

SUMMARY
The syndrome of inappropriate antidiuretic hormone secretion (SIADH) is a common cause of hyponatremia. We describe two infants whose clinical and laboratory evaluations were consistent with the presence of SIADH, yet who had undetectable arginine vasopressin (AVP) levels. We hypothesized that they had gain-of-function mutations in the V2 vasopressin receptor (V2R). DNA sequencing of each patient’s V2R gene (AVPR2) identified missense mutations in both, with resultant changes in codon 137 from arginine to cysteine or leucine. These novel mutations cause constitutive activation of the receptor and are the likely cause of the patients’ SIADH-like clinical picture, which we have termed “nephrogenic syndrome of inappropriate antidiuresis.”
Fluid homeostasis depends on proper water intake, governed by an intact thirst mechanism, and on urinary excretion of free water, mediated by appropriate secretion of arginine vasopressin (AVP) (also known as antidiuretic hormone).1 AVP exerts its antidiuretic action by binding to the V2 vasopressin receptor (V2R), a G protein-coupled receptor, on the basolateral membrane of epithelial cells in the collecting duct of the kidney. Ligand binding activates the V2R, stimulating adenylate cyclase by means of Gs proteins. The resulting increase in intracellular cyclic AMP (cAMP) promotes shuttling of intracellular vesicles containing the water channel aquaporin-2 to the apical membrane of the collecting-duct cells, thereby increasing water permeability and inducing antidiuresis.
Clinical disorders of water balance are common, and alterations in many steps of this pathway have been described.1 Urinary concentrating defects associated with diabetes insipidus may result from a deficiency of AVP or from nephrogenic causes, such as X-linked, inactivating mutations in the V2R or autosomal recessive or autosomal dominant lesions in aquaporin-2.2 Conversely, the syndrome of inappropriate antidiuretic hormone secretion (SIADH) manifests as an inability to excrete a free water load, with inappropriately concentrated urine and resultant hyponatremia, hypo-osmolality, and natriuresis. SIADH occurs in the setting of euvolemia, without evidence of renal disease or thyroxine or cortisol deficiency. Though usually transient, SIADH may be chronic; it is often associated with drug use or a lesion in the central nervous system or lung. When the cardinal features of SIADH were defined by Bartter and Schwartz,3 AVP levels could not be measured. Subsequently, radioimmunoassays have revealed that SIADH is usually associated with measurably elevated serum levels of AVP.
We describe two unrelated male infants whose clinical presentation was consistent with the presence of chronic SIADH but who had undetectable AVP levels. We postulated that novel activating mutations of the V2R might account for their unique presentation. Evaluation revealed novel activating mutations of the V2R leading to what we term “nephrogenic syndrome of inappropriate antidiuresis” (NSIAD).
CASE REPORTS
Patient 1 presented at 3 months of age with irritability, and Patient 2 presented at 2.5 months of age with two generalized seizures. Both children had had unremarkable early neonatal courses. Both were exclusively bottle-fed formula (7 mmol of sodium per liter). Both infants had mild systolic hypertension with otherwise normal physical examinations. Initial laboratory evaluations demonstrated hyponatremia with normal serum levels of potassium and bicarbonate (Table 1). Both children had serum hypo-osmolality with inappropriately elevated urinary osmolality and urinary sodium levels. Both had low blood urea nitrogen and low or low- normal serum creatinine levels, low or suppressed plasma renin activity, and normal aldosterone levels, indicating euvolemia. Serum cortisol, thyroid- function tests, and coagulation studies were all normal. Imaging studies of the head and chest were unremarkable, with the exception of a small pars intermedia cyst in Patient 2. Despite clinical and laboratory presentations consistent with the presence of SIADH, serum AVP levels were undetectable in both patients (Quest Diagnostics Nichols Institute). Both children were initially treated with fluid restriction, followed by the administration of an osmotic agent (urea), resulting in increased urinary output and normalization of the serum sodium level.
Table 1.
Characteristics of the Two Patients.*
| Characteristic | Patient 1 | Patient 2 | Age-Matched Controls |
|---|---|---|---|
| Age at presentation (mo) | 3.0 | 2.5 | — |
| Clinically significant findings | Irritability | Generalized seizures | — |
| Blood pressure (mm Hg) | |||
| Systolic | 80–128 | 93–118 | 108† |
| Diastolic | 34–92 | 37–57 | 66† |
| Serum and plasma studies | |||
| Sodium (mmol/liter) | 123 | 118 | 134–143 |
| Potassium (mmol/liter) | 4.6 | 4.7 | 3.4–4.9 |
| Chloride (mmol/liter) | 91 | 86 | 98–107 |
| Bicarbonate (mmol/liter) | 28 | 21 | 23–32 |
| Creatinine (mg/dl) | <0.3 | 0.3 | 0.3–0.7 |
| Urea nitrogen (mg/dl) | <5 | 3 | 8–23 |
| Aldosterone (ng/dl) | 10 | 24‡ | 6–68 |
| Plasma renin activity (ng of angiotensin 1/ml/hr) | 3.8 | <1‡ | <15 |
| Osmolality (mOsm/kg) | 252 | 247 | 285–293 |
| AVP (pg/ml) | <1 | <1‡ | 1.0–13.3 |
| Thyrotropin (mIU/liter) | 0.77 | 4.58 | 1.7–9.1 |
| Free thyroxine (ng/dl) | 1.09 | 0.97 | 0.8–1.8 |
| Cortisol (μg/dl) | |||
| Baseline | 13.3 | 10.3, 43§ | 4–20 |
| 60 Min after intravenous synthetic corticotropin (15μg/kg) | 36.5 | — | >20 |
| Coagulation studies | |||
| von Willebrand factor antigen (%) | 104 | 100 | 46–155 |
| Ristocetin cofactor (%) | 108 | 65 | 56–155 |
| Factor VIII activity (%) | 153 | 123 | 50–150 |
| Fibrinogen (mg/dl) | 198 | 360 | 170–435 |
| D-Dimer fragment | Negative | Negative | Negative |
| Prothrombin time (sec) | 11.7 | 10.9 | 9.2–11.9 |
| Partial-thromboplastin time (sec) | 24.3 | 32.5 | 21.3–34.8 |
| Urine studies | |||
| Osmolality (mOsm/kg) | 284 | 390‡ | 300–900 |
| Sodium (mmol/liter) | 35 | 75‡ | — |
| Magnetic resonance image of the head | Normal | Pars intermedia cyst, otherwise normal | — |
| Chest radiograph | Normal | Normal | — |
To convert values for creatinine to micromoles per liter, multiply by 88.4. To convert values for urea nitrogen to millimoles per liter, multiply by 0.357. To convert values for aldosterone to nanomoles per liter, multiply by 0.0277. To convert values for AVP to picomoles per liter, multiply by 0.923. To convert values for free thyroxine to picomoles per liter, multiply by 12.87. To convert values for cortisol to nanomoles per liter, multiply by 27.59. To convert values for fibrinogen to micromoles per liter, multiply by 0.0294.
Measurement is given for the 95th percentile.
The value was obtained during an episode of hyponatremia after initial presentation.
Two baseline levels were obtained on separate occasions.
Neither child had a family history of hyponatremia or of an SIADH-like syndrome. Both patients had no siblings. The mother of Patient 1, subsequently found to be heterozygous for an activating mutation of AVPR2 (see below), had normal simultaneous serum sodium levels (140 mmol per liter) and serum and urine osmolality (293 and 795 mOsm per kilogram of water, respectively).
The parents of the patients provided written informed consent for the publication of the case reports through a protocol approved by the institutional review board of the University of California at San Francisco.
METHODS
MUTATION ANALYSIS
Genomic DNA from the two patients and their mothers was isolated from whole blood with the use of the Puregene Blood Kit (Gentra Systems). The entire coding region of the V2R gene, AVPR2, was amplified as described previously.4 The resulting amplicons were sequenced with multiple forward and reverse primers with the use of Big Dye (version 3.1) sequencing chemistry and an ABI Prism sequencer (model 3100, Applied Biosystems) according to the manufacturer’s protocols. SeqScape software (Applied Biosystems) was used to assemble the sequence data and compare the results with the AVPR2 reference sequence (GenBank accession number NT 025965).
CONSTRUCTION OF VASOPRESSIN EXPRESSION CONSTRUCTS
pCDNA3 (Invitrogen) was used as the control plasmid. Human wild-type V2R complementary DNA (cDNA),5 subcloned into pCDNA3, was used as a normal control and template for mutagenesis. Site- directed mutagenesis was performed with the use of a QuickChange II site-directed mutagenesis kit (Stratagene). Mutations were confirmed by direct sequencing.
CELL CULTURE AND TRANSIENT TRANSFECTION
For functional studies, COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium with 10 percent fetal-calf serum and antibiotics. Cells were plated in six-well plates (Falcon 3046, Becton Dickinson) at approximately 95 percent confluence 24 hours before transfection with the use of Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Each well received 3 μg of plasmid DNA and 500 ng of a cAMP-responsive luciferase reporter plasmid (pCREluc) containing 16 copies of the consensus cAMP response element.6 To control for transfection efficiency, cells were cotransfected with 50 ng of renilla luciferase reporter plasmid (pRL-CMV, Promega) per well. Cells were incubated at 37°C in 5 percent carbon dioxide for 24 hours after transfection and were then lysed and assayed for luciferase activity with the use of the Dual Luciferase Reporter Assay System (Promega), as described previously.7,8 Data are presented as mean (±SE) luciferase activity expressed in arbitrary units and adjusted for renilla luciferase activity in three experiments, each performed in triplicate. All five types of samples (pCDNA3 vector alone and vector with wild-type V2R, the R137C mutant, the R137L mutant, or the R137H mutant) were run simultaneously under the same conditions during each of the three experiments.
RESULTS
Genomic DNA was isolated from both boys and their mothers, and the V2R gene, AVPR2, was sequenced directly. AVPR2 is X-linked, and therefore, paternal DNA is not informative. Each patient carried a mutation in codon 137 of AVPR2. In Patient 1, nucleotide 770 was mutated from cytosine to thymine, changing arginine to cysteine at codon 137 (R137C); in Patient 2, nucleotide 771 was mutated from guanine to thymine, changing arginine to leucine at codon 137 (R137L) (Fig. 1A). Arginine 137 maps to the predicted second cytoplasmic loop, near the cytoplasmic boundary of the third transmembrane domain; this same amino acid is mutated to histidine (R137H) in a form of familial nephrogenic diabetes insipidus (Fig. 1B). The mother of Patient 1 was heterozygous for the R137C mutation, whereas the mother of Patient 2 was homozygous for wild-type AVPR2, suggesting that Patient 2 had a spontaneous mutation.
Figure 1. Nucleotide Sequence of the Wild-Type and Two Mutant AVPR2 Genes in the Affected Region (Panel A) and Diagram of V2R (Panel B).
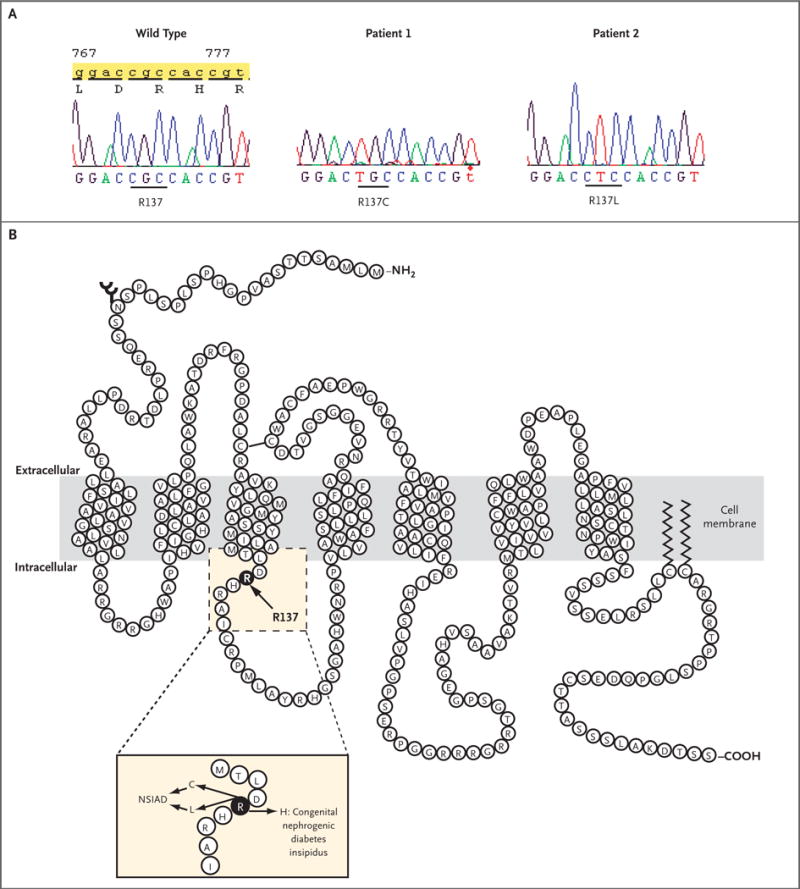
In Panel A, the normal CGC sequence encoding R137 is changed to TGC (R137C) in Patient 1 and to CTC (R137L) in Patient 2. In Panel B, R137 is indicated. The inset highlights changes in R137 that result in either congenital nephrogenic diabetes insipidus or NSIAD.
To evaluate the effect of these novel mutations on V2R function, we developed a functional assay for V2R. Production of cAMP has previously been used to assess many G protein-coupled receptors, including V2R9; we adopted our previous procedures8 to use with V2R. COS-7 cells transiently transfected with the vector alone, wild-type V2R, or the R137H nephrogenic diabetes insipidus mutant induced low levels of cAMP (Fig. 2). However, basal levels of cAMP production in cells expressing V2R with the R137C mutation were four times the levels in cells expressing wild-type V2R (P=0.01), and cells expressing the R137L mutant had 7.5 times the level of activity of cells expressing wildtype V2R (P<0.004) (Fig. 2). These results indicate that these novel mutations create a constitutively active V2R and provide an explanation for the hyponatremia with increased urinary osmolality in our patients. The condition in both patients is clinically similar to SIADH, despite the fact that AVP levels were undetectable.
Figure 2. Basal Levels of cAMP Production in Cells Expressing Wild-Type and Mutant V2R.
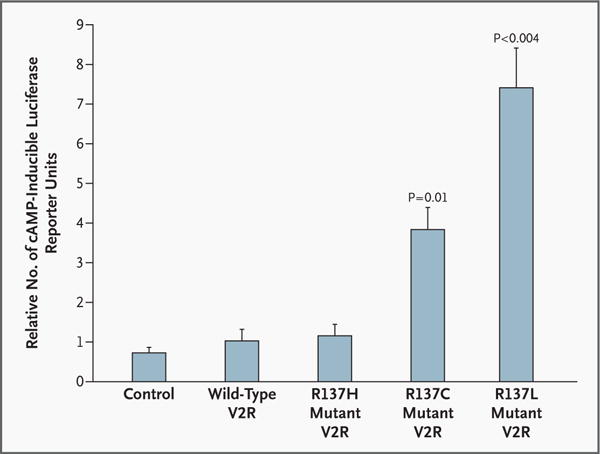
The ability of the mutant V2R from each patient to induce cAMP, as measured by a cAMP-inducible luciferase reporter, was evaluated. Each bar represents cells transfected with the renilla luciferase expression vector, the cAMP-inducible luciferase reporter plasmid, and pCDNA3 either alone (control), reflecting basal cAMP levels in the cells, or with the following inserts subcloned into the pCDNA3 plasmid: wild-type V2R; R137H mutant V2R, found in some patients with nephrogenic diabetes insipidus; R137C mutant V2R, found in Patient 1; or R137L mutant V2R, found in Patient 2. Each value represents the mean (±SE) of three independent experiments with all five samples run simultaneously, each performed in triplicate.
DISCUSSION
G protein-coupled receptors constitute the largest gene family of receptors involved in signal transduction and are responsible for regulating many physiological processes.10 Many diseases are caused by mutations in G protein-coupled receptors.11,12 For some G protein-coupled receptors, a particular disease state has been ascribed to inactivating mutations that render the receptors unresponsive to ligand, whereas a converse condition has been linked to gain-of-function mutations, resulting in constitutive activation.
Although many different inactivating mutations of the X-linked V2R have been described that cause nephrogenic diabetes insipidus,2 to our knowledge, no naturally occurring activating mutations of the V2R have been reported previously. The two cases described here are characterized by chronic SIADH but with undetectable AVP levels, constituting a novel example of gain-of-function mutations caused by a hemizygous V2R mutation. In the light of these findings, we suggest referring to all SIADH-like conditions as syndromes of inappropriate antidiuresis (SIAD) and that these two case reports constitute a subtype, NSIAD. To our knowledge, these cases are the only reported examples in which mutations affecting the same amino acid cause two different genetic diseases: R137H causes nephrogenic diabetes insipidus, and R137L and R137C cause NSIAD (Fig. 1B).
The mechanism by which these missense mutations constitutively activate the V2R requires further investigation. V2R, a class 1b G protein-coupled receptor,12,13 exists in the plasma membrane in equilibrium with inactive and active conformations.10 The binding of ligand shifts the equilibrium to the active state, permitting coupling with intracellular G proteins and activation of intracellular effectors. With activation, V2R is desensitized through phosphorylation by specific G protein-coupled receptor kinases. Subsequent recruitment of β arrestin to the phosphorylated receptor terminates the signal by blocking further interaction with G proteins and also initiates receptor internalization through its ability to bind clathrin and other en- docytic adapters.14
By comparison with other class 1 G proteincoupled receptors, the highly conserved motif of aspartic acid, arginine, and tyrosine or histidine (DRY/H) in V2R at the junction of the third transmembrane domain and second intracellular loop appears to be critical for receptor function. The arginine residue in the DRY/H motif appears to be invariant, though the aspartic acid and tyrosine may be replaced by other amino acids without altering the function (e.g., from aspartic acid to glutamic acid).15 The arginine residue in the DRY/H motif corresponds to R137, the site of the inactivating R137H mutation in nephrogenic diabetes insipidus and the activating R137C and R137L mutations in NSIAD. The R137H mutant behaves as a constitutively desensitized receptor, since it is phosphorylated, binds to β arrestin (thereby blocking its ability to activate G proteins), and is sequestered in intracellular vesicles.16
Constitutively activated mutant G protein-coupled receptors have also been created by altering this DRY/H motif17—for example, through in vitro mutation of aspartate to alanine at codon 136 in the V2R.18 The NSIAD mutations found in this domain may stabilize the receptor in an active conformation, activating G proteins and downstream signaling events in the absence of ligand. In theory, such a gain of function could also affect many other aspects of V2R biology, including phosphorylation, internalization, down-regulation, and recycling to the cell membrane.
The effects of the constitutive activation of V2R may not be limited to the kidney. V2R is also expressed in endothelial cells, where it appears to mediate vasodilation after the administration of the vasopressin analogue desmopressin19,20 and to mediate the rise in circulating levels of von Willebrand factor and tissue plasminogen activator.21 We thought that these responses, which are absent in patients with nephrogenic diabetes insipidus,22 might be constitutively activated in our patients with NSIAD. However, we found no clinical or laboratory evidence of coagulopathy in either patient (Table 1).
The frequency of NSIAD is not known, but it may not be rare. Previous studies of patients with SIADH have noted variable patterns of AVP secretion, particularly in response to water loading and water restriction.23 As many as 10 to 20 percent of affected patients have AVP levels at or below the limits of detection by radioimmunoassay. Thus, some of these patients may in fact have NSIAD due to V2R-activating mutations. Such patients would probably have clinical presentations similar to those in our patients, although the severity and clinical course may depend on the nature of the mutation. Patients with NSIAD would be expected to have low AVP levels. However, since the AVP assay is not optimized to identify low values, we recommend sequencing the V2R gene to identify specific mutations before the diagnosis of NSIAD is finalized. Other patients may be identified who have the NSIAD phenotype but without V2R mutations, suggesting the presence of additional defects in this signaling cascade. One such possibility would be an activating mutation in aquaporin-2.
Treatment of NSIAD poses a challenge. Water restriction improved serum sodium levels and osmolality in both infants but limited calorie intake in these formula-fed infants. Agents that act downstream from the V2R, such as demeclocycline or lithium, might antagonize the constitutively activated receptors, but they have potentially limiting adverse effects. AVP antagonists are under clinical development but would probably be ineffective, given the ligand-independent nature of the lesion.24 Ideally, one might be able to use an inverse agonist that would suppress receptor activity in the absence of agonist; two potential nonpeptide V2R inverse agonists have been studied in vitro.18 In the absence of a definitive therapy, we have successfully treated our patients with urea to induce an osmotic diuresis.25 This approach has occasionally been used for the treatment of chronic SIADH in adults.26
Study of the V2R has been important for understanding the physiology of water balance and has served as a prototype for G protein-coupled receptor biology. Further characterization of NSIAD may offer additional insights into fluid homeostasis and clinical disease, as well as expand our understanding of G protein-coupled receptor signaling.
Acknowledgments
Supported in part by a training grant (T32DK07161, to Drs. Feldman and Huang) and a grant (M01RR01271, to the Pediatric Clinical Research Center) from the National Institutes of Health.
Dr. Rosenthal reports having received grant support and speaking honoraria from Pfizer and having served on an advisory board for Tercica and Pfizer. Dr. Vargas reports having received grant support from Wyeth and Janssen. Dr. Lustig reports having received lecture fees from Novo Nordisk and grant support from Novartis. Dr. Mathias reports having equity interest in AstraZeneca, Pfizer, Merck, and SIRNA Therapeutics; receiving grant support from Satellite Healthcare and Genentech; and receiving speaking honoraria from Shire. Dr. Fenwick is employed by Quest Diagnostics, owns stock in the company, and developed the clinical assay used for sequencing AVPR2 in this study.
We are indebted to Ian Ocrant and Kamer Tezcan for referring the patients; to Mariel Birnbaumer (National Institute of Environmental Health Sciences, Division of Intramural Research) for sharing the V2R cDNA; to Christian Vaisse (University of California at San Francisco) for the cAMP-responsive luciferase reporter plasmid; to Mark von Zastrow (University of California at San Francisco), Jon Nakamoto (Quest Diagnostics), and Hillel Gitelman for thoughtful discussions and review of the manuscript; and to Izabella Damm (University of California at San Francisco) for technical support.
References
- 1.Robertson GL. Antidiuretic hormone: normal and disordered function. Endocrinol Metab Clin North Am. 2001;30:671–94. doi: 10.1016/s0889-8529(05)70207-3. [DOI] [PubMed] [Google Scholar]
- 2.Morello JP, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001;63:607–30. doi: 10.1146/annurev.physiol.63.1.607. [DOI] [PubMed] [Google Scholar]
- 3.Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42:790–806. doi: 10.1016/0002-9343(67)90096-4. [DOI] [PubMed] [Google Scholar]
- 4.Bichet DG, Arthus MF, Lonergan M, et al. X-linked nephrogenic diabetes insipidus mutations in North America and the Hopewell hypothesis. J Clin Invest. 1993;92:1262–8. doi: 10.1172/JCI116698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birnbaumer M, Gilbert S, Rosenthal W. An extracellular congenital nephrogenic diabetes insipidus mutation of the vasopressin receptor reduces cell surface expression, affinity for ligand, and coupling to the Gs/adenylyl cyclase system. Mol Endocrinol. 1994;8:886–94. doi: 10.1210/mend.8.7.7984150. [DOI] [PubMed] [Google Scholar]
- 6.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocor-tin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253–62. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stables J, Scott S, Brown S, et al. Development of a dual glow-signal firefly and Renilla luciferase assay reagent for the analysis of G-protein coupled receptor signalling. J Recept Signal Transduct Res. 1999;19:395–410. doi: 10.3109/10799899909036660. [DOI] [PubMed] [Google Scholar]
- 8.Fluck CE, Martens JW, Conte FA, Miller WL. Clinical, genetic, and functional characterization of adrenocorticotropin receptor mutations using a novel receptor assay. J Clin Endocrinol Metab. 2002;87:4318–23. doi: 10.1210/jc.2002-020501. [DOI] [PubMed] [Google Scholar]
- 9.Rosenthal W, Antaramian A, Gilbert S, Birnbaumer M. Nephrogenic diabetes insipidus: a V2 vasopressin receptor unable to stimulate adenylyl cyclase. J Biol Chem. 1993;268:13030–3. [PubMed] [Google Scholar]
- 10.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–50. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 11.Spiegel AM, Weinstein LS. Inherited diseases involving G proteins and G proteincoupled receptors. Annu Rev Med. 2004;55:27–39. doi: 10.1146/annurev.med.55.091902.103843. [DOI] [PubMed] [Google Scholar]
- 12.Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wildtype receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- 13.Bockaert J, Pin JP. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 1999;18:1723–9. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–65. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 15.Probst WC, Snyder LA, Schuster DI, Brosius J, Sealfon SC. Sequence alignment of the G-protein coupled receptor superfamily. DNA Cell Biol. 1992;11:1–20. doi: 10.1089/dna.1992.11.1. [DOI] [PubMed] [Google Scholar]
- 16.Barak LS, Oakley RH, Laporte SA, Caron MG. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc Natl Acad Sci U S A. 2001;98:93–8. doi: 10.1073/pnas.011303698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parnot C, Miserey-Lenkei S, Bardin S, Corvol P, Clauser E. Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol Metab. 2002;13:336–43. doi: 10.1016/s1043-2760(02)00628-8. [DOI] [PubMed] [Google Scholar]
- 18.Morin D, Cotte N, Balestre MN, et al. The D136A mutation of the V2 vasopressin receptor induces a constitutive activity which permits discrimination between antagonists with partial agonist and inverse agonist activities. FEBS Lett. 1998;441:470–5. doi: 10.1016/s0014-5793(98)01585-3. [DOI] [PubMed] [Google Scholar]
- 19.Hirsch AT, Dzau VJ, Majzoub JA, Creager MA. Vasopressin-mediated forearm vasodilation in normal humans: evidence for a vascular vasopressin V2 receptor. J Clin Invest. 1989;84:418–26. doi: 10.1172/JCI114182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaufmann JE, Iezzi M, Vischer UM. Desmopressin (DDAVP) induces NO production in human endothelial cells via V2 receptor- and cAMP-mediated signaling. J Thromb Haemost. 2003;1:821–8. doi: 10.1046/j.1538-7836.2003.00197.x. [DOI] [PubMed] [Google Scholar]
- 21.Mannucci PM, Ruggeri ZM, Pareti FI, Capitanio A. 1-Deamino-8-d-arginine vasopressin: a new pharmacological approach to the management of haemophilia and von Willebrand’s diseases. Lancet. 1977;1:869–72. doi: 10.1016/s0140-6736(77)91197-7. [DOI] [PubMed] [Google Scholar]
- 22.Bichet DG, Razi M, Lonergan M, et al. Hemodynamic and coagulation responses to 1-desamino[8-D-arginine] vasopressin in patients with congenital nephrogenic diabetes insipidus. N Engl J Med. 1988;318:881–7. doi: 10.1056/NEJM198804073181403. [DOI] [PubMed] [Google Scholar]
- 23.Zerbe R, Stropes L, Robertson G. Vasopressin function in the syndrome of inappropriate antidiuresis. Annu Rev Med. 1980;31:315–27. doi: 10.1146/annurev.me.31.020180.001531. [DOI] [PubMed] [Google Scholar]
- 24.Verbalis JG. Vasopressin V2 receptor antagonists. J Mol Endocrinol. 2002;29:1–9. doi: 10.1677/jme.0.0290001. [DOI] [PubMed] [Google Scholar]
- 25.Huang EA, Geller DH, Gitelman SE. The use of oral urea in the treatment of chronic syndrome of inappropriate antidiuretic hormone secretion (SIADH) in children. Pediatr Res. 2004;55:161A. abstract. [Google Scholar]
- 26.Decaux G, Prospert F, Penninckx R, Namias B, Soupart A. 5-Year treatment of the chronic syndrome of inappropriate secretion of ADH with oral urea. Nephron. 1993;63:468–70. doi: 10.1159/000187255. [DOI] [PubMed] [Google Scholar]