

Abstract
Open sharing of clinical genetic data promises to both monitor and eventually improve the reproducibility of variant interpretation among clinical testing laboratories. A significant public data resource has been developed by the NIH ClinVar initiative, which includes submissions from hundreds of laboratories and clinics worldwide. We analyzed a subset of ClinVar data focused on specific clinical areas and we find high reproducibility (>90% concordance) among labs, although challenges for the community are clearly identified in this dataset. We further review results for the commonly tested BRCA1 and BRCA2 genes, which show even higher concordance, although the significant fragmentation of data into different silos presents an ongoing challenge now being addressed by the BRCA Exchange. We encourage all laboratories and clinics to contribute to these important resources.
1. Background
1.1. Clinical genetic testing
Clinical genetic tests of germline DNA are routinely used to direct patient care in oncology, cardiology, neurology, pediatrics, obstetrics, and other clinical specialties. Excitement surrounds the future of medical genetics, which will likely involve routine and proactive sequencing of patient genomes or exomes. However, even today genetics is used pervasively: over one million clinical genetic tests will be performed in 2016 to inform various pressing medical decisions facing doctors and patients. This number is considerably larger if tests for infectious disease and tumors (somatic testing) are included. Such testing is regulated, often paid for by private insurance and public health systems, and written into many current clinical care guidelines established by payers and medical professional societies.
It is not glib to say that many of these tests are ordered in life-or-death situations. One example is BRCA1 and BRCA2 (collectively, BRCA1/2) tests, where erroneous results can have substantial deleterious consequences for patients. With a false positive, a radical preventative procedure such as prophylactic bilateral oophorectomy may be indicated, thereby causing an otherwise healthy woman to enter premature menopause and to face the multiple health risks associated with that procedure and with the hormone replacement therapy that often follows. Prophylactic chemotherapy (specifically, tamoxifen) is another option offered to some healthy BRCA1/2 carriers, with significant side effects. Conversely a false negative could eliminate the chance to prevent a fatal early-onset carcinoma. Such errors are either analytic (reporting a variant to be present in a patient when it is not, or vice versa) or interpretive (concluding that a variant is pathogenic [disease causing] when it is not, or vice versa). This paper focuses on the latter subject.
1.2. Clinical variant interpretation
In response to concerns about reproducibility among laboratories, the American College of Medical Genetics (ACMG) and the Association for Molecular Pathology (AMP) jointly developed revised guidelines for clinical variant interpretation [Richards 2015]. These guidelines require laboratory directors to scrutinize the literature and all other available evidence for each variant observed in a patient. The guidelines provide a structured framework for which evidence is weighed in final interpretations. Under these guidelines, variants are classified as pathogenic (P), likely pathogenic (LP), variants of uncertain significance (VUS), likely benign (LB), or benign (B). Despite the significant improvement in standardization that these new guidelines represent compared with their predecessor, laboratory directors must still use a significant degree of expert judgment, which can result in different classifications from different laboratories for the same variant. Date also matters: classifications that pre-date availability of an important piece of evidence should indeed be different than those that post-date it.
1.3. Data sharing and clinical genetics
Of course, the first step toward achieving reproducibility is measuring reproducibility, which requires data to be shared among clinical labs. The sharing of genetic data from research projects has long been accepted and encouraged (despite being incompletely implemented). Unfortunately, the open sharing of de-identified clinical genetic data has been far less common owing to a combination of informed consent issues, the commercial interests of certain healthcare providers, and the lack of a community mechanism for doing so.
Recently, the National Institutes of Health established ClinVar, “a freely available archive for interpretations of clinical significance of variants for reported conditions” [Landrum 2016]. By storing only individual variants and classifications, the re-identification of patients whose genotypes are submitted to ClinVar becomes essentially impossible, at least without an independent test of the same variant in the same patient for comparison (in which case, the patient's genotype is already known). Thus, fully de-identified clinical genetic data can be disclosed publicly under US laws and regulations. The American Medical Association (AMA) and National Society of Genetic Counselors (NSGC), among others, have issued recommendations urging laboratories to share such data.
Some commercial and academic laboratories have, unfortunately, declined to participate. Most famously, Myriad Genetics, the largest BRCA1/2 testing laboratory in the world, has maintained its large genetic database as a proprietary asset [Cook-Deegan 2013]. Moreover, Myriad claims that by leveraging this database, it can deliver superior variant classifications compared to other labs [Angrist 2014]. This stands in sharp contrast with the American Medical Association and the National Society for Genetic Counselors recommendations. It also is inconsistent with accepted practice in many non-genetics medical fields in which data sharing is common. Thankfully thousands of de-identified Myriad reports have been submitted to ClinVar by ordering clinicians through the Sharing Clinical Reports Project [SCRP website].
2. ClinVar
Since its inception in 2013, ClinVar has grown rapidly, and as of August 2016 contains more than 186,000 records from 560 submitters, most of which are clinical genetic testing laboratories [ClinVar website]. Importantly, three of the top eight submitters to ClinVar are commercial laboratories (GeneDx, Invitae, and Ambry). Another three are large academic laboratories (Harvard Partners Laboratory for Molecular Medicine, Emory Genetics Laboratory, and the University of Chicago Genetic Services Laboratories), and two are academic efforts that aggregate literature-based information (OMIM and GeneReviews) These submitters account for more than half of the data in ClinVar, although the many smaller submitters provide key data as well. This high degree of industry–academic collaboration is encouraging and critical given the degree of privatization in the American healthcare system.
2.1. Data set used for analysis
We extracted variant classifications from ClinVar (May 2016 XML download, which remains archived online [ClinVar website]). We included data for genes in six different clinical specialties that our laboratory (Invitae) offered for clinical testing at the time and with which we were thus familiar (Supplemental Data). For simplicity, when one gene may be tested by multiple specialties, we used the most common one. Because variant-phenotype assertions are inconsistently populated in ClinVar these were ignored. We further limited our data set to classifications of germline (not somatic) variants from licensed clinical diagnostic laboratories. Thus data submitted by literature curation efforts (e.g. OMIM), expert panels (e.g., ENIGMA, InSiGHT) and research were also excluded, as these do not reflect actual clinical test reports provided to physicians. Finally, we required that variant classifications be on the 5-class ACMG system and be asserted by at least two submitters. Our data set contained 9875 variants in 409 genes (Table 1, Supplemental Data). We note that many of these classifications pre-date the 2015 ACMG guidelines mentioned above.
Table 1.
ClinVar-based data set used in this analysis.
| Variants | Genes | Classifications | Variants/Gene | Classifications/Variant | |
|---|---|---|---|---|---|
| Cancer | 4802 | 55 | 12,703 | 87.3 | 2.7 |
| Cardiology | 3289 | 163 | 7611 | 20.2 | 2.3 |
| Epilepsy | 739 | 58 | 1659 | 12.7 | 2.2 |
| Metabolic | 383 | 56 | 850 | 6.8 | 2.2 |
| Neurology | 662 | 77 | 1376 | 8.6 | 2.1 |
| Total | 9875 | 409 | 24,199 | 24.1 | 2.5 |
Overall, variants considered benign (B or LB) by most or all submitters composed the largest group (44.5%). Pathogenic variants (P or LP) made up 17.9% of the data set. Many variants (26.9%) were considered VUS, and 10.7% had no consensus (as defined below) for any category. This distribution varied significantly by clinical area (Figure 1).
Figure 1.
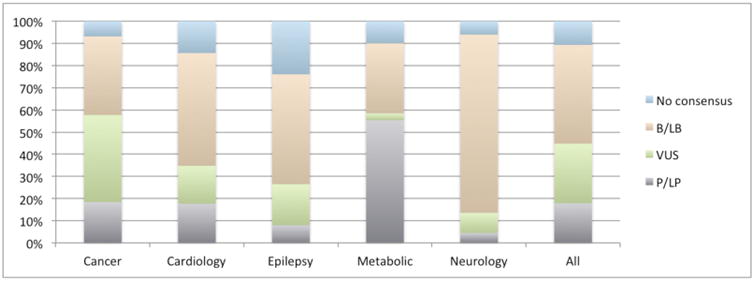
Fraction of variants in ClinVar for each clinical area by consensus pathogenicity.
2.2. Rarity of clinically observed variants
Because our data set was limited to variants from two or more submitters, it was naturally biased away from the rarest of variants. Nevertheless, this data set was predominantly composed of rare variants (Figure 2). Most (62%) of the ClinVar variants that also appear in ExAC [Lek, 2016] had population allele frequencies less than 0.001, and for 36%, that frequency was less than 0.0001. Another 22.8% of the ClinVar variants were not in ExAC at all, either because they are very rare or because they lie outside of ExAC's well-covered regions. This rarity also manifests itself in the number of submitters who have classified each variant: Most variants had been classified by only two or three of the 23 submitters in this data set (Table 1). Even in the case of BRCA1/2, one of the most common clinically tested genes, the average was only 2.9 classifications per variant. Rare variants comprise an even larger fraction of ClinVar overall, particularly variants with only a single submitter which were excluded from this data set.
Figure 2.
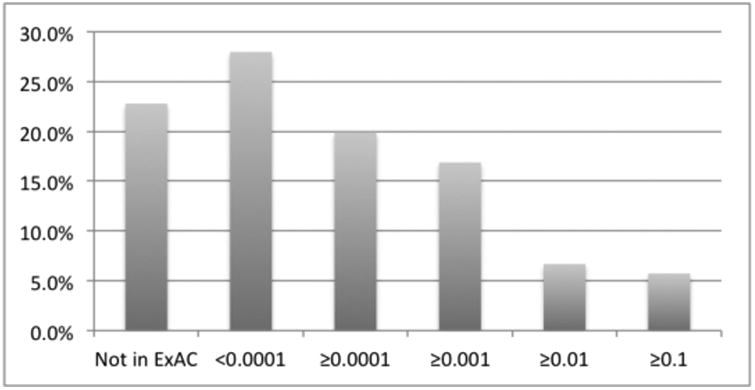
Histogram of allele frequency in ExAC for all ClinVar variants in our analysis regardless of pathogenicity. Note that the vast majority of ClinVar variants are in or near exons.
2.3. Concordance of variant classifications
We compared variant classifications in ClinVar to assess the degree of agreement among clinical testing laboratories (Figure 3). We first focused on differences between positive (P or LP) classifications, which are potentially clinically actionable, as opposed to findings that are not actionable (VUS, B, or LB). We refer to this analysis as the P-NP (positive versus not positive) comparison. Counting each of the 9875 variants as a data point, concordance among laboratories was high: 96.1% of variants agreed across all (two or more) submitters. For an additional 0.9% of variants, there was a consensus among a majority of the submitters. We defined consensus as agreement in two-thirds of the submissions (i.e., consensus required two of two submissions to agree, or 2/3, 3/4, 4/5, 4/6, etc.). In 3% of variants, there were only two submitters who disagreed, and only one variant had four submitters with a 2–2 tie. Clinical care guidelines generally state that patients with only VUS should be managed according to their personal and family histories and not their genetic test results [e.g. NCCN 2016]. Thus the P-NP comparisons correlate most with the impact of interpretation discordance on patient care decisions.
Figure 3.
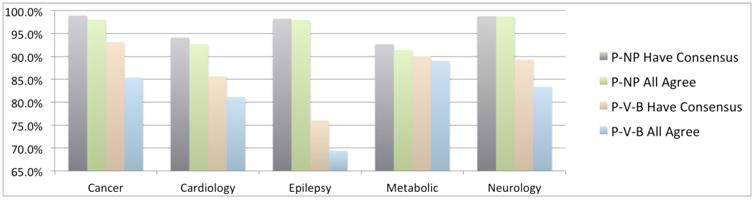
Concordance among labs measured in different ways. See text.
When the comparison was performed on a different basis—not combining VUS with B/LB classifications—concordance was, of course, lower. We refer to this analysis as the P-V-B (pathogenic versus VUS versus benign) comparison. In this evaluation, only 83% of variants agreed among all submitters. A further 6% achieved consensus but with some submitter(s) in dissent. This much lower rate indicates that the criteria for discriminating between VUS and B/LB variants varies among laboratories, more so than criteria for establishing pathogenicity.
Concordance varied considerably among clinical areas. On a P-NP basis, variants in cardiology and metabolic genes had concordances lower that those in the other areas, although in all cases concordance was greater than 90%. On a P-V-B basis, epilepsy genes fared the worst, followed by cardiology. The gap between P-NP and P-V-B is particularly large in epilepsy genes, suggesting that evidence against pathogenicity is used quite inconsistently by labs. Cursory analysis suggests that classification date, as expected, plays a significant role in discordance (Supplemental Data). A detailed analysis of the basis for discordance is important future work.
3. BRCA1/2
BRCA1/2 had the largest number of variants of any gene(s) in our ClinVar data set (1771 combined) for several reasons: BRCA1 and BRCA2 are not only among the most commonly tested genes in clinical practice today, but also have been clinically tested for more than 20 years. Moreover, a significant international effort has focused on adding BRCA1/2 variants to ClinVar, whereas data sharing efforts for some other commonly tested genes center on previously established databases (e.g. the CFTR2 database for cystic fibrosis). Finally, compared with most human genes, BRCA1/2 have relatively large coding sequences and thus can harbor an atypically large number of variants. Thus, the “long tail” of BRCA1/2 variants is particularly long, and new variants needing classification are continually uncovered, as shown by our own data (Figure 4). This conclusion is consistent with unpublished reports from Myriad Genetics, which claims to encounter >50 new variants per week despite offering testing for 20 years [Myriad 2015]
Figure 4.
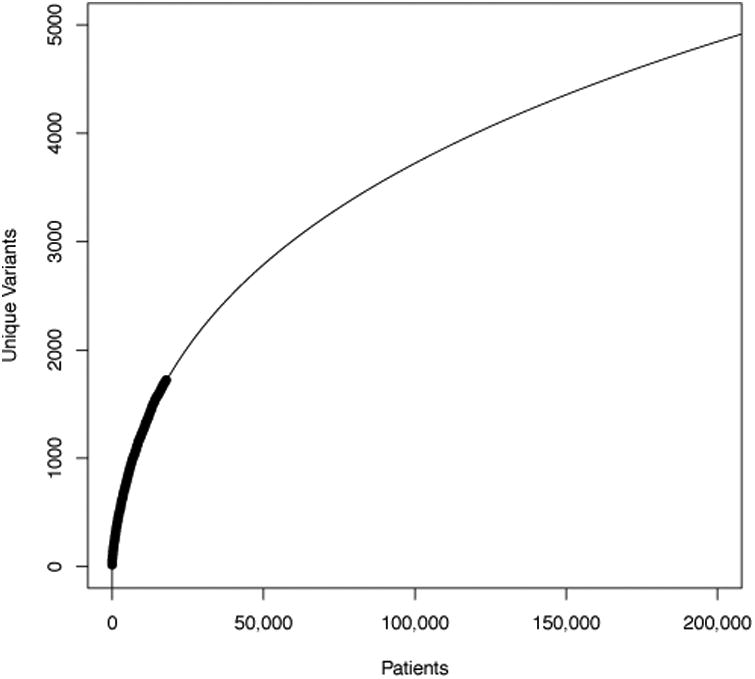
The relationship of number of unique BRCA1/2 variants to number of patients tested at Invitae (dark curve). The extrapolation (light curve) was fit in R using the formula poly(log(Patients), 3). We chose the polynomial degree empirically by minimizing the Akaike Information Criteria [Sakamoto 1986].
3.1. Concordance among BRCA1/2 variant classifications
In a separate study, we performed a much more detailed comparison of ClinVar data for BRCA1/2 using a ClinVar data set of more than 2000 comparable variants [Lincoln 2016]. This analysis considered only classifications from clinical labs with significant experience (as evidenced by submitting 200 or more variants to ClinVar) and excluded submitters where most classifications were >5 years old. On a P-NP basis, 98.5% of variants showed no disagreement among submitters—a concordance higher than that observed in ClinVar overall. This previous study also showed that variants with classification discordance were rare (allele frequencies were always less than 0.0005 and usually were immeasurably low). Although they are numerous, rare variants by definition appear in very few patients: less than 15% of the 30,000 patients studied carried any rare variants in BRCA1 or BRCA2, and most of those were concordantly classified. In this prior study, concordance per patient (not per variant) was thus estimated to be 99.8%.
3.2. Variants of Uncertain Significance (VUS) in BRCA1/2
VUS can present a challenge in day-to-day clinical decision-making, and the most prevalent type of VUS are rare missense changes. VUS rates are traditionally defined as the fraction of patients with one or more VUS and no positive findings. Major U.S. laboratories report VUS rates in the range of 3–5% for BRCA1/2, although this rate varies considerably with ethnic mix and with the fraction of cancer-affected versus unaffected patients [Lincoln 2015]. On a per-variant (rather than per-patient) basis, the VUS rate is much higher: 31.4% of BRCA1/2 variants in our data set (Table 1) were VUS, although most are very rare and thus appear in very few patients.
The evidence suggests that the majority of VUS are actually benign variants that have inadequate evidence to demonstrate that fact. This is supported by our own experience that most VUS, when reclassified, are “downgraded” to LB or B. We also observed this in a sequential analysis of ClinVar releases from the past 2 years (available at [ClinVar website]) in which roughly 95% of BRCA1/2 VUS reclassifications were downgrades. Others have also observed this in Myriad data [Murray 2011]. In terms of clinical impact, a rough approximation is that if 4% of patients have a VUS, and if 5% of those findings are truly pathogenic variants lacking evidence of pathogenicity, then 1/500 BRCA1/2-positive patients may currently be missed.
BRCA1/2 tests are increasingly being replaced by multi-gene panels that assay additional genes that significantly increase the risk of various cancers. By virtue of testing more genes, the VUS rate in these panels is substantially larger. For example, VUS rates of roughly 40% have been reported by 25-29 gene panels [Lincoln 2015; Desmond 2015; Tung 2015], although again, experience suggests that the majority of these VUS will ultimately be classified as benign.
3.3. The BRCA Exchange
As of August 2016, ClinVar contains more than 9000 variants in BRCA1/2, many of which are either unclassified or are considered VUS. Most of these variants have been reported by only a single submitter. These data still represent only a fraction of the known human variation in BRCA1/2, much of which is either not submitted to ClimVar or is not appropriate for ClinVar (yet is useful to have linked). In an effort to collect a more comprehensive view of BRCA1/2 variation, the BRCA Exchange project has been initiated under the auspices of the Global Alliance for Genomics and Health's BRCA Challenge. European laboratory data, coordinated by the Leiden Open Variation Database (LOVD), population databases, and other data sources are being combined with ClinVar in this BRCA1/2-specific public database. In its current preliminary form, the BRCA Exchange describes more than 13,000 variants, many of which originate from only a single source database (Figure 5). Not only is the BRCA exchange database open, but the code that populates it is open source. Future analyses of the type described in this paper could and should leverage this code in order to further improve reproducibility of such research.
Figure 5.
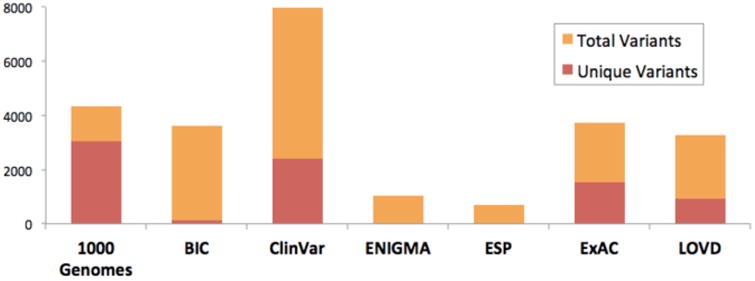
Sources of data in the pre-release BRCA exchange. Many variants not in ClinVar and indeed many are unique to a single database. For details and references see brcaexchange.org.
4. Discussion
4.1. Summary: Most variant classifications agree, but …
In the analysis described above, we examined nearly 10,000 variants from ClinVar in more than 400 genes across six clinical areas and found generally high (>90%) variant classification concordance among clinical laboratories in terms of potential effect on clinical management (our P-NP comparison). In separate prior studies, we examined BRCA1/2 in particular detail found higher concordance on both a per-variant (99.0%) and a per-patient (99.8%) basis than is seen in the broader gene list. It is reassuring, at least for geneticists, to note that this level of concordance is higher than that observed among pathologists reading breast biopsies or radiologists reading mammograms [Elmore 2015(a,b); Elmore 2016; Sprague 2016]. Nevertheless, resolving differences in variant classification is critical to doctors and patients. Moreover, many variants (30.1%) have classifications that are concordantly VUS and much more work is required to classify these variants definitively even though laboratories agree.
Public databases such as ClinVar play critical roles in the identification of both disagreements and uncertainties, and these databases can facilitate collaborative interactions that will resolve many such issues. The value of such collaboration in improving variant classifications has recently been demonstrated by multiple groups [Amendola 2016]. Efforts are now organized into disease-specific working groups by the ClinGen consortium [Rehm 2015; Pfimister 2015] and support preexisting efforts such as ENIGMA and InSiGHT. Those with interest and expertise in these areas should certainly consider joining and contributing.
Public databases can also play a critical role in laboratory quality control by allowing detailed independent peer scrutiny of all variant classifications by the global community. In our opinion, no laboratory could (or probably would) mount such an effort alone, and publication peer review processes can not provide this type of ongoing quality assessment. In our opinion, laboratory directors who are both confident in their quality yet continually working to improve should have no reservations about unrestricted public data submission of their data.
4.2. Considerations when using public clinical databases
Our analysis highlights important considerations users must keep in mind when accessing public databases such as ClinVar. Foremost is that it is a fallacy to say, for example, “ClinVar says that variant X is pathogenic.” ClinVar itself generates no assertions; it only collects them from submitters. Database users must pay careful attention to the original source of each classification, which may be a reputable clinical laboratory rigorously following accepted classification guidelines, or it may not be. Dates are important, as submissions to these databases can become outdated, which results in false discrepancies.
It is important that users understand the biology and medical practice considerations for each gene they examine in a public database. Consider three examples of the rates of variant pathogenicity (Figure 1) which we find unsurprising: (a) In some genes (e.g., most hereditary cancer genes) loss-of-function variants are pathogenic, and nature provides many means of disabling genes or their proteins. In other cases (e.g., some neurology and cardiology genes), gain-of-function mutations are clinically more important, and these, by their very nature, are less numerous, reducing the fraction of pathogenic variants in ClinVar. (b) The large fraction of pathogenic variants and small fraction of VUS in metabolic genes reflect the fact that experimental confirmation of pathogenicity (e.g., through blood chemistry and urinalysis) is relatively straightforward and standard clinical practice. However, the relatively low concordance in metabolic genes (Figure 3) suggests that these procedures are imperfect. (c) In cardiology, complexities in both phenotyping and penetrance are well known to increase the complexity of variant classification [Van Driest].
Deliberate (and not nefarious) submission biases also affect ClinVar. Notably, laboratory policies vary as to whether and when B/LB variants are reported to patients/physicians or to ClinVar (even though benign polymorphisms are frequently observed). Similarly, practices for the detection and reporting of non-coding variants vary. Although many routine tests detect copy number variants, these variants are less commonly reported to ClinVar for logistical reasons (a situation we hope will change). Furthermore, a test may or may not be sensitive to complex alterations such as copy-neutral inversions, Alu insertions, or variants in low complexity or highly conserved regions. Although ClinVar can record the observed prevalence of any variant, this field is rarely filled in. Finally, ClinVar submissions generally represent laboratory patient series, which are subject to many undocumented ascertainment biases. For these reasons, ClinVar cannot be used to evaluate the spectrum of disease-causing or benign variation in any gene.
4.3. Whither data sharing
Although sharing of clinical genetic data has been successful, and clearly impactful, challenges remain. For example, during our various analyses of ClinVar, we uncovered a number of out of date and erroneous submissions, which are an obvious concern. A bigger problem is the multiple laboratories who do not contribute. In addition to not contributing, Myriad Genetics has updated its terms of service to, in theory, prohibit ordering clinicians from sharing data with ClinVar [Robinson 2016]. A further challenge is the fragmentation of data into multiple silos. Although the BRCA Exchange aims to address this problem for BRCA1/2, this is a considerable effort and only applies to these two genes, not the many others of clinical relevance.
In environmental policy, the term “greenwashing” has emerged to describe the characterization of various activities as environmentally friendly when in fact they are not. Activities can occur in our field that one might perhaps call “sharewashing”. For example two large commercial labs (Labcorp and Quest) currently contribute variants only to BRCAShare, a database whose terms effectively prohibit either incorporation of the data into a common repository (like the BRCA Exchange) or its use in comparisons such as those described here. We hope this changes, but at present these data are not available in unrestricted form. The BRCAShare terms also prohibit use of the data by other commercial labs without paying a significant fee (unlike ClinVar). Separately, Myriad has tried to argue that its participation in the PROMPT patient registry comprises data sharing. PROMPT is indeed valuable, but serves a very different purpose than ClinVar. We encourage all groups to support and contribute to open, unrestricted, public databases, particularly ClinVar.
Footnotes
Supplement: The dataset upon which this analysis is based is available at: https://drive.google.com/drive/folders/0B79LNgCdve9BSWN0VHhodFFsMmM
Contributor Information
Shan Yang, Invitae, San Francisco, California, USA.
Melissa Cline, University of California Santa Cruz, Santa Cruz, California, USA.
Can Zhang, University of California Santa Cruz, Santa Cruz, California, USA.
Benedict Paten, University of California Santa Cruz, Santa Cruz, California, USA.
Stephen e. Lincoln, Invitae, San Francisco, California, USA
References
- Amendola LM, et al. Am J Hum Genet. 2016 10.1016/j.ajhg.2016.03. [Google Scholar]
- Angrist M, Cook-Deegan R. Appl Transl Genom. 2014;3(4):124–127. doi: 10.1016/j.atg.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinVar website: www.clinvar.com
- Cook-Deegan R, et al. Eur J Hum Genet. 2013;21(6):585–8. doi: 10.1038/ejhg.2012.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond A, et al. JAMA Oncol. 2015;1(7):943–51. doi: 10.1001/jamaoncol.2015.2690. [DOI] [PubMed] [Google Scholar]
- ENIGMA website: enigmaconsortium.org
- Elmore JG, et al. JAMA. 2015;313(11):1122–32. doi: 10.1001/jama.2015.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore JG, et al. JAMA. 2015;314(1):83–4. doi: 10.1001/jama.2015.6239. [DOI] [PubMed] [Google Scholar]
- Elmore JG, et al. Ann Intern Med. 2016;164(10):649–55. doi: 10.7326/M15-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereviews website: www.ncbi.nlm.nih.gov/books/NBK1116/
- InSiGHT website: insight-group.org
- Landrum MJ, et al. Nucleic Acids Res. 2016;44(D1):D862–8. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, et al. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln 2016: Lincoln SE, ACMG 2016 platform presentation, copy available at www.invitae.com; manuscript in review.
- Lincoln SE, et al. J Mol Diagn. 2015;17(5):533–44. doi: 10.1016/j.jmoldx.2015.04.009. [DOI] [PubMed] [Google Scholar]
- Murray ML, et al. Genet Med. 2011;13(12):998–1005. doi: 10.1097/GIM.0b013e318226fc15. [DOI] [PubMed] [Google Scholar]
- Myriad Analyst Day Presentation, September 2015 from www.myriad.com
- NCCN (National Comprehensive Cancer Network) NCCN Practice Guidelines in Oncology. Genetic/Familial High Risk Assessment: Breast and Ovarian, Version 2. 2016 www.nccn.org.
- OMIM website: omim.org
- Phimister EG. New Engl J Med. 2015;372(23):2227–8. doi: 10.1056/NEJMe1506276. [DOI] [PubMed] [Google Scholar]
- Richards S, et al. Genet Med. 2015;17(5):405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm HL, et al. New Engl J Med. 2015;372(23):2235–42. doi: 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson L Robinson, genetic counselor, UT Southwestern; personal communication 2016 [Google Scholar]
- Sakamoto Y, et al. Akaike Information Criterion Statistics. D Reidel Publishing; 1986. [Google Scholar]
- SCRP website: www.clinicalgenome.org/data-sharing/sharing-clinical-reports-project-scrp/
- Sprague BL, et al. Ann Intern Med. 2016 10.7326/M15-2934. [Google Scholar]
- Tung N, et al. Cancer. 2015;121:25e33. [Google Scholar]
- Van Driest SL, et al. JAMA. 2016;315(1):47–57. doi: 10.1001/jama.2015.17701. [DOI] [PMC free article] [PubMed] [Google Scholar]