

Abstract
Aims
Mitochondria in adult cardiomyocytes exhibit static morphology and infrequent dynamic changes, despite the high abundance of fission and fusion regulatory proteins in the heart. Previous reports have indicated that fusion proteins may bear functions beyond morphology regulation. Here, we investigated the role of fission protein, dynamin-related protein 1 (DRP1), on mitochondrial respiration regulation in adult cardiomyocytes.
Methods and results
By using genetic or pharmacological approaches, we manipulated the activity or protein level of fission and fusion proteins and found they mildly influenced mitochondrial morphology in adult rodent cardiomyocytes, which is in contrast to their significant effect in H9C2 cardiac myoblasts. Intriguingly, inhibiting endogenous DRP1 by dominant-negative DRP1 mutation (K38A), shRNA, or Mdivi-1 suppressed maximal respiration and respiratory control ratio in isolated mitochondria from adult mouse heart or in adult cardiomyocytes from rat. Meanwhile, basal respiration was increased due to increased proton leak. Facilitating mitofusin-mediated fusion by S3 compound, however, failed to inhibit mitochondrial respiration in adult cardiomyocytes. Mechanistically, DRP1 inhibition did not affect the maximal activity of individual respiratory chain complexes or the assembly of supercomplexes. Knocking out cyclophilin D, a regulator of mitochondrial permeability transition pore (mPTP), abolished the effect of DRP1 inhibition on respiration. Finally, DRP1 inhibition decreased transient mPTP-mediated mitochondrial flashes, delayed laser-induced mPTP opening and suppressed mitochondrial reactive oxygen species (ROS).
Conclusion
These results uncover a novel non-canonical function of the fission protein, DRP1 in maintaining or positively stimulating mitochondrial respiration, bioenergetics and ROS signalling in adult cardiomyocyte, which is likely independent of morphological changes.
Keywords: Mitochondrial respiration, Dynamin related protein 1, Mitochondrial morphology, Mitochondrial permeability transition pore, Adult cardiomyocyte
1. Introduction
Mitochondria play a central role in cellular bioenergetics and signalling transduction.1 Meanwhile, mitochondrial function and morphology are controlled by intracellular signalling pathways.2 Besides being the powerhouse of the cell, mitochondria also carry out other functions ranging from reactive oxygen species (ROS) generation and ion homeostasis regulation to redox regulation and cell fate determination.3–5 Mitochondrial respiration through electron flow along electron transport chain (ETC) fundamentally supports other functions of the mitochondria such as maintaining a negative inner membrane potential for Ca2+ uptake. Conversely, mitochondrial respiration is being modulated by the other functions or processes, including intermediate metabolism, ATP hydrolysis, Ca2+, and ROS.6 Recently, we showed that transient openings of the mitochondrial permeability transition pore (mPTP) also stimulate respiration in individual mitochondrion as visualized by monitoring mitochondrial flash activities in the adult heart.7,8 Thus, a sophisticated and intertwined network exists, which connects and integrates the various functions of mitochondria.6 Identifying novel mechanisms or signalling pathways for mitochondrial respiration regulation will provide new insights into the role of mitochondria in health and disease and new targets for the treatment of mitochondrial dysfunction related disorders.
Mitochondrial dynamics has become an emerging field of research.9 Numerous studies have indicated that mitochondrial form and function are closely interrelated.10 For instance, it has been proposed that mitochondrial respiration and energy production is generally enhanced when the morphological balance is tilted towards fusion (i.e. filamentous and interconnected mitochondria) and vice versa.11 However, recent studies also show that the fission and fusion proteins may modulate mitochondrial respiration through unique mechanisms that are not directly related to morphological changes. For instance, the inner membrane fusion protein, optic atrophy 1 (OPA1) helps maintain the cristae structure of the inner membrane and through which promotes mitochondrial respiration.12 The outer membrane fusion protein, mitofusin (MFN1/2) tethers mitochondria with endoplasmic reticulum, which facilitates mitochondrial Ca2+ uptake and metabolism.13,14 The master regulator of fission, dynamin related protein 1 (DRP1) has also been shown to promote mitochondrial respiration, since knockdown of DRP1 leads to decreased oxygen consumption rate (OCR).15 However, no change in mitochondrial respiration is found in a mouse model of inducible DRP1 knockout.16 It remains unclear whether endogenous DRP1 can directly modulate mitochondrial respiration.
This study is designed to address the above question and to further explore the potential mechanisms by which DRP1 modulates mitochondrial respiration. The potential role of fission–fusion proteins in mitochondrial respiration is particularly important in the heart. Cardiac mitochondria are very abundant and provide majority of the energy for the continued beating of the heart. However, they exhibit a fragmented and static morphology with infrequent dynamic changes despite high levels of the fission and fusion proteins.17 These unique features suggest that fission and fusion proteins may bear novel functions in the adult heart beyond morphology regulation. Indeed, cardiac specific knockout of DRP1 causes deranged mitophagy, apoptosis, decreased energetics, and cardiomyopathy.15 However, the near complete and chronic ablation of DRP1 in the heart may stimulate compensatory responses or other pathways, which could complicate the interpretation of these results. Therefore, in this study, we employed in vitro approach and acutely inhibited the guanosine triphosphate hydrolase (GTPase) activity of DRP1 in adult cardiomyocytes to monitor its role in mitochondrial oxygen consumption. We provide evidence to show that endogenous DRP1 maintains mitochondrial respiration independent of mitochondrial fission. We further determined that this effect is likely mediated by transient openings of mPTP, and may contribute to bioenergetics, ROS, and Ca2+ signalling in adult cardiomyocytes.
2. Methods
2.1 Animals and reagents
All the animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Washington and conform to the NIH guidelines (Guide for the care and use of laboratory animals). The mitochondrial targeted-circularly permuted yellow fluorescent protein (mt-cpYFP) transgenic mice were described previously.7 The cyclophilin D knockout (Ppif−/−) mice were purchased from Jackson Lab. The adenoviruses containing dominant-negative mutation of DRP1 (K38A), wild type (WT) DRP1 (plasmid purchased from Addgene and virus made by Vector Biolabs), PeriCam, DRP1 short hairpin RNA (shRNA) (shDRP1, a kind gift from Dr Junichi Sadoshima at the Rutgers University), mitochondrial targeted green fluorescent protein (mGFP), or mt-cpYFP were amplified in 293 cells (∼1 × 1011 viral particles per mL), divided into aliquots, and stored at −80°C.
2.2 Adult cardiomyocyte and H9C2 cell culture
Ventricular myocytes were enzymatically isolated from the heart of female Sprague-Dawley rat (200–250 g, Harlan) as described previously.7 Briefly, the rat was anaesthetized by intraperitoneal injection of pentobarbital (100 mg/kg). The heart was quickly removed, cannulated through aorta and perfused with oxygenated modified Tyrode’s solution (in mM: 118 NaCl, 25 HEPES, 11 D-glucose, 4.8 KCl, 1.2 MgCl2, 1.2 KH2PO4, and 1 CaCl2, pH 7.4) for 5 min. The heart was perfused with low Ca2+ solution containing type II collagenase (80 U/mL, Worthington) and hyaluronidase (0.15 mg/mL, Sigma) at 37°C for 30 min. The ventricle was cut into small pieces and further digested under gentle agitation. Rod shaped adult cardiomyocytes were collected by brief centrifugation and used freshly or plated in 96 well plates for XF96 Extracellular Flux Analyzer (Seahorse Bioscience) or on glass coverslips for confocal imaging. Laminin (10 μg/mL) was used to coat the surface. The density was 2–3 × 104 cells per coverslip or 250–3000 cells per well in 96 well plates. The cells were cultured in M199 medium (Sigma) supplemented with 10 mM glutathione, 26.2 mM sodium bicarbonate, 0.02% bovine serum albumin, 50 U/mL penicillin-streptomycin, and 5% fetal bovine serum (FBS). Two hours after the plating, the medium was changed to serum-free M199 and myocytes were cultured for up to 72 h. The rat cardiac myoblast cell line, H9C2 cells were cultured in high glucose DMEM medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin. Adenovirus-mediated gene expression was done at a multiplicity of infection of 50–100 for up to 3 days.
2.3 Confocal imaging
For confocal imaging, we used modified Tyrode’s solution (in mM: 138 NaCl, 0.5 KCl, 20 HEPES, 1.2 MgSO4, 1.2 KH2PO4, 1 CaCl2, 5 Glucose, pH 7.4). We used Zeiss LSM510 (Zeiss, Germany) or Leica TCS SP8 (Leica, Germany) inverted confocal microscopes with a 40 × 1.3 NA oil-immersion objective for confocal imaging at room temperature.7,8,18 Images were taken at 1024 resolution. The pinhole was adjusted to keep the thickness of optical slices at ∼2 μm. For mitochondrial morphology, cells were loaded with JC-1 (5 μM for 15 min, 37 °C, Invitrogen) or overexpressing mGFP. JC-1 was excited by 488 nm laser and emission collected at 510–545 and 570–650 nm. mGFP was excited by 488 nm laser and emission collected at >505 nm. For mitochondrial Ca2+, cells expressing PeriCam19 were excited at 405 and 488 nm and the emissions were collected at 505–765 nm. For mitochondrial superoxide, MitoSOX red (5 μM, loading for 15 min at 37 °C) was excited at 405 and 514 nm and emissions collected at >530 nm. For mitochondrial flashes, mt-cpYFP expressing cells were excited at 405 and 488 nm and emissions collected at >505 nm. Time-lapse 2D images were collected at a speed of 1 s per frame. Laser-induced membrane potential dissipation followed a protocol reported previously.20 Briefly, tetramethylrhodamine methyl ester (TMRM) (20 nM) was loaded to cells without washing and linescan confocal image (1000 lines) was taken by exciting the cells at 543 nm and emission collected at >560 nm at a scanning speed of 100 ms/line.
2.4 Mitochondrial morphology analysis
Digital images were analysed using ImageJ software (NIH) and followed a reported procedure with modifications.21,22 The parameters obtained are mitochondrial size (area), the aspect ratio of the length of major and minor axes, and circularity. Form factor is calculated as 1/circularity to evaluate mitochondrial length and network branching.
2.5 Mitochondria isolation
The mitochondria from adult mouse heart were isolated following a protocol reported previously.23 Briefly, the mouse was anesthetized by intraperitoneal injection of pentobarbital (150 mg/kg) and the heart was quickly removed and put on ice in mitochondria isolation buffer (300 mM sucrose, 10 mM Na+-HEPES, and 0.2 mM EDTA, pH 7.4). The heart was rinsed, minced, and digested with 0.1 mg/mL trypsin for 10 min before adding 0.1% BSA and 0.5 mg/mL soybean trypsin inhibitor to stop the digestion. The tissues were centrifuged at 800 g for 1 min, resuspended and homogenized by using a motorized Dounce homogenizer with a Teflon pestle and centrifuged again at 800 g for 10 min. The supernatant, which contains mitochondria, was collected in a new tube, centrifuged at 8000 g for 15 min twice. The pellet was resuspended in mitochondria isolation buffer and kept on ice before use.
2.6 OCR measurement
For measuring OCR in isolated mitochondria or permeabilized adult cardiomyocytes, we used a Clark-type oxygen electrode (Hansatech Instruments) as previously described.8 Briefly, after probe calibration, mitochondria (250 µg/mL) or adult cardiomyocytes (1×105, permeabilized with 15 µg/mL digitonin) were incubated in a chamber containing 1–2 mL respiration buffer (in mM: 125 KCl, 20 HEPES, 3 MgCl2, 0.4 EGTA, 0.3 DTT, and 5 KH2PO4, pH 7.2). Substrate mediated respiration (State 2) was initiated by the addition of 2.5 mM malate and 5 mM glutamate or 2 mM succinate and 1 μM rotenone. adenosine diphosphate (ADP) (500 µM) was added to obtain maximal respiration (State 3). The respiration control ratio (RCR) was calculated by dividing the State 3 respiration rate with that of State 2. For measuring OCR in intact cardiomyocyte, we used the XF96 Extracellular Flux Analyzer (Seahorse Bioscience). The cardiomyocytes were plated on laminin coated 96 well plates and cultured in M199 medium for 3 days. When measuring OCR, DMEM with 5 mM glucose and 1 mM pyruvate was used and 2.5 µM oligomycin A, 1 µM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and 2.5 µM antimycin A plus 1 µM rotenone were added in three sequential injections.
2.7 Evaluation of supercomplexes assembly
To evaluate the assembly of ETC supercomplexes, we used the blue native gel electrophoresis (Invitrogen). Isolated mitochondria from adult mouse heart (100 µg) were treated with 50 µM Mdivi-1 or dimethyl sulphoxide (DMSO) (control) for 30 min. The samples were solubilized in cold 4× NativePAGE Sample Buffer containing 5% Digitonin and 5% Coomassie Blue G-250 sample additive and centrifuged. The samples were loaded on NativePAGE Novex 3–12% Bis-Tris Protein Gel and run at 100 V for 1 h, then at 300 V for another 2 h. The gel was stained with Coomassie Blue for overnight and destained for >10 h (buffer changed every ∼2 h) before imaging (Bio-Rad imaging system).
2.8 Mitochondrial ETC complexes activity assay
The activity of ETC complexes were measured in permeabilized cardiomyocytes as described previously.24 We used the Evolution 220 UV-visible spectrophotometer (Thermo Scientific) to obtain the absorption of cytochrome C (at 540 and 550 nm) for Complex IV and III, DCPIP (at 600 and 750 nm) for Complex II, NADH (at 340 and 380 nm) for Complex I and V, and DTNB (at 412 nm) for citrate synthase.
2.9 Confocal imaging of Langendorff perfused mouse heart
Adult mt-cpYFP transgenic mouse (20–30 g) was anesthetized with pentobarbital (150 mg/kg). The heart was removed, cannulated via ascending aorta, and put on a modified perfusion system and in a custom made chamber on the confocal stage as previously reported.25,26 The perfusion was maintained under a constant flow (2.8 mL/min) with O2/CO2-bubbled KHB solution (in mM: 118 NaCl, 0.5 EDTA, 10 D-glucose, 5.3 KCl, 1.2 MgCl2, 25 NaHCO3, 0.5 Pyruvate, and 2 CaCl2, pH 7.4) at 37 °C. To minimize motion artifact during imaging, 10 μM (-)-Blebbistatin (Toronto Research Chemicals) were included in the perfusion solution. We also included TMRM (150 nM) for mitochondrial membrane potential. During imaging, the left ventricle was gently pressed against the coverslip at the bottom of the chamber to further suppress motion artifact. Serial 2D confocal images were done following the same procedure described above for mitochondrial flash measurement.
2.10 Statistics
Data are shown as mean ± standard error (SEM). One-way analysis of variance (ANOVA) was used for experiments with more than two groups and followed by Tukey’s post hoc analysis. P < 0.05 was considered statistically significant.
3. Results
3.1 Fission/fusion manipulation altered mitochondrial morphology in adult cardiomyocytes
Previously, very few reports have studied mitochondrial morphology in cultured adult cardiomyocytes. Here, we combined confocal imaging with mitochondria-targeted fluorescent probes and imaging processing by ImageJ to establish a protocol and quantified mitochondrial morphology in cultured adult cardiomyocytes (see Supplementary material online, Figure S1). The morphology of mitochondria in adult cardiomyocytes was altered towards more fission by overexpressing WT DRP1 or more fusion by overexpressing dominant negative DRP1 mutation (K38A) or DRP1 shRNA (shDRP1) (Figure 1A–C). Incubating with S3 compound, which facilitates the interaction between mitofusin1/2 and induces fusion,27,28 also increased mitochondrial size (single mitochondrial area) and length to width ratio (aspect ratio) in adult cardiomyocytes. Mdivi-1 (50 μM, 30 min), a widely used DRP1 inhibitor that inhibits the GTPase activity of DRP1, showed no effect on mitochondrial morphology in adult cardiomyocytes, while it has been shown to induce significant fusion in other cell types and neonatal cardiomyocytes.15,29 The mitochondrial networking parameter, form factor, was not significantly changed by these treatments (data not shown). These results suggest that acute manipulations of fission and fusion proteins can achieve modest mitochondrial morphology changes in adult cardiomyocytes.
Figure 1.

DRP1 inhibition altered mitochondrial morphology in adult cardiomyocytes. (A) Representative images of mitochondrial targeted JC1 showing the morphology of adult cardiomyocytes with or without the indicated treatments. (B–C) Quantitative analysis of mitochondrial size (B, area) and aspect ratio (C) with ImageJ software. N = 357–1774 mitochondria from 17–65 cells and 4–9 rats. *P < 0.05 vs. Control.
Despite high abundance of fission and fusion proteins in the heart, the above morphological changes in cardiac mitochondria are relatively small as compared to the dramatic changes in H9C2 cardiac myoblast cells with the same manipulations (Figure 2 and Supplementary material online, Figure S2). Thus, acute manipulation of fission and fusion proteins induced significant morphological changes in H9C2 cells but only mild changes in adult cardiomyocytes.
Figure 2.

DRP1 inhibition caused mitochondrial fusion in H9C2 myoblast cells. (A) Representative images showing the morphology of mitochondria in H9C2 cells with manipulations of fission and fusion proteins. (B–E) Quantitative analysis of mitochondrial morphology showing mitochondrial size, aspect ratio, and form factor. N = 1689–7786 mitochondria from 13–36 cells and 3–4 independent experiments. *, #: P < 0.05 or 0.01 vs. Control, respectively.
3.2 Inhibiting endogenous DRP1 activity suppressed cardiac mitochondrial respiration
Next, we tested whether the above manipulations that induced mild morphological changes have any effect on mitochondrial respiration in adult cardiomyocytes. Surprisingly, Mdivi-1 inhibited mitochondrial respiratory control ratio (RCR, the ratio of State 3 OCR over State 2 OCR, which is the most reliable parameter for mitochondrial respiratory capacity30) in isolated mitochondria from adult mouse heart (Figure 3A). We further used permeabilized adult rat cardiomyocytes and found that Mdivi-1 (50 µM, for 30 min) also inhibited State 3 respiration and RCR without affecting State 2 respiration (Figure 3B). Finally, we overexpressed K38A, WT DRP1, or shDRP1 in adult cardiomyocytes (Figure 3C and D) and used Seahorse XF96 Extracellular Flux Analyzer to monitor OCR and extracellular acidification rate (ECAR) (Figure 3F–G and Supplementary material online, Figure S3). From the titration curve, we chose 500 myocytes per well for subsequent experiments (Figure 3F). The results showed that inhibiting DRP1 by K38A, shDRP1, or Mdivi-1 all significantly suppressed RCR. The maximal (FCCP induced) OCR was decreased, while basal OCR was increased largely due to increased proton leak (after oligomycin A, Supplementary material online, Figure S3) as reported previously.31–33 Since Mdivi-1 may have DRP1-independent effects,15 we used Mdivi-1 on top of genetic DRP1 inhibitions. As shown in Figure 3G, Mdivi-1 plus K38A or Mdivi-1 plus shDRP1 showed slightly more inhibition on respiration, but the effect is much smaller than the additive effects of individual treatments. This indicates that Mdivi-1’s effect on respiration may overlap with genetic inhibitors of DRP1 or at least in part, depend on DRP1. Because inhibiting endogenous DRP1 suppressed mitochondrial respiration and mildly promoted fusion in adult cardiomyocytes, we conclude that endogenous DRP1 plays a role in maintaining mitochondrial bioenergetics. Interestingly, S3 compound, which also induced fusion, had no effect on respiration. This suggests that morphological changes may not underlie the effect of DRP1 in mitochondrial respiration.
Figure 3.

Inhibiting endogenous DRP1 activity suppressed cardiac mitochondrial respiration. (A) Mdivi-1 dose-dependently inhibited respiratory control ratio (RCR, State 3/State 2 ratio) in isolated mitochondria from adult mouse heart. N = 3 independent experiments. (B) Mdivi-1 (50 µM) suppressed State 3 respiration and RCR in permeabilized adult cardiomyocytes. N = 6 from three rats. (C–E) Representative images and summarized data showing adenovirus mediated overexpression of the dominant negative mutation K38A (C), WT DRP1 (D), or DRP1 shRNA (shDRP1) in adult cardiomyocytes. N = 3. *P < 0.05 vs. Control (Scrambled shRNA). (F) Titration of the number of adult cardiomyocytes for OCR measurement by Seahorse system (XF96). N = 5 in each group. (G) Effects of manipulating DRP1 on RCR of intact adult cardiomyocytes measured by Seahorse system. Mdivi-1 (50 µM) added for 30 min and S3 compound (2 µM) for 24 h before measurement. N = 4–12. The RCR was calculated by the ratio between maximal OCR (induced by 1 µM FCCP) and basal OCR. *, #: P < 0.05 or 0.01 vs. Control.
3.3 DRP1 inhibition on ETC supercomplex assembly and individual complex activity
Next, we asked how DRP1, which is a cytosolic protein and binds with proteins on the outer membrane of mitochondria, can modulate mitochondrial respiration in a fission independent manner. We first tested whether the assembly of ETC complexes and supercomplexes is affected by DRP1 inhibition. Isolated mitochondria were treated with Mdivi-1 (50 µM) and the extracted proteins were loaded on blue native gels. As shown in Figure 4A, no significant difference in respiratory chain supercomplexes assembly was detected. Furthermore, we determined the activity of individual ETC complexes and found no change in the maximal activities of Complex I–V and citrate synthase (Figure 4B). In gel Complex I activity assay also revealed no difference between Mdivi-1 treated and vehicle (DMSO) treated mitochondria (see Supplementary material online, Figure S4). These results suggest that the role of endogenous DRP1 on respiration regulation is not through direct modulation of individual ETC complexes, but more likely through an indirect mechanism and affect the activity of the whole ETC.
Figure 4.

DRP1 inhibition had no effect on ETC supercomplexes assembly and activity. (A) Blue native gel image showing Mdivi-1 did not cause significant changes in ETC supercomplexes assembly. N = 3 mice. (B) Effects of Mdivi-1 and K38A on the enzyme activity of Complexes I–V and citrate synthase in adult rat cardiomyocytes. N = 6–11 rats.
3.4 Role of mPTP in DRP1 regulation of respiration
Previous reports have suggested that Mdivi-1 or K38A may inhibit mPTP opening in the heart and other tissues.16,21,34 However, whether mPTP is involved in DRP1 regulation of respiration is not known. To test this, we evaluated the effect of Mdivi-1 in cardiac mitochondria from cyclophilin D knockout (CypD KO) mouse (Ppif−/−). CypD is a known regulator of mPTP and located in the matrix. Mitochondria lacking CypD showed significantly less mPTP openings.35 Interestingly, CypD KO slightly inhibited State 3 respiration, and completely abolished the inhibitory effect of Mdivi-1 on State 3 respiration (Figure 5A). Similar effect was observed in cardiomyocytes from WT mice after incubation with the CypD inhibitor, cyclosporine A (Figure 5A). These results imply that mPTP may be needed for endogenous DRP1 to regulate respiration. To directly test whether DRP1 modulates mPTP, we monitored laser-induced membrane potential dissipation in intact myocytes as an indication for oxidative stress-induced mPTP opening.20 Overexpression K38A delayed the onset of membrane potential dissipation (loss of TMRM signal) indicating decreased mPTP opening (Figure 5B). K38A overexpression also increased Ca2+ levels in mitochondrial matrix while maintaining the membrane potential (Figure 5C and D) supporting that decreased mPTP openings lead to stable membrane potential and matrix Ca2+ retention. Thus, DRP1 inhibition suppressed mPTP openings in adult cardiomyocytes.
Figure 5.

DRP1 stimulated mPTP and through which regulated mitochondrial respiration. (A) The effect of Mdivi-1 (50 μM) on respiration inhibition was abolished by mPTP inhibitor, cyclosporine A (CsA, 1 μM) in permeabilized adult cardiomyocyte from WT mice, and was missing in permeabilized adult cardiomyocytes from mice lacking the mPTP regulator, cyclophilin D (Ppif−/−). N = 3–4 mice in each group. *P < 0.05 vs. Control or WT. (B) Overexpression of K38A suppressed laser-induced mPTP openings (as indicated by loss of TMRM signal) in intact adult cardiomyocytes. N = 92–100 mitochondria from 7–8 cells and three rats. #P < 0.01 vs. Control. (C) Increased mitochondrial Ca2+ in K38A overexpressed myocytes. N = 11–31 cells from three rats. *P < 0.05 vs. Control. (D) Mitochondrial membrane potential in K38A overexpressed myocytes was maintained. N = 16–27 cells from three rats.
3.5 DRP1 inhibition suppressed mitochondrial flash and ROS
To further determine the physiological significance of DRP1 on mPTP and respiration regulation, we monitored mitochondrial flashes, which are triggered by transient mPTP openings and fueled by mitochondrial respiration.18,36 First, in intact and Langendorff perfused hearts from mt-cpYFP transgenic mice, we found significantly decreased flash activity by Mdivi-1 indicating suppressed transient mPTP opening and mitochondrial respiration (Figure 6A–D). In adult cardiomyocyte from rats, Mdivi-1 suppressed basal and pyruvate-induced flash activity (Figure 6E). Overexpression of K38A or shDRP1 also inhibited flash in adult cardiomyocytes (Figure 6F). These results support that inhibiting endogenous DRP1 activity decreased transient mPTP openings and respiration in individual mitochondrion. Since mitochondrial respiration is the major source of ROS in the heart, we monitored steady state ROS in mitochondria matrix by using MitoSOX red and found decreased mitochondrial ROS by Mdivi-1 or K38A in adult cardiomyocytes (Figure 6G). Taken together, endogenous DRP1 modulates transient mPTP opening and impacts ROS production.
Figure 6.

DRP1 modulated mitochondrial flash and ROS. (A, B), Representative images (A) and summarized data (B) showing that Mdivi-1 inhibited the frequency of mitochondrial flash in perfused beating heart from mt-cpYFP transgenic mouse. The white boxes in (A) indicate the location of flashes during a 100 s serial scanning. N = 3 mice. #P < 0.01 vs. Control. (C, D) Enlarged serial images (C) and traces (D) showing the fluorescence changes during a typical mitochondrial flash observed in the perfused heart. The intervals between each images in C is 2 s, the colour coding indicates relative fluorescence intensity. (E) Summarized data showing the frequency of mitochondrial flash in Midivi-1 treated adult cardiomyocytes from rat in the presence or absence of respiration substrate, pyruvate (1 mM). N = 16–29 cells from 3–5 rats. *, †: P < 0.05 or 0.001 vs. Control, respectively. (F) Summarized data showing K38A or shDRP1 inhibited mitochondrial flash in adult cardiomyocytes from rats. N = 26–29 cells from four rats. *, #: P < 0.05 or 0.01 vs. Control, respectively. (G) Mdivi-1 or DRP1-K38A decreased constitutive mitochondrial ROS in adult cardiomyocytes monitored by the slop of fluorescence increase of MitoSox. N = 11–16 cells from 3–4 rats. *P < 0.05 vs. Control.
4. Discussion
In this study, we report a novel function of the fission protein, DRP1 on maintaining mitochondrial respiration in adult cardiomyocytes (Figure 7). We further showed that this effect depends on CypD, a soluble matrix protein that binds to membrane proteins and sensitizes the mPTP to matrix Ca2+. We also explored the physiological significance of this function on cell bioenergetics and ROS signalling. Moreover, this effect is largely independent of the role of DRP1 in promoting mitochondrial fission, since acute manipulations of DRP1 yielded modest (K38A, shDRP1, or WT DRP1 overexpression) or no change (Mdivi-1) in mitochondrial morphology while significantly altered mitochondrial respiration in adult cardiomyocytes. Our findings are in line with recent reports on cardiac specific DRP1 knockdown models, which showed compromised mitochondrial respiration.15,37 This study provides new insights into the multi-functional roles of mitochondrial fission and fusion regulators in the adult heart.
Figure 7.
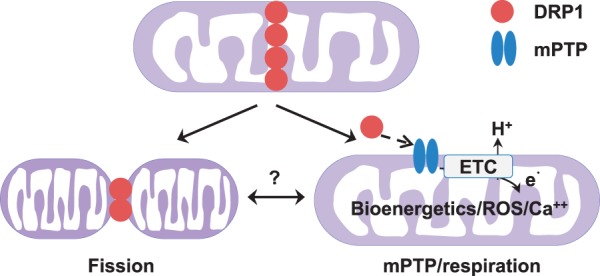
Schematic diagram showing the canonical and non-canonical roles of endogenous DRP1 in adult cardiomyocytes. The recruitment of DRP1 to mitochondria (upper panel) can induce fission (lower right panel), which is the canonical function of DRP1. At the same time, DRP1 also positively regulates mitochondrial respiration in adult cardiomyocytes (lower left panel). This novel non-canonical function of DRP1 may involve transient mPTP and contribute to bioenergetics, Ca2+ and ROS signalling in adult cardiomyocytes. Whether the canonical and non-canonical functions are connected or independent to each other needs further study. ETC, electron transport chain.
The major contribution of this work is the strong evidence showing a causal role of endogenous DRP1 in mitochondrial respiration regulation in adult cardiomyocytes. Recent reports have shown inconsistent results that inhibiting or knocking out DRP1 may or may not affect mitochondrial respiration.15,16 Here, with in vitro and acute manipulation approaches, we were able to specifically inhibit, but not abolish, the activity of endogenous DRP1 in adult cardiomyocytes. This approach can avoid or minimize the compensatory or none-specific responses in the in vivo models where DRP1 protein is significantly and chronically decreased. Furthermore, we monitored mitochondrial respiration through OCR by different methods including the Clark electrode and the Seahorse system and under different conditions from isolated mitochondria to permeabilized cells and intact cells. In all these experiments, maximal respiration is decreased after DRP1 inhibition. In the Seahorse measurements, basal respiration is also increased after DRP1 inhibition, likely due to increased proton leak. These data show, for the first time, that endogenous DRP1 indeed maintains mitochondrial respiration in adult cardiomyocytes. Since S3 acutely induced fusion but had no effect on OCR in adult cardiomyocytes, it suggests that morphological change per se cannot underlie mitochondrial respiration regulation by DRP1. Finally, overexpression of WT DRP1 had a tendency to increase respiration. It is possible that under resting conditions the overexpressed DRP1 may mainly exist in cytosol rather than being recruited to the mitochondria. Previously, the role of OPA1 and MFN1/2 on mitochondrial respiration has been linked to specific mechanisms, such as cristae formation and mitochondrial-ER tethering, respectively.12,14 Our results extend the current knowledge of the novel roles of fission and fusion proteins on mitochondrial respiration by showing the fission protein, DRP1, also positively modulates respiration through a mechanism independent of morphological regulation.
Another contribution of this work is the potential link between DRP1, transient mPTP, and mitochondrial respiration. DRP1 is mainly a cytosolic protein and can be recruited to mitochondrial outer membrane by Ca2+, phosphorylation, and oxidation.22,38,39 In resting adult cardiomyocytes, a significant amount of DRP1 can be detected in the mitochondrial fraction.28 Since inhibiting DRP1 suppressed laser induced mPTP and mitochondrial flash events, DRP1 may modulate mPTP. Moreover, the effect of Mdivi-1 on State 3 respiration is abolished by cyclosporine A or absent in CypD KO myocytes suggesting a role of mPTP in mediating DRP1’s effect on mitochondrial respiration. Previous reports from us and others have shown that transient mPTP openings, as monitored by mitochondrial flashes, are physiologically important individual mitochondrial events and coupled to the respiration status of mitochondria.7,8,18,20 In this study, we further showed that DRP1 inhibition decreased maximal respiration and transient mPTP openings. However, basal respiration is either unchanged or increased in the whole mitochondrial population. This is probably because the random, low frequency and transient mPTP openings occurring in individual mitochondrion under resting conditions may not significantly impact the basal respiration of the mitochondrial population. Proton leak may play a more important role in basal respiration regulation. During respiration stimulation, the transient mPTP openings are significantly increased and may contribute to the increased maximal respiration. In addition, unlike the permanent or prolonged mPTP openings, transient mPTP openings will not deplete co-factors such as cytochrome C from the mitochondria and thus, will not lead to compromised respiration. These notions are consistent with recent reports regarding the role of CypD in mPTP regulation,40,41 the decreased maximal respiration in CypD KO cardiomyocytes,42 and the partial rescue of DRP1 KO-induced cardiomyopathy by CypD KO.16 Thus, it is likely that DRP1 modulates transient mPTP opening and through which maintains respiration in cardiomyocytes. In addition, other mechanisms, such as proton leak, may also play a role in mediating the effect of DRP1 on respiration regulation. It remains elusive; however, how the outer membrane protein, DRP1 can positively modulate respiration through transient mPTP. A potential answer could be through an indirect mechanism or protein on the outer membrane. A number of outer membrane proteins can bind DRP1 acting as its receptors.43 One of them is Bak/Bax, the pro-apoptotic Bcl-2 family proteins, which also modulate outer membrane permeability.29,44 Bak/Bax has been shown as components of the mPTP and the mPTP may involve outer membrane permeabilization and inner membrane permeabilization.45,46 Therefore, we may tentatively hypothesize that DRP1 could modulate mPTP through interacting with Bak/Bax and outer membrane permeability. Definitive evidence of the molecular targets of DRP1 on outer membrane that can link DRP1 with mPTP awaits future studies.
This study also advances our understanding of the role of DRP1 in cardiac physiology and pathology. Recent reports using inducible and heart specific DRP1 knockout models15,16 showed that chronic ablation of DRP1 in the heart leads to severe cardiomyopathy and significantly shortened lifespan. One potential mechanism is altered mitophagy.47 However, what remains unknown is whether the observed phenotypes and the accompanying pathological changes, such as mitophagy, apoptosis, mPTP opening, are secondary or independent to morphological defects (e.g. suppressed fission). The current study provides some clues by showing a novel role of DRP1 in maintaining mitochondrial respiration independent of morphological change. It is possible that DRP1 inhibition acutely compromises mitochondrial respiration, in parallel to suppressing fission, and contributes to energetic decompensation, mitophagy and apoptosis in the long run. In addition, since DRP1 inhibition facilitated proton leak in adult cardiomyocytes, it may also contribute to the phenotypes of chronic DRP1 ablation. Moreover, findings of the current study may also help explain pathological roles of DRP1 in human disease. One example is the increased mitochondrial translocation of DRP1 and/or fission upon stress such as high glucose and ischemia.21,48 It will be interesting to test the hypothesis that increased DRP1 is a compensatory response to maintain mitochondrial respiration but eventually causes detrimental effects such as fragmentation or excessive mPTP opening. Another example is the protective role of DRP1 inhibition in cardiac ischemia reperfusion injury.34 It is possible that DRP1 inhibition may suppress mitochondrial respiration during early reperfusion, and through which prevent oxidative stress-induced cell death. DRP1 inhibition also ameliorated diabetes-induced liver oxidative stress through uncoupling respiration with ATP production.32 Finally, accumulating evidence supports that DRP1 may play multiple roles in cell biology and disease. How to differentiate these effects is critical for developing function-oriented approaches to regulate DRP1. For example, Mdivi-1 is known to have multiple targets and its protective effects on stress-induced cell death may not be DRP1-dependent,15 while its role in respiration and fission may be more dependent on DRP1.
In summary, in addition to its canonical role in mitochondrial fission, endogenous DRP1 also plays a novel and non-canonical role in maintaining mitochondrial respiration. Mechanistically, this effect is mediated by mPTP, dissociated from morphological changes, and does not affect the assembly or activity of individual ETC complexes. This new pathway of DRP1 exemplifies the multi-functional roles of DRP1 in regulating both mitochondrial function and morphology in the heart under physiological and pathological conditions.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Acknowledgements
We thank J.S., H.C., R.M., B.M.W., B.M.D., P.M., and G.G. for technical support and helpful discussions.
Conflict of interest: none declared.
Funding
This work was supported by American Heart Association (10SDG3450009 to W.W.) and the National Institutes of Health (HL114760 to W.W. and S.-S.S.).
References
- 1.Chandel NS. Mitochondria as signaling organelles. BMC Biol 2014;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheffler IE. Mitochondria. J. New York, NY USA: Wiley and Sons, Inc; 2008. [Google Scholar]
- 3.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Rourke B, Cortassa S, Aon MA. Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda) 2005;20:303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008;27:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004;287:C817–833. [DOI] [PubMed] [Google Scholar]
- 7.Gong G, Liu X, Wang W. Regulation of metabolism in individual mitochondria during excitation-contraction coupling. J Mol Cell Cardiol 2014;76:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong G, Liu X, Zhang H, Sheu SS, Wang W. Mitochondrial flash as a novel biomarker of mitochondrial respiration in the heart. Am J Physiol Heart Circ Physiol 2015;309:H1166–H1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 2012;46:265–287. [DOI] [PubMed] [Google Scholar]
- 10.Friedman JR, Nunnari J. Mitochondrial form and function. Nature 2014;505:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galloway CA, Lee H, Yoon Y. Mitochondrial morphology-emerging role in bioenergetics. Free Radic Biol Med 2012;53:2218–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013;155:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008;456:605–610. [DOI] [PubMed] [Google Scholar]
- 14.Eisner V, Csordas G, Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca(2)(+) and reactive oxygen species signaling. J Cell Sci 2013;126:2965–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 2015;116:264–278. [DOI] [PubMed] [Google Scholar]
- 16.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 2015;21:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall AR, Burke N, Dongworth RK, Hausenloy DJ. Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Br J Pharmacol 2014;171:1890–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell 2008;134:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc Natl Acad Sci U S A 2001;98:3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000;192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res 2008;79:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hom J, Yu T, Yoon Y, Porter G, Sheu SS. Regulation of mitochondrial fission by intracellular Ca2+ in rat ventricular myocytes. Biochim Biophys Acta 2010;1797:913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcu R, Neeley CK, Karamanlidis G, Hawkins BJ. Multi-parameter measurement of the permeability transition pore opening in isolated mouse heart mitochondria. J Vis Exp 2012;67:e4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benit P, Goncalves S, Philippe Dassa E, Briere JJ, Martin G, Rustin P. Three spectrophotometric assays for the measurement of the five respiratory chain complexes in minuscule biological samples. Clin Chim Acta 2006;374:81–86. [DOI] [PubMed] [Google Scholar]
- 25.Gong G, Wang W. Confocal imaging of single mitochondrial superoxide flashes in intact heart or in vivo. J Vis Exp 2013;81:e50818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, Shang W, Zhang X, Gu J, Wang X, Zheng M, Wang Y, Zhou Z, Cao JM, Ji G, Zhang R, Cheng H. β-Adrenergic-stimulated L-type channel Ca2+ entry mediates hypoxia-induced Ca2+ overload in intact heart. J Mol Cell Cardiol 2013;65:51–58. [DOI] [PubMed] [Google Scholar]
- 27.Yoon Y, Pitts KR, McNiven MA. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell 2001;12:2894–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yue W, Chen Z, Liu H, Yan C, Chen M, Feng D, Wu H, Du L, Wang Y, Liu J, Huang X, Xia L, Liu L, Wang X, Jin H, Wang J, Song Z, Hao X, Chen Q. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res 2014;24:482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008;14:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 2011;435:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galloway CA, Lee H, Brookes PS, Yoon Y. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol 2014;307:G632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galloway CA, Lee H, Nejjar S, Jhun BS, Yu T, Hsu W, Yoon Y. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes 2012;61:2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, Yu T, Lee H, O'Brien DK, Sesaki H, Yoon Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res 2015;106:272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010;121:2012–2022. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005;434:652–658. [DOI] [PubMed] [Google Scholar]
- 36.Hou T, Wang X, Ma Q, Cheng H. Mitochondrial flashes: new insights into mitochondrial ROS signalling and beyond. J Physiol 2014;592:3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 2014;33:2798–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 2013;17:491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta 2013;1833:1256–1268. [DOI] [PubMed] [Google Scholar]
- 40.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434:658–662. [DOI] [PubMed] [Google Scholar]
- 41.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 2010;120:3680–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shang W, Gao H, Lu F, Ma Q, Fang H, Sun T, Xu J, Ding Y, Lin Y, Wang Y, Wang X, Cheng H, Zheng M. Cyclophilin D regulates mitochondrial flashes and metabolism in cardiac myocytes. J Mol Cell Cardiol 2016;91:63–71. [DOI] [PubMed] [Google Scholar]
- 43.Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 2013;24:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 2002;159:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH, Robbins J, Kinnally KW, Molkentin JD. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2013;2:e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P, Martinou JC. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 2010;142:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorn GW., 2nd. Gone fission…: diverse consequences of cardiac Drp1 deficiency. Circ Res 2015;116:225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J 2014;28:316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.