstream signals and then coordinates these signals into specific biological responses (for a review, see reference 65). Biochemically, Ras is a small GTPase that functions as a GDP-GTP-regulated switch. Stimulus-mediated activation of guanine nucleotide exchange factors (GEFs) facilitates GDP-GTP exchange and promotes the formation of active Ras-GTP (54), whereas GTPase-activating proteins (GAPs) promote formation of inactive, GDP-bound Ras (6). VEGF stimulation of endothelial cells increases the amount of activated Ras (18, 48, 66), and introduction of dominant negative Ras into endothelial cells inhibits VEGF-induced endothelial cell DNA synthesis, migration, and tube formation (48). Moreover, Ras signaling is sufficient to promote an angiogenic phenotype in primary endothelial cells in culture (49), suggesting that regulation of endothelial Ras signaling is essential for blood vessel formation. However, the mechanism by which VEGF regulates Ras and the precise role of Ras activation in mediating VEGF function in endothelial cells are presently not well understood.
Genetic experiments in vivo are consistent with a role for Ras in vascular development. Genetic ablation of Son of Sevenless (SOS) RasGEF, neurofibromatosis 1 (NF1) RasGAP, or RasGAP results in cardiovascular defects (7, 36, 76). Partial functional overlap exists within the ras family itself, as mice deficient for N-ras, H-ras, or both are normal (27, 74). K-ras has a unique and required function, as mice deficient for K-ras die progressively during development due to multiple defects, including defects in the hematopoietic and cardiovascular systems (40, 45). Although expression of Ras and these Ras regulators is not restricted to blood vessels, targeted deletion of NF1 RasGAP in endothelial cells leads to multiple cardiovascular defects (34). This result indicates that NF1 is a required gene product in endothelial cells during blood vessel development and suggests that regulation of Ras signaling is important in angiogenesis in vivo. However, the role of Ras activation in blood vessel formation remains poorly understood.
We identified Ras guanine-releasing protein 3 (RasGRP3) as an endothelial cell-expressed GEF in an embryonic stem (ES) cell-based gene trap screen for genes expressed in blood vessels (37, 67). RasGRP3 is one of four members of the RasGRP family of RasGEFs that couple diacylglycerol (DAG) signaling to Ras activation (12, 22, 41, 46, 47, 55, 56, 73, 78, 81). While the different RasGRP proteins share similar mechanisms of regulation, they exhibit distinct patterns of tissue expression and specificity for Ras and Rap GTPases. We determined that vascular expression of RasGRP3 was restricted to sites of active blood vessel formation developmentally and in the adult. The Rasgrp3 gene trap locus (Rasgrp3gt) is a loss-of-function allele, yet homozygous Rasgrp3gt/gt mice and embryos displayed no obvious vascular defects. However, wild-type ES cell-derived vessels responded to the phorbol ester phorbol myristate acetate (PMA), which mimics DAG activity, with extensive aberrant morphogenesis. These changes required RasGRP3 function, since Rasgrp3gt/gt vessels were completely refractory to the phorbol ester-mediated effects. The aberrant morphogenesis induced by phorbol ester in wild-type vessels closely resembled the phenotype of vessels with up-regulated VEGF signaling, and we showed that RasGRP3 expression is up-regulated by VEGF stimulation of endothelial cells. Taken together, these results show that RasGRP3 is a VEGF-responsive vascular GEF that is essential for the endothelial response to phorbol esters. RasGRP3 functions in a pathway that is redundant developmentally, but its stimulation perturbs normal angiogenesis, suggesting that it may exacerbate DAG-mediated vascular phenotypes in diseases such as diabetes.
MATERIALS AND METHODS
Generation of Rasgrp3+/gt ES cells.
ES cells were maintained and differentiated as previously reported (4, 42, 43). ES cells with a gene trap vector in the Rasgrp3 locus (Rasgrp3+/gt) were identified by analysis of β-galactosidase (β-Gal) expression upon differentiation as previously described (37, 67). Briefly, the PT1-ATG vector, which contains a promoterless lacZ gene with its own start codon, was introduced into ES cells by electroporation, and selection was performed with G418. Drug-resistant colonies were picked, expanded, differentiated, and analyzed for β-Gal expression in blood vessels.
β-Gal detection.
Differentiated ES cell cultures, embryos, and adult tissues were processed for β-Gal detection as previously described (42). Briefly, ES cell cultures were differentiated to day 6 to 8 and then fixed in glutaraldehyde fixative (0.2% glutaraldehyde, 5 mM EGTA [pH 7.3], 2 mM MgCl2 in 0.1 M phosphate buffer [pH 7.3]) for 5 min at room temperature. After three washes with phosphate buffer, cultures were incubated overnight at 37°C in 5-bromo-4-chloro-3-indolyl-ā-d-galactopyranoside (X-Gal; Sigma) staining solution (0.625 mg of X-Gal/ml, 5 mM potassium ferrocyanide and 5 mM potassium ferricyanide in ES cell wash buffer). ES cell wash buffer consisted of 2 mM MgCl2 and 0.02% NP-40 in 0.1 M sodium phosphate buffer (pH 7.3). After incubation, cultures were rinsed and stored in wash buffer at 4°C.
Embryos and adult tissues were rinsed in 1× phosphate-buffered saline (PBS) and fixed in either glutaraldehyde fixative or 4% paraformaldehyde (PFA)-PBS for 20 min at room temperature. After two 10-min washes in embryo wash buffer (0.1 M phosphate buffer [pH 7.3], 0.1% sodium deoxycholate, 0.02% NP-40, 0.05% bovine serum albumin [Sigma]), tissues were incubated overnight at 37°C in X-Gal staining solution (wash buffer containing 1 mg of X-Gal/ml [Sigma], 5 mM ferrocyanide, and 5 mM ferricyanide). Tissues were then postfixed in the appropriate fixative and stored at 4°C until embedded. Adult tissues were frequently dissected into smaller pieces prior to fixation for better penetration.
RACE cloning.
Rapid amplification of cDNA ends (RACE) cloning was done as previously described (67). Briefly, RNA was prepared from differentiated ES cell cultures with Trizol (Roche Applied Sciences, Indianapolis, Ind.) per the manufacturer's protocol. RACE cloning was performed with a 5′ RACE kit (GIBCO/BRL, Grand Island, N.Y.) according to the manufacturer's instructions. RACE products were subcloned into the pCRII vector (Invitrogen, Carlsbad, Calif.).
Nucleic acid isolation and analysis.
For Southern blot analysis, genomic DNA was digested with EcoRI, separated on a 0.8% agarose gel, and transferred to a nylon membrane. Hybridization was performed with a RasGRP3-specific cDNA probe generated by RACE cloning. This probe encompassed exons 2 to 4 of RasGRP3 and part of exon 1. Primer sequences for PCR genotyping were 5′ ATC TGG AGC CCA TTG AGT TG 3′ and 5′ CAG GCC AGA ACC AAG AGG TC 3′, flanking exon 3 of RasGRP3, and 5′ TGC CCT TTC TCC TCC ATG AC 3′ and 5′ GCG GGC CTC TTC GCT ATT AC 3′, which are specific for the gene trap (see Fig. 2A).
FIG. 2.

The gene trap construct integrated into the Rasgrp3 locus. (A) Schematic of the mouse Rasgrp3 locus. The locus is approximately 63 kb and consists of 18 exons. The region encompassing exons 2 to 7 is enlarged to show the restriction fragments analyzed by Southern blot analysis. E, EcoRI sites. Arrows denote PCR primers for genotyping Rasgrp3 mice. (B) Southern blot analysis of EcoRI-digested genomic DNA from Rasgrp3+/+(lane 1), Rasgrp3+/gt (lane 2), and Rasgrp3gt/gt (lane 3) mice. The blot was hybridized with a cDNA probe consisting of exons 2 to 4. Arrows denote hybridizing fragments in Rasgrp3+/+ DNA. Arrowheads denote the hybridizing fragments inRasgrp3gt/gt DNA. (C) Genomic PCR with primers specific for exons 2 to 4 and the gene trap. Rasgrp3+/+(lane 1), Rasgrp3+/gt (lane 2), and Rasgrp3gt/gt (lane 3) are shown.
Total RNA was isolated from cells or tissues with Trizol Reagent, per the manufacturer's protocol, for Northern blot analysis. RNA was separated on a 1.2% formaldehyde-agarose gel and transferred to nitrocellulose. The membrane was hybridized in Quickhyb hybridization buffer (Stratagene, La Jolla, Calif.) with [32P]dCTP-labeled cDNAs and then exposed to film. All radioactive probes were generated by random priming with the Prime-It RmT Random Primer labeling kit (Stratagene). Template for the mouse RasGRP3 Northern probe was generated using the primers 5′ GCT CGG GAA AGC GGC AAC AC 3′ and 5′ ATT GGC GGT CTG GCT TTG G ′3, corresponding to nucleotides 344 to 1575 of mouse RasGRP3. Human RasGRP3 probe was generated using a 772-bp HindIII fragment from a human RasGRP3 cDNA clone, KIAA0846 (51). This probe corresponds to nucleotides 2643 to 3415 of KIAA0846. The platelet endothelial cell adhesion molecule (PECAM) probe was generated from a 660-bp reverse transcriptase PCR (RT-PCR) product that corresponds to nucleotides 1244 to 1904 of PECAM.
Generation, maintenance, and genotyping of mice.
Chimeric mice were produced from Rasgrp3+/gt ES cells by injection of ES cells into the blastocoel cavity of mouse blastocysts (32). The Rasgrp3gt strain was established using three independent chimeras that demonstrated germ line transmission when bred to wild-type CD1 mice. For timed pregnancies, day 0.5 of gestation (E0.5) was noon of the day of vaginal plug observation. Genotyping was performed using DNA isolated from tail biopsies or embryonic yolk sac by Southern blotting or PCR analysis. MMTV-PyVT mice (the gift of W. Muller and T. Van Dyke) were crossed to Rasgrp3gt mice through two rounds of matings to obtain MMTV-PyVT;Rasgrp3+/gt and MMTV-PyVT;Rasgrp3gt/gt mice.
In situ hybridization.
In situ hybridization on whole-mount embryos was performed by a modified protocol (61). The template for the RasGRP3 probe was the same template used for mouse Northern blot analysis. Briefly, mid-gestation embryos were isolated and fixed in 4% PFA-PBS overnight. Embryos were then dehydrated in a methanol-PBS series, followed by rehydration in a methanol-PBT (PBS with 0.1% Tween) series. Embryos were bleached in 6% H2O2 for 1 h, treated with 10 μg of proteinase K/ml in PBT for 15 min, and then washed with fresh 2 mg of glycine in PBT/ml. Refixation was performed using 0.2% glutaraldehyde-4% PFA in PBT for 20 min. Embryos were incubated in prehybridization solution (50% formamide, 5× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate] [pH 4.5], 0.1% Tween 20, and 0.05 mg of heparin/ml) for 1 h at 70°C prior to adding heat-shocked digoxigenin-labeled probe (1 μg/ml). Hybridization was performed overnight at 70°C.
Following hybridization, embryos were washed according to protocol and then incubated with 100 μg/ml RNase A for 30 min at 37°C. Blocking was performed using 10% lamb serum in Tris-buffered saline plus 0.1% Tween 20 (TBST) for 2.5 h at room temperature, and embryos were incubated with an alkaline phosphatase-conjugated anti-digoxigenin antibody (Boehringer Mannheim, Indianapolis, Ind.) at a 1:2,000 dilution overnight at 4°C. After antibody incubation, embryos were washed in TBST, and detection was with 50 mg of nitroblue toluene/ml and 30 mg of 5′-bromo-4-chloro-3-indolyl phosphate (BCIP)/ml (both from Promega, Milwaukee, Wis.) in 100 mM NaCl, 100 mM Tris-HCl (pH 9.5), 50 mM MgCl2, 0.1% Tween 20, and 2 mM levamisole. When detection was complete, embryos were washed in PBT (pH 5.5) and then postfixed in 4% PFA. Embryos were stored in PBS at 4°C.
RNA in situ hybridization of differentiated ES cell cultures was performed as previously reported (5) and as described above for embryos except for the following: day 8 cultures were fixed in 4% PFA for 1 h and dehydrated through a methanol-PBS series. Wells were rehydrated and then incubated in 6% H2O2 in PBT for 30 min, in proteinase K (3 μg/ml in PBT) for 10 min, and then in fresh glycine (2 mg/ml in PBT) for 4 min. Following the PBT washes, wells were refixed in 0.2% glutaraldehyde and 4% PFA in PBT for 8 min. Wells were hybridized as above except that an initial 3-h incubation was performed at 80°C prior to addition of probe. RNase treatment, blocking incubation, antibody reaction, and substrate development were all as previously described.
Antibody staining.
Antibody staining of ES cell cultures was as previously described (5, 42). Rat anti-mouse PECAM was used at a 1:1,000 dilution (Mec 13.3; BD Pharmingen, San Diego, Calif.), and donkey anti-rat tetramethyl-rhodamine isothiocyanate (Jackson Immunoresearch, West Grove, Pa.) was used at a 1:200 dilution. Cultures were stored at 4°C in PBS. Antibody staining of mid-gestation embryos was also as previously described (1, 2). Embryos were incubated with rat anti-mouse PECAM (Mec 13.3; BD Pharmingen) at a 1:200 dilution, followed by incubation with horseradish peroxidase-conjugated goat anti-rat secondary antibody (Pierce, Rockford, Ill.) at a 1:200 dilution. Detection was with 0.5 mg of 3′,3′-diaminobenzidine tetrahydrochloride (Sigma)/ml, 3 mg of nickel sulfate/ml, and 0.03% hydrogen peroxide in PBS.
HUVEC stimulation.
Human umbilical vein endothelial cells (HUVECs) were purchased from Cambrex Bio Science Walkersville, Inc. (Walkersville, Md.) and maintained in endothelial growth medium 2 supplemented with the CC-4176 bullet kit per the manufacturer's protocol. HUVECs were utilized between passages 4 and 8. For RasGRP3 regulation experiments, HUVECs were grown to near-confluency, then serum starved overnight in M199 supplemented with 2% fetal bovine serum. HUVECs were then stimulated with various concentrations of VEGF or basic fibroblast growth factor (bFGF) for 24 h, and isolated RNA was analyzed on Northern blots. Recombinant human VEGF164 (R&D Systems) was used at 5 to 60 ng/ml, and recombinant human bFGF (Sigma) was used at 5 to 80 ng/ml.
Production of Rasgrp3gt/gt ES cells.
Rasgrp3+/gt ES cells were seeded at 5 × 104 cells per 10-cm plate in ES cell medium (4, 43), and 1 day later, 640 μg of G418/ml was added to the medium. After 10 to 14 days of G418 selection, colonies were picked, expanded, and screened for homozygosity at the Rasgrp3 locus by PCR and Southern blot analysis. Three different Rasgrp3gt/gt ES cell clones were characterized.
PMA treatment of differentiating ES cell cultures.
PMA (Calbiochem, San Diego, Calif.) was resuspended in dimethyl sulfoxide at 10 mM. PMA (10 to 1,000 nM) was added to differentiating ES cell cultures starting on day 5 of differentiation. Fresh PMA was then added daily. At day 8, cultures were fixed and processed for antibody staining as previously described.
RESULTS
Identification of a gene trap insertion in the Rasgrp3 locus.
We carried out a gene trap screen in ES cells to identify novel genes expressed in developing blood vessels or blood cells (37, 67). The approach was to induce random insertion into ES cells of a vector that both lacked a promoter/enhancer and contained a splice acceptor site. Thus, expression of the reporter should be facilitated by insertion into an intron, and the expression pattern is predicted to reflect the expression pattern of the insertion locus. ES cells can be induced to undergo a programmed differentiation in vitro that recapitulates developmental processes and results in the formation of multiple tissues, including primitive blood vessels (4). The combination of gene trap insertion and ES cell differentiation to primitive vessels is a powerful approach for the identification of new genes and/or genes not previously associated with blood vessel formation.
One such gene trap ES cell line selectively expressed the β-Gal reporter gene in developing ES cell-derived blood vessels (Fig. 1A). Initial RACE cloning combined with analysis of expressed sequence tag databases and the mouse genome sequence indicated that the gene trap insertion occurred at the 5′ end of a gene encoding the small GTPase activator RasGRP3 (Fig. 2A). To confirm that the insertion occurred in this locus, we performed Southern blot analysis of DNA from ES cells that were wild type (Rasgrp3+/+), heterozygous (Rasgrp3+/gt), or homozygous (Rasgrp3gt/gt) for the gene trap insertion (Fig. 2B). A band shift was detected in Rasgrp3+/gt and Rasgrp3gt/gt DNA with a RasGRP3 probe, confirming that the gene trap insertion did indeed occur in the Rasgrp3 locus.
FIG. 1.

RasGRP3 is expressed in ES-cell derived blood vessels. (A) ES cells with a lacZ-containing gene trap integration in the Rasgrp3 locus (Rasgrp3+/gt) were differentiated to day 8 (day 8 ES cell cultures) and stained for β-Gal. β-Gal expression was detected in ES cell-derived blood vessels. (B) Wild-type (Rasgrp3+/+) day 8 ES cell cultures were hybridized with an in situ hybridization probe specific for RasGRP3. Arrows point to several blood vessels that reacted with the probe.
Further analysis of the genomic DNA showed three hybridizing EcoRI fragments in wild-type DNA, but only two such bands in Rasgrp3gt/gt DNA (Fig. 2B). Although other possibilities exist, the simplest explanation for this result is that the gene trap insertion deleted one of the Rasgrp3 exons. To test this hypothesis, we performed PCR experiments to assay for the presence of exons 2, 3, and 4 in genomic DNA (Fig. 2C). All three primer sets amplified fragments from wild-type and heterozygous DNA, and exon 2 and 4 primer sets amplified fragments from Rasgrp3gt/gt DNA. However, a PCR product corresponding to exon 3 was not amplified from Rasgrp3gt/gt DNA, suggesting that exon 3 was deleted by the gene trap insertion (Fig. 2A). This result is consistent with the fragment sizes detected by Southern blot analysis and suggests that the Rasgrp3gt locus is a null allele, since exon 3 contains the AUG start codon of RasGRP3.
Analysis of the mouse genome sequence predicted that the mouse RasGRP3 transcript was approximately 4.2 kb and contained 18 exons. We cloned the predicted open reading frame of this transcript by RT-PCR from the mouse endothelial cell line Py-4-1 (21), and sequenced the transcript (Fig. 3A). The mouse RasGRP3 protein is predicted to be 691 amino acids, with four conserved domains shared by all RasGRP proteins: a REM (Ras exchange motif) domain, a CDC25 homology domain, a pair of atypical EF hands, and a regulatory C1 domain (Fig. 3B). The RasGRPs activate Ras and other small GTPases via the CDC25 homology catalytic domain, and they are activated in turn by the formation of DAG, which resides in the membrane. The C1 domain interacts with DAG, causing recruitment of RasGRP to the plasma membrane, where membrane-bound Ras is stimulated (Fig. 3C) (for a review, see reference 17). Phospholipase C-γ (PLC-γ) generates DAG by cleaving PIP2, and PLC-γ is downstream of receptor tyrosine kinases such as the VEGF receptor (69). Mouse RasGRP3 shares extensive nucleotide and protein homology to human RasGRP3 and to the other three RasGRP family members from both species (data not shown).
FIG. 3.

Cloning and analysis of the RasGRP3 cDNA. (A) Full-length RasGRP3 was cloned from the mouse endothelial cell line Py-4-1 by RT-PCR and sequenced. Underlined sequences represent conserved domains. (B) Schematic of RasGRP3 protein. REM, RasGEF N-terminal domain; CDC25, catalytic RasGEF domain; EF, atypical EF hand domain; C1, PKC region 1 domain that binds DAG. (C) Model of RasGRP3 activation and signaling. Upon ligand-receptor engagement, PLC-γ is activated, which leads to the production of DAG from PIP2. DAG is localized to the plasma membrane. C1 domain binding of DAG recruits RasGRP3 to the membrane where Ras activation occurs.
The expression of RasGRP3 in ES cell-derived blood vessels suggested that this activator might be involved in angiogenesis. Three criteria were considered important in assigning RasGRP3 a role in vascular processes: (i) vascular expression during embryonic development, (ii) endothelial cell expression during neoangiogenesis in the adult, and (iii) altered blood vessel formation upon perturbation of RasGRP3 function. These criteria were assessed with molecular, genetic, and pharmacological tools.
RasGRP3 is expressed in blood vessels during mouse development.
Because Ras is a critical transducer of numerous signals important for cell proliferation and differentiation, we asked if RasGRP3 was expressed developmentally. To do this, we generated Rasgrp3+/gt mice from Rasgrp3+/gt ES cells using standard techniques, and analyzed expression of RasGRP3 via the β-Gal reporter gene. We focused on a developmental window from E7.5 to E10.5 because this is when vascular development is most prominent. During development, β-Gal expression from the Rasgrp3 locus was predominately found in blood vessels. In E9.5 and E10.5 embryos, β-Gal was strongly expressed in the developing intersomitic vessels and the dorsal aorta (Fig. 4A and B). The majority of head vessels and small vessels along the sides of the embryo also faintly expressed β-Gal. β-Gal expression in developing vessels was not uniform at a given stage, suggesting that RasGRP3 expression is modulated developmentally. β-Gal from the Rasgrp3 locus was also expressed in a limited number of nonvascular embryonic cells such as the pericardium and the myotome of the somite (Fig. 4A and B and data not shown). Examination of transverse sections at E9.5 revealed β-Gal expression in the dorsal aortae, cardinal veins, and the perineural vascular plexus that surrounds the developing spinal cord (Fig. 4C).
FIG. 4.

RasGRP3 is expressed in blood vessels developmentally. (A and B) Embryos at E9.5 (A) and E10.5 (B) with lacZ inserted into the Rasgrp3 locus (Rasgrp3+/gt) were stained for β-Gal. β-Gal expression was detected in the dorsal aorta, the intersomitic vessels, and many of the smaller vessels throughout the embryo. The black arrow points to RasGRP3 β-Gal expression in the dorsal aorta, black arrowheads indicate expression in the intersomitic vessels, the white arrowhead indicates somitic myotome expression, and the white arrow indicates expression in the pericardium. (C) Transverse section of an E9.5 embryo stained for RasGRP3 β-Gal. NT, neural tube; PNVP, perineural vascular plexus; CV, cardinal veins; DA, dorsal aortae. (D) Whole-mount in situ hybridization of Rasgrp3+/+ wild-type E9.5 embryo for RasGRP3. RasGRP3 mRNA was detected in the dorsal aorta, intersomitic vessels, and several smaller vessels throughout the embryo. The arrow designates expression in the dorsal aorta, whereas arrowheads indicate expression in the intersomitic vessels.
To determine if β-Gal expression from the Rasgrp3 locus faithfully recapitulated expression of the endogenous gene, we utilized whole-mount in situ hybridization with a probe for RasGRP3 that did not recognize other family members. RasGRP3 RNA was detected in ES cell-derived vessels, the dorsal aorta, and intersomitic vessels and faintly in the small vessels of the head (Fig. 1B and 4D). This expression pattern closely resembles the whole-mount β-Gal expression pattern, confirming that the β-Gal reporter recapitulates vascular expression of endogenous RasGRP3.
RasGRP3 expression is modulated during angiogenesis and by VEGF.
Although RasGRP3 was expressed in blood vessels of the developing embryo, the majority of vessels in the adult mouse lacked RasGRP3 β-Gal expression (Fig. 5A and B). Additionally, there was only rare expression of RasGRP3 β-Gal in adult nonvascular cells in organs such as the brain and spleen (data not shown). Since RasGRP3 was expressed in embryonic vessels undergoing angiogenesis, we asked whether vascular RasGRP3 expression is up-regulated during adult angiogenesis. During pregnancy, vessels of the female reproductive tract undergo extensive expansion to provide nutrients and oxygen to the developing fetus. RasGRP3 β-Gal was expressed in maternal vessels of the decidua that surrounds the murine embryo, suggesting that RasGRP3 is reexpressed in these adult angiogenic vessels (Fig. 5C).
FIG. 5.

RasGRP3 expression is up-regulated during normal and pathological adult angiogenesis. (A) Brain section from a Rasgrp3+/gt mouse stained for β-Gal. Note the absence of β-Gal staining in the blood vessels. The insert shows a vessel (black arrow) at higher magnification that does not express β-Gal. (B) Equivalent brain section from a flt-1+/− mouse with lacZ inserted in the flt-1 locus. Note the β-Gal expression in the brain blood vessels. (C) Rasgrp3gt/gt decidua stained for β-Gal. Note expression of β-Gal in decidual vessels. (D) Spontaneous tumor from a Rasgrp3gt/gt mouse, stained for β-Gal. Note expression of β-Gal in tumor vessels. (E) Section of a mammary tumor from a MMTV-PyVT;Rasgrp3gt/gt mouse stained for β-Gal. Note the expression of β-Gal in tumor vessels. The insert shows a β-Gal-positive vessel at a higher magnification. The arrow points to the lumenized vessel.
As tumors proliferate, they recruit host-derived blood vessels via angiogenesis. To address regulation of RasGRP3 in tumor vessels, we first took advantage of a Rasgrp3gt/gt mouse that developed spontaneous sarcomas. We found that RasGRP3 β-Gal was reexpressed in these tumor blood vessels (Fig. 5D). To further extend this analysis, we obtained MMTV-PyVT transgenic mice, and bred this transgene into the Rasgrp3gt background. MMTV-PyVT mice rapidly develop mammary tumors as a consequence of PyVT oncogene expression in the mammary glands, and the resulting tumors are highly angiogenic (35). Analysis of MMTV-PyVT;Rasgrp3gt/gt mice revealed that RasGRP3 β-Gal was also reexpressed in the vessels of these tumors (Fig. 5E). Taken together, these results indicate that vascular RasGRP3 expression is up-regulated during both normal and pathological angiogenesis.
Although most adult vessels did not express RasGRP3, RasGRP3 was expressed constitutively in the kidney glomerulus (Fig. 6A and B) as was previously reported (78). VEGF expression is also maintained in adult kidney glomeruli (9, 25); the VEGF receptor, flk-1 (VEGFR-2), is also expressed at this site (Fig. 6C and D) (58), indicating that VEGF signaling is intact in the glomerulus. Thus, VEGF expression is associated with neoangiogenesis in the embryo and the adult, and it is maintained in the glomerulus of the kidney, all sites of RasGRP3 expression. This coincidence suggested that VEGF signaling through flk-1 may induce RasGRP3 expression in vivo.
FIG. 6.

RasGRP3 and flk-1 are both expressed in the glomerulus of the kidney. Kidneys from Rasgrp3+/gt mice and flk-1+/− mice with lacZ inserted into the flk-1 locus were dissected and stained for β-Gal. (A and C) Whole-mount view of β-Gal expression in Rasgrp3+/gt (A) and flk-1+/− (C) kidneys. Note the peripheral, punctate expression of β-Gal in both backgrounds. (B and D) Kidney sections from Rasgrp3+/gt (B) and flk-1+/− (D) mice stained for β-Gal. Note that the β-Gal reporter is expressed in the glomerulus in both genetic backgrounds.
We tested this hypothesis by treating primary endothelial cells in culture (HUVECs) with increasing concentrations of recombinant VEGF. RasGRP3 RNA was barely detectable in serum-starved HUVECs by Northern blot analysis; however, VEGF stimulation resulted in up-regulation of RasGRP3 transcription in a dose-dependent manner, indicating that Rasgrp3 is a VEGF-responsive gene (Fig. 7A). This effect was not universal to growth factor stimulation, as HUVEC stimulated with another angiogenic growth factor, bFGF, did not show increased RasGRP3 RNA levels (Fig. 7B).
FIG. 7.
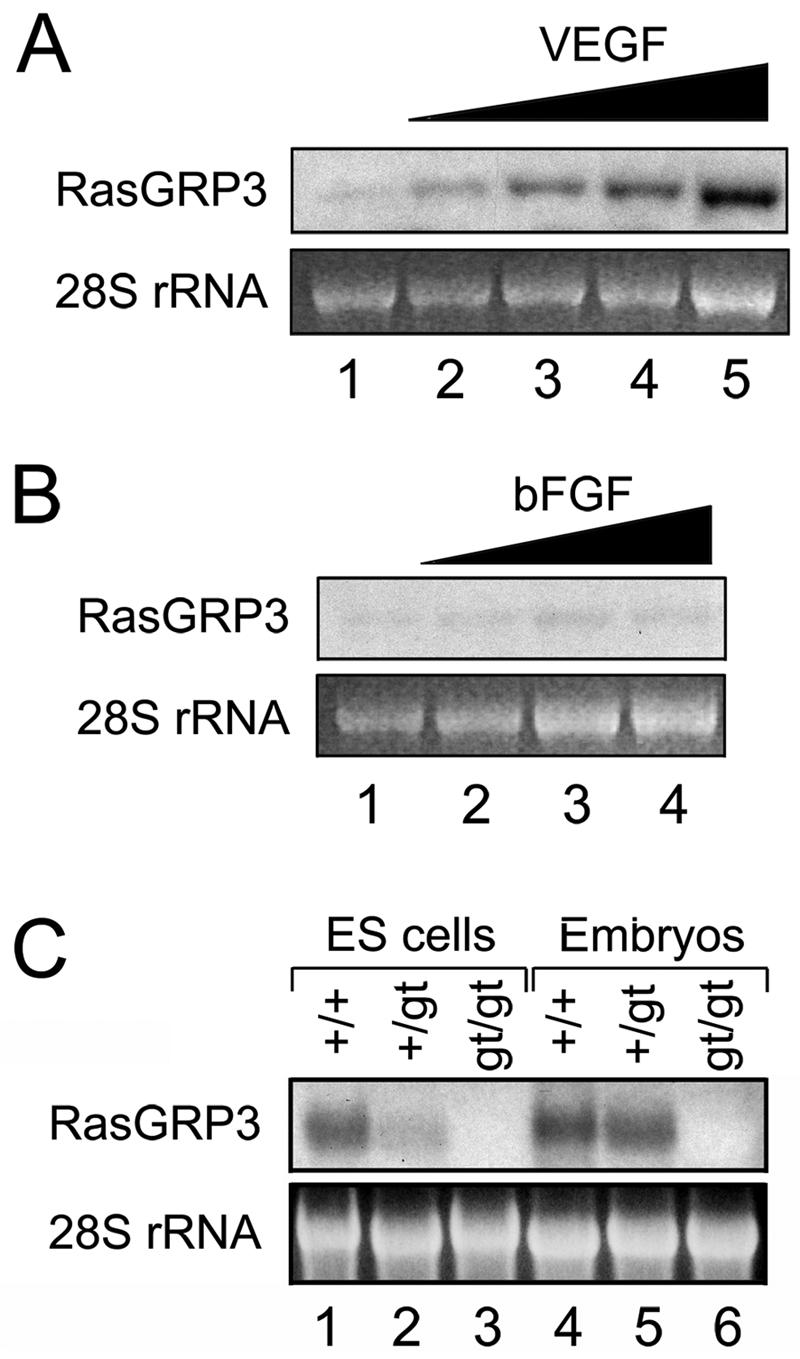
Rasgrp3 is a VEGF-responsive gene and a loss-of-function mutation. (A and B) HUVECs were incubated with increasing concentrations of VEGF (5 to 60 ng/ml) (A) or bFGF (5 to 80 ng/ml) (B). After 24 h, RNA was collected and subjected to Northern blot analysis. (C) RasGRP3 Northern blot analysis was performed on RNA isolated from Rasgrp3+/+, Rasgrp3+/gt, and Rasgrp3gt/gt E9.5 embryos and day 8 ES cell cultures.
RasGRP3 is a phorbol ester receptor that functions in endothelial cells.
The RasGRP3 gene trap insertion most likely deleted exon 3 of Rasgrp3, which contains the predicted start codon for the RasGRP3 protein, so the gene trap insertion is likely to have resulted in a null mutation (Fig. 2A). To further investigate this possibility, we analyzed total RNA isolated from Rasgrp3+/+, Rasgrp3+/gt, and Rasgrp3gt/gt E9.5 embryos and day 8 ES cell cultures for RasGRP3 mRNA by Northern blot analysis (Fig. 7C). In all cases, RasGRP3 mRNA was readily detectable in Rasgrp3+/+ and Rasgrp3+/gt samples; however, no RasGRP3 transcript could be detected in Rasgrp3gt/gt samples. This result strongly indicates that the gene trap insertion resulted in a null mutation in Rasgrp3, although we cannot exclude the formal possibility that it is a severe hypomorphic mutation. Moreover, we were unable to detect expression of RasGRP3 in Rasgrp3gt/gt embryos and day 8 ES cell cultures by in situ hybridization (data not shown), further supporting the conclusion that functional RasGRP3 RNA is not made from the Rasgrp3gt locus.
To determine if loss of RasGRP3 affected development, we intercrossed Rasgrp3+/gt mice and analyzed the progeny. Rasgrp3gt/gt mice were born at the expected Mendelian ratio, and the life spans of these mice were comparable to those of littermate controls, indicating that RasGRP3 is not required for development or viability (data not shown). Furthermore, no differences in reproductive ability were observed. The blood vessels of Rasgp3gt/gt mice also appeared normal, as no hemorrhage or morphological defects were detected (data not shown). Because RasGRP3 expression is regulated developmentally, we asked whether Rasgrp3gt/gt embryos had a subtle vascular phenotype that was rescued prior to birth. To address this issue, we isolated mid-gestation embryos from Rasgrp3+/gt intercrosses and stained them with the vascular marker PECAM. Compared to blood vessels of littermate controls, the blood vessels of Rasgrp3gt/gt embryos appeared normal (Fig. 8A to C). Consistent with these in vivo results, the vasculature of differentiated Rasgrp3gt/gt ES cell cultures also appeared normal (Fig. 8D to F). The Rasgrp3gt/gt vessels were the proper thickness and branched appropriately, with normal variation in those parameters as seen in wild-type controls.
FIG. 8.

Rasgrp3gt/gt embryos and ES cell cultures do not have an obvious vascular phenotype. Rasgrp3+/+, Rasgrp3+/gt, and Rasgrp3gt/gt E9.5 embryos and day 8 ES cell cultures were incubated with anti-PECAM antibody and reacted with 3′,3′-diaminobenzidine tetrahydrochloride (A to C) or an immunofluorescent secondary antibody (D to F). Rasgrp3+/+ (A and D), Rasgrp3+/gt (B and E), and Rasgrp3gt/gt (C and F) embryos or cultures are shown. Note the lack of differences among the genotypes.
Since RasGRP3 was up-regulated during tumor angiogenesis, the consequences of RasGRP3 deficiency during tumor development were investigated. Compared to littermate controls, the kinetics and frequency of mammary tumor development in MMTV-PyVT;Rasgrp3gt/gt mice were not affected (data not shown). We also examined the kidneys of Rasgrp3gt/gt mice by histology and found no obvious lesions or abnormalities (data not shown). Taken together, these results indicate that RasGRP3 deficiency does not result in an obvious vascular phenotype at the stages and sites analyzed.
RasGRP3 localization and activity is regulated by DAG, and this interaction is mimicked by the phorbol ester PMA. Thus, PMA is predicted to increase RasGRP3 activity and Ras activation (47). Normally, ES cell-derived blood vessels form a vascular plexus consisting of thin vessels that branch at regular intervals (Fig. 9A to C); however, wild-type ES cell-derived vessels treated with PMA underwent extensive alterations and formed large sheets with little branching (Fig. 9D). Formation of these endothelial sheets required RasGRP3 activity, since RasGRP3gt/gt cultures treated with PMA did not form vascular sheets, but instead had morphologically normal vessels (Fig. 9F). This was true of three independent isolates of homozygous Rasgrp3gt/gt ES cells. RasGRP3+/gt cultures treated with PMA displayed an intermediate phenotype (Fig. 9E), suggesting a dose response to the PMA-induced morphological changes mediated by RasGRP3. PMA-induced morphological changes in vessels occurred over a range of PMA concentrations from 10 to 1,000 nM (data not shown).
FIG. 9.

PMA-induced morphological alteration of blood vessels requires RasGRP3 function. Day 5 to 8 ES cell cultures were treated with PMA, fixed on day 8, and reacted with anti-PECAM antibody. (A to C) Untreated; (D to F) 100 nM PMA. Rasgrp3+/+ (A and D), Rasgrp3+/gt (B and E), Rasgrp3gt/gt (C and F), and flt-1−/− (G) cultures are shown. Note the formation of PECAM-positive endothelial sheets in the PMA-treated Rasgrp3+/+ cultures and the intermediate structures in Rasgrp3+/gt cultures. Cultures lacking flt-1 also formed endothelial sheets.
The alteration of vessels observed following PMA addition to wild-type ES cell cultures closely resembles the vascular phenotype resulting from genetic ablation of the VEGF receptor, flt-1 (Fig. 9G), which up-regulates VEGF signaling. Because the PMA-induced vascular changes required RasGRP3 activity, we asked whether RasGRP3 was up-regulated in the flt-1−/− background. Northern blot analysis revealed that, when normalized to the amount of vasculature, RasGRP3 RNA levels were increased 1.5-fold in flt-1−/− cultures (Fig. 10). Taken together, these results suggest that increased RasGRP3 activity may contribute to the aberrant morphology of flt-1−/− vessels.
FIG. 10.

RasGRP3 is upregulated in flt-1−/−blood vessels. (A) Northern blot analysis was performed on RNA isolated from flt-1−/− cultures (lane 3). RNA was also isolated from Rasgrp3+/+ and Rasgrp3gt/gt cultures treated with PMA (lanes 2 and 5) or left untreated (lanes 1 and 4). The blot was probed for RasGRP3 and then reprobed for the vascular marker PECAM. (B) Graph showing relative levels of RasGRP3 RNA in ES cell cultures. RasGRP3 RNA levels were normalized to the amount of PECAM RNA.
DISCUSSION
This study identifies RasGRP3 as a GEF expressed in the vasculature. Its expression is associated with developing vessels in the embryo and in the adult, suggesting that Ras activation through RasGRP3 is utilized for angiogenesis. Indeed, RasGRP3 expression is directly regulated by VEGF signaling, and the VEGF pathway is critical for angiogenesis. Moreover, RasGRP3 is required to mediate the aberrant endothelial morphogenesis that results from PMA exposure, a treatment that phenocopies aspects of up-regulated VEGF signaling. Taken together, our results indicate that RasGRP3 is a phorbol ester receptor that functions in endothelial cells, where it transduces signals that modulate angiogenic activity.
Endothelial RasGRP3 expression is regulated by angiogenesis and VEGF.
RasGRP3 belongs to a GEF subfamily of four highly related proteins that contain a C1 domain. This domain binds DAG, a membrane-localized second messenger, and brings the GEF into close proximity to membrane-localized GTPases to effect its activation function. Mouse RasGRP3 shares extensive sequence homology with human RasGRP3 and with the other family members (78). RasGRP family members, including RasGRP3, are expressed in hematopoietic cells: RasGRP1 is expressed in T cells, RasGRP4 is expressed in myeloid cells, and RasGRP3 is expressed in B cells (19, 23, 53, 56, 81). RasGRP2 has a more ubiquitous hematopoietic expression profile (G. Reuther and C. Der, personal communication), and RasGRP1 to RasGRP3 are expressed in some nonhematopoietic tissues such as brain and kidney (78). Given that endothelial cells share expression of numerous genes with hematopoietic cells, it is not surprising that we identified murine RasGRP3 in our gene trap screen to uncover novel vascular genes. B cells do not mature from ES cells in our differentiation protocol, and they are not present in mid-gestation embryos, so we did not score B-cell expression of RasGRP3. We did score expression of RasGRP3 using the β-Gal reporter readout in embryonic tissues such as the somitic myotome and the pericardium and in rare nonendothelial adult cells. However, RasGRP3 expression was most consistently found in blood vessels undergoing angiogenesis in both embryos and adults, suggesting that Ras activation through RasGRP3 accompanies angiogenesis.
The up-regulation of RasGRP3 expression during angiogenesis suggested that angiogenic signaling may regulate expression of this locus. VEGF signaling is a prominent feature of angiogenesis throughout life, and RasGRP3 expression is up-regulated upon VEGF stimulation of primary endothelial cells. VEGF-dependent up-regulation of RasGRP3 was also documented in a microarray analysis (80). Although VEGF signaling is dramatically down-regulated in quiescent vessels, the pathway remains active in the glomerulus of the kidney, where it is required to establish and maintain endothelial fenestrations for filtration (25, 26, 60). Likewise, RasGRP3 expression is maintained at this site, suggesting that Ras activation via RasGRP3 is involved in this process. RasGRP3 expression is also up-regulated in the maternal vessels of the decidua and in vessels that form in response to tumor angiogenic signals. These findings are consistent with a model in which RasGRP3 expression is downstream of VEGF signaling and VEGF stimulation increases the amount of RasGRP3 RNA in HUVECs. It will be interesting to determine the intracellular pathways downstream of VEGF that mediate up-regulation of RasGRP3 expression.
RasGRP3 is not necessary for angiogenesis but mediates the endothelial response to phorbol esters.
Numerous studies implicate Ras signaling in endothelial function and vascular development (34, 36, 76). Generation of a Rasgrp3 loss-of-function allele allowed us to assess the effects of RasGRP3 deficiency in developing vessels. Homozygous Rasgrp3gt/gt ES cell-derived vessels formed normally, and Rasgrp3gt/gt mutant embryos and mice had no obvious phenotype. RasGRP2 is required for proper platelet aggregation (13) and RasGRP1 is required for T-cell maturation in vivo (19), but we did not observe similar defects in Rasgrp3gt/gt endothelial cells. This is not surprising, because redundancy for RasGRP3 function is predicted. It is possible that expression of other RasGRPs and/or other RasGEFs provides a compensatory activating function. We detected RasGRP2 but not RasGRP1 or RasGRP4 in a murine endothelial cell line by RT-PCR analysis, suggesting that RasGRP2 function may compensate for loss of RasGRP3 (D. Roberts and V. Bautch, unpublished results). However, RasGRP2 is a Rap1 activator and not a Ras activator, whereas RasGRP3 is an activator of Ras and Rap1. Therefore, other RasGEFs may also compensate for the loss of RasGRP3 regulation of Ras. There are multiple RasGEFs expressed in most cells, and other GEFs such as the Drosophila melanogaster RhoGEF GEF64C do not exhibit a loss-of-function phenotype (3). One prominent RasGEF that is expressed ubiquitously is the mammalian Son-of-Sevenless (SOS) homologue (20). This GEF is complexed with the adaptor protein Grb2, and the SOS-Grb2 complex translocates to the plasma membrane via Grb2 association with the cytoplasmic domain of activated receptor tyrosine kinases, thus promoting SOS activation of Ras. Studies with B cells suggest that SOS and RasGRP3 may activate Ras coordinately (53); therefore, SOS may be able to partially compensate for the lack of RasGRP3 activity in endothelial cells (Fig. 11). Ras activation can also occur independently of RasGEFs. Protein kinase C (PKC) activation of extracellular signal-regulated kinases 1 and 2 in endothelial cells is insensitive to Ras-N17, a dominant negative form of Ras that sequesters and inactivates RasGEFs (18, 66, 70). In this case, PKC is thought to activate sphingosine kinase (SPK), and SPK may activate Ras by RasGAP inhibition (Fig. 11). However, PKC also phosphorylates RasGRP3, and this phosphorylation event is highly correlated with membrane localization of RasGRP3 and Ras activation in B cells (8, 71). This finding suggests that PKC may affect cellular functions at least in part via activation of RasGRP3.
FIG. 11.

Model of Ras activation downstream of VEGF signaling. Several potential pathways downstream of VEGF receptor activation may lead to Ras activation, and some involve RasGRP3, as follows. (i) Activation of PLC-γ leads to DAG production and RasGRP3 activation. (ii) DAG also recruits PKC which can phosphorylate RasGRP3. (iii) PKC may also activate Ras through SPK. (iv) VEGF signaling through SOS/Grb2 may lead to Ras activation. (v) DAG production can occur independently of VEGF signaling through aberrant glucose metabolism.
We thus asked whether a gain-of-function manipulation of RasGRP3 produced a vascular phenotype. Other GEFs up-regulate their target pathways when they are constitutively localized to the membrane, so developing ES cell-derived vessels were incubated with PMA, a phorbol ester that mimics DAG and is predicted to localize RasGRP3 to the membrane (47). RasGRP3-expressing wild-type vessels responded dramatically to PMA, exhibiting extensive morphogenetic changes that resulted in the formation of sheets rather than branched vessels. A preliminary analysis indicates that both increased endothelial proliferation and aberrant cell-cell interactions contribute to the phenotypic response to PMA (Roberts and Bautch, unpublished). PMA has complex effects on endothelial cells, since it also potently activates PKC and increases vascular permeability (39, 52, 75). However, despite the potential complexity of the endothelial response to PMA, it is dependent on RasGRP3, since Rasgrp3gt/gt vessels were completely refractory to the PMA-induced morphogenetic changes. This result shows that RasGRP3 functions in endothelial cells and confirms that the Rasgrp3gt allele is a loss-of-function mutation. Future work will delineate the components downstream of the response of newly formed vessels to PMA and how these components interact with RasGRP3.
How does RasGRP3 function in endothelial cells? One clue comes from the phenotype of wild-type vessels incubated with PMA, which in many aspects resembles the phenotype of vessels with up-regulated VEGF signaling. Although the VEGF receptor flt-1 (VEGFR-1) can activate downstream signaling pathways in some contexts (82), flt-1 is thought to act developmentally primarily as a decoy receptor to bind excess VEGF (42, 44, 59). Thus, deletion of flt-1 in ES cell cultures leads to aberrant vascular morphogenesis and large endothelial sheets, and these perturbations are consistent with up-regulation of VEGF signaling (42). This phenotype results from both increased proliferation and compromised migration of affected endothelial cells (42, 44). RasGRP3 expression is up-regulated in the flt-1 mutant ES cell cultures, consistent with the possibility that excess Ras activation through RasGRP3 contributes to the flt-1 mutant phenotype. In addition, data suggests that RasGRP3 also activates other Ras-related GTPases such as Rap (55, 68, 78), so RasGRP3-mediated activation of Rap may also contribute to the vascular phenotype seen in the flt-1 mutants and upon exposure to PMA. However, since PMA stimulation of primary endothelial cells results in robust Ras activation (75), Ras signaling is likely to contribute extensively to PMA-mediated vascular effects. Moreover, preliminary results indicate that the farnesyltransferase inhibitor FTI-2153 partially rescues the aberrant vascular phenotype induced by PMA (Roberts and Bautch, unpublished), a finding consistent with a model in which Ras is a downstream target of RasGRP3. Whether PMA stimulation also activates Rap in endothelial cells is yet to be determined. It will be interesting to determine which pathways are activated downstream of the flt-1 mutation, and which components of VEGF signaling are mediated by Ras activation in vivo.
Our data demonstrate that endothelial cell expression of a GEF, RasGRP3, is downstream of VEGF signaling during blood vessel formation in vivo. RasGRP3 functions in endothelial cells, as it is required for PMA-induced vascular morphogenetic defects, but it does not appear to be essential for vascular development or normal angiogenesis. We suggest that this mode of Ras activation downstream of VEGF signaling is important in situations of pathway activation where redundant pathways are not up-regulated. RasGRPs uniquely utilize DAG to localize to the membrane and activate Ras, and DAG is produced by several intracellular pathways. Thus, disease states that lead to excess DAG production may depend on RasGRP3-mediated Ras activation (Fig. 11). Several studies have correlated aberrant glucose metabolism and concomitant increases in DAG with diabetes (for reviews, see references 10 and 77). Thus, RasGRP3 may be involved in the vascular complications of diabetic patients, such as nephropathy and retinopathy, and it may provide a novel target for therapeutics aimed at ameliorating diabetic symptoms.
Acknowledgments
We thank Guo-Hua Fong for flt-1−/− ES cells and mice, Rebecca Rapoport and Sheena Waters for technical help, William Muller and Terry Van Dyke for MMTV-PyVT mice, members of the Bautch lab and the Carolina Cardiovascular Development Group for stimulating discussion, and Adrienne Cox, Channing Der, Mark Majesky, and Mark Peifer for critical comments on the manuscript.
This work was supported by grants from the NIH (HL43174 and HL71993) to V.L.B., CIHR (FRN36653) to W.L.S., and a NSF predoctoral fellowship and a DOD predoctoral fellowship to D.M.R.
REFERENCES
- 1.Ambler, C. A., J. L. Nowicki, A. C. Burke, and V. L. Bautch. 2001. Assembly of trunk and limb blood vessels involves extensive migration and vasculogenesis of somite-derived angioblasts. Dev. Biol. 234:352-364. [DOI] [PubMed] [Google Scholar]
- 2.Ambler, C. A., G. M. Schmunk, and V. L. Bautch. 2003. Stem cell-derived endothelial cells/progenitors migrate and pattern in the embryo using the VEGF signaling pathway. Dev. Biol. 257:205-219. [DOI] [PubMed] [Google Scholar]
- 3.Bashaw, G. J., H. Hu, C. D. Nobes, and C. S. Goodman. 2001. A novel Dbl family RhoGEF promotes Rho-dependent axon attraction to the central nervous system midline in Drosophila and overcomes Robo repulsion. J. Cell Biol. 155:1117-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bautch, V. L. 2002. Embryonic stem cell differentiation and the vascular lineage. Methods Mol. Biol. 185:117-125. [DOI] [PubMed] [Google Scholar]
- 5.Bautch, V. L., S. D. Redick, A. Scalia, M. Harmaty, P. Carmeliet, and R. Rapoport. 2000. Characterization of the vasculogenic block in the absence of vascular endothelial growth factor-A. Blood 95:1979-1987. [PubMed] [Google Scholar]
- 6.Bernards, A. 2003. GAPs galore! A survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. Biochim. Biophys. Acta 1603:47-82. [DOI] [PubMed] [Google Scholar]
- 7.Brannan, C. I., A. S. Perkins, K. S. Vogel, N. Ratner, M. L. Nordlund, S. W. Reid, A. M. Buchberg, N. A. Jenkins, L. F. Parada, and N. G. Copeland. 1994. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 8:1019-1029. [DOI] [PubMed] [Google Scholar]
- 8.Brodie, C., R. Steinhart, G. Kazimirsky, H. Rubinfeld, T. Hyman, J. N. Ayres, G. M. Hur, A. Toth, D. Yang, S. H. Garfield, J. C. Stone, and P. M. Blumberg. 2004. PKCδ associates with and is involved in the phosphorylation of RasGRP3 in response to phorbol esters. Mol. Pharmacol. 66:76-84. [DOI] [PubMed] [Google Scholar]
- 9.Brown, L. F., B. Berse, K. Tognazzi, E. J. Manseau, L. Van de Water, D. R. Senger, H. F. Dvorak, and S. Rosen. 1992. Vascular permeability factor mRNA and protein expression in human kidney. Kidney Int. 42:1457-1461. [DOI] [PubMed] [Google Scholar]
- 10.Brownlee, M. 2001. Biochemistry and molecular cell biology of diabetic complications. Nature 414:813-820. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet, P., V. Ferreira, G. Breier, S. Pollefeyt, L. Kieckens, M. Gertsenstein, M. Fahrig, A. Vandenhoeck, K. Harpal, C. Eberhardt, C. Declercq, J. Pawling, L. Moons, D. Collen, W. Risau, and A. Nagy. 1996. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380:435-439. [DOI] [PubMed] [Google Scholar]
- 12.Clyde-Smith, J., G. Silins, M. Gartside, S. Grimmond, M. Etheridge, A. Apolloni, N. Hayward, and J. F. Hancock. 2000. Characterization of RasGRP2, a plasma membrane-targeted, dual specificity Ras/Rap exchange factor. J. Biol. Chem. 275:32260-32267. [DOI] [PubMed] [Google Scholar]
- 13.Crittenden, J. R., W. Bergmeier, Y. Zhang, C. L. Piffath, Y. Liang, D. D. Wagner, D. E. Housman, and A. M. Graybiel. 2004. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 10:982-986. [DOI] [PubMed] [Google Scholar]
- 14.Cross, M. J., J. Dixelius, T. Matsumoto, and L. Claesson-Welsh. 2003. VEGF-receptor signal transduction. Trends Biochem. Sci. 28:488-494. [DOI] [PubMed] [Google Scholar]
- 15.Damert, A., L. Miquerol, M. Gertsenstein, W. Risau, and A. Nagy. 2002. Insufficient VEGFA activity in yolk sac endoderm compromises haematopoietic and endothelial differentiation. Development 129:1881-1892. [DOI] [PubMed] [Google Scholar]
- 16.Daniel, T. O., and D. Abrahamson. 2000. Endothelial signal integration in vascular assembly. Annu. Rev. Physiol. 62:649-671. [DOI] [PubMed] [Google Scholar]
- 17.Di Fiore, P. P. 2003. Signal transduction: life on Mars, cellularly speaking. Nature 424:624-625. [DOI] [PubMed] [Google Scholar]
- 18.Doanes, A. M., D. D. Hegland, R. Sethi, I. Kovesdi, J. T. Bruder, and T. Finkel. 1999. VEGF stimulates MAPK through a pathway that is unique for receptor tyrosine kinases. Biochem. Biophys. Res. Commun. 255:545-548. [DOI] [PubMed] [Google Scholar]
- 19.Dower, N. A., S. L. Stang, D. A. Bottorff, J. O. Ebinu, P. Dickie, H. L. Ostergaard, and J. C. Stone. 2000. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat. Immunol. 1:317-321. [DOI] [PubMed] [Google Scholar]
- 20.Downward, J. 1996. Control of ras activation. Cancer Surv. 27:87-100. [PubMed] [Google Scholar]
- 21.Dubois, N. A., L. C. Kolpack, R. Wang, R. G. Azizkhan, and V. L. Bautch. 1991. Isolation and characterization of an established endothelial cell line from transgenic mouse hemangiomas. Exp. Cell Res. 196:302-313. [DOI] [PubMed] [Google Scholar]
- 22.Ebinu, J. O., D. A. Bottorff, E. Y. Chan, S. L. Stang, R. J. Dunn, and J. C. Stone. 1998. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science 280:1082-1086. [DOI] [PubMed] [Google Scholar]
- 23.Ebinu, J. O., S. L. Stang, C. Teixeira, D. A. Bottorff, J. Hooton, P. M. Blumberg, M. Barry, R. C. Bleakley, H. L. Ostergaard, and J. C. Stone. 2000. RasGRP links T-cell receptor signaling to Ras. Blood 95:3199-3203. [PubMed] [Google Scholar]
- 24.Eliceiri, B. P., and D. A. Cheresh. 2001. Adhesion events in angiogenesis. Curr. Opin. Cell Biol. 13:563-568. [DOI] [PubMed] [Google Scholar]
- 25.Eremina, V., M. Sood, J. Haigh, A. Nagy, G. Lajoie, N. Ferrara, H. P. Gerber, Y. Kikkawa, J. H. Miner, and S. E. Quaggin. 2003. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 111:707-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esser, S., K. Wolburg, H. Wolburg, G. Breier, T. Kurzchalia, and W. Risau. 1998. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J. Cell Biol. 140:947-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esteban, L. M., C. Vicario-Abejón, P. Fernández-Salguero, A. Fernández-Medarde, N. Swaminathan, K. Yienger, E. Lopez, M. Malumbres, R. McKay, J. M. Ward, A. Pellicer, and E. Santos. 2001. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell. Biol. 21:1444-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrara, N., K. Carver-Moore, H. Chen, M. Dowd, L. Lu, K. S. O'Shea, L. Powell-Braxton, K. J. Hillan, and M. W. Moore. 1996. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380:439-442. [DOI] [PubMed] [Google Scholar]
- 29.Ferrara, N., H. P. Gerber, and J. LeCouter. 2003. The biology of VEGF and its receptors. Nat. Med. 9:669-676. [DOI] [PubMed] [Google Scholar]
- 30.Folkman, J. 2003. Fundamental concepts of the angiogenic process. Curr. Mol. Med. 3:643-651. [DOI] [PubMed] [Google Scholar]
- 31.Fong, G. H., J. Rossant, M. Gertsenstein, and M. L. Breitman. 1995. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376:66-70. [DOI] [PubMed] [Google Scholar]
- 32.Galli-Taliadoros, L. A., J. D. Sedgwick, S. A. Wood, and H. Korner. 1995. Gene knock-out technology: a methodological overview for the interested novice. J. Immunol. Methods 181:1-15. [DOI] [PubMed] [Google Scholar]
- 33.Gerhardt, H., M. Golding, M. Fruttiger, C. Ruhrberg, A. Lundkvist, A. Abramsson, M. Jeltsch, C. Mitchell, K. Alitalo, D. Shima, and C. Betsholtz. 2003. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161:1163-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gitler, A. D., Y. Zhu, F. A. Ismat, M. M. Lu, Y. Yamauchi, L. F. Parada, and J. A. Epstein. 2003. Nf1 has an essential role in endothelial cells. Nat. Genet. 33:75-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guy, C. T., R. D. Cardiff, and W. J. Muller. 1992. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 12:954-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henkemeyer, M., D. J. Rossi, D. P. Holmyard, M. C. Puri, G. Mbamalu, K. Harpal, T. S. Shih, T. Jacks, and T. Pawson. 1995. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 377:695-701. [DOI] [PubMed] [Google Scholar]
- 37.Hidaka, M., G. Caruana, W. L. Stanford, M. Sam, P. H. Correll, and A. Bernstein. 2000. Gene trapping of two novel genes, Hzf and Hhl, expressed in hematopoietic cells. Mech. Dev. 90:3-15. [DOI] [PubMed] [Google Scholar]
- 38.Hood, J. D., R. Frausto, W. B. Kiosses, M. A. Schwartz, and D. A. Cheresh. 2003. Differential αv integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J. Cell Biol. 162:933-943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang, Q., and Y. Yuan. 1997. Interaction of PKC and NOS in signal transduction of microvascular hyperpermeability. Am. J. Physiol. 273:H2442-H2451. [DOI] [PubMed] [Google Scholar]
- 40.Johnson, L., D. Greenbaum, K. Cichowski, K. Mercer, E. Murphy, E. Schmitt, R. T. Bronson, H. Umanoff, W. Edelmann, R. Kucherlapati, and T. Jacks. 1997. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 11:2468-2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawasaki, H., G. M. Springett, S. Toki, J. J. Canales, P. Harlan, J. P. Blumenstiel, E. J. Chen, I. A. Bany, N. Mochizuki, A. Ashbacher, M. Matsuda, D. E. Housman, and A. M. Graybiel. 1998. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. USA 95:13278-13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kearney, J. B., C. A. Ambler, K. A. Monaco, N. Johnson, R. G. Rapoport, and V. L. Bautch. 2002. Vascular endothelial growth factor receptor Flt-1 negatively regulates developmental blood vessel formation by modulating endothelial cell division. Blood 99:2397-2407. [DOI] [PubMed] [Google Scholar]
- 43.Kearney, J. B., and V. L. Bautch. 2003. In vitro differentiation of mouse ES cells: hematopoietic and vascular development. Methods Enzymol. 365:83-98. [DOI] [PubMed] [Google Scholar]
- 44.Kearney, J. B., N. C. Kappas, C. Ellerstrom, F. W. DiPaola, and V. L. Bautch. 2004. The VEGF receptor flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood 103:4527-4535. [DOI] [PubMed] [Google Scholar]
- 45.Koera, K., K. Nakamura, K. Nakao, J. Miyoshi, K. Toyoshima, T. Hatta, H. Otani, A. Aiba, and M. Katsuki. 1997. K-ras is essential for the development of the mouse embryo. Oncogene 15:1151-1159. [DOI] [PubMed] [Google Scholar]
- 46.Lorenzo, P. S., M. Beheshti, G. R. Pettit, J. C. Stone, and P. M. Blumberg. 2000. The guanine nucleotide exchange factor RasGRP is a high-affinity target for diacylglycerol and phorbol esters. Mol. Pharmacol. 57:840-846. [PubMed] [Google Scholar]
- 47.Lorenzo, P. S., J. W. Kung, D. A. Bottorff, S. H. Garfield, J. C. Stone, and P. M. Blumberg. 2001. Phorbol esters modulate the Ras exchange factor RasGRP3. Cancer Res. 61:943-949. [PubMed] [Google Scholar]
- 48.Meadows, K. N., P. Bryant, and K. Pumiglia. 2001. Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J. Biol. Chem. 276:49289-49298. [DOI] [PubMed] [Google Scholar]
- 49.Meadows, K. N., P. Bryant, P. A. Vincent, and K. M. Pumiglia. 2004. Activated Ras induces a proangiogenic phenotype in primary endothelial cells. Oncogene 23:192-200. [DOI] [PubMed] [Google Scholar]
- 50.Miquerol, L., B. L. Langille, and A. Nagy. 2000. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development 127:3941-3946. [DOI] [PubMed] [Google Scholar]
- 51.Nagase, T., K. Ishikawa, M. Suyama, R. Kikuno, M. Hirosawa, N. Miyajima, A. Tanaka, H. Kotani, N. Nomura, and O. Ohara. 1998. Prediction of the coding sequences of unidentified human genes. XII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 5:355-364. [DOI] [PubMed] [Google Scholar]
- 52.Nagpala, P. G., A. B. Malik, P. T. Vuong, and H. Lum. 1996. Protein kinase C β 1 overexpression augments phorbol ester-induced increase in endothelial permeability. J. Cell. Physiol. 166:249-255. [DOI] [PubMed] [Google Scholar]
- 53.Oh-hora, M., S. Johmura, A. Hashimoto, M. Hikida, and T. Kurosaki. 2003. Requirement for Ras guanine nucleotide releasing protein 3 in coupling phospholipase C-γ2 to Ras in B cell receptor signaling. J. Exp. Med. 198:1841-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quilliam, L. A., J. F. Rebhun, and A. F. Castro. 2002. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog. Nucleic Acid Res. Mol. Biol. 71:391-444. [DOI] [PubMed] [Google Scholar]
- 55.Rebhun, J. F., A. F. Castro, and L. A. Quilliam. 2000. Identification of guanine nucleotide exchange factors (GEFs) for the Rap1 GTPase. Regulation of MR-GEF by M-Ras-GTP interaction. J. Biol. Chem. 275:34901-34908. [DOI] [PubMed] [Google Scholar]
- 56.Reuther, G. W., Q. T. Lambert, J. F. Rebhun, M. A. Caligiuri, L. A. Quilliam, and C. J. Der. 2002. RasGRP4 is a novel Ras activator isolated from acute myeloid leukemia. J. Biol. Chem. 277:30508-30514. [DOI] [PubMed] [Google Scholar]
- 57.Risau, W. 1997. Mechanisms of angiogenesis. Nature 386:671-674. [DOI] [PubMed] [Google Scholar]
- 58.Robert, B., X. Zhao, and D. R. Abrahamson. 2000. Coexpression of neuropilin-1, Flk1, and VEGF164 in developing and mature mouse kidney glomeruli. Am. J. Physiol. Renal Physiol. 279:F275-F282. [DOI] [PubMed] [Google Scholar]
- 59.Roberts, D. M., J. B. Kearney, J. H. Johnson, M. P. Rosenberg, R. Kumar, and V. L. Bautch. 2004. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am. J. Pathol. 164:1531-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roberts, W. G., and G. E. Palade. 1995. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J. Cell Sci. 108:2369-2379. [DOI] [PubMed] [Google Scholar]
- 61.Rosen, B., and R. S. Beddington. 1993. Whole-mount in situ hybridization in the mouse embryo: gene expression in three dimensions. Trends Genet. 9:162-167. [DOI] [PubMed] [Google Scholar]
- 62.Ruhrberg, C., H. Gerhardt, M. Golding, R. Watson, S. Ioannidou, H. Fujisawa, C. Betsholtz, and D. T. Shima. 2002. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 16:2684-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shalaby, F., J. Rossant, T. P. Yamaguchi, M. Gertsenstein, X. F. Wu, M. L. Breitman, and A. C. Schuh. 1995. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376:62-66. [DOI] [PubMed] [Google Scholar]
- 64.Shibuya, M. 2003. Vascular endothelial growth factor receptor-2: its unique signaling and specific ligand, VEGF-E. Cancer Sci. 94:751-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shields, J. M., K. Pruitt, A. McFall, A. Shaub, and C. J. Der. 2000. Understanding Ras: ‘it ain’t over ‘til it’s over. Trends Cell Biol. 10:147-154. [DOI] [PubMed] [Google Scholar]
- 66.Shu, X., W. Wu, R. D. Mosteller, and D. Broek. 2002. Sphingosine kinase mediates vascular endothelial growth factor-induced activation of Ras and mitogen-activated protein kinases. Mol. Cell. Biol. 22:7758-7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stanford, W. L., G. Caruana, K. A. Vallis, M. Inamdar, M. Hidaka, V. L. Bautch, and A. Bernstein. 1998. Expression trapping: identification of novel genes expressed in hematopoietic and endothelial lineages by gene trapping in ES cells. Blood 92:4622-4631. [PubMed] [Google Scholar]
- 68.Stope, M. B., F. vom Dorp, D. Szatkowski, A. Böhm, M. Keiper, J. Nolte, P. A. Oude Weernink, D. Rosskopf, S. Evellin, K. H. Jakobs, and M. Schmidt. 2004. Rap2B-dependent stimulation of phospholipase C-ε by epidermal growth factor receptor mediated by c-Src phosphorylation of RasGRP3. Mol. Cell. Biol. 24:4664-4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takahashi, T., and M. Shibuya. 1997. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-γ pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene 14:2079-2089. [DOI] [PubMed] [Google Scholar]
- 70.Takahashi, T., H. Ueno, and M. Shibuya. 1999. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 18:2221-2230. [DOI] [PubMed] [Google Scholar]
- 71.Teixeira, C., S. L. Stang, Y. Zheng, N. S. Beswick, and J. C. Stone. 2003. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood 102:1414-1420. [DOI] [PubMed] [Google Scholar]
- 72.Terman, B. I., and M. Dougher-Vermazen. 1996. Biological properties of VEGF/VPF receptors. Cancer Metastasis Rev. 15:159-163. [DOI] [PubMed] [Google Scholar]
- 73.Tognon, C. E., H. E. Kirk, L. A. Passmore, I. P. Whitehead, C. J. Der, and R. J. Kay. 1998. Regulation of RasGRP via a phorbol ester-responsive C1 domain. Mol. Cell. Biol. 18:6995-7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Umanoff, H., W. Edelmann, A. Pellicer, and R. Kucherlapati. 1995. The murine N-ras gene is not essential for growth and development. Proc. Natl. Acad. Sci. USA 92:1709-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Verin, A. D., F. Liu, N. Bogatcheva, T. Borbiev, M. B. Hershenson, P. Wang, and J. G. Garcia. 2000. Role of ras-dependent ERK activation in phorbol ester-induced endothelial cell barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 279:L360-L370. [DOI] [PubMed] [Google Scholar]
- 76.Wang, D. Z., V. E. Hammond, H. E. Abud, I. Bertoncello, J. W. McAvoy, and D. D. Bowtell. 1997. Mutation in Sos1 dominantly enhances a weak allele of the EGFR, demonstrating a requirement for Sos1 in EGFR signaling and development. Genes Dev. 11:309-320. [DOI] [PubMed] [Google Scholar]
- 77.Way, K. J., N. Katai, and G. L. King. 2001. Protein kinase C and the development of diabetic vascular complications. Diabet. Med. 18:945-959. [DOI] [PubMed] [Google Scholar]
- 78.Yamashita, S., N. Mochizuki, Y. Ohba, M. Tobiume, Y. Okada, H. Sawa, K. Nagashima, and M. Matsuda. 2000. CalDAG-GEFIII activation of Ras, R-ras, and Rap1. J. Biol. Chem. 275:25488-25493. [DOI] [PubMed] [Google Scholar]
- 79.Yancopoulos, G. D., S. Davis, N. W. Gale, J. S. Rudge, S. J. Wiegand, and J. Holash. 2000. Vascular-specific growth factors and blood vessel formation. Nature 407:242-248. [DOI] [PubMed] [Google Scholar]
- 80.Yang, S., K. Toy, G. Ingle, C. Zlot, P. M. Williams, G. Fuh, B. Li, A. de Vos, and M. E. Gerritsen. 2002. Vascular endothelial growth factor-induced genes in human umbilical vein endothelial cells: relative roles of KDR and Flt-1 receptors. Arterioscler. Thromb. Vasc. Biol. 22:1797-1803. [DOI] [PubMed] [Google Scholar]
- 81.Yang, Y., L. Li, G. W. Wong, S. A. Krilis, M. S. Madhusudhan, A. Sali, and R. L. Stevens. 2002. RasGRP4, a new mast cell-restricted Ras guanine nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Identification of defective variants of this signaling protein in asthma, mastocytosis, and mast cell leukemia patients and demonstration of the importance of RasGRP4 in mast cell development and function. J. Biol. Chem. 277:25756-25774. [DOI] [PubMed] [Google Scholar]
- 82.Zeng, H., H. F. Dvorak, and D. Mukhopadhyay. 2001. Vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF) receptor-1 down-modulates VPF/VEGF receptor-2-mediated endothelial cell proliferation, but not migration, through phosphatidylinositol 3-kinase-dependent pathways. J. Biol. Chem. 276:26969-26979. [DOI] [PubMed] [Google Scholar]