

Abstract
Background:
The addition of anti-human epidermal growth factor receptor 2 (HER2)-targeted drugs, such as trastuzumab, lapatinib, and trastuzumab emtansine (T-DM1), to chemotherapy significantly improved prognosis of HER2-positive breast cancer patients. However, it was confused that metastatic patients vary in the response of targeted drug. Therefore, methods of accurately predicting drug response were really needed. To overcome the spatial and temporal limitations of biopsies, we aimed to develop a more sensitive and less invasive method of detecting mutations associated with anti-HER2 therapeutic response through circulating-free DNA (cfDNA).
Methods:
From March 6, 2014 to December 10, 2014, 24 plasma samples from 20 patients with HER2-positive metastatic breast cancer who received systemic therapy were eligible. We used a panel for detection of hot-spot mutations from 50 oncogenes and tumor suppressor genes, and then used targeted next-generation sequencing (NGS) to identify somatic mutation of these samples in those 50 genes. Samples taken before their first trastuzumab administration and subsequently proven with clinical benefit were grouped into sensitive group. The others were collected after disease progression of the trastuzumab-based therapy and were grouped into the resistant group.
Results:
A total of 486 single-nucleotide variants from 46 genes were detected. Of these 46 genes, phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), proto-oncogene c-Kit (KIT), and tumor protein p53 (TP53) were the most common mutated genes. Seven genes, including epidermal growth factor receptor (EGFR), G protein subunit alpha S (GNAS), HRas proto-oncogene (HRAS), mutL homolog 1 (MLH1), cadherin 1 (CDH1), neuroblastoma RAS viral oncogene homolog (NRAS), and NOTCH1, that only occurred mutations in the resistant group were associated with the resistance of targeted therapy. In addition, we detected a HER2 S855I mutation in two patients who had persistent benefits from anti-HER2 therapy.
Conclusion:
Targeted NGS of cfDNA has potential clinical utility to detect biomarkers from HER2-targeted therapies.
Keywords: Biomarkers, Breast Neoplasms, DNA, Drug Resistance, High-throughput Nucleotide Sequencing, ErbB-2
Introduction
Breast cancer with human epidermal growth factor receptor 2 (HER2) positive is characterized by high malignant degree and poor prognosis. Available data suggest that the incidence of breast cancer for HER2 overexpression is approximately 20–25%.[1,2] Fortunately, clinical studies data have shown that in these subgroup's patients, the addition of anti-HER2-targeted drugs, such as trastuzumab, pertuzumab, trastuzumab emtansine (T-DM1), and lapatinib, to chemotherapy significantly improves both progression-free survival and overall survival in primary and metastatic settings.[3,4,5] Despite effectiveness, it is confusing that different metastatic patients vary in the response to those targeted therapies. What is more, it is inevitable for them to develop resistant to anti-HER2 treatment.[4] Therefore, accurately predicting response of anti-HER2 treatment is necessary. The ideal way is a genomic scan which can reflect the overall tumor's information clearly.[6] Although tumor tissue through biopsies is the gold standard for clinical and investigational genomic scans, they have significantly inherent limitations. Biopsies, as the invasive procedure for patients, are inconvenient, uncomfortable, and accompanied with clinical complications.[7] All of those contributed to the difficulty of tissues acquisition. In addition, the repeatability of tissue biopsy remains a problem. Furthermore, as tumor tissue is a single snapshot in time and space, there are spatial and temporal limitations due to heterogeneity of tumors.[8,9] To overcome these barriers, a less invasive technique capable of capturing tumor heterogeneity and molecular changes of cancer cells when they are exposed to systemic therapy is needed.[9,10] With the advantages of being less invasive, real-time, and dynamic, circulating-free DNA (cfDNA) detection has become the promising method for predicting therapeutic response and is expected to be an alternative of tissue biopsies.[10] The development of next-generation sequencing (NGS) has enabled the detection of cfDNA which represents somatic mutations of individual tumors.[11,12,13,14,15]
Methods
Patient selection and plasma sample collection
Eligible patients were 18–75 years old women with HER2-positive metastatic breast cancer currently undergoing anti-HER2-targeted treatment at Affiliated Hospital of Academy of Military Medical Sciences. Other eligibility criteria included the presence of measurable disease (RECIST criteria), performance status of 0–2 (World Health Organization criteria), and normal coagulation function. Patients were excluded from the study if they had clinical evidence of active brain metastasis or a clinically serious concurrent illness and history of previous cancer. HER2 positive was defined as either gene amplification by fluorescence in situ hybridization or protein overexpression by immunohistochemistry. All treatments and radiographic examinations were performed as part of standard clinical care. All participants provided written informed consent to participate in the study.
Ten milliliters of peripheral blood was collected from prepatient into ethylenediaminetetraacetic acid-containing tubes. Blood samples were immediately stored in a 4°C environment within 2 h after collection and then centrifuged at 3200 ×g for 15 min at 4°C. Plasma was carefully removed and stored at −80°C in 1 ml aliquots in 1.5 ml tubes for cfDNA detection of further processing.
DNA extraction and library preparation
Briefly, DNA was extracted from 1 ml of plasma samples using the QIAamp circulating nucleic acid kit (Qiagen, Hilden, Germany) and 200 µl of peripheral blood leukocytes (PBL) of patients using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's recommendations. Then, the extracted DNA was eluted into 50 µl buffer AVE (Qiagen, Hilden, Germany) and stored at −20°C. DNA quantity was measured using Qubit 2.0 Fluorometer (Life Technologies, CA, USA) according to the manufacturer's instructions. The concentration of cfDNA per milliliter of plasma and DNA per milliliter of PBL was calculated. The targeted sequencing libraries were generated using DNA Seq NGS Library Preparation kit for Amplicon Sequencing-Illumina Compatible (Gnomegen, San Diego, USA) according to the manufacturer's protocol. The starting material consisted of 10 ng DNA for PBL samples which represented for germline DNA and of 0.5 ng for the cfDNA samples.
Sequencing assay
We used a commercialized and already validated panel which has been designed to amplify 207 amplicions covering approximately 2800 COSMIC mutations from 50 oncogenes and tumor suppressor genes.[16] Samples passing quality control (QC) were then bidirectionally sequenced on the Hiseq2000 (Illumina, CA, USA) instrument, including the germline cfDNA that was derived from leukocyte lysis and the somatic cfDNA. Those sequencing data were expected to achieve at least 1000 coverage per target amplicon for PBL samples and 10,000 coverage per target amplicon for plasma samples.
Sequencing data analysis and variant calling
Sequencing data after QC and filtrating were aligned independently to the human genome (hg19) by Burrows-Wheeler Alignment tool (Sanger Institute, Cambridge, UK) in pair-end mode. Single-nucleotide polymorphisms (SNPs) were identified using VarScan (Genome Institute, St. Louis, USA), and a receivable mutation was called after filtration if it met all the following conditions: coverage ≥100×; no strand bias; P < 0.1 by binomial test; SNP frequency ≥0.1%.
Single-nucleotide variants (SNVs) are quantitated as mutation allele frequency (AF) which is the number of cfDNA fragments divided by the number of wild-type DNA fragments that overlap the same mutated nucleotide base position. A somatic mutation was called if the AF was ≥2% with minimum coverage up to 1000× in plasma and the AF was ≤10% with minimum coverage to 100× in PBL.
Results
Patient characteristics
From March 6, 2014 to December 10, 2014, 24 plasma samples from 20 patients fulfilled the study criteria. Of these 24 samples, eight were grouped into sensitive group and the others were grouped into the resistant group. Four blood samples were collected twice from the resistant cases. They were collected after either the disease progression of trastuzumab or both trastuzumab and lapatinib. The clinical details and results of all imaging performed during the study were collected. All 24 plasma samples’ demographics and treatment details at the time of collection are shown in Table 1.
Table 1.
Characteristics of 24 plasma samples from 20 patients with HER2-positive metastatic breast cancer
| Parameters | Sensitive group (n = 8) | Resistant group (n = 16) |
|---|---|---|
| Age (years), median (range) | 48.5 (27.0–61.0) | 46.5 (38.0–71.0) |
| Hormone receptor status, n (%) | ||
| ER or PgR positive | 3 (37.5) | 12 (75.0) |
| ER and PgR negative | 5 (62.5) | 4 (25.0) |
| Metastatic sites, n (%) | ||
| Visceral | 8 (100.0) | 11 (68.8) |
| Nonvisceral | 0 (0.0) | 5 (31.2) |
| Lines of therapy, n (%) | ||
| 1 | 8 (100.0) | 0 (0.0) |
| 2–3 | 0 (0.0) | 4 (25.0) |
| >3 | 0 (0.0) | 12 (75.0) |
| Prior anti-HER2 therapy | ||
| Trastuzumab alone | 0 (0.0) | 8 (50.0) |
| Trastuzumab and lapatinib | 0 (0.0) | 8 (50.0) |
HER2: Human epidermal growth factor receptor 2; ER: Estrogen receptor; PgR: Progesterone receptor.
The mutational prevalence of 50 genes
We compared SNVs from cfDNA with matched lymphocytes of patients as a source of germline DNA. A total of 486 somatic nonsynonymous mutations of 46 genes were detected in the 24 blood samples [Figure 1]. No mutations were identified in G protein subunit alpha 11 (GNA11), colony-stimulating factor 1 receptor, anaplastic lymphoma receptor tyrosine kinase (ALK), or nucleophosmin genes. There were 26 genes which were detected containing less than 10 SNVs (2%). These 26 genes contained fibroblast growth factor receptor 1, G protein subunit alpha q, RB transcriptional corepressor 1, ABL proto-oncogene 1, B-Raf proto-oncogene, fibroblast growth factor receptor 3, guanine nucleotide-binding protein alpha stimulating (GNAS), HRas proto-oncogene (HRAS), fms-related tyrosine kinase 3, AKT serine/threonine kinase 1, ret proto-oncogene, cadherin 1 (CDH1), cyclin-dependent kinase inhibitor 2A, HER2, enhancer of zeste 2 polycomb repressive complex 2 subunit, isocitrate dehydrogenase 1, isocitrate dehydrogenase 2, MET proto-oncogene, mutL homolog 1 (MLH1), neuroblastoma RAS viral oncogene homolog (NRAS), catenin beta 1, janus kinase 2, NOTCH1, protein tyrosine phosphatase nonreceptor type 11, janus kinase 3, and MPL proto-oncogene. Interestingly, the percentage of all these 26 genes’ SNVs was 27%, which was twice as much as the most common mutated phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene (11%).
Figure 1.
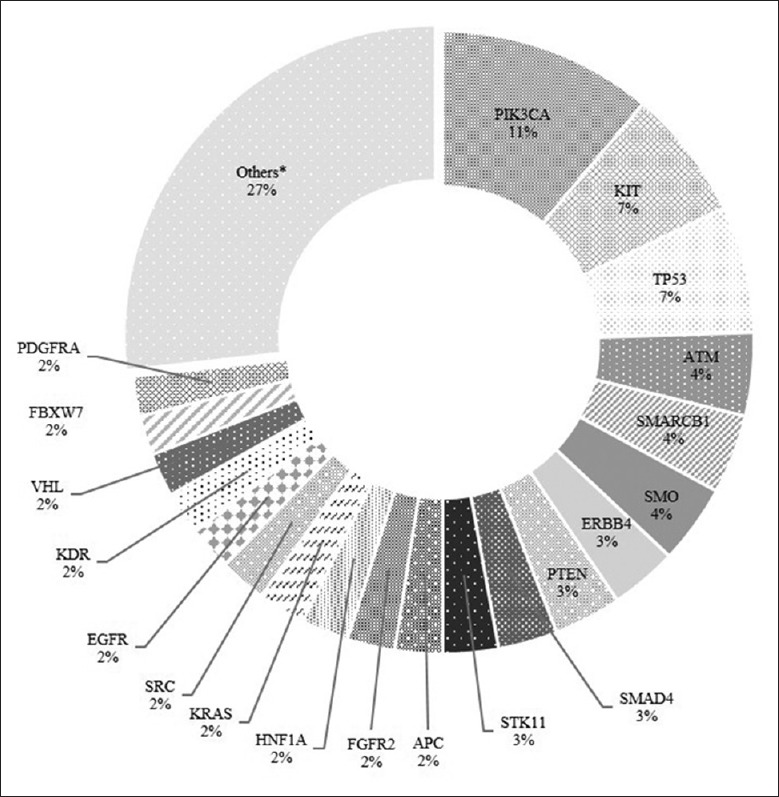
The constitution ratio of 486 somatic mutations detected from 24 plasma samples. *Genes that owe less than 10 single-nucleotide variants (2%), those genes contain fibroblast growth factor receptor 1 (FGFR1), G protein subunit alpha q (GNAQ), RB transcriptional corepressor 1 (RB1), ABL proto-oncogene 1 (ABL1), B-Raf proto-oncogene (BRAF), fibroblast growth factor receptor 3 (FGFR3), guanine nucleotide-binding protein alpha stimulating (GNAS), HRas proto-oncogene (HRAS), fms-related tyrosine kinase 3 (FLT3), AKT serine/threonine kinase 1 (AKT), ret proto-oncogene (RET), cadherin 1 (CDH1), cyclin-dependent kinase inhibitor 2A (CDKN2A), ERBB2, enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), isocitrate dehydrogenase 1 (IDH1), isocitrate dehydrogenase 2 (IDH2), MET proto-oncogene (MET), mutL homolog 1 (MLH1), neuroblastoma RAS viral oncogene homolog (NRAS), catenin beta 1 (CTNNB1), janus kinase 2 (JAK2), NOTCH1, protein tyrosine phosphatase nonreceptor type 11 (PTPN11), janus kinase 3 (JAK3), and MPL proto-oncogene (MPL).
All samples harbored at least one mutation (mean 20, range 1–51) and the mean detected AF was 3.9% (range 2.0–31.7%). The most frequent alteration identified were PIK3CA, followed by proto-oncogene c-Kit (KIT), and tumor protein p53 (TP53). The mutational prevalence of 50 genes identified varied widely from 100% to 0% and reflected a long tail of biomarkers showing substantial genetic differentiation [Figure 2].
Figure 2.

Mutational prevalence of 24 human epidermal growth factor receptor 2-positive breast cancer patients’ plasma samples from 46 genes.
Targeted therapy response-associated alterations
We next addressed whether the repertoire of somatic genetic alterations identified in the analysis of cfDNA would reflect anti-HER2-targeted therapy response in advanced breast cancer patients.
Comparing the SNV from sensitive group with that from resistant group, we found seven genes’ mutations that only occurred in the resistant group. These 7 resistance-associated genes included epidermal growth factor receptor (EGFR), GNAS, HRAS, MLH1, CDH1, NRAS, and NOTCH1 [Figure 3]. Seven resistant samples (43.7%) were found to harbor eleven EGFR mutations: four in exon 7 (p.V292M, p.K284E, p.V292E, and p.G288D), four in exon 18 (p.I706T, p.R705G, p.E711K, and p.G696E), two in exon 19 (p.L760F and p.P741S), and one in exon 21 (p.A822V). Four samples (25%) harbored seven GNAS mutations: three in exon 7 (p.R186H, p.D181G, and p.N203S) and four in exon 8 (p.R216L, p.M206V, p.R216C, and p.D214N). Four samples (25%) contained seven HRAS mutations: five in exon 2 (p.S17N, p.G12S, p.T2A, p.N26S, and p.V9A) and two in exon 3 (p.D54N and p.Q61X). We detected four different CDH1 mutations in three samples (19%): two in exon 8 (p.V345A and p.A348V) and two in exon 3 (p.R74Q and p.R90Q). Three resistant samples harbored four MLH1 mutations in exon 11 (p.F155S, p.Q168K, p.V143D, and p.S160N). In addition, we also detected three NOTHC1 mutations in two resistant samples: p.V1599M in exon 26 and p.S1689P and p.V1676A in exon 27 as well as four NRAS mutations in two samples: two in exon 2 (p.F28S and p.V14A) and two in exon 3 (p.F78L and p.A66T). The potential genes’ function and the potential therapeutic strategies while harboring those genetic mutations are shown in Table 2.
Figure 3.
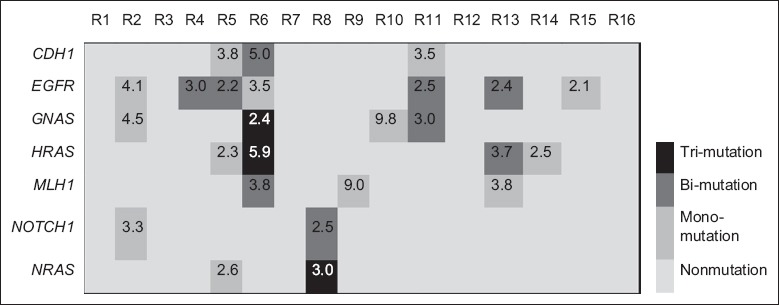
The landscape of the identified mutations in seven anti-human epidermal growth factor receptor 2-targeted therapy-resistant genes. The number shown in the box meant the mean allele frequency of the identified mutations in that gene. R: Resistant group. EGFR: Epidermal growth factor receptor; GNAS: Guanine nucleotide-binding protein alpha stimulating; HRAS: HRas proto-oncogene; MLH1: mutL homolog 1; CDH1: Cadherin 1; NRAS: Neuroblastoma RAS viral oncogene homolog.
Table 2.
Seven anti-HER2 therapy resistance-associated genes and its potential function
| Gene | Resistant group, n (%) | Potential gene function | Potential therapeutic strategies |
|---|---|---|---|
| CDH1 | 3 (19.0) | The CDH1 gene encodes a transmembrane glycoprotein E-cadherin that maintains Ca2+-dependent cell–cell adhesion in epithelial tissues[17] | None |
| EGFR | 7 (43.7) | This gene encodes a protein that is a receptor for members of the epidermal growth factor family. Binding of the protein to a ligand induces receptor dimerization and tyrosine autophosphorylation and leads to cell proliferation | TKI |
| GNAS | 4 (25.0) | Inhibit the adenylyl cyclase-stimulating activity of guanine nucleotide-binding protein G(s) subunit alpha[18,19] | None |
| HRAS | 4 (25.0) | It relates to the transforming genes of mammalian sarcoma retroviruses. Ras proteins bind GDP/GTP and possess intrinsic GTPase activity | MET inhibitors |
| MLH1 | 3 (19.0) | It may play a role to recruit the DNA polymerase III to the site of the MMR[20] | PARP inhibitors |
| NOTCH1 | 2 (13.0) | It encodes a member of the notch family that play a role in a variety of developmental processes by controlling cell fate decisions[21] | None |
| NRAS | 2 (13.0) | It encodes a membrane protein that shuttles between the Golgi apparatus and the plasma membrane. Ras proteins bind GDP/GTP and possess intrinsic GTPase activity | MET inhibitors |
HER2: Human epidermal growth factor receptor 2; CDH1: Cadherin 1; EGFR: Epidermal growth factor receptor; GNAS: Guanine nucleotide-binding protein alpha stimulating; HRAS: HRas proto-oncogene; MLH1: mutL homolog 1; NRAS: Neuroblastoma RAS viral oncogene homolog; MMR: Measles, Mumps, Rubella; TKI: Tyrosine kinase inhibitor; PARP: Poly ADP-ribose polymerase.
In addition, as all the samples we collected were HER2 overexpressed, we paid special attention to the mutation of this gene. Four samples harbored three different HER2 mutations, and among those mutational samples, two had the same p.S855I mutation which was never reported previously [Figure 4]. It was encouraging to see that these two patients received longest time of effective therapy combined with anti-HER2-targeted agents in this trial.
Figure 4.

The gene map of human epidermal growth factor receptor 2 p.S855I mutation identified in circulating-free DNA.
Patient R2 was diagnosed with metachronous bilateral invasive breast cancer on October, 2008 and immediately received systemic treatment. Unfortunately, 13 months after operation, she underwent a local recurrence. Subsequently, she received therapy of lapatinib (1250 mg, PO, daily) combined with capecitabine (1500 mg, PO, twice daily days 1–14 cycled every 21 days) and reached to clinical complete response (CR) 8 months later. We collected the plasma sample during this line therapy for cfDNA detection. It was 48 months later that she got disease progression (new metastatic bone lesion) [Figure 5a].
Figure 5.

The clinical timeline for two metastatic breast cancer patients with the HER2 p.S855I mutation in cfDNA. (a and b) The clinical treatment history and the dramatic effect of containing anti-HER2 agents’ regimen in the Patient R2 and S2 with HER2 activating mutation, respectively. Values within each circle represent mutation frequency. Positron emission tomography/computed tomography scan showed the clinical complete response in the right breast tumor of patient S8 after eight cycles’ therapy. *TPH: Docetaxel, carboplatin, trastuzumab; RT: Radiotherapy; ANA: Anastrozole; G: Goserelin. †LX: Lapatinib, capecitabine. ‡TXH: Docetaxel, capecitabine, trastuzumab. §The pathological images showed the pathological complete response in the breast tumor of patient S8 after eight cycles’ therapy. HER2: Human epidermal growth factor receptor 2; cfDNA: Circulating-free DNA.
Patient S8 was first diagnosed with stage IV breast cancer. She achieved clinical CR in primary and live lesions and subsequently was proved to achieve pathological complete response in breast after 8 cycles’ therapy combined with trastuzumab (440 mg, IV, day 1, every 21 days). Later, we collected the plasma sample during her following first-line maintenance therapy of trastuzumab (440 mg, IV, day 1, every 21 days) combined with capecitabine (1500 mg, PO, twice daily days 1–14 cycled every 21 days). Encouragingly, she got 35-month response to trastuzumab therapy until her lung lesion mildly growing up [Figure 5b].
Discussion
The characteristics of breast cancer tumors are critical in determining appropriate treatment options and predicting patient prognosis. Currently, the hormone receptor status and HER2 expression work as major biomarkers to direct drug treatments. Yet, there is increasingly more evidence showing that gene mutations play a role in predicting breast cancer progression and response to treatment. Herein, we demonstrated that targeted NGS of cfDNA had potential clinical utility to detect biomarkers and gene alternations which were correlative with HER2-targeted therapies, and blood sample could be used as an alternative specimen source for targeted gene screen in molecular screening programs. We detected the nucleotide alterations of cfDNA in 20 advanced breast cancer patients’ plasma samples through targeted NGA aiming to direct targeted therapy.
The mutational prevalence of 50 genes identified varies widely from 100% to 0%, which reflected a long tail of markers showing substantial genetic differentiation. Although PIK3CA, TP53, and KIT were the most common mutational genes in HER2 positive patients, the therapies targeted these genes were controversial. Investigators from PEGGY, OPPORTUNE, and FERGI reported little or no association between PIK3CA mutation status and clinical outcome of the PI3k family-inhibitor, pictilisib.[22,23,24,25] It means that those high-frequency mutations may not be driven genes which mediate tumors development, and the therapy targeted those genes obtained no benefits. However, those low-frequency mutations which were uncommon event in breast cancer probably predicted the response of targeted therapy, such as ALK in nonsmall cell lung cancer.
Here, we detected seven mutated genes occurred only in the resistant group and were considered to be correlative with anti-HER2 therapy resistance. Particularly, we detected 11 different EGFR mutations from 7 resistant samples. Ritter et al.[26] have reported that trastuzumab-resistant cells exhibited higher levels of EGFR and EGFR/HER2 heterodimers. Moreover, the phosphorylated EGFR protein could be inhibited by the EGFR tyrosine kinase inhibitors (TKIs) lapatinib, erlotinib, or gefitinib. Therefore, those EGFR-mutated patients who are resistant to standard anti-HER2-targeted treatment may benefit from TKI.[27]
Another resistance-associated gene we detected, NOTCH1, is a well-established mediator of cell-cell communication that plays a critical role in stem cell survival, self-renewal, cell fate decisions, tumorigenesis, invasion, metastasis, and drug resistance in a variety of cancers.[28,29] It has been reported that resistance of human breast cancer cell lines to HER2/neu inhibition can be mediated by activation of Notch signaling in vitro[30,31,32] and in xenograft models.[33] It indicated that drugs targeting the Notch pathway may be potential therapeutic strategies for some patients who are resistant to current anti-HER2 treatment. The potential gene function and therapeutic strategies of the other 5 resistance-associated genes are shown in Table 2.
In our trial, two patients harbored the same HER2 p.S855I mutation obtained more benefit from anti-HER2 therapy. Bose et al.[34] had reported that most of HER2 mutations are likely to be driver events in the breast cancer which suggested that HER2 mutation positive patients constitute a breast cancer subpopulation who would likely benefit from HER2-targeted drugs. However, it does not mean that all the mutations were able to drive a breast cancer cell progression. Park et al.[35] suggested that in HER2-amplified breast cancer patients, HER2 mutations contribute to resistance to anti-HER2-directed therapy. It appeared that protein structure and conformation makes a large contribution to the functional effect of the mutation, with conservative substitutions occurring in critical locations for the protein have a large effect of HER2.[34] Therefore, whether it is an accidental phenomenon or a truth that patient harboring this mutation preforms extremely sensitive to anti-HER2 therapy required further preclinical researches.
Moreover, the mutational prevalence of 46 genes that detected somatic mutations in this study is higher than previously reported though the rates reported vary widely by study.[36,37] The higher prevalence of these mutations in our study could be attributed to the following reasons: (1) the mean sequencing depth is up to 40,000× of cfDNA samples which enables us to detect the mutation AF as low as 2%; (2) the patients undergoing cfDNA testing are in advanced state with relatively heavy tumor burden; and (3) the patients met the enrollment criteria in this study are all HER2-positive breast cancer characterized by high malignant degree and poor prognosis. This study had several limitations so far. First, the limited number of samples for cfDNA detection makes the interpretation of results should be cautiously given. Second, it was a cohort study but not self-control design, which restricted us to observe the biomarker difference between sensitive and resistant of anti-HER2 target treatment. Finally, it was unable for mutation calling using tissue-based NGS as a comparison.
Although tissue-based NGS was the gold standard for clinical and investigational sequencing, cfDNA relied on its unique advantages was hopeful to be an alternative. Compared with tissue-based NGS, cfDNA sequencing was a less invasive technique capable of capturing tumor heterogeneity and the molecular changes cancer cells.[10] On the other hand, cfDNA tests may be sensitive enough to detect some low-frequency mutations that tissue-based NGS could not be identified. Further studies will be needed to perform in-depth analyses of the concordance between tissue and cfDNA molecular results to better understand the observations from each test.[37]
In conclusion, we demonstrate that targeted NGS of cfDNA has potential clinical utility to detect biomarkers and gene alternations which are correlative with HER2-targeted therapies, and blood sample could be used as an alternative specimen source for targeted gene screen in molecular screening programs.
Financial support and sponsorship
This work was supported by a grant from the National Science Foundation of China (No. 81472477).
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We are grateful to Dr. Yan Wang for providing technical help. We also acknowledge Dr. Xiao-Di Wang for language editing, and Dr. Feng-Rui Xu and Jian-Bin Li for data analysis.
Footnotes
Edited by: Ning-Ning Wang
References
- 1.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 2.Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN. The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–68. doi: 10.1634/theoncologist.2008-0230. doi: 10.1634/theoncologist.2008-0230. [DOI] [PubMed] [Google Scholar]
- 3.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–72. doi: 10.1056/NEJMoa052306. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 4.Marty M, Cognetti F, Maraninchi D, Snyder R, Mauriac L, Tubiana-Hulin M, et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: The M77001 study group. J Clin Oncol. 2005;23:4265–74. doi: 10.1200/JCO.2005.04.173. doi: 10.1200/JCO.2005.04.173. [DOI] [PubMed] [Google Scholar]
- 5.Gianni L, Eiermann W, Semiglazov V, Manikhas A, Lluch A, Tjulandin S, et al. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): A randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet. 2010;375:377–84. doi: 10.1016/S0140-6736(09)61964-4. doi: 10.1016/S0140-6736(09)61964-4. [DOI] [PubMed] [Google Scholar]
- 6.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–60. doi: 10.1038/nature11143. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Overman MJ, Modak J, Kopetz S, Murthy R, Yao JC, Hicks ME, et al. Use of research biopsies in clinical trials: Are risks and benefits adequately discussed? J Clin Oncol. 2013;31:17–22. doi: 10.1200/JCO.2012.43.1718. doi: 10.1200/JCO.2012.43.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 10.Diaz LA, Jr, Bardelli A. Liquid biopsies: Genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–86. doi: 10.1200/JCO.2012.45.2011. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90. doi: 10.1038/nm.1789. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2:20ra14. doi: 10.1126/scitranslmed.3000702. doi: 10.1126/scitranslmed.3000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–54. doi: 10.1038/nm.3519. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw JA, Page K, Blighe K, Hava N, Guttery D, Ward B, et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 2012;22:220–31. doi: 10.1101/gr.123497.111. doi: 10.1101/gr. 123497.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra68. doi: 10.1126/scitranslmed.3003726. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 16.Frenel JS, Carreira S, Goodall J, Roda D, Perez-Lopez R, Tunariu N, et al. Serial next-generation sequencing of circulating cell-free DNA evaluating tumor clone response to molecularly targeted drug administration. Clin Cancer Res. 2015;21:4586–96. doi: 10.1158/1078-0432.CCR-15-0584. doi: 10.1158/1078-0432.CCR-15-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang R, Ding P, Yang F. Clinicopathological significance and potential drug target of CDH1 in breast cancer: A meta-analysis and literature review. Drug Des Devel Ther. 2015;9:5277–85. doi: 10.2147/DDDT.S86929. doi: 10.2147/DDDT.S86929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiele S, de Sanctis L, Werner R, Grötzinger J, Aydin C, Jüppner H, et al. Functional characterization of GNAS mutations found in patients with pseudohypoparathyroidism type Ic defines a new subgroup of pseudohypoparathyroidism affecting selectively Gsa-receptor interaction. Hum Mutat. 2011;32:653–60. doi: 10.1002/humu.21489. doi: 10.1002/humu.21489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linglart A, Mahon MJ, Kerachian MA, Berlach DM, Hendy GN, Jüppner H, et al. Coding GNAS mutations leading to hormone resistance impair in vitro agonist- and cholera toxin-induced adenosine cyclic 3’,5’-monophosphate formation mediated by human XLalphas. Endocrinology. 2006;147:2253–62. doi: 10.1210/en.2005-1487. doi: 10.1210/en.2005-1487. [DOI] [PubMed] [Google Scholar]
- 20.Harkness EF, Barrow E, Newton K, Green K, Clancy T, Lalloo F, et al. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: A cohort study. J Med Genet. 2015;52:553–6. doi: 10.1136/jmedgenet-2015-103216. doi: 10.1136/jmedgenet-2015-103216. [DOI] [PubMed] [Google Scholar]
- 21.Groth C, Fortini ME. Therapeutic approaches to modulating Notch signaling: Current challenges and future prospects. Semin Cell Dev Biol. 2012;23:465–72. doi: 10.1016/j.semcdb.2012.01.016. doi: 10.1016/j.semcdb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothé F, Laes JF, Lambrechts D, Smeets D, Vincent D, Maetens M, et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol. 2014;25:1959–65. doi: 10.1093/annonc/mdu288. doi: 10.1093/annonc/mdu288. [DOI] [PubMed] [Google Scholar]
- 23.Schmid P, Pinder SE, Wheatley D, Macaskill J, Zammit C, Hu J, et al. Phase II randomized preoperative window-of-opportunity study of the pi3k inhibitor pictilisib plus anastrozole compared with anastrozole alone in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2016;34:1987–94. doi: 10.1200/JCO.2015.63.9179. doi: 10.1200/JCO.2015.63.9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17:811–21. doi: 10.1016/S1470-2045(16)00106-6. doi: 10.1016/S1470-2045(16)00106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vuylsteke P, Huizing M, Petrakova K, Roylance R, Laing R, Chan S, et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: Interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann Oncol. 2016;27:2059–66. doi: 10.1093/annonc/mdw320. doi: 10.1093/annonc/mdw320. [DOI] [PubMed] [Google Scholar]
- 26.Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13:4909–19. doi: 10.1158/1078-0432.CCR-07-0701. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- 27.Ni J, Zhang L. Evaluation of three small molecular drugs for targeted therapy to treat nonsmall cell lung cancer. Chin Med J. 2016;129:332–40. doi: 10.4103/0366-6999.174484. doi: 10.4103/0366-6999.174484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker AT, Zlobin A, Osipo C. Notch-EGFR/HER2 bidirectional crosstalk in breast cancer. Front Oncol. 2014;4:360. doi: 10.3389/fonc.2014.00360. doi: 10.3389/fonc.2014.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu BF, Qin YY, Zhang SL, Quan ZW, Zhang MD, Bi JW. Downregulation of notch-regulated ankyrin repeat protein exerts antitumor activities against growth of thyroid cancer. Chin Med J. 2016;129:1544–52. doi: 10.4103/0366-6999.184465. doi: 10.4103/0366-6999.184465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene. 2008;27:5019–32. doi: 10.1038/onc.2008.149. doi: 10.1038/onc. [DOI] [PubMed] [Google Scholar]
- 31.Abravanel DL, Belka GK, Pan TC, Pant DK, Collins MA, Sterner CJ, et al. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J Clin Invest. 2015;125:2484–96. doi: 10.1172/JCI74883. doi: 10.1172/JCI74883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han M, Deng HY, Jiang R. Effect of trastuzumab on notch-1 signaling pathway in breast cancer SK-BR3 cells. Chin J Cancer Res. 2012;24:213–9. doi: 10.1007/s11670-012-0213-9. doi: 10.1007/s11670-012-0213-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pandya K, Meeke K, Clementz AG, Rogowski A, Roberts J, Miele L, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer. 2011;105:796–806. doi: 10.1038/bjc.2011.321. doi: 10.1038/bjc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–37. doi: 10.1158/2159-8290.CD-12-0349. doi: 10.1158/2159-8290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park YH, Shin HT, Jung HH, Choi YL, Ahn T, Park WY, et al. Role of HER2 mutations in refractory metastatic breast cancers: Targeted sequencing results in patients with refractory breast cancer. Oncotarget. 2015;6:32027–38. doi: 10.18632/oncotarget.5184. doi: 10.18632/oncotarget.5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Wang H, Zhang L, Tang C, Jones L, Ye H, et al. Rapid detection of genetic mutations in individual breast cancer patients by next-generation DNA sequencing. Hum Genomics. 2015;9:2. doi: 10.1186/s40246-015-0024-4. doi: 10.1186/s40246-015-0024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwaederle M, Husain H, Fanta PT, Piccioni DE, Kesari S, Schwab RB, et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget. 2016;7:9707–17. doi: 10.18632/oncotarget.7110. doi: 10.18632/oncotarget.7110. [DOI] [PMC free article] [PubMed] [Google Scholar]