

Abstract
Blockade of the coinhibitory checkpoint molecule PD-1 has emerged as an effective treatment for many cancers, resulting in remarkable responses. However, despite successes in the clinic, most patients do not respond to PD-1 blockade. Metabolic dysregulation is a common phenotype in cancer, but both patients and tumors are metabolically heterogeneous. We hypothesized that the deregulated oxidative energetics of tumor cells present a metabolic barrier to antitumor immunity through the generation of a hypoxic microenvironment and that normalization of tumor hypoxia might improve response to immunotherapy. We show that the murine tumor lines B16 and MC38 differed in their ability to consume oxygen and produce hypoxic environments, which correlated with their sensitivity to checkpoint blockade. Metformin, a broadly prescribed type II diabetes treatment, inhibited oxygen consumption in tumor cells in vitro and in vivo, resulting in reduced intratumoral hypoxia. Although metformin monotherapy had little therapeutic benefit in highly aggressive tumors, combination of metformin with PD-1 blockade resulted in improved intratumoral T-cell function and tumor clearance. Our data suggest tumor hypoxia acts as a barrier to immunotherapy, and that remodeling the hypoxic tumor microenvironment has potential to convert patients resistant to immunotherapy into those that receive clinical benefit.
Introduction
Immunotherapy has emerged as a viable and effective treatment for a variety of cancer types. One of the major successes in cancer immunotherapy involves the antibody-mediated blockade of coinhibitory ‘checkpoint’ molecules, negative regulators highly upregulated on the surface of tumor-infiltrating T cells (1). The goal of these treatments is to relieve cell-intrinsic inhibition of a patient’s own immune response to the cancer treatment. Antibodies targeting cytotoxic lymphocyte antigen 4 (CTLA-4) and programmed death 1 (PD-1) signaling, alone or in combination, have resulted in durable antitumor immunity and remarkable clinical responses (1).
It is now accepted that tumors generate an suppressive microenvironment which acts to evade and inhibit immune responses by a number of distinct factors, including recruitment of suppressive populations like regulatory T cells as well as secretion of suppressive cytokines (2). However, it is now becoming clear that the metabolic nature of the tumor microenvironment also contributes to suppression of antitumor immunity (3). Carrying out T-cell effector function is metabolically demanding, requiring intermediates necessary for proliferation, cytokine synthesis, and cytotoxicity(4). The tumor microenvironment has low concentrations of glucose and other metabolites, an acidic interstitial pH, and low oxygen tension (5). This is due to altered blood supply as well as deregulated energetics of tumor cells themselves (5). Thus, in addition to being inhibited through immunosuppressive mechanisms, tumor-infiltrating T cells also may lack the fuel required for effector function.
Hypoxia is a well-known component of the tumor microenvironment and has been rigorously studied in a variety of experimental systems and patient samples (6). Hypoxia is generally considered to be immunosuppressive, although previous studies employing HIF1α, VHL-, and PHD-deficient T cells have revealed that the role of these proteins in T-cell differentiation and function is likely complex (7,8). Studies with ‘true’ hypoxia remain unclear, as apparent roles for oxygen tension in differentiation versus effector function can be disparate (9). Still, oxidative phosphorylation (OXPHOS) is required for many aspects of T-cell function (10). Thus, we examined how oxygen tension plays a role in responses to immunotherapy using murine models of cancer coupled with metabolic analysis and pharmacologic modulation of the tumor microenvironment.
Here we show mitigation of tumor hypoxia using the mitochondrial complex 1 inhibitor metformin. Remodeling of the tumor microenvironment this way resulted in increased sensitivity to PD-1 blockade, increased intratumoral T-cell function, and tumor regression.
Materials and Methods
Mice
All animal work was done in accordance with the Institutional Animal Care and Use Committee of the University of Pittsburgh. All mice were housed in specific pathogen free conditions. 6–8 week mice of similar weight and mixed genders were randomized prior to experimentation. C57/BL6, SJ/L (Thy1.1), and OT-I mice were obtained from The Jackson Laboratory.
Reagents and cell lines
B16-F10 were obtained from ATCC. MC38 were a gift from Dario Vignali. B16OVA (MO5) was obtained from Per Basse and Lou Falo. Both MC38 and B16OVA have not been authenticated, but OVA expression was verified on B16OVA by immunoblot and flow cytometry. Cell lines were obtained in 2014 and Mycoplasma testing was performed June 2014. Cell lines were not passaged more than 3 times before experimentation. Antibodies to CD8 (53-6.7), CD4 (GK1.5), PD-1 (29F.1812), Tim-3 (RMT3-23), CD44 (IM7), CD62L (MEL-14), TNFα (MP6-XT22), IFNγ (XMG1.2), CD11b (M1/70), CD11c (N4180), Ki67 (16A8), CD45 (30-F11), F4/80 (BM8), Ly6C (HK1.4), and propidium iodide were from BioLegend. Hypoxia staining was detected with an antibody to pimonidazole (Hypoxyprobe). Antibody to PD-1 (J43) and its hamster IgG control were obtained from Bio-X-Cell. CellTrace Violet was purchased from eBioscience. 2-NBD-glucose and metformin were purchased from Cayman Chemical. In all in vivo studies, mice were injected with 2.5 × 105 tumor cells intradermally.
Metabolism Assay
Using a Seahorse XFe96 Bioanalyzer (Agilent), B16 (50,000/well), MC38 (50,000/well), direct ex vivo sorted CD8+ LN cells and tumor-infiltrating lymphocytes (TILs) (100,000/well), or in vitro cultured OT1 CD8+ T cells (100,000/well) were plated on Seahorse culture plates in media consisting of minimal, unbuffered DMEM supplemented with 1% BSA and 25 mM glucose, 1mM pyruvate, and 2 mM glutamine. Basal oxygen consumption rates were taken for 30 minutes. Cells were stimulated with 2 μM oligomycin, 0.5 μM FCCP, 100 mM 2-deoxyglucose and 100 μM rotenone/antimycin A to obtain maximal respiratory and control values.
In vitro T-cell functional assays
Spleen and lymph node preparations from OT-I mice were stimulated with SIINFEKL peptide (250 ng/mL, Anaspec) and IL2 (25 U/mL, Peprotech) for 24 h. Cells were washed, expanded 10X into fresh media with IL2, and cultured for 7 days to generate previously-activated cytotoxic T cells. To measure proliferation, OT-I T cells were labeled with the proliferation dye CellTrace Violet and stimulated for 72 h with peptide and APCs. To measure cytokine production, cells were stimulated overnight with C57/B6 antigen-presenting cells plus SIINFEKL. Cell supernatants were analyzed by ELISA. To measure cytotoxicity, OT-I T cells were cocultured with B16 or B16.OVA cells for 16h at ratios ranging from 10:1 (effector:target) to 1:2, then stained with propidium iodide to detect apoptosis in the target cells. For each assay, cells were culture in either ambient normoxic or hypoxic conditions (1.5% O2, BioSpherix).
Tumor infiltrating lymphocyte analysis
When tumors reached ≥ 6 mm, mice were injected intraperitoneally with 50 metformin (mg/kg, Cayman Chemical) on days −3 and −1. On day 0, mice were injected intravenously with pimonidazole (80mg/kg, Hypoxyprobe) in PBS 1.5 hours before sacrifice. Nondraining and draining lymph nodes were harvested and manually disrupted to single cell suspension. Explanted tumors were injected whole with a mixture of collagenase, dispase, and DNaseI (Fisher), incubated at 37 degree for 15–30 min, then dissociated between two frosted glass slides to single-cell suspension. Suspensions of LN and tumor were filtered and vortexed at high speed for 1 min prior to downstream analyses. Pimonidazole was visualized using anti-pimonidazole antibodies after 1% PFA fixation and 0.1% Triton X-100 permeabilization. Ki67 and cytokine staining was performed using the eBioscience Fix/Perm Kit. Cytokine staining was completed after 18h stimulation with PMA and ionomycin (Sigma) (the final 4 h in the presence of a protein transport inhibitor).
Histology
In some experiments, after tumor growth, metformin treatment, and pimonidazole pulsing, tumors were dissected and frozen at −80° in OCT (Tissue-Tek) and sectioned (Cryostat microtome). Tissue was fixed in histology-grade acetone (Fisher) at −20°, then rehydrated in staining buffer, stained with hypoxyprobe (Hypoxyprobe) and DAPI (Life Technologies), and mounted with ProLong Diamond Antifade Mountant (Life Technologies). Sections were imaged with an Olympus IX83 microscope and analyzed with ImageJ.
Immunoblotting analysis
Direct ex vivo sorted CD8+ LN cells and TILs were lysed in lysis buffer (1% NP40, 150 mM NaCl, 20 mM Tris, 2 mM EDTA, 2 mM EGTA). Lysates were denatured with lithium dodecyl sulfate and dithiotreitol. Lysates were loaded into 4–12% Bis-Tris Plus Bolt PAGE Gels (Life Technologies). Gels were transferred to membranes using NuPAGE Bolt electroblotting in the presence of 2mM Tris, 192 mM glycine, and 10% methanol. Membranes were blocked in 3% bovine serum albumin in Tris-buffered saline with 0.1% TWEEN20 (TBST). Primary antibodies to Hif1α (Signaling Technology) or actin (Santa Cruz) were diluted in 3% BSA/TBST, and incubated with membranes overnight. After three washes with TBST, secondary antibodies (Jackson Immunoresearch) diluted in 3% BSA/TBST were incubated with membranes for 1 h. After three TBST washes, membranes were incubated with Western Lightning ECL substrate and exposed to film. Digitally captured films were analyzed densitometrically by ImageJ software.
Real time PCR
Direct ex vivo sorted CD8+ LN cells and TILs, in vitro activated CD8+ T cells, CD45-depleted direct ex vivo B16 cells, or in vitro cultured MC38 cells were lysed in Trizol and RNA extracts were made. cDNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time PCR was performed with primers for Slc22a1 and Ppib (cyclophilin b) and quantitation was performed using the ΔΔCt method.
Metformin plus anti-PD1 therapy
On day 5, when there were 1–10 mm2 palpable tumors, mice were started on either 0.2mg anti–PD-1 or hamster IgG isotype control (Bio X Cell), injected every 4 days intraperitoneally, and metformin (50 mg/kg, Cayman Chemical) or PBS, injected every 2 days intraperitoneally. Cohorts were sacrificed when control mouse tumors reached 15 mm in any direction measured. For metformin drinking water cohorts, mice with 1–10mm2 palpable tumors started on 1g/L metformin drinking water, and were injected with anti–PD-1 every 4 days intraperitoneally. Cohorts were sacrificed when control mouse tumors reached 15 mm in any direction measured.
Results
Tumor hypoxia is variable between tumor types and inhibits T-cell function
The C57/BL6 tumor line B16-F10 (referred to here as B16) melanoma, and MC38 colon adenocarcinoma, are common transplantable tumor models used in murine cancer immunology, and have different degrees of immunogenicity (11). To determine if these tumor lines also have different metabolic characteristics, we used a Seahorse Bioanalyzer to analyze the tumor cells’ ability to perform oxidative phosphorylation, known as oxygen consumption rate (OCR). We found that B16 has a higher baseline OCR compared to MC38, as well as higher spare respiratory capacity (mitochondrial reserve induced by the uncoupling reagent FCCP) (Fig. 1A). B16 and MC38 had minimal glycolytic differences, as measured by extracellular acidification rate (ECAR) (Fig. 1B). The different OCR of these lines has in vivo effects, as CD8+ tumor-infiltrating lymphocytes (TIL) display a higher degree of hypoxia in B16 tumors, as measured by binding of pimonidazole (an injectable, irreversible hypoxia tracer), than in MC38 tumors examined directly ex vivo (Fig. 1C). Thus, the capacity of a tumor cell to consume oxygen has effects on the TIL hypoxia phenotype.
Figure 1. Tumor hypoxia is variable between tumor types and inhibits T-cell function.
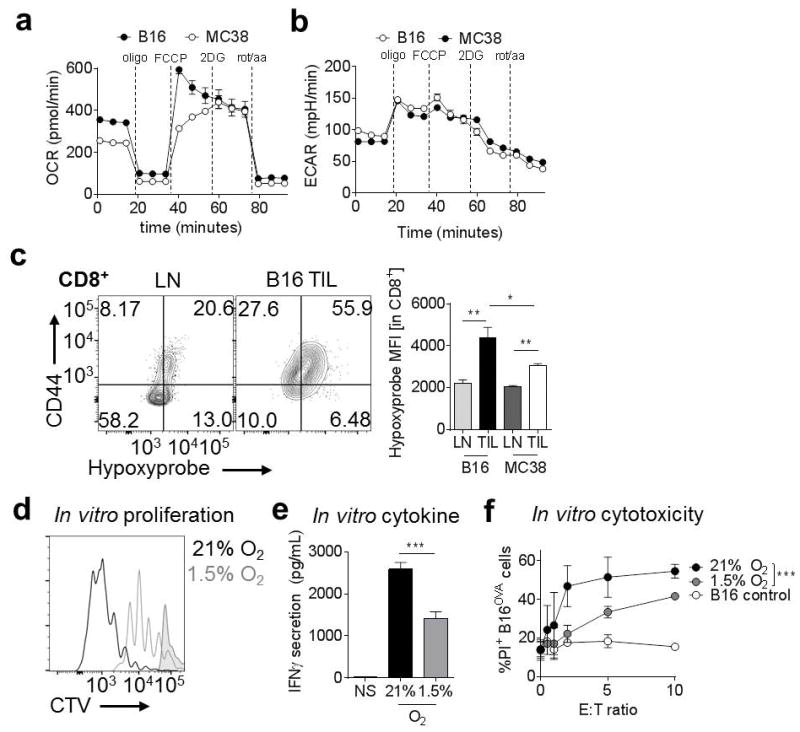
(A) Oxygen consumption rate (OCR) trace of B16 and MC38 cells (50,000 cells/well) interrogated for mitochondrial activity in the Seahorse instrument. (B), Extracellular acidification rate (ECAR) trace of B16 and MC38 cells interrogated for glycolytic activity in the Seahorse instrument. (C) Hypoxyprobe staining of T cells isolated from B16 and MC38 tumors. Results are tabulated to the right. (D) CellTrace Violet (CTV) dye dilution showing proliferation of OT-I T cells activated with peptide in ambient normoxia (20%) or hypoxic (1.5%) conditions. Shaded histogram shows unstimulated cells. (E) Cytokine production of CD8+ T cells stimulated as in b overnight. (F) Cytotoxicity (PI staining) of parental or OVA-expressing B16 tumor cells incubated with previously activated, effector OT-I T cells overnight under conditions of normoxia or hypoxia. * P < 0.05, ** P < 0.01, *** P < 0.001 by unpaired t-test (C, E) or two-way ANOVA with repeated-measures (F). Results represent three independent experiments.
Since hypoxia has been shown to have various effects on T cells in culture (10), we tested the effects of hypoxia on antigen-specific (OT-I) T cells previously activated and rested in normoxia, then restimulated in vitro using a hypoxia chamber set at average tumor hypoxia (1.5%). We found that T cells proliferated less compared to T cells cultured in normoxic conditions (Fig. 1D). Cytotoxic T cells generated via peptide stimulation and expansion in normoxic conditions but restimulated in hypoxic conditions also synthesized significantly lower levels of IFNγ than their normoxic counterparts (Fig. 1E) and had lower cytolytic activity against antigen-expressing tumor targets (Fig. 1F). Thus, cytotoxic T cells in acutely hypoxic conditions are functionally impaired compared to T cells in normoxia.
Metformin treatment acts as an inhibitor of tumor oxygen consumption
To determine if tumor hypoxia can be pharmacologically modulated, we used the mitochondrial complex I inhibitor metformin on B16 and MC38 tumor cells in metabolic flux assays, revealing that metformin decreases tumor cell OCR in both B16 and MC38 cells in vitro (Fig. 2A). B16 tumors isolated from mice treated with metformin, depleted of CD45+ cells, and assayed directly ex vivo had similar phenotypes (Fig. 2B). Immunofluorescent analysis of metformin-treated tumor-bearing mice of hypoxyprobe revealed that, in agreement with previously published data (12), metformin treatment decreased overall tumor hypoxia (Fig. 2C). Thus, metformin acting on tumor cells induced a decrease in overall tumor hypoxia.
Figure 2. Metformin treatment acts as an inhibitor of tumor oxygen consumption.
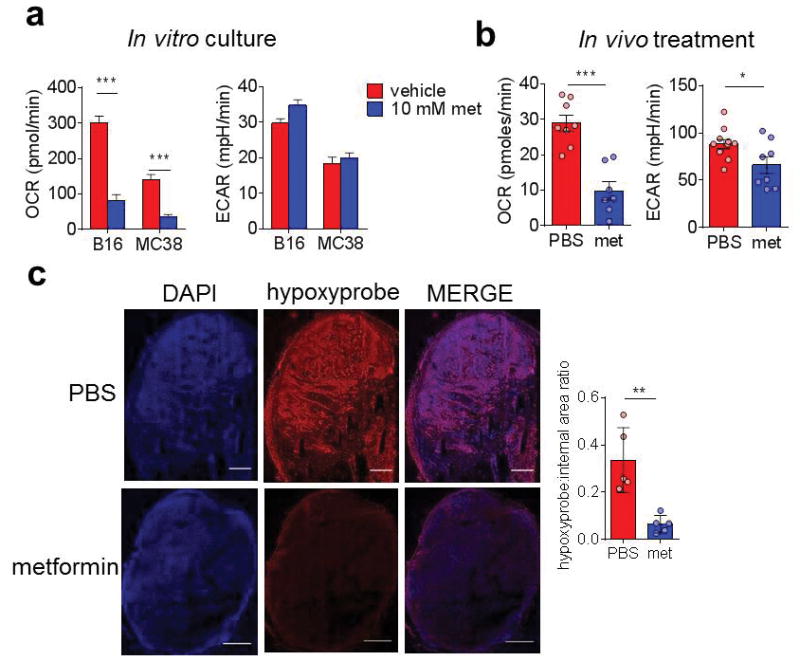
(A) Oxygen consumption rate of B16 or MC38 cells (50,000 cells/well) treated overnight in the presence or absence of 10 mM metformin. (B) OCR of B16 tumor cells (CD45-depleted) plated directly ex vivo from mice bearing small tumors treated with PBS or metformin (50 mg/kg) for 3 days. (C) Pimonidazole staining of full tumor sections (stitched from 300–500 individual panels) from mice bearing B16 tumors receiving PBS or metformin treatment for 3 days as in B. Tabulated results quantify the internal hypoxyprobe signal from a set threshold normalized for each day of imaging. Scale bar = 1mm. ** P < 0.01, *** P < 0.001 by unpaired t-test. Data represent the mean (A, B, C tabulation) or are representative (C images) of at least three independent experiments.
Metformin treatment alters hypoxia-driven changes in T-cell phenotypes
After establishing that metformin decreased tumor hypoxia, in part by inhibiting cancer cell oxygen consumption, we sought to determine the effect of metformin treatment on tumor-infiltrating T cells in vivo. We have previously shown that T cells in the tumor microenvironment are at a metabolic disadvantage and repress oxidative metabolism (13). Seahorse analysis of tumor cells and T cells sorted directly from untreated and metformin-treated tumors revealed that, in untreated animals, tumor cell oxidative metabolism dwarfs that of T cells (Fig. 3A). In contrast, metformin-induced inhibition of tumor cell metabolism results in similar oxygen consumption rates for both tumor and tumor-infiltrating T cells: repression of tumor cell OCR and an increase in T-cell OCR (Fig. 3A). This opposite effect on T cells was intriguing, and suggested T cells from the tumor microenvironment were affected by metformin indirectly. Metformin requires active transport by the organic cation transporter (OCT1, encoded by Slc22a1). Tumor cells express more of this transporter than T cells, suggesting that, especially in the tumor microenvironment, tumor cells preferentially take up metformin and are specifically inhibited by it (Supplementary Fig. S1A). Metformin treatment in vitro does impact T-cell OCR, as high doses of metformin decreased T-cell OCR and increased ECAR (Supplementary Fig. S1B). Metformin can cause increases in glucose uptake, so we also analyzed the glucose uptake of tumor-infiltrating T cells by 2NBDG uptake, a fluorescent glucose tracer, and found that metformin did not induce increased glucose uptake in vivo, further suggesting the effect of metformin on tumor-infiltrating T cells was indirect (Supplementary Fig. S1C). We additionally found that metformin treatment of mice significantly decreased hypoxia experienced by TIL T cells, with no significant effect on LN-resident populations (Fig. 3B). In order to determine whether metformin-induced changes in TIL hypoxia would impact tumor control or clearance, mice with small, palpable B16 tumors were treated therapeutically with metformin. Mice treated with metformin had no significant difference in tumor size compared to PBS-treated mice (Fig. 3C).
Figure 3. Metformin treatment reduces intratumoral T-cell hypoxia.
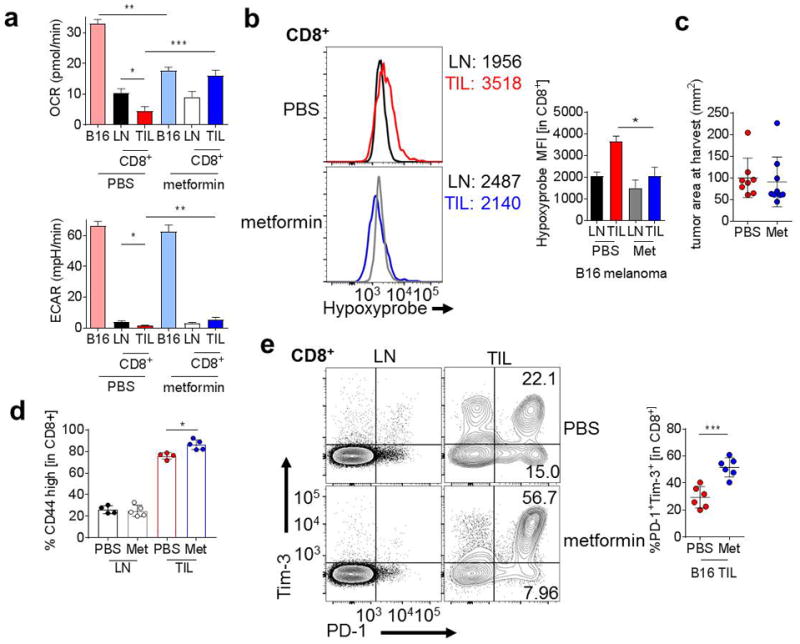
(A) Oxygen consumption rate (top) or extracellular acidification rate (bottom) from B16-bearing mice treated with metformin or vehicle for 3 days. CD45+CD8+ T cells were sorted by flow cytometry and assayed directly ex vivo, whereas B16 tumor cells were CD45-depleted before assaying. (B) Flow cytogram (left) and tabulation (right) of pimonidazole staining in T cells from B16-bearing mice treated with metformin or vehicle for 3 days. (C) Tumor area at 21 days for mice treated during tumor progression with PBS or metformin. (D) Quantification of CD44hi CD8+ T cells from B16-bearing mice treated with metformin or vehicle for 3 days (E) PD-1 and Tim-3 expression in CD8+ T cells from mice treated as in D. * P < 0.05, *** P < 0.001 by unpaired t-test. Results are representative of three (A, B, D, E) or four (C) independent experiments.
To investigate why T cells in more oxygenated environments were still unable to decrease tumor burden, we characterized the infiltrate from the tumors of metformin-treated mice. Metformin did not enhance the expression of any coinhibitory molecules on antigen-presenting populations, nor did it change the percentage of tolerogenic myeloid-derived suppressor cells (MDSCs) in the tumor (Supplementary Fig. S1D–F). However, tumor-infiltrating T cells from metformin-treated tumor-bearing mice had a small but significant increase in the number of activated T cells (CD44hi) (Fig. 3D), suggesting that metformin treatment may allow for increased T-cell activation. To supplement our hypoxia studies, we also examined the direct ex vivo protein quantity of HIF1α. Intriguingly, even though in vivo metformin treatment decreased tumor and T-cell hypoxia (Fig. 2C, 3B), HIF1α protein in TIL was unaltered or increased (Supplementary Fig. S1G). HIF1α expression is usually associated with hypoxia, but T cells also upregulate HIF1α upon activation (14), so we believe the trend towards increased HIF1α in metformin-treated TIL may be a readout of increased T-cell activation (Fig. 3D and Supplementary Fig. S1G). As chronic activation can lead to T-cell dysfunction, we then assessed the expression of coinhibitory ‘exhaustion’ molecules, and found coinhibitory molecules PD-1 and Tim-3 were elevated compared to control mice (Fig. 3E). Thus, although metformin treatment alone does not affect tumor burden in these aggressive models, metformin treatment may allow for increased activation of T cells, resulting in further upregulation of immunologic checkpoints on tumor-infiltrating CD8+ T cells.
Metabolic remodeling synergizes with checkpoint blockade to unleash antitumor immunity
Anti-PD1 immunotherapy can cause tumor clearance in 40% of C57/BL6 mice with MC38 tumors, but has little beneficial effect on reducing tumor burden or causing tumor clearance in B16 tumors (11). We hypothesized that normalization of tumor hypoxia with metformin might generate a microenvironment more permissive to anti-PD1 immunotherapy. We treated mice with metformin or PBS in combination with anti-PD1 or isotype control when mice developed palpable tumors (1–10mm2) and continued therapy throughout the course of the experiment. We found that whereas anti-PD1 alone or metformin alone had no impact on tumor burden, the combination of anti-PD1 and metformin induced regressions in 80% of mice, and tumor clearance in 70% of the mice with B16 (Fig 4A). Analysis of the TILs in these mice revealed that CD8+ T cells produced more effector cytokines (Fig. 4B, C) and are substantially more proliferative (Fig. 4D) than T cells from the single treatment or control groups. Although our study is a therapeutic model, sequencing studies in the B16 model have revealed that, like many immunotherapeutic strategies, a synergistic metformin and anti-PD1 effect had a size threshold; once B16 tumors were large (>10 mm2) prior to treatment, they lost sensitivity to this treatment strategy (data not shown).
Figure 4. Metabolic remodeling synergizes with checkpoint blockade to effect antitumor immunity.
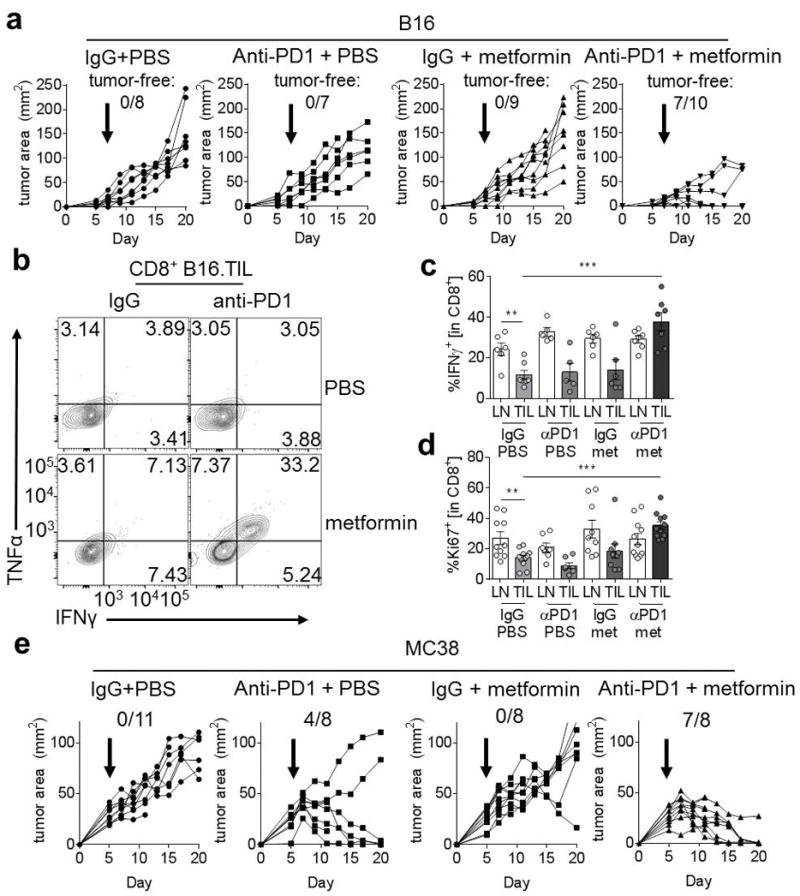
(A), Tumor measurements of C57/BL6 mice inoculated with B16 melanoma. Mice began receiving treatment on d5 as indicated, receiving 0.2mg anti-PD1 or its isotype control every 4 days, and either metformin or vehicle. Number of mice tumor-free of the total inoculated is reported. (B) Representative flow cytogram depicting IFNγ and TNFα production from CD8+ tumor infiltrating T cells from B16-bearing mice treated as in A. (C) Tabulated IFNγ staining from multiple mice; each dot represents one animal. (D) Ki67 expression in CD8+ T cells from mice treated as in a as indicated. (E) As in A, but mice were inoculated with MC38. * P < 0.05, ** P < 0.01, *** P < 0.001 by unpaired t-test. Results represent the mean (A, C, D, E) or are representative of (B) three to five independent experiments.
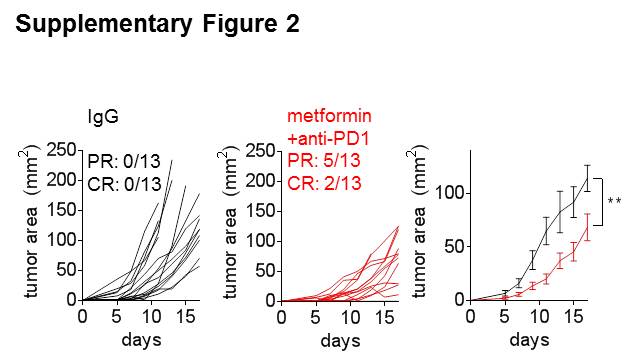
Our studies were mostly conducted using intraperitoneal (IP) injections of metformin, but we also utilized metformin delivered in the drinking water (1 g/L). We observed similar synergy between these two treatments in B16 melanoma, although the effect was not as striking as what was observed using IP injections (Supplementary Fig. S2). Given that our data suggest that tumor cells may be preferentially taking up metformin, the continually administrated nature of the drinking water treatment may start to have inhibitory effects on T cells.
MC38 is partially sensitive to PD-1 therapy, which we hypothesize is at least partially due to their less oxidative and thus hypoxic nature. We then asked whether the existing response rate could be improved by further hypoxia mitigation by metformin treatment. Indeed, mice with MC38 tumors fared even better with metformin and anti-PD1 treatment than with anti-PD1 alone; 88% of mice treated with anti-PD1 and metformin showed complete tumor clearance, and every mouse experienced tumor regression (Fig. 4E). This suggests that tumor oxidative metabolism exists as a common barrier to immunotherapy. Thus, anti-PD1 and metformin combination therapy shows synergistic effects on tumor clearance when compared to single therapy treatment, increasing TIL T-cell activation and effector function.
Discussion
Our study links the hypoxic nature of the tumor microenvironment with resistance to immunotherapy. We have revealed, as many others have shown, that oxygen is a vital metabolite for T-cell function, which is limiting in the tumor microenvironment. Remodeling the tumor microenvironment through inhibition of tumor cell oxidative metabolism resulted in increased sensitivity to immunotherapy, which unleashed the antitumor immune response to promote cancer regression.
Our data adds to an increasing number of reports suggesting that the metabolic makeup of the tumor microenvironment is a critical inhibitory factor for T-cell function. Although T cells can be metabolically plastic, and are primed to utilize glucose in oxygen-poor conditions, glucose, too, can be limiting in the tumor microenvironment (15,16). Thus, although immunotherapies like checkpoint blockade may allow for optimal activation, microenvironmental deficiencies may prevent T cells from generating enough energy to carry out effector function (13).
It is established that patients taking metformin for type II diabetes have a decreased risk of cancer (17,18) and that much of the antitumor metformin effect can be attributed to T cells (19). However, the precise causes and extent of metformin’s antitumor effect have been elusive, undoubtedly complicated by the fact that the mechanism of action of metformin is still unclear (20). Whether through inhibition of complex I or activation of the energy-charge sensor AMPK, however, it is clear that metformin treatment can inhibit the oxygen consumption of tumors, and consequent generation of hypoxia, in agreement with prior studies (12). Although metformin likely has pleiotropic effects on a wide array of cellular functions, its effect on tumor hypoxia serves as an important proof-of-concept study, suggesting pharmacologic inhibition of tumor cell oxidative metabolism or oxygen-diffusing drugs may serve as critical potentiators of antitumor immunity. An alternative approach, utilizing hyperoxygenation of mice, indeed suggests that antitumor immune function is improved when tumor hypoxia is mitigated (21). Our data build on this platform, suggesting that metformin-induced remodeling of microenvironment oxygen tension acts to create a ‘level playing field’ for T- cell activation in response to immunotherapy.
Metformin can have immunosuppressive effects in a number of models: graft-versus-host disease, lupus, and graft rejection (22–24). In these scenarios, T cells are the ‘dominant force’ at the site of activation, and may be experiencing either the systemic hypoglycemic or lactic acidotic effects of metformin. In the tumor microenvironment, where T cells are competing metabolically with cancer cells, metformin may simply be preferentially taken up by tumor cells. Our data showing that T cells demonstrate relatively low expression of the metformin transporter OCT-1 (Slc22a1) compared to tumor cells support this notion. Along these lines, our drinking water experiments suggest continual administration may mediate a less synergistic effect; translating these observations to the clinic will require dosing and sequencing experimentations to find optimal synergy with immunotherapeutic treatments.
Previous studies exploring the role of the HIF pathway have revealed somewhat surprising data regarding this oxygen sensing pathway in T-cell function. T cells lacking VHL, the upstream inhibitor of HIF1α, show increased T-cell production of effector molecules like granzyme B and had enhanced antitumor activity, suggesting the hypoxia response might be advantageous for T cells (25). Hypoxia may function to promote aerobic glycolysis, which is tied to effector function epigenetically and post-transcriptionally (26–28). However, in the tumor microenvironment, as glucose is limiting, only cells able to effectively compete for glucose would experience an enhanced response (15,16). Hypoxia and HIF are not always linked in T cells, as HIF1α is also stabilized upon T-cell activation and other downstream pathways (14). Indeed, our data showing that tumor-infiltrating T cells from metformin-treated animals had similar amounts of HIF1α, even though they experienced less hypoxia, as shown by pimonidazole staining, support the notion that HIF1α, especially in T cells, is not always directly linked to true hypoxia.
As we identified hypoxia as a barrier to tumor immunity, our data also suggest that the degree of tumor hypoxia may predict the response to immunotherapy. This is in agreement with a report examining transcriptomes from patients receiving PD-1 blockade, identifying a hypoxia signature in innate resistance to immunotherapy (29).
Our study employing metformin as a method to modulate the oxygen tension of the tumor microenvironment showed substantial effects on the efficacy of PD-1 blockade immunotherapy. However, we hypothesize that, as hypoxia acts as a general barrier to T-cell function, many forms of immunotherapy that aim to act by reinvigorating T cells at the tumor site will be improved through modulation of this aspect of the microenvironment. Future studies will explore how blockade of other checkpoints, stimulatory antibody treatment, or adoptive cell therapy might be improved by pharmacologic modulation of hypoxia.
Our data support a model in which a common phenotype of cancer cells, metabolic dysregulation, can mediate immune evasion and resistance to immunotherapy through the generation of a nutrient poor, hypoxic microenvironment. Remodeling the tumor microenvironment through modulation of cancer cell metabolism may have the potential to convert non-responder patients into those that can receive the benefits of immunotherapeutic cancer treatment.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This project used the UPCI Animal and Cytometry Facilities, supported in part by award P30CA047904.
This work was supported by the Sidney Kimmel Foundation for Cancer Research (SKF-015-039), the Samuel and Emma Winters Foundation, the University of Pittsburgh Cancer Institute Skin Cancer SPORE (P50CA121973-08), the University of Pittsburgh Cancer Institute Head and Neck Cancer SPORE (P50 CA097190), a Stand Up To Cancer-AACR Innovative Research Grant (SU2C-AACR-IRG-04-16) and startup funds to G.M.D.
Footnotes
Conflict of interest statement
The authors have no conflicts of interest to disclose.
References
- 1.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell. 2015;27:450–61. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Current opinion in immunology. 2013;25:268–76. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends in immunology. 2015;36:257–64. doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delgoffe GM, Powell JD. Feeding an army: The metabolism of T cells in activation, anergy, and exhaustion. Molecular immunology. 2015;68:492–6. doi: 10.1016/j.molimm.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Justus CR, Sanderlin EJ, Yang LV. Molecular Connections between Cancer Cell Metabolism and the Tumor Microenvironment. International journal of molecular sciences. 2015;16:11055–86. doi: 10.3390/ijms160511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends in cell biology. 2014;24:472–8. doi: 10.1016/j.tcb.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tao JH, Barbi J, Pan F. Hypoxia-inducible factors in T lymphocyte differentiation and function. A Review in the Theme: Cellular Responses to Hypoxia. American journal of physiology Cell physiology. 2015;309:C580–9. doi: 10.1152/ajpcell.00204.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Ertl HC. Starved and Asphyxiated: How Can CD8(+) T Cells within a Tumor Microenvironment Prevent Tumor Progression. Frontiers in immunology. 2016;7:32. doi: 10.3389/fimmu.2016.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, et al. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. Journal of immunology (Baltimore, Md : 1950) 2001;167:6140–9. doi: 10.4049/jimmunol.167.11.6140. [DOI] [PubMed] [Google Scholar]
- 10.Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. American journal of physiology Cell physiology. 2015;309:C569–79. doi: 10.1152/ajpcell.00207.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer research. 2012;72:917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:6741–50. doi: 10.1158/1078-0432.CCR-13-1787. [DOI] [PubMed] [Google Scholar]
- 13.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity. 2016;45:374–88. doi: 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phan AT, Goldrath AW. Hypoxia-inducible factors regulate T cell metabolism and function. Molecular immunology. 2015;68:527–35. doi: 10.1016/j.molimm.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–41. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–28. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bodmer M, Meier C, Krahenbuhl S, Jick SS, Meier CR. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes care. 2010;33:1304–8. doi: 10.2337/dc09-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ (Clinical research ed) 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1809–14. doi: 10.1073/pnas.1417636112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He H, Ke R, Lin H, Ying Y, Liu D, Luo Z. Metformin, an old drug, brings a new era to cancer therapy. Cancer journal (Sudbury, Mass) 2015;21:70–4. doi: 10.1097/PPO.0000000000000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Science translational medicine. 2015;7:277ra30. doi: 10.1126/scitranslmed.aaa1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park MJ, Lee SY, Moon SJ, Son HJ, Lee SH, Kim EK, et al. Metformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between T helper 17 and Tregs. Translational research: the journal of laboratory and clinical medicine. 2016 doi: 10.1016/j.trsl.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Science translational medicine. 2015;7:274ra18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CF, Lo YC, Cheng CH, Furtmuller GJ, Oh B, Andrade-Oliveira V, et al. Preventing Allograft Rejection by Targeting Immune Metabolism. Cell reports. 2015;13:760–70. doi: 10.1016/j.celrep.2015.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nature immunology. 2013;14:1173–82. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (New York, NY) 2016 doi: 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nature immunology. 2013;14:1064–72. doi: 10.1038/ni.2687. [DOI] [PubMed] [Google Scholar]
- 28.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–51. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.