

Preface
Autophagy is a fundamental cell biological pathway affecting immunity. Whereas autophagy is an antimicrobial effector of conventional pattern recognition receptors (PRRs), autophagic adaptors termed SLRs represent a new subset of PRRs and provide the mechanistic basis for autophagic elimination of intracellular microbes. Autophagy controls inflammation via regulatory interactions with innate immunity signalling, by removing endogenous inflammasome agonists, and thorough effects on secretion of immune mediators. Autophagy contributes to antigen presentation, T cell homeostasis, and affects T cell repertories and polarization including Th17 inflammation. Here, we review the above relationships organized into four principal roles of autophagy in infection, inflammation, and immunity.
Introduction
Autophagy as an immunological process can be organized in four principal manifestations illustrated in Fig. 1: direct elimination of microbes, control of inflammation, antigen presentation and lymphocyte homeostasis, and secretion of immune mediators. Immunological autophagy fits but in some aspects exceeds the scope of autophagy as a cytoplasmic quality and quantity control process that also provides nutrients through cytosol autodigestion at times of starvation1. The word autophagy (“self-eating”) refers to a collection of diverse processes enabling cells to digest their cytoplasmic constituents in lysosomes. This definition includes macroautophagy, microautophagy, chaperone mediated autophagy and noncanonical autophagy2,3. Several non-autophagic roles, including immunological effects, of individual or subsets of autophagy factors have also been recognized4,5. In this review we cover only the sensu stricto autophagy – macroautophagy. We refer to it by name as autophagy and by function as a defined cell biological pathway that depends on specialized Atg factors1 (Box 1). This pathway is distinct from other cytoplasmic digestive processes, including proteasomal degradation, by its ability to capture and eliminate large targets such as toxic protein aggregates, defunct or disused organelles, and invading microbes.
Figure 1. Four principal roles of autophagy in immunity.
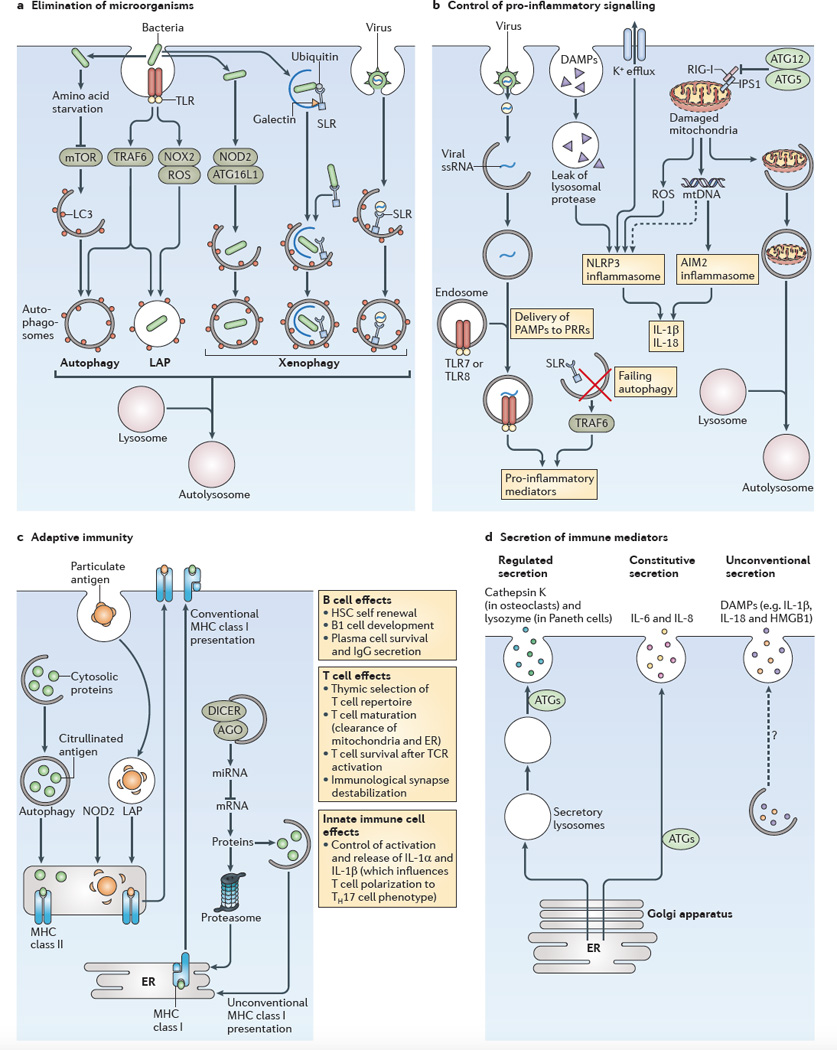
a | The role of autophagy in the elimination of microorganisms is shown. An incoming microorganism can induce autophagy by competing for nutrients or by stimulating innate immune receptors, such as Toll-like receptors (TLRs). When the microorganism is taken up by phagocytosis and remains in an intact vacuole, an autophagic process termed LC3-associated phagocytosis (LAP) can promote the maturation of autophagosomes into autolysosomes. Xenophagy of pathogens that enter the cytosol can be initiated by sequestosome 1-like receptors (SLRs) or other mechanisms, including nucleotide-binding oligomerization domain-containing protein 2 (NOD2)–autophagy-related protein 16-like 1 (ATG16L1) interactions. b Several examples of the role of autophagy in the control of pro-inflammatory signalling (see also FIG. 3) are shown. Failure to remove SLRs by autophagy can increase the levels of these receptors and the levels of pro-inflammatory signalling. Autophagy can deliver cytoplasmic pathogen-associated molecular patterns (PAMPs) to endocytic TLRs and can stimulate their activity. NOD-like receptors (NLRs; such as NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3)) and RIG-I-like receptors (RLRs; such as absent in melanoma 2 (AIM2)) show complex positive and negative co-regulation with autophagy: the ATG5–ATG12 complex inhibits retinoic acid-inducible gene I (RIG-I) signalling, and autophagy limits inflammasome activation by removing damaged mitochondria, which then release the inflammasome activators reactive oxygen species (ROS) and mitochondrial DNA. c The role of autophagy in adaptive immunity is shown. Autophagy can increase the MHC class II presentation of cytoplasmic antigens, including self or viral antigens, as well as promoting the citrullination of antigens. LAP can enhance the processing of particulate antigens for MHC class II presentation. NOD2 enhances autophagic antigen presentation. Autophagy may directly or indirectly affect MHC class I presentation by competing with the proteasome for substrates, by influencing the peptidome pools through the control of levels of components of microRNA (miRNA) machinery (for example, argonaute (AGO) and DICER), or by supporting unconventional MHC class I presentation. In addition, autophagy affects the self-renewal of haematopoietic stem cells (HSCs), B1 cell development, plasma cell survival and IgG secretion. Autophagy affects T cell survival following T cell receptor (TCR) activation, and it destabilizes the immunological synapse. It also controls innate immune cell (such as macrophage) signalling through the release of interleukin-1α _(IL-1α)_ _and IL-1β, which influence the polarization of T cells into T helper 17 (TH17) cells. Autophagy also affects naive T cell repertoire selection in the thymus and the survival and function of maturing T cells by removing the mitochondria and endoplasmic reticulum (ER), thus ensuring calcium homeostasis. d The role of autophagy in the secretion of immune mediators is shown. Autophagy affects the quality of regulated secretion from pre-stored granules. Autophagy affects the quality and the quantity of the output of the constitutive secretory pathway (which is the conventional pathway of protein secretion via the ER, the Golgi apparatus and the plasma membrane). Autophagy supports a form of unconventional secretion that captures cytoplasmic proteins for extracellular release. Note that secretory protein cargo in the regulated and constitutive secretory pathways contains conventional leader peptides for co-translational import into the ER lumen, whereas protein cargo that enters the unconventional secretory pathway lacks leader peptides and does not enter the ER. Dashed arrow indicates that this pathway remains to be defined. DAMPs, damage-associated molecular patterns; HMGB1, high-mobility group box 1 protein; IPS1, IFNβ _promoter stimulator protein 1; mtDNA, mitochondrial DNA; mTOR, mammalian target of rapamycin; NOX2, NADPH oxidase 2; PRRs, pattern recognition receptors; ssRNA, single-stranded RNA; TRAF6, TNF receptor-associated factor 6.
Box 1. Autophagy pathway.
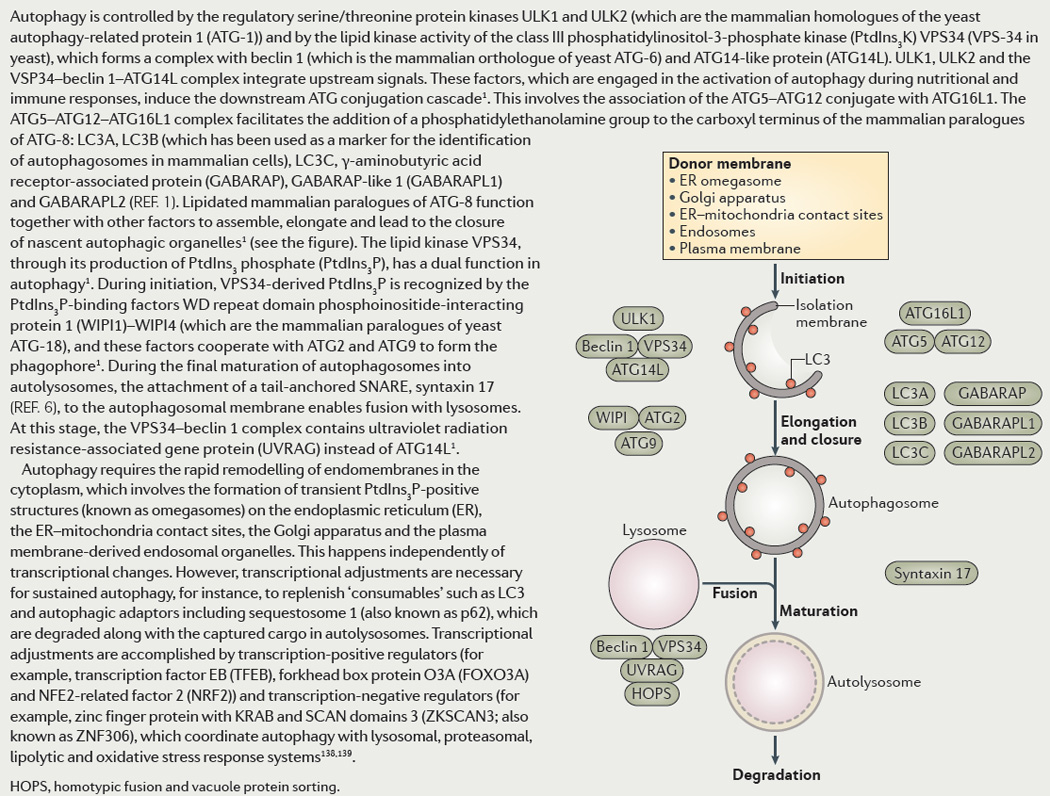
The Atg nomenclature is based on yeast genetics. It includes the Ser/Thr kinase Atg1 (mammalian Ulk1 and Ulk2), Atg6 (mammalian Beclin 1) in complexes with class III phosphatidylinositol 3-phosphate kinase (PI3K) Vps34 (mammalian hVPS34), Atg5-Atg12 conjugate in complex with Atg16 (mammalian Atg16L1), and Atg8 (mammalian orthologs, mAtg8s: LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2). Historically, LC3B has been used as a marker for identification of autophagosomes in mammalian cells. A hierarchical organization of mAtg8s has begun to emerge in conjunction with autophagic adaptors and recognition of intracellular microbes. Autophagy is controlled by the regulatory protein kinases Ulk1/2 plus the lipid kinase hVPS34, the latter being in a complex with Beclin 1 and Atg14L. They integrate upstream signals, and set in motion the downstream Atg conjugation cascade. This involves Atg5-Atg12/Atg16L1 assembly, which then acts as a functional equivalent of E3 ligases from the ubiquitin system. This results in a hallmark event of mAtg8s C-terminal lipidation with phosphatidylethanolamine, the basis for a key assay in measuring autophagy. Lipidated mAtg8s, in cooperation with other factors, assemble, elongate, and lead to closure of nascent autophagic organelles. The lipid kinase hVPS34 participates once again during the final maturation stages of autophagosomes into autolysosomes. For this, it is still complexed with Beclin 1 but now contains UVRAG instead of Atg14L. Autophagy requires rapid remodeling of endomembranes in the cytoplasm independently of transcriptional changes. Appropriate transcriptional adjustments are necessary for sustained autophagy, e.g. to replenish “consumables” such as LC3 and autophagic adaptors including Seqestosome 1/p62, degraded along with the captured cargo in autolysosomes. Transcriptional adjustments are accomplished via global positive (TFEB, FoxO3A, NRF2) and negative (ZKSCAN3/ZNF306) regulators coordinating autophagy with lysosomal, proteasomal, and oxidative stress response systems.
Autophagy may be the most primordial form of innate immunity against invading microbes. The present view of autophagy as an immune defense has its mechanistic beginnings in the recognition of autophagy as a downstream effector of PKR, a pivotal antiviral protein in mammalian cells6,7 and in the reports of autophagic elimination of intracellular streptococci8 and Mycobacterium tuberculosis9. The latter two examples epitomize the most primal immune manifestation of autophagy - direct capture of intracellular microbes for elimination in autophagosomal organelles (Fig. 1a). The appreciation of immunological functions of autophagy has increased in volume and scope10 since the initial overviews of connections with innate and adaptive immunity11 and today innate immunity manifestations of autophagy are considered as one of its principal roles. One of the tenets of this review is that autophagic capture and elimination of intracellular microbes has been a key contributing driver in the evolution of the autophagy pathway as a whole. In the present day eukaryotes, in particular in mammals, these primordial roots have evolved and become incorporated into multiple layers of innate and adaptive immunity. Here we dissect these relationships organized in four principal categories (Fig. 1) following the escalating complexity of the immunological systems with which autophagy has been integrated.
Autophagy pathway and immune signalling
Autophagy as an Atg-dependent pathway
The principal morphological feature of autophagy is the formation of endomembranous organelles called autophagosomes (Box 1). An ensemble of Atg factors1 (Box 1), with Atg6/Beclin 1, Atg1/Ulk1, and Atg8/LC3 being key regulators, drives the formation in the cytosol of the autophagic isolation membrane (phagophore). A phagophore, often rendered in models as a stand alone crescent in the cytoplasm, is transiently connected to and derived from phosphatidylinositol 3-phosphate (PI3P)-positive domains of the endoplasmic reticulum (ER) termed omegasomes1. Other compartments such as the Golgi, mitochondria, and plasma membrane-derived endocytic organelles contribute as well1. The latest studies demonstrate that the ER-derived autophagosomes form at the ER-mitochondria contact sites12. A phagophore sequesters the captured cytoplasmic cargo destined for autophagic disposal, and upon elongation and closure, an autophagosome is formed. At this stage, the corresponding endomembranes are commonly visualized as double membrane structures1. The degradation of the captured cargo begins when autophagosomes mature into autolysosomes, delimited by a single membrane, through acidification and acquisition of lysosomal hydrolases1.
A shared maturation pathway between autophagy and phagocytosis
Autophagy is often morphologically equated with the formation of double membrane autophagosomes in the cytoplasm. This is the result of a geometrical necessity requisite when a phagosome is derived from a pre-existing membranous intracellular compartment such as ER1 rather then a requirement for double membrane per se (Box. 1). One well-studied exception to the double membrane precursors is the role of Atg factors in maturation of the standard phagolysosome. This process has been called LC3-associated phagocytosis (LAP)13,14 and occurs following uptake of various extracellular targets: particles coated with Toll-like receptors (TLR) agonists15,16, phagocytosed dead cells17, live epithelial cells engulfed via entotisis by neighboring cells18, and FcγR-dependent uptake of immune complexes19. If a microbe or targets that can activate TLRs are taken up by conventional phagocytosis, the autophagic machinery enhances maturation of the conventional phagosomes indistinguishably from how it works with the internally formed autophagosomes. LAP utilizes Beclin 1-hVPS34 and LC3-conjugation systems. As expected, LAP is independent of Ulk1, since Ulk1 is needed to generate autophagosomes from internal ER membranes during starvation18,19. The recapitulation of autophagic maturation steps in LAP is informative about the role of autophagy-associated membrane transactions such as PI3P generation, long known to be key for phagosomal maturation20, whereas the role of LC3 may be a manifestation of the tethering and fusogenic properties of LC321,22.
Starvation and autophagy: IKKs and nutrient depletion as an alarmin
Competition for intracellular nutrients might have been one of the most primordial alarmin or danger signals available to the eukaryotic cell to detect microbial invasion and eliminate them through autophagy. Nutritional signalling has been experimentally linked to antimicrobial autophagy in response to bacterial invasion23. (Fig. 1a-(v)). The metabolic regulation via mTOR (inhibiting autophagy) and AMPK (activating autophagy), covered in detail elsewhere1, merges with immune signalling through IKKα and IKKβ24,25 (Box 2). The role of IKKs in autophagy initiation in response to starvation is independent of NFκB action24 and instead is associated with events involving TAB2/3-TAK1 (the upstream regulators of IKKs) and Beclin 124,25. TAB2 and TAB3 in the resting state bind Beclin 1 and repress its activity. TAB2 interacts with members of the Ulk complex and is phosphorylated by Ulk125. Following autophagy induction, TAB2 and TAB3 dissociate from Beclin 1, thus activating it, and also bind to TAK124. TAK1 in turn phosphorylates and activates AMPK. In sum, this triggers both branches of autophagy regulatory (protein and lipid) kinases - Ulk1/2 (via AMPK) and hVPS34 (via Beclin 1).
Box2. Immune signaling and autophagy.
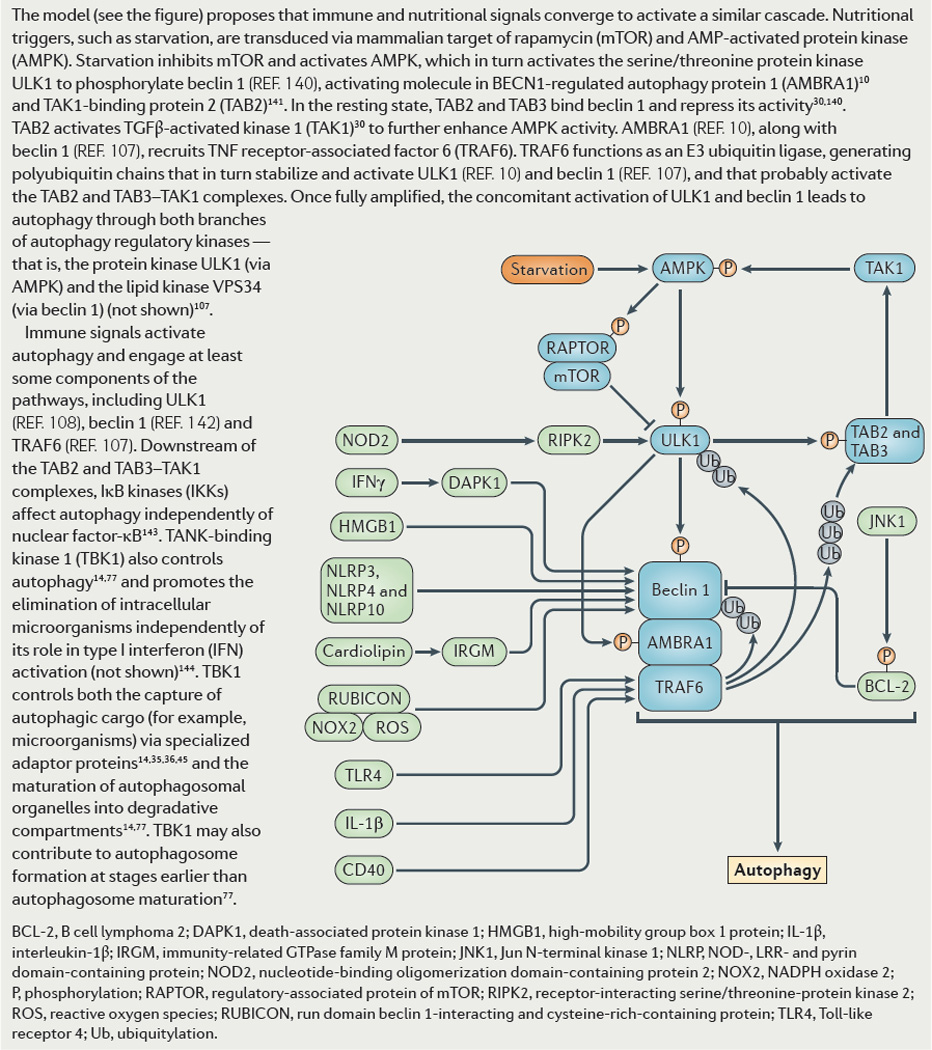
IKK signaling and its upstream regulators TAB2, TAB3 and TAK1 control autophagy. TAB2 and TAB3 repress Beclin 1. Upon induction, TAB2 and TAB3 dissociate from Beclin 1, allowing it to activate autophagy. They also bind to TAK1. TAK1 in turn activates AMPK, which helps induce autophagy. PAMPs and PRRs can set in motion autophagosome formation. TLR4-MyD88 signaling leads to a direct ubiquitination of Beclin 1 by TRAF6, which in turn dissociates Bcl-2 from Beclin 1 and disinhibits it. Whether signalling from TLRs engages IKKs and TBK-1 has not been experimentally addressed. HMGB1, an alarmin, derepresses Beclin 1 by displacing its negative regulator Bcl-2. It can additionally activate autophagy extracellularly via RAGE signalling. Other DAMPs/alarmins such as DNA complexes and ATP can activate autophagy. IL-1β, a key pro-inflammatory cytokine induces autophagy in autophagic control of M. tuberculosis in infected macrophages. IL-1β does it through MyD88 downstream of IL-1R and may involve TRAF6-dependent Beclin 1 ubiquitination. Th1 cytokines such as IFN-γ induce autophagy in effector cells including macrophages where they restrict mycobacteria. IFN-γ activation of autophagy involves a death associated protein kinase (DAPK)-dependent phosphorylation of Beclin 1 on Thr 119 located within its BH3 domain, which (congruently with other processes lading to Beclin 1 activation) dissociates its inhibitor Bcl-2. TNF-α stimulates Sequestosome 1/p62 adaptor-guided autophagy to restrict intracytosolic bacteria such as Shigella and Listeria. Th2 cytokines, such as IL-4 and IL-13 inhibit autophagy. IL-10 can also inhibit autophagy via Akt signalling. STAT3, which transduces signals downstream of various inputs including IL-6, can inhibit autophagy. This occurs not via transcriptional signalling, but rather by binding of the cytosolic STAT3 to protein kinase R (PKR). T cell receptor engagement and CD28 co-stimulation induce autophagy in T cells. CD40L-CD40 engagement between activated T cells and macrophages induces autophagy via TRAF6 to help eliminate Toxoplasma gondii parasitizing phagocytes.
TBK-1 controls autophagy
Another immunologically significant IKK-related kinase, TBK-1, affects autophagy26–29. It is involved in eliminating intracellular microbes independently of its role in type I interferon activation30. TBK-1 controls both the capture of autophagic cargo (e.g. microbes) via specialized adaptors26–29 and the maturation of autophagosomal organelles into degradative compartments29 (Box 2). Thus, there is a sequential engagement of IKKs, with IKKα/β acting at the initiating stages and TBK-1 in the completion of autophagy. The exocyst, a dynamic multiprotein platform best known for its role in vesicular trafficking and TBK-1-STING localization and activity31,32, also regulates autophagy33. When centered on the component Exo84, exocyst assembles autophagy initiation-relevant complexes containing Ulk1, Beclin 1, Atg14L and VPS3433. When centered on the component Sec5, exocyst controls STING and TBK-1 relocalization from the ER to sites important for activation of immune processes, i.e. IRF3 and type I IFN stimulation31,32. The role of IKK and TBK-1 signalling in autophagy is of growing significance and in keeping with a key position of TBK-1 that now includes interactions with autophagic factors26–29, as further highlighted in systems biology analyses of innate immunity interactome for type I interferon34.
Autophagy is controlled by PRRs, cytokines, and reactive nitrogen and oxygen species
Although is an ancient defense mechanism. However, the present-day antimicrobial autophagy in mammalian cells is well integrated with other innate and adaptive immune regulatory systems as summarized in Box 2. Furthermore, whereas autophagy is a built-in self-defense available in principle to any eukaryotic cell, the magnitude of its contribution in complex organisms is cell type dependent, as illustrated by the neuron-specific significance of autophagy against HSV-1 but a lack of its role in keratinocytes of the vaginal mucosa35. Autophagy is controlled by nearly all classes of PRRs and responds to PAMPs and DAMPs. It is also regulated by cytokines and receptors modulating innate and adaptive immunity. These relationships are summarized in Box 2. Reactive oxygen species (ROS) and systems that generate them affect autophagy13,36–39. NADPH oxidase and ROS are engaged in LAP during Salmonella phagocytosis13. NADPH oxidase and autophagic machinery are connected via a regulatory protein Rubicon. By jumping from the autophagy inhibitory complex to NADPH oxidase activating complex, Rubicon activates both bactericidal (autophagy and ROS) mechanisms simultaneously38. Nitric oxide inhibits autophagy by inactivating JNK-1 and IKK-β via their direct S-nitrosylation39. This prevents JNK-1-dependent Bcl-2 phosphorylation39 and its dissociation from the BH3 domain of Beclin 1,40,41 and inhibits IKK-β-associated24 AMPK-dependent autophagy initiation39,42.
First principal role: direct elimination of microbes
Autophagy intercepts microbes at different stages of invasion
Antimicrobial autophagy is a sequential set of barriers against incoming microbes (Fig. 1a). To deploy autophagy, mammalian cells utilize guidance systems detecting the presence, location and extent of the cytoplasmic penetration by a pathogen. This begins with conventional pattern recognition receptors (PRRs)43 acting at any stage, and, if microbe penetrates the cytosol, ends with autophagic adaptors44 involved in elimination of microbes. In their antimicrobial role, these autophagic adaptors are referred to as Sequestosome 1/p62-like receptors (SLRs)45. SLRs physically recruit the autophagic machinery while recognizing the molecular tags (ubiquitin, galectin, membrane phospholipid modifications) found on the invading microbe or on damaged host membranes associated with the microbe26,27,46–49 (Fig. 1a–vi).
PRRs can elicit autophagic responses at different stages of host-microbe encounters (Fig. 1a). With bacteria, this includes early TLR- and Nod-dependent detection of the released microbial products (e.g. PAMPs). Autophagy stimulation can occur before (Fig. 1a-i)50,51, during adhesion and pathogen-induced uptake of bacteria by the host cell (Fig. 1a-ii), or during active phagocytosis of microbes by macrophages (Fig. 1a-iii)15,52,53. At stages past bacterial uptake, autophagy is elicited via pathogen-induced damage to the newly formed parasitophorous vacuoles (Fig. 1a-iv)26–28,49,54,55 and upon escape of bacteria into the cytosol (Fig. 1a-v)47,56–58. The engagement of autophagy with viruses involves various stages of their life cycle (Fig. 1a-vi)7,16,35,37,59–65. Two terms have been coined to refer to the aspects of the above continuum of antimicrobial autophagy: xenophagy refers to the uptake of intracellular microbes into double membrane autophagosomes10, whereas LC3-asociated phagocytosis (LAP)13,14 refers to an engagement of the autophagic machinery while the bacterium is confined within the nascent phagosome.
Microbes defend themselves
The significance of autophagy in protecting the cytosol from microbial invasion is underscored by microbial countermeasures (Box. 3)66. These mechanisms often involve targeting of Beclin 1 by microbial factors to block autophagy67 59, prevent autophagosomal maturation61,65 or even in some cases activate autophagy to generate nutrients for microbes68. In other instances, an autophagosomal membrane can be perforated to prevent acidification55 or mAtg8s are proteolytically cleaved to irreversibly remove C-terminal lipid modifications69, whereas if all else fails, epitopes or tags for recognition by SLRs can be masked56,57,70.
Box 3. Microbial countermeasures against autophagy.
Microbes show a wide range of mechanisms to prevent, counter, or commandeer autophagy. Listeria avoids autophagic capture of bacteria via AktA and InlK, and Shigella via IcsB. These processes mask bacterial epitopes or host ubiquitin tags that can be recognized by the autophagic machinery, either directly or by recruiting cytosolic proteins. For the fraction of bacteria that do not escape autophagic capture, Listeria can block autophagosome acidification via its pore-forming toxin LLO and thus prevent its maturation into autolysosomes. Legionella RavZ, an effector injected into the host cytoplasm via bacterial type IV secretion system, inhibits host autophagy through irreversible mAtg8 deconjugation. This happens by proteolysis of the mAtg8 C-terminal tails removing the C-terminal glycine along with its PE modification. Some of the viral defense examples include HSV-1 protein ICP34.5, influenza M2 protein, and HIV Nef, all of which target Beclin 1 to inhibit autophagy altogether or just to block autophagosomal maturation. Nef binds to the evolutionary conserved domain of Beclin 1 at the same 267–299 region where the host protein GAPR-1 (Golgi-associated plant pathogenesis-related protein 1) binds to act as an endogenous inhibitor of Beclin 1. Mouse herpesvirus 68 encoded protein M11 (viral Bcl-2) inhibits Beclin 1 via its BH3 domain whereas KSHV FLIP blocks Atg3 in the LC3 lipidation cascade. HIV Nef, Hepatitis C virus NS3, and Measles virus Mev3 proteins interact, with a yet to be fully understood consequences, with IRGM, a factor that controls antimicrobial autophagy and confers genetic risks in Crohn’s disease and tuberculosis. Finally, several microbes can also actively utilize autophagy for their life cycle based on an array of effects including membrane platforms for replication and metabolic effects, as previously reviewed. As a mechanistic example of the latter, the obligatory intracellular pathogen, Anaplasma phagocytophilum, uses its type IV secretion effector Ats-1 to bind Beclin 1 and activate autophagy to generate nutrients.
TLRs and autophagy cooperate in responses to PAMPs
TLR-ligands activate autophagy via specific molecular cascades (Box 2). Stimulation of TLRs induces autophagy, revving up this defense in advance of microbial invasion50,51 (Fig. 2a(a)), possibly promoting generation of antimicrobial peptides for eventual delivery to the parasitophorous vacuoles71, or focusing autophagic response to the points of microbial entry15 (Fig. 2) as during LAP15 (Fig. 2a). Autophagy stimulated via TLRs enhances antigen presentation in conjunction with LAP16,72. Autophagy scoops cytosolic PAMPs for delivery to endosomal lumens where they can come in contact with the correct end of endosomal TLRs and stimulate other responses, e.g. type I IFN, as in the case of TLR7 recognition of cytosolic ssRNA virus replication intermediates in pDCs60 (Fig. 2a-(c) – or/and Fig. 1b(viii-1)).
Figure 2. Autophagy-mediated clearance of intracellular pathogens.
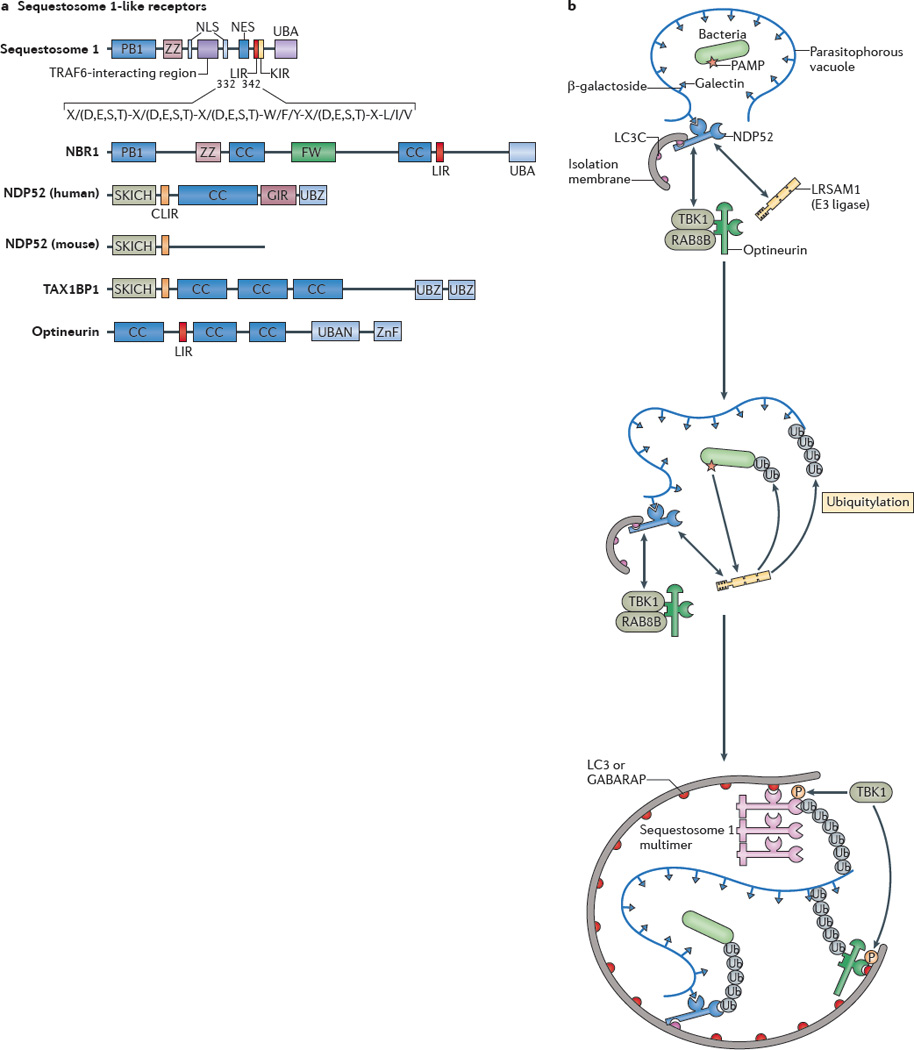
a| Protein domains of sequestosome 1-like receptors (SLRs) are shown. The LC3-interacting region (LIR) motif of an SLR binds to autophagy-related LC3 proteins through its consensus sequence at amino acids 332–342 in sequestosome 1 (also known as p62). The conserved residues are shown. X/(D,E,S,T) indicates that any amino acid (X) is allowed but that acidic (D,E) or phosphorylatable amino acids (S,T) are often present (usually at least one or more within the entire consensus sequence). The core LIR motif residues are aromatic pocket-filling W (or F or Y) residues and aliphatic pocket-filling L (or I or V). They form an intermolecular parallel β-_sheet with LC3 proteins or γ-aminobutyric acid receptor-associated proteins (GABARAPs). The CLIR motif, which is a LIR motif that is specific for LC3C, lacks the aromatic residue found in the LIR motif and, instead, uses hydrophobic contacts provided by additional aliphatic residues located between the W and L position anchors to stabilize interactions with LC3C. All human SLRs also contain a ubiquitin-binding domain (UBD): UBA (as found in sequestosome 1 and NBR1) is a three-helix bundle UBD that has affinity for monoubiquitin and K63 ubiquitin linkages; UBAN (as found in optineurin) is a parallel coiled-coiled dimer UBD that has specificity for linear ubiquitin chains; and UBZ (as found in nuclear dot protein 52 (NDP52)) is a zinc finger ββα-fold UBD that binds to monoubiquitin and polyubiquitin. b A model of cooperative action between different SLRs and E3 ligases in bacterial targeting for xenophagy is shown. The schematic shows a parasitophorous vacuole with glycosylated molecules (in this case β-galactosides) facing the lumen of the vacuole that contains a bacterium and that is experiencing membrane damage. This membrane tear exposes β-galactosides to galectins (for example, galectin 8) which in turn bind to the galectin-interacting region (GIR) motif of NDP52. NDP52 also directly interacts with the E3 ligase LRSAM1 and indirectly with the serine/threonine protein kinase TANK-binding kinase 1 (TBK1), which interacts with optineurin. The CLIR motif of NDP52 binds to LC3C, which is a proposed initiator in the LC3 and GABARAP cascade during bacterial xenophagy. LRSAM1 or other E3 ubiquitin ligases polymerize ubiquitin at molecular targets that are yet to be identified. The hypothetical model includes the putative recognition of bacterial pathogen-associated molecular patterns (PAMPs) by LRSAM1 through its leucine-rich repeat domain. Ubiquitin tags are recognized by UBDs of NDP52, optineurin and sequestosome 1. The LIR motif of optineurin is phosphorylated by TBK1 and this improves LC3 and GABARAP binding. The UBA of sequestosome 1 is also phosphorylated by TBK1 and this improves ubiquitin chain binding. As a consequence, the autophagic isolation membrane initiates at the appropriate location and grows to capture the bacterium and to eliminate it through autophagy. CC, coiled-coil domain; FW, four W domain (also known as the NBR1 domain); KIR, KEAP1 interacting region; NES, nuclear export signal; NLS, nuclear localization signal; P, phosphorylation; PB1, protein-binding domain 1; SKICH, skeletal muscle and kidney enriched inositol phosphatase carboxyl homology domain; TAX1BP1, TAX1-binding protein 1; Ub, ubiquitylation; ZnF, zinc finger domain; ZZ, ZZ-type ZnF domain.
NLRs interact with Atgs to localize autophagy
The cooperation in antimicrobial defense between Nod like receptors (NLRs) and autophagy is conserved from flies 73 to humans 52,53. Atg16L1 and Nod2, encoded by Crohn’s disease (CD) risk loci74, interact52. Atg16L1 also interacts with Nod152 (Fig. 2b). Nod1 and Nod2 are necessary to induce autophagy in response to the presence of muramyl peptides in the cytosol52. Nod1 and Nod2 direct autophagic machinery by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry52. The Nod2-assisted localization of Atg16L1 at the plasma membrane fits with the general role of Atg16L1 in formation of autophagic precursors from the plasma membrane75. Cells from donors homozygous for the CD-conferring ATG16L1*300T allele have diminished capacity for autophagy induction in response to Nod2 agonist muramyl dipeptide. A Nod2 truncation in CD, which renders it completely cytosolic, retains Atg16L1 in the cytosol instead of facilitating its recruitment to bacterial entry sites thus precluding timely and site-specific autophagic control. NLRs other than Nods interact with the autophagic machinery. NLRX1 and its interacting partner mitochondrial Tu translation elongation factor, which associates with Atg5-Atg12 and Atg16L1, promote autophagy76. NLRC4, NLRP3, NLRP4, and NLRP10 interact with Beclin 177. In the case of NLRP4, recruitment of NLPR4 to the plasma membrane during phagocytosis of group A streptococci leads to its transient dissociation from Beclin 177. This permits initiation of Beclin 1-mediated autophagic responses77. Collectively, NLRs may induce autophagy by gathering autophagy factors in the vicinity of the incoming microbe (or resident mitochondria) followed by autophagy activation events.
Cyclic-di-GMP sensors, cytosolic DNA sensing, RLRs and autophagy
The conserved signaling molecules cyclic dinucleotides (e.g. cyclic di-GMP) released by bacteria can directly bind STING to induce TBK-1 signaling and type I IFN production78. When cyclic di-GMP or di-AMP are introduced into cytosol, they also activate autophagy28 (Fig. 2c(i)). STING does transduce signals from cytosolic DNA receptors to induce autophagy. Infection with dsDNA viruses (HSV-1, HCMV) induces autophagy79,80 dependent on STING80. Akin to the findings with viruses79,80 (Fig. 2c(ii)), bacterial DNA released during infection can induce autophagy via STING28 (Fig. 2c(iii)). It remains to be tested whether cGAPM (cyclic GMP-AMP)81 transduces the recognition of cytosolic bacterial and host DNA to induce autophagy. This second messenger, cGAMP, is generated by DNA-receptor/cGAMP syntase to activate type I IFN pathway via STING82, and may in parallel promote autophagy. Bacterial DNA leaches out from a subpopulation of M. tuberculosis phagosomes through pores introduced by ESX-128. Targeting of the DNA by autophagy occurs in a manner associated with ubiquitin tags and was shown to depend on SLRs28. As its side effect, the pathogen’s cytosolic DNA elicits, via STING and TBK-1, type I IFN response that suppresses production of the key anti-tuberculosis cytokine IL-1β and leads to pathology during later stages of tuberculosis83. DNA-dependent clearance of bacteria may be associated with an effort by the host cell to clear DNA mistaken for viral, whereas the release of DNA by the bacteria may be a pathogen’s strategy to elicit an unproductive (in the context of tuberculosis) type I IFN inflammation (Fig. 2c(iii)). In keeping with this, only a minor fraction of all M. tuberculosis phagosomes becomes labeled with LC3+ 28 whereas bacterial DNA is targeted via autophagy to autolysosomes for clearance28 (Fig 2c(iv)).
RIG-I-like receptors (RLRs) signaling is a prominent exception to the rule that autophagy is induced by PRRs. A typical observation with RLRs and autophagy is one of a negative regulatory relationship, with a pattern of reciprocal activation and repression between autophagy and RLR-driven type I IFN responses76,84.
SLRs clear invading microbes from the cytosol
When a pathogen makes it past the autophagy barriers directed by conventional PRRs, it can still be captured in the cytosol. These processes depend on specialized adaptors called SLRs (Fig. 3a)45. SLRs (Fig. 3a) are defined as autophagic receptors/adaptors for capture of microbes in contact with the cytosol. SLRs have been named as a group45,85 after the archetypical protein termed Sequestosome 1/p62. In addition to Seqestosome 1/p6286 and NBR187, which have similar domain organization, the extended definition of SLRs include Optineruin27 and NDP5226. SLRs (i) act as adaptors in antimicrobial autophagy26–28,46,47,57,88, (ii) contain one or more cargo recognition domains (CRDs)45 with ubiquitin26–28,46,47,57,88 and galectin49,89 thus far identified as tags associated with microbial targets, (iii) have one of the LC3/Atg8 interacting region motifs LIR (D/E-D/E-D/E-W/F/Y-X-X-L/I/V)44 or CLIR (LVV, thus far identified in NDP52)90; (iv) can affect inflammation47,91,92; (v) can be consumed during autophagy44; and (vi) are subject to modulation of their autophagic activities by phosphorylation of LIRs or CRDs27,29,93.
Figure 3. Autophagy controls inflammatory processes.
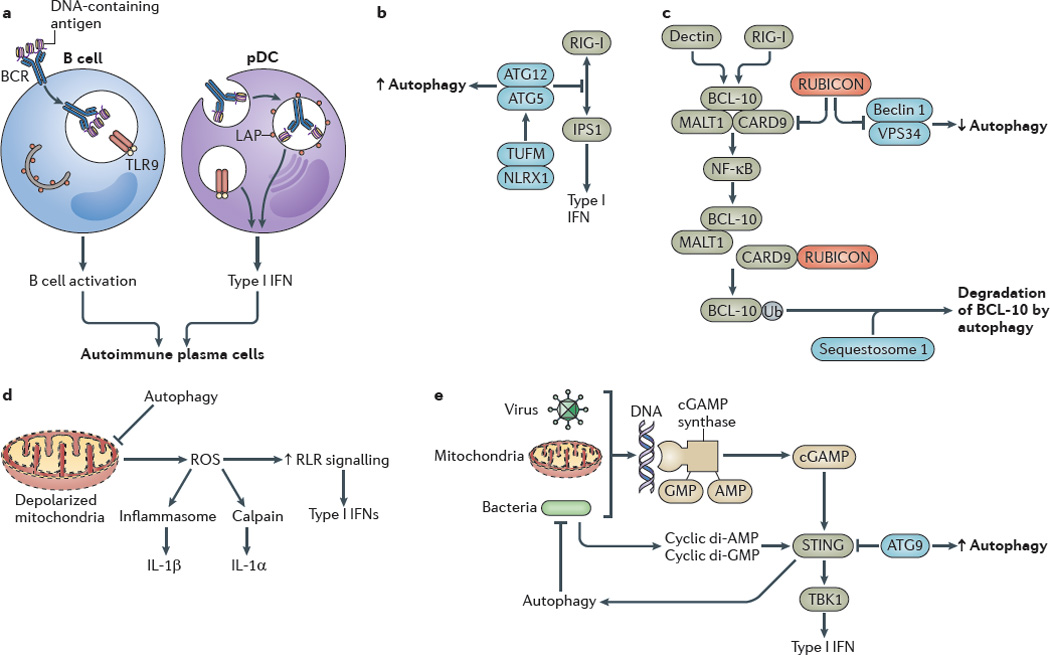
a Autophagy promotes Toll-like receptor 9 (TLR9) signalling in B cells and type I interferon (IFN) production by plasmacytoid dendritic cells (pDCs). b The autophagy protein complex autophagy-related protein 5 (ATG5)–ATG12 inhibits RIG-I-like receptor (RLR) signalling by binding to the caspase recruitment domains of retinoic acid-inducible gene I (RIG-I) and IFNβ _promoter stimulator protein 1 (IPS1), which is the mitochondrial adaptor of RIG-I signalling. The NOD-like receptor X1 (NLRX1)-interacting partner mitochondrial Tu elongation factor (TUFM) associates with the ATG5–ATG12 complex to promote autophagy while inhibiting RLR-dependent type I IFN activation. c Autophagy factors negatively regulate the caspase recruitment domain-containing protein 9 (CARD9)–B cell lymphoma 10 (BCL-10)– mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) complex. RUBICON (run domain beclin 1-interacting and cysteine-rich-containing protein), which is a binding partner and a negative regulator of beclin 1, inhibits CARD9, whereas sequestosome 1 leads to the degradation of BCL-10. d Excessive production of reactive oxygen species (ROS) by depolarized mitochondria that are not cleared by autophagy enhance RLR signalling. e Viral, mitochondrial or bacterial DNA lead to the activation of stimulator of IFN genes protein (STING), probably through cGAMP synthase and cyclic GMP–AMP (cGAMP) production, which increases the type I IFN response. Autophagy removes sources of agonists that stimulate STING, whereas autophagic factors (for example, ATG9) inhibit the activation of STING by affecting its cytoplasmic translocation. Bacterial cyclic dinucleotides (di-AMP and di-GMP) can activate autophagy, thereby functioning as a regulatory loop that amplifies the removal of infectious or endogenous irritants. BCR, B cell receptor; IL, interleukin; LAP, LC3-associated phagocytosis; NK-κB, nuclear factor-κB; TBK1, TANK-binding kinase 1; Ub, ubiquitylation.
The tags on microbial targets come in different varieties with ubiquitin and galectin being the best characterized (Fig. 3b). All known SLRs contain ubiquitin-binding domains (UBDs)94 with different specificities for ubiquitin chains (UBA, UBZ, UBAN)94 which decorate invading microbes (Fig. 3b-i). CRDs in SLRs can bind tags other than ubiquitin. A hook-like CRD in NDP52 enables it to interact with Galectin 889 (Fig2b-ii). Galectin 8 bridges NDP52 with cytoplasmically exposed β-galactoside glycans on pathogen-damaged host membranes via Galectin 8’s carbohydrate binding domains49. These compact domains are shaped as bent β-sandwich structures consisting of two antiparallel β-sheets with 6 and 5 strands, presenting its concave surface to β-galactoside glycans whereas the convex surface binds the hook-like CRD of NDP5289 (Fig. 3b-ii). The number of SLRs and the type of unique or repetitive structures recognized as tags may expand as research continues.
Cooperative processes in clearance of cytosolic microbes
The process of autophagic clearance of microbes penetrating into the cytosol is a tight cooperation between several components (Fig. 3c). This includes host DAMPs, microbial PAMPs, and multiple SLRs. DAMPs are exemplified by freshly exposed glycans upon damage to host membranes as in the case of Salmonella49,89. A PAMP may be a microbial polymer such as DNA in contact with the cytosol as in the case of M. tuberculosis28. As a part of the process, ubiquitin chains are generated around the microbe26–28,46,47,57,88 (Fig. 3b-i). This may involve specialized E3 ligases such as LRSAM195 (Fig. 3b). LRSAM1 has the ability to recognize Salmonella by the virtue of its LRR (leucine rich repeat), a domain often recognizing microbial ligands95. LRSAM1 self-ubiquitinates, and ubiquitinates yet to be defined microbial targets in vitro95. In vivo, E3 ligases may alternatively or in addition ubiquitinate host molecules associated with microbes.
A further cooperation exists between recognition of the initial tears in microbe-harboring vacuoles and ubiquitination, as exemplified in the case of Salmonella (Fig. 3c). In addition to LRSAM1’s auto-ubiquitination or ubiquitination of bacterial targets, LRSAM1 interacts directly with NDP5295. NDP52 (Fig. 3a) is an SLR with triple specificity recruited to the site of microbial penetration via: (i) binding of its hook-like CRD to Galectin-8-β-galactoside complex on perforated host membranes49,89; (ii) association of the NDP52 region 127–255 to the LRR domain of LSRAM195; and (iii) binding of its BUZ domain to ubiquitin chains26 placed on the target by E3 ligases. The downstream autophagic machinery is brought in via the NDP52 CLIR motif that binds to LC3C, requisite for acquisition of other mAtg8s and autophagy of Salmonella90. NDP52 action is nonredundatly strengthened by other SLRs recruited to Salmonella: Optineurin (partitioning with NDP52 in microdomains surrounding Salmonella) and Sequestosome 1/p62 (not partitioning with NDP52)27. These SLRs bind ubiquitin through their UBD domains with somewhat varied specificities for ubiquitin chains94, and bring via their conventional LIR motifs other mAtg8s (e.g. the remaining LC3s and GABARAPs) to execute autophagosome formation. The links of SLRs to LC3 and ubiqutin are further enhanced by phosphorylation of the LIR in Optineruin and the UBD in Seqeustosome 1/p62 by TBK-1. TBK-1 is recruited to these sites via binding to NDP5226, Optienurin27, and to Rab8b29, a small GTPases regulating membrane trafficking. To complete the gestalt, the cargo capture and autophagy is coordinated with maturation into autolysosomes by Rab8b29.
Other methods of targeting cytosolic microbes
Autophagy factors can directly target microbial proteins as in the case of Shigella56,58. Atg5 binds directly to a bacterial surface protein VirG (Fig. 3d-i). The Atg5-VirG interaction occurs only when the VirG recognition epitope is not masked by another Shigella protein – IcsB (Box 3), and requires an Atg5-binding partner Tecpr1 (Tectonin domain-containing protein) that interacts with WIPI-258. WIPI-2 is one of the four mammalian PI3P-binding Atg18 paralogs, and plays a role in phagophore formation. Of note, the control of icsB-mutant Shigella still requires Nod action (Nod1) to induce autophagy and limit intracellular growth52, illustrating the cooperative nature of different layers of autophagic defenses against a microorganism52,58. This is complemented by ubiquitin and SLR contribution to Shigella clearance47,88. In addition, cardiolipin unique to bacteria and mitochondria is targeted directly by autophagy factors96. Cardiolipin binds IRGM96, Beclin 197, and LC398 (Fig. 3d-ii).
SMURF1, a HECT domain E3 ligase, is required for antiviral response to Sindbis virus and HSV-199. Similarly to another E3 ligase Parkin, SMURF1 is involved in mitophagy99. Surprisingly, the SMURF1 HECT domain (and hence its E3 ligase activity) is dispensable for these activities (tested in mitophagy), and instead its phospholipid-binding domain C2 is necessary (Fig. 3d-iii). In keeping with membrane phospholipids or their derivatives as potential tags or signaling intermediates, diacylglycerol has also been implicated in bacterial autophagy48. Thus, the presence of an E3 ligase domain in an autophagy-targeting factor does not necessarily guarantee that it places ubiquitin tags on the cargo.
Second principal role: control of inflammation
Genetics links with inflammatory, autoimmune, and immunodeficiency disorders
The role of autophagy in inflammatory diseases has been initially established through genome-wide association studies (GWAS)74. The GWAS studies have exposed links between ATG16L1 and IRGM with Crohn’s disease (CD), a common form of inflammatory bowel disease (IBD)74, and have been replicated with exceptional reproducibility in numerous follow-up population studies. A recent GWAS analysis of 75,000 cases indicates an overlap between susceptibility loci for IBD and mycobacterial infections100. This expands a potential lead that autophagy as a process and IRGM as a genetic susceptibility locus play a role in both CD and tuberculosis (ref___). Other links have been reported between additional autophagy loci and genetic predispositions for chronic inflammatory disorders and autoimmune diseases (Fig. 1b(i)). A link between CD and a newly identified autophagy targeting factor for viral and mitochondrial clearance, SMURF199, has been reported100. Polymorphisms in the ULK1 gene have been linked to CD101. In addition to single nucleotide polymorphisms, IRGM is an example of gene dosage (copy number variants) correlation with predisposition to CD in human populations102. One of the common IRGM polymorphisms in CD leads to an escape from negative regulation of IRGM expression by a microRNA103. IRGM may be a factor in systemic lupus erythematosus (SLE)104. GWAS data link ATG5 variants to SLE105,106. Rheumatoid arthritis (RA) is associated with variations in the PRDM1-ATG5 intergenic region107. In a relationship to clinical markers of RA, autophagy promotes presentation of citrullinated antigens108, correlating with the presence of autoantibodies against citrullinated proteins (e.g. vimentin) as a hallmark of RA. Citrullin is enzymatically generated from arginine residues by peptidylarginine deaminase, an enzyme that may be enriched in auytophagosomes108 (Fig. 1c(v)). A new human autophagy locus, first functionally identified in C. elegans screens as epg5, has been linked to the complex Vici syndrome that includes immunodeficiency109. Thus, autophagy shows clinicall relevance through genetic links with immunological and inflammatory disorders.
Autophagy regulates innate immune response mediated by nucleic acid-sensing TLRs
Autophagic machinery can amplify PAMP-TLR signalling. For example, autophagy enhances delivery of cytosolic PAMPs to TLRs, as shown for TLR7, enabling recognition of cytosolic viral replication intermediates and IFN-α production by plasmacytoid dendritic cells (pDCs)60. The role of autophagy in PAMP-PRR amplification loops can backfire and lead to aberrations and autoimmunity. In the case of TLR9, normally responding to microbial unmethylated CpG DNA, autophagic machinery can enhance aberrant self-DNA presentation. This occurs via intracellular trafficking events following B cell receptor (BCR) stimulation with self DNA-containing antigens110. Autophagy promotes a merger of the DNA-containing antigen-loaded BCR compartment with the TLR9-containing endosomes and leads to B cell hyper-responses110 (Fig. 1b-viii-1). This phenomenon is mirrored in pDCs by a similar process akin to LAP, whereby the autophagic machinery enhances particulate self DNA-immune complexes and TLR9 trafficking to signaling-competent compartments resulting in increased IFN-α production19 (Fig. 1b-viii-2). These anomalies, spurred by autophagic machinery in two key cell types (B cells and pDCs) could synergize and lead to differentiation of autoimmune plasma and memory cells (Fig. 1b-viii 1+2).
Autophagy factors can downregulate RLR and type I IFN signalling
There are many examples (Fig. 1b-ii) where autophagy factors directly inhibit formation or suppress activation of proinflammatory protein complexes: Atg5-Atg12 complex negatively regulates RLR signalling by direct binding to CARD domains of RIG-I and IPS-1 (also known as MAVS, VISA, or CARDIF)84 (Fig. 1b-ii). Rubicon, a Beclin 1-hVPS34 interacting protein, inhibits CARD9, BCL10, and MALT1 (the CBM complex) by binding to CARD9, thus terminating RIG-I or Dectin-1 induced pro-inflammatory signaling111 (Fig. 1b-iii). Atg9 negatively controls trafficking of STING and suppresses activation of TBK-1 in type I interferon signalling in response to double stranded DNA112 (Fig. 1b-iv). Absence of autophagy amplifies RLR signaling via increased IPS-1 levels due to accumulation of mitochondria, whereas increased pools of depolarized mitochondria in the absence of mitophagy are a source of ROS that also enhance RLR outputs37 (Fig. 1b-v). NLRX1 promotes autophagy and inhibits RLR-dependent induction of type I IFN signalling and inflammation76 (Fig. 1b-vi). These phenomena may represent feedback loops whereby autophagy is positioned to downregulate type I IFN responses following a period of their productive induction or elevating the threshold for their activation (Fig. 1b-vii-1). Alternatively, autophagic interference with RLR signalling could reflect competition for limited resources such as TBK-1, shared between type I IFN signalling and autophagy pathways27–29,34 (Fig. 1b-vii-2). These phenomena, in addition to other independent factors not discussed here, could explain why autophagy paradoxically enhances replication of certain viruses.
Autophagy suppresses inflammasome activation
The functional recognition of anti-inflammatory function of autophagy has its roots in the findings of increased IL-1β and IL-18 production, and associated intestinal inflammation in a mouse model of CD with defective Atg16L1113. The inflammasomes are a group of cytosolic innate immunity complexes responding to PAMPs and endogenous alarmins/DAMPs by triggering proteolytic processing and secretion of pivotal pro-inflammatory mediators such as IL-1β and IL-18114. An inflammasome consists of pro-caspase 1, ASC adaptor, and one of the sensor proteins from the NLR family (e.g. NLRP1, NLRP3, NLRC4/IPAF, NLRP6 or NLRP12) or the PYHIN family (AIM2 or IF116)114. Upon recognition of an agonist (which can be microbial, endogenous, or an environmental agent), inflammasome components coalesce leading to capase-1-dependent pro-IL-1β processing in the cytosol into mature IL-1β and its secretion114. A number of convergent reports115–119 show that autophagy plays a negative role in inflammasome activation. Under sterile conditions, autophagy cleanses cytoplasm ridding the cellular interior of debris, aggregates, and defunct organelles that can act as endogenous inflammasome agonists. It turns out that basal level of autophagy control the set point for inflammasome activation115,116. If autophagy is blocked, this leads to accumulation of depolarized mitochondria that leak endogenous inflammasome agonists such as mitochondrial DNA (detected at least in part by AIM2) and ROS (activating NLRP3 inflammasome)115 116 (Fig. 1b-***iii). Additional studies suggest that pro-IL-1β or other inflammasome components may be substrates for autophagic degradation possibly as a failsafe mechanism to block unwarranted inflammasome activation117 (Fig. 1b-x ****). Importantly, autophagy may, following a period of physiological inflammasome activation, downregulate the process by removing aggregated inflammasome components119. This depends on Sequestosome 1/p62 recruitment to K63-ubiquitinated ASC119 (Fig. 1b-xi ****). A role for exocyst has been implicated in the overall process119, but it could be self-starting, since Sequestosome 1/p62 associates with the E3 ligase TRAF691 that ubiquitinates Beclin 1 to initiate autophagy120, although this has not been explored (Fig. 1b-xi ****). In sum, basal autophagy protects cells from inadvertent inflammasome activation as a source of damaging sterile inflammation whereas deficient autophagy causes increased IL-1β levels, in keeping with the IL-1β-based inflammation in an Atg16L1 murine model of CD (Fig. 1b-xii ****)113.
Autophagy suppresses calpain-dependent IL-1 α activation
Just like IL-1β, IL-1α is synthesized as a cytosolic proform. It is processed by calpain or other proteases and secreted actively from cells or released passively upon cell death121. Activation and secretion of IL-1α involve caspase 1-dependent and caspase 1-independent processes122. Autophagy-defective (Atg5fl/fl LysM-Cre+) macrophages generate excess IL-1α121. This phenotype has been observed both in vitro and in vivo28,121. Surplus active IL-1α secreted by autophagy deficient macrophage is inflammasome-independent and calpain-dependent121. Calpain-processing of pro-IL-1α is triggered by ROS released from accumulated depolarized mitochondria in autophagy-deficient cells121 (Fig. 1-xiii). The role of ROS from depolarized mitochondria is similar to inflammasome activation and IL-1β secretion in autophagy-deficient cells115. However, excess IL-1β released by Atg5fl/fl LysM-Cre+ macrophages is independent of caspase 1, NLRP3 or ASC121. Thus ROS in autophagy-defective cells leads to activation of both inflammasome and calpain pathways that lead to excessive release of IL-1β and IL-1α, respectively115 121. IL-1α released from Atg5-defective macrophages has important consequences in vivo, as it leads to enhanced and prolonged Th17 response in the context of M. tuberculosis infection, contributing to lung tissue damage in the murine model of tuberculosis121 (Fig 1c-(iii)).
Autophagy and degradation of pro-inflammatory signaling factors
Autophagy factors inhibit Bcl10-conaining complexes111 whereas autophagy degrades Bcl10123. Bcl10 is a CARD-domain member of L-CBM and M-CBM tripartite complexes transducing signals from ITAM-coupled receptors in lymphoid and myeloid immune cells. Autophagy degrades Bcl10 to reduce NFκB activation following TCR activation in effector T cells123. This occurs via Sequestosome 1/p62 association with K63-polyubiquitinated Bcl10 (Fig. 1b-iii-2). Bcl10-containing complexes are also inhibited upon binding of the autophagy regulator Rubicon to CARD9111 (Fig. 1b-iii-1). Since Rubicon is a negative regulator of autophagic degradation, its translocation to Bcl10 complexes accomplishes two things: it directly inhibits M-CBM signalling and at the same time may enhance their autophagic degradation (Fig. 1b-iii-3). NFκB signalling may also be downregulated by autophagy via p47/NSFL1C, a protein that possesses a UBD (UBA domain) and whose ortholog in yeast binds Atg8. This potential adaptor acts a negative regulator of IKK through lysosomal (presumably autophagic) degradation of polyubiquitinated NEMO (Fig. 1b-iii-4)124.
Third principal role: antigen presentation and lymphocyte homeostasis
Autophagy promotes MHC II presentation of cytosolic and phagocytosed antigens
One of the unique capabilities of autophagy is that it acts as a bulk “topological inverter”, by transporting proteins from the cytosol into the lumen of antigen processing compartments63,125,126 (Fig. 1c-(i)). This property can be further artificially enhanced by generating protein fusions (e.g. influenza matrix protein 1) with LC3 to enhance MHC II presentation to CD4 T cells127. The topological inversion principle is more general as it feeds cytosolic ligands to endosomal receptors such as viral RNA replication intermediates to lumenal TLR7 (Fig. 1b (viii-1) or Fig. 2a-(c)).
The contributions of autophagy to MHC II presentation are not restricted to cytosolic proteins and extend via LAP to exogenous antigens. Autophagy augments MHC II processing and presentation of phagocytosed extracellular particulate antigens, as shown with PRR (via LPS)-stimulated LAP of OVA-latex beads in dendritic cells (DCs)16 (Fig. 1c (ii)-a). PRR-enhancement of these process is also reflected in NOD2 stimulation with bacterial MDP, which induces autophagy and increases MHC class II presentation and generation of antigen-specific CD4+ T cell responses53 (Fig. 1c(ii)-b).
Autophagy role in MHC II presentation affects tolerance and adaptive immunity
Physiological effects of autophagy on MHC II presentation are broad in scope. An overarching role of autophagy in MHC II presentation is its proposed role in selection of naïve T cell repertoires (Fig. 1c (iii))126. In a comprehensive study addressing this topic126, both positive and negative selection of CD4 T cells but not CD8 T cells were affected in the autophagy-deficient thymus, with consequences for selection of appropriate TCR repertoires and elimination of autoreactive T cells. Transplantation of Atg5-defficient thymi caused autoreactive T cell infiltration of multiple organs and autoimmune colitis reminiscent of issues in CD126. These effects have been ascribed to defective cytosolic antigen presentation by autophagy-deficient thymic epithelial cells126. However, autophagy-dependent antigen presentation is also defective in DCs from CD patients expressing ATG16L1 or NOD2 disease risk variants53. All of these issues may be compounding the defective direct autophagic control of intestinal bacteria52,53. Contributions of autophagy to MHC II presentation in antimicrobial defense are experimentally detectable in the context of adaptive immunity against viral63,125 and bacterial72 infections. Related to the enrichment of peptidylarginine deaminase in autophagosomes108, autophagy promotes presentation of citrullinated antigens of relevance for autoreactivity (Fig. 1c(iv)). This process is constitutive in DCs and macrophages, but in B cells occurs only after BCR stimulation108 (Fig. 1c(iv)).
Autophagy downregulates signalling during antigen presentation
Autophagy helps disassemble immunological synapses (Fig. 1c-(v))128. Knocking down ATG16L1 and IRGM in DCs leads to hyperstable interactions with T cells and increase in T cell activation (Fig. 1c(v))128. This may be complemented at the T cell end by autophagic degradation of Bcl10 to downregulate TCR activation (Fig. 3b(iii)-2)123. Thus, although autophagy initially promotes MHC II antigen processing it later on may downregulate the response. Autophagy negatively regulates MHC I presentation by competing with proteasome for degradation of newly synthesized cytosolic proteins (Fig. 1c-(vi))129.
Autophagy role in T cell homeostasis
Autophagy in T cells affects them in both general homeostatic and specialized functional aspects (Fig. 1c(vii)-a). Autophagy and clearance of mitochondria are needed for normal hematopoietic stem cell maintenance and function130 affecting primarily lymphocytes but not the myeloid lineage16,60. Upon exit from the thymus, naïve T cells depend on autophagy for maturation including mitochondrial content reduction131. T cells require maintenance of an ER capable of Ca2+ homeostasis132. In the absence of autophagy, Ca2+ is sequestered in ER-like structures building up in the Atg7-deficient T cells. This curtails Ca2+ fluxes in response to physiological stimulation, e.g. TCR engagement132.
Autophagy and Th17 cell polarization
Autophagy affects cooperation between key immune cell types that lead to T cell polarization. Th17 phenotype is promoted by excessive secretion of IL-1 from autophagy-deficient macrophages121 (Fig. 1c(ix)). IL-17 is increased in the lungs of mice with Atg5 defect in myeloid cells during M. tuberculosis infection121. CD4+ T cells show increased IL-17 expression when lung leukocytes from mice with Atg5 deletion in the myeloid lineage are re-stimulated with mycobacterial antigens ex vivo121. This is based at least in part on excessive IL-1α release from autophagy-defective macrophage, and IL-1α’s ability combination with IL-16 and TGF-β to drive IL-17 production in CD+ T cells in121. Th17 response can aslo be enhanced by the increased durability of immunological synapses between T cells and autophagy-defective DCs121,128 (Fig. 1c(iii)).
Autophagy in plasma cells and humoral immunity
Although autophagy appears not to be important for survival of the majority of mature B cells133, lymphoid precursor stages are affected134. Autophagy is needed for survival of a subset of self-renewing cells (called B1) that cannot be replenished from the adult bone marrow133 (Fig. 1c(x)). Upon differentiation of B cell into plasma cells, autophagy plays a role in ER maintenance under conditions of high secretory demands. Somewhat paradoxically, absence of autophagy leads to runaway immunoglobulin secretion135. Autophagy is important in survival and homeostasis of plasma cells (Fig. 1c(xi)), especially the preservation of the bone marrow plasma cell pool, and thus is of significance for the maintenance of long-lived humoral immunity135.
Emerging fourth principal role: autophagy in secretion of immune mediators
Further afield from its intracellular domain of action, autophagy influences extracellular release of immunologically significant substrates (Fig. __). Autophagy supports regulated secretion of extracellular innate immunity effectors and mediators136–138, some of which are stored as pre-made secretory granules cargo. This is mirrored by the role of autophagy factors and LC3 in exocytosis of secretory lysosomes during osteoclastic bone resorption, possibly representing a form of LAP whereby LC3 helps lysosomes to fuse with the plasma membrane of the ruffled border139 in the resorption lacuna in place of the fully formed phagosomes. Defective secretion from Paneth cells in Atg16L1 hypomorph mice correlates with the corresponding intestinal phenotype in CD patients136. Autophagic processes in senescent cells affect secretion of IL-6 and IL-8 exported via the constitutive ER-to-Golgi secretory pathway140. Autophagy reduces immunoglobulin secretion from plasma cells, and thus appears to be a mechanism that prevents excessive antibody production in the body135.
Autophagy may contribute to unconventional secretion of cytosolic proteins with extracellular immune functions. The exact mechanism of how this occurs is not fully understood. IL-1β and IL-18 do not have signal peptides to enter the ER and follow the conventional secretory pathway (ER-Golgi-plasma membrane). Instead, they are delivered upon inflammasome activation to the extracellular environment via unconventional secretion. Although autophagy suppresses inflammasome activation under normal, e.g. nutrient-rich, conditions, autophagy can contribute to the unconventional secretion of IL-1β under stress, e.g. nutrient starvation118. It is possible that this represents a double-latch mechanism to repress inflammasome under basal conditions but increase temporarily its physiological output in response to infection or DAMPs/PAMPs. Autophagy contributes to the unconventional secretion of IL-1β118. This also extends to IL-18 and HMGB1118. The unconventional secretion of these immune mediators depends on Atg factors and on a specialized unconventional secretion regulator GRASP (GRASP55). GRASP is important not only in unconventional secretion but also affects the canonocal starvation-induced autophagy in mammalian cells118.
The secretory role of autophagy is still in its embryonic phase regarding understanding both the molecular and cellular mechanisms and its full biological scope. It nevertheless extends the sphere of autophagy influence from the intracellular space to the extracellular milieu, where it affects tissue organization, remodelling, and delivery of extracellular mediators of immunity and inflammation.
Conclusions
Autophagy is a bona fide immunological process permeating many aspects of innate and adaptive immunity. Autophagy may have indeed evolved as one of the first antimicrobial defenses available to eukaryotic cells, shaped early on in evolution from what may have arisen as a metabolic and quality control pathway. Autophagy has its own set of PRRs, the SLR adaptors, to eliminate invading microbes whereas pathogens have evolved strategies to evade autophagic capture. As evolution progressed, nearly all innate immunity systems such as conventional PRRs and inflammasomes have become integrated with autophagy. In the chordate lineage, this has further extended to adaptive immunity as best documented in mammalian systems. In humans, a failure in parts of the autophagic apparatus can lead to inflammatory, autoimmune or general immunity disorders. The present knowledge of immunological autophagy is still in its infancy, and many interesting puzzles and important questions remain. As IKKs and TBK-1 regulate autophagy, why is this often at cross-purposes with type I interferon inflammation? Is ubiqutin the primary tag in recognizing incoming microbes by SLRs, or is it a result of failure to detect them early on by some other means? How are the steps in recognizing microbial entry and their targeting for autophagy integrated via host membrane damage, E3 ligases, and protein and lipid kinases? Have eukaryotic cells trained on mitochondria to learn how to deal with microbes, or are mitochondria and the present day mitophagy the result of an evolutionary battle between autophagy and the bacterium endosymbiont precursor to mitochondria? The expansion of autophagy role via its effects on secretion to the extracellular space in inflammation and tissue remodelling is an important development. In this context, the relationship of autophagy with inflammasome and unconventional secretion needs further work. Finally, while early progress has been made in adaptive immunity roles of autophagy, this area beckons more study. Some of the questions recently opened in this area are at the interface between innate and adaptive immunity and include networks between different types of immune cells. This includes the role of autophagy in Th17 polarization and possibly other T cell subsets along with general lymphocyte homeostasis and differentiation. In summary, autophagy and immunity are fully integrated and our continuing study of this interface will be a fruitful area of scientific inquiry for many yers to come. In translational terms, autophagy is undoubtedly a target of opportunity for developing new treatments in inflammatory disorders and autoimmunity, and possibly as an anti-infective mechanism.
References
- 1.Mizushima N, Yoshimori T, Ohsumi Y. The role of atg proteins in autophagosome formation. Annual review of cell and developmental biology. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 2.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 3.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 4.Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013 doi: 10.1038/embor.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Talloczy Z, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Talloczy Z, Virgin HWt, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–29. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 8.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 9.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 10.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. doi:nature09782 [pii] 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deretic V. Autophagy in innate and adaptive immunity. Trends Immunol. 2005;26:523–528. doi: 10.1016/j.it.2005.08.003. doi:S1471-4906(05)00206-1 [pii] 10.1016/j.it.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Hamasaki M, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013 doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 13.Huang J, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106:6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol. 2012;22:R540–R545. doi: 10.1016/j.cub.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Sanjuan MA, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 16.Lee HK, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez J, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13:1335–1343. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henault J, et al. Noncanonical Autophagy Is Required for Type I Interferon Secretion in Response to DNA-Immune Complexes. Immunity. 2012;37:986–997. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vergne I, Chua J, Singh SB, Deretic V. Cell biology of mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol. 2004;20:367–394. doi: 10.1146/annurev.cellbio.20.010403.114015. [DOI] [PubMed] [Google Scholar]
- 21.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 22.Weidberg H, et al. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell. 2011;20:444–454. doi: 10.1016/j.devcel.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Tattoli I, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. 2012;11:563–575. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 24.Criollo A, et al. Inhibition of autophagy by TAB2 and TAB3. The EMBO journal. 2011;30:4908–4920. doi: 10.1038/emboj.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takaesu G, Kobayashi T, Yoshimura A. TGFbeta-activated kinase 1 (TAK1)-binding proteins (TAB) 2 and 3 negatively regulate autophagy. J Biochem. 2012;151:157–166. doi: 10.1093/jb/mvr123. [DOI] [PubMed] [Google Scholar]
- 26.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 27.Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilli M, et al. TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity. 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radtke AL, Delbridge LM, Balachandran S, Barber GN, O'Riordan MX. TBK1 protects vacuolar integrity during intracellular bacterial infection. PLoS Pathog. 2007;3:e29. doi: 10.1371/journal.ppat.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chien Y, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–170. doi: 10.1016/j.cell.2006.08.034. doi:S0092-8674(06)01155-X [pii] 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 32.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bodemann BO, et al. RalB and the Exocyst Mediate the Cellular Starvation Response by Direct Activation of Autophagosome Assembly. Cell. 2011;144:253–267. doi: 10.1016/j.cell.2010.12.018. doi:S0092-8674(10)01436-4 [pii] 10.1016/j.cell.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li S, Wang L, Berman M, Kong YY, Dorf ME. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity. 2011;35:426–440. doi: 10.1016/j.immuni.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe. 2012;12:334–345. doi: 10.1016/j.chom.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scherz-Shouval R, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. The EMBO journal. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tal MC, et al. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang CS, et al. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe. 2012;11:264–276. doi: 10.1016/j.chom.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarkar S, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maiuri MC, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. The EMBO journal. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. 2010;189:925–935. doi: 10.1083/jcb.201002021. doi:jcb.201002021 [pii] 10.1083/jcb.201002021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Current opinion in immunology. 2012;24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng YT, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 47.Dupont N, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–149. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 48.Shahnazari S, et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010;8:137–146. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Y, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. The EMBO journal. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 53.Cooney R, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. doi:nm.2069 [pii] 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 54.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–11383. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 55.Birmingham CL, et al. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature. 2008;451:350–354. doi: 10.1038/nature06479. [DOI] [PubMed] [Google Scholar]
- 56.Ogawa M, et al. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 57.Yoshikawa Y, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 58.Ogawa M, et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe. 2011;9:376–389. doi: 10.1016/j.chom.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 59.Orvedahl A, et al. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host and Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 60.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 61.Kyei GB, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. doi:jcb.200903070 [pii] 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gannage M, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blanchet FP, et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010;32:654–669. doi: 10.1016/j.immuni.2010.04.011. doi:S1074-7613(10)00160-3 [pii] 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Orvedahl A, et al. Autophagy Protects against Sindbis Virus Infection of the Central Nervous System. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shoji-Kawata S, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. doi:S1931-3128(09)00183-8 [pii] 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ku B, et al. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog. 2008;4:e25. doi: 10.1371/journal.ppat.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc Natl Acad Sci U S A. 2012;109:20800–20807. doi: 10.1073/pnas.1218674109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choy A, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012;338:1072–1076. doi: 10.1126/science.1227026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dortet L, et al. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog. 2011;7:e1002168. doi: 10.1371/journal.ppat.1002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ponpuak M, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010;32:329–341. doi: 10.1016/j.immuni.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jagannath C, et al. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15:267–276. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- 73.Yano T, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011;146:303–317. doi: 10.1016/j.cell.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lei Y, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. 2012;36:933–946. doi: 10.1016/j.immuni.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jounai N, et al. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol. 2011;186:1646–1655. doi: 10.4049/jimmunol.1001654. [DOI] [PubMed] [Google Scholar]
- 78.Burdette DL, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McFarlane S, et al. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J Virol. 2011;85:4212–4221. doi: 10.1128/JVI.02435-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rasmussen SB, et al. Activation of autophagy by alpha-herpesviruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on stimulator of IFN genes. J Immunol. 2011;187:5268–5276. doi: 10.4049/jimmunol.1100949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu J, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium Tuberculosis Activates the DNA-Dependent Cytosolic Surveillance Pathway within Macrophages. Cell Host Microbe. 2013;11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jounai N, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deretic V. Autophagy: an emerging immunological paradigm. J Immunol. 2012;189:15–20. doi: 10.4049/jimmunol.1102108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bjorkoy G, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kirkin V, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 88.Mostowy S, et al. p62 and NDP52 Proteins Target Intracytosolic Shigella and Listeria to Different Autophagy Pathways. J Biol Chem. 2011;286:26987–26995. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li S, Randow F. A crystal structure of an autophagy receptor with a ligand reveals how Salmonella is specifically targeted for degradation. Science Signaling. 2013 INn press. [Google Scholar]
- 90.von Muhlinen N, et al. LC3C, Bound Selectively by a Noncanonical LIR Motif in NDP52, Is Required for Antibacterial Autophagy. Mol Cell. 2012;48:329–342. doi: 10.1016/j.molcel.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang JQ, Liu H, Diaz-Meco MT, Moscat J. NBR1 is a new PB1 signalling adapter in Th2 differentiation and allergic airway inflammation in vivo. The EMBO journal. 2010;29:3421–3433. doi: 10.1038/emboj.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 Phosphorylation of p62/SQSTM1 Regulates Selective Autophagic Clearance of Ubiquitinated Proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 94.Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- 95.Huett A, et al. The LRR and RING Domain Protein LRSAM1 Is an E3 Ligase Crucial for Ubiquitin-Dependent Autophagy of Intracellular Salmonella Typhimurium. Cell Host Microbe. 2012;12:778–790. doi: 10.1016/j.chom.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Singh SB, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol. 2010;12:1154–1165. doi: 10.1038/ncb2119. doi:ncb2119 [pii] 10.1038/ncb2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang W, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012;22:473–489. doi: 10.1038/cr.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chu C, Kagan V. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in mammalian cells. Nat Cell Biol. 2013 doi: 10.1038/ncb2837. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Orvedahl A, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jostins L, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Henckaerts L, et al. Genetic variation in the autophagy gene ULK1 and risk of Crohn's disease. Inflammatory bowel diseases. 2011;17:1392–1397. doi: 10.1002/ibd.21486. [DOI] [PubMed] [Google Scholar]
- 102.Craddock N, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. doi:nature08979 [pii] 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brest P, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. doi:ng.762 [pii] 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 104.Ramos PS, et al. A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS genetics. 2011;7:e1002406. doi: 10.1371/journal.pgen.1002406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Harley JB, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Han JW, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 107.Raychaudhuri S, et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet. 2009;41:1313–1318. doi: 10.1038/ng.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med. 2011;208:2625–2632. doi: 10.1084/jem.20110640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cullup T, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013;45:83–87. doi: 10.1038/ng.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang CS, et al. The autophagy regulator Rubicon is a feedback inhibitor of CARD9-mediated host innate immunity. Cell Host Microbe. 2012;11:277–289. doi: 10.1016/j.chom.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saitoh T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. doi:0911267106 [pii] 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 114.Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012;13:333–332. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 116.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Harris J, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dupont N, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. The EMBO journal. 2011;30:4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42. doi: 10.1126/scisignal.2000751. doi:3/123/ra42 [pii] 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Castillo EF, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109:E3168–E3176. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gross O, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 123.Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity. 2012;36:947–958. doi: 10.1016/j.immuni.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shibata Y, et al. p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nat Commun. 2012;3:1061. doi: 10.1038/ncomms2068. [DOI] [PubMed] [Google Scholar]
- 125.Paludan C, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 126.Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400. doi: 10.1038/nature07208. [DOI] [PubMed] [Google Scholar]
- 127.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wildenberg ME, et al. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology. 2012;142:1493–1503. e1496. doi: 10.1053/j.gastro.2012.02.034. [DOI] [PubMed] [Google Scholar]
- 129.Wenger T, et al. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI-restricted presentation. Autophagy. 2012;8:350–363. doi: 10.4161/auto.18806. [DOI] [PubMed] [Google Scholar]
- 130.Mortensen M, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–467. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 132.Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol. 2011;186:1564–1574. doi: 10.4049/jimmunol.1001822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Miller BC, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 134.Arsov I, et al. A role for autophagic protein beclin 1 early in lymphocyte development. J Immunol. 2011;186:2201–2209. doi: 10.4049/jimmunol.1002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pengo N, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. 2013 doi: 10.1038/ni.2524. [DOI] [PubMed] [Google Scholar]
- 136.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ushio H, et al. Crucial role for autophagy in degranulation of mast cells. The Journal of allergy and clinical immunology. 2011;127:1267–1276. e1266. doi: 10.1016/j.jaci.2010.12.1078. [DOI] [PubMed] [Google Scholar]
- 138.Michaud M, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 139.DeSelm CJ, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21:966–974. doi: 10.1016/j.devcel.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Narita M, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–970. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]