

Abstract
(2S,3R)-2-Amino-3-methyl-4-phosphono-butanoic acid (Pmab) is a phosphatase-stable analog of phosphothreonine (pThr), which has been used in a variety of biological contexts. Among these applications are peptidomimetic ligands that bind to the polo-box domain (PBD) of polo-like kinase 1 (Plk1) with affinities approaching that of the corresponding pThr-containing peptides. However, Pmab is not widely used, because there are no direct, high-yield preparations of suitably protected reagent. We have now achieved an efficient synthesis of protected Pmab, as well as variants with different substituents at the 3R-center. When incorporated into our peptidomimetic scaffold, these new Pmab analogs exhibit Plk1 PBD-binding affinities that are several-fold higher than Pmab, yet retain good selectivity for Plk1 relative to the PBDs of Plk2 and Plk3. These findings will significantly impact the future development of PBD-binding inhibitors, as well as ligands directed against a broad spectrum of pThr-dependent processes.
Keywords: Phosphothreonine, phosphatase, polo-like kinase 1, polo-box domain
Foiling Phosphatases
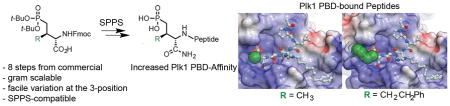
The use of phosphoamino acids can be limited by their hydrolytic lability to cellular phosphatases. We report an efficient synthesis of phosphatase-stable mimics of phosphothreonine that enables facile variation at the C3 position. These analogs provide significant enhancements of inhibitory potency when incorporated into peptidomimetic inhibitors of the polo-like kinase 1 polo-box domain.
Post-translational modification by phosphorylation is an integral component of cellular physiology. Phosphoserine (pSer, 1), phosphothreonine (pThr, 2) and phosphotyrosine (pTyr) residues often have the capacity to serve as molecular switches that control protein-protein interactions (PPIs).1–3 The phosphoryl groups often provide key elements of ligand interactions and mimicry of phosphorylated epitopes by short peptides can result in high affinity inhibitors against these PPIs.4 However, inhibitors that directly utilize phosphoryl groups can be susceptible to reduced cell-based activity due to the potential hydrolytic lability of the phosphoryl ester linkage in the presence of cellular phosphatases. For this reason, a significant body of work has been devoted to producing hydrolytically-stable surrogates of phosphoamino acids.5–7 Replacement of the phosphoryl ester oxygen of pThr with a methylene group provides the phosphonic acid-containing (2S,3R)-2-amino-3-methyl-4-phosphono-butanoic acid (Pmab, 3, Figure 1), which can serve as a phosphatase-stable isostere in pThr-containing epitopes.
Figure 1.
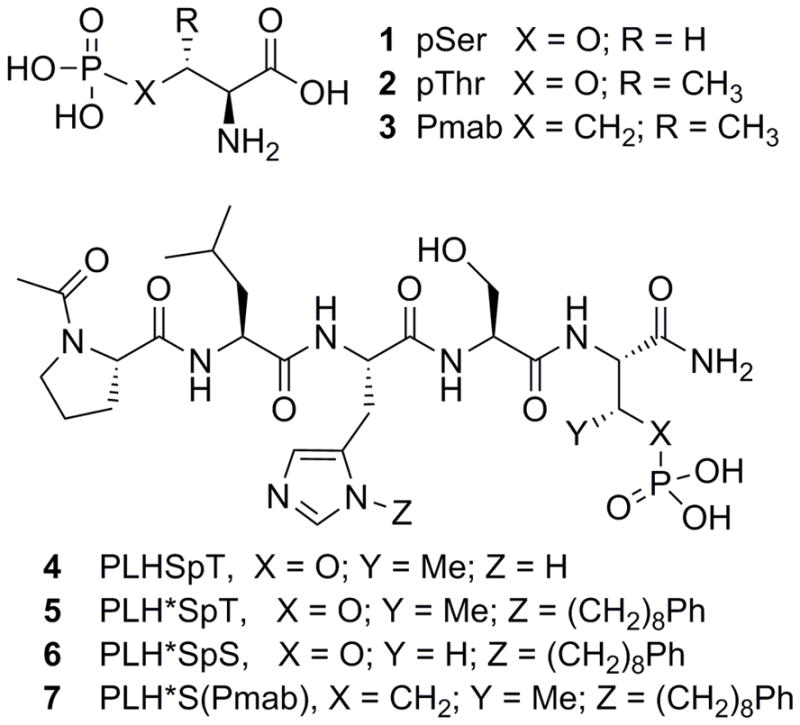
Structures of phosphoamino acids (1–3) and peptidomimetics (4–7).
The cell cycle regulatory serine/threonine-specific polo-like kinase 1 (Plk1) contains a C-terminal polo-box domain (PBD) that is responsible for intracellular localization through PPIs that target pSer and pThr-containing sequences.8,9 Plk1 is recognized as an oncogenic target, for which PBD-binding ligands are being developed as competitive antagonists designed to inhibit Plk1 function by disrupting its localization to mitotic structures.10 To date, our most advanced PBD-binding ligands are based on a modified variant of the polo-box interacting protein 1 (PBIP1)-derived pentapeptide sequence “Pro-Leu-His-Ser-pThr” (4), in which the His residue contains a Ph(CH2)8–group at its N3 (π) nitrogen (designated as His* or H*).11 This peptide (5) displays low-nanomolar IC50 values in competitive ELISA assays against Plk1. We have shown that pThr provides a 5-fold enhancement in binding affinity versus the pSer-containing peptide (6).12,13 Presumably, this affinity enhancement is due to the 3R-methyl group restricting the side chain rotation (or χ1 angle) in a favorable binding orientation.14 More importantly, we have also shown that the pThr residue in 5 can be replaced with Pmab to produce a phosphatase-stable peptidomimetic (peptide 7) having complete retention of inhibitory potency.13,15,16 Unfortunately, the use of Pmab in further ligand development is significantly hindered by relatively inefficient synthetic access to orthogonally-protected Pmab constructs that are compatible with solid-phase peptide synthesis (SPPS). In this work, we have developed a more direct synthetic route to SPPS-compatible Pmab analogs. In light of the importance of the 3R-methyl group for PBD-binding affinity, we specifically utilized chemistry that is compatible with facile introduction of other functional groups at the C3-position. Finally, we incorporated these new phosphonic acid-containing intermediates into our high-affinity PBD-binding scaffold and determined their effects on Plk1 PBD binding affinity and selectivity versus the PBDs of Plks 2 and 3.
The first reported stereoselective syntheses of Pmab, including SPPS-compatible N-Fmoc-O, O-diallyl-Pmab, utilized the Michael addition of lithiated Schöllkopf’s bis-lactim ethers into (E)-prop-2-enyl-phosphonate esters.17,18 This method can selectively generate each diastereomer at the C2- and C3-positions with high selectivity, and it was later demonstrated that this method can incorporate functionality at the C3-position through Michael addition to a scope of vinyl phosphonates.19,20 In order to produce a Pmab analog for SPPS that contains acid-labile protection, we previously reported a procedure for the stereoselective synthesis of N-Fmoc-O, O-di-tert-butyl-Pmab (13a, Scheme 1). This synthesis utilized a stereospecific alkylation to introduce the key 3R-methyl group and required a minimum of 15 steps (10–12% overall yield).21 Envisioning a more direct approach to this stereochemistry, we took note of progress that has been made in the application of asymmetric aminocatalytic Mannich reactions for the synthesis of amino acid intermediates having chiral centers in their side chains.22,23 Gianelli and co-workers reported the first examples of a synthetic proline catalyst to effect anti-selective Mannich reactions, which proceede in high diastero- and enantioselectivity using N-Cbz or N-Boc protected imines, generated in situ from α-amido sulfones.24,25 Subjecting commercially available aldehydes to this protocol provides efficient access to (2S)-amino-(3R)-alkyl precursors, which are central for the synthesis of orthogonally-protected Pmab, as well as of other pThr mimetics bearing different substituents at the C3 position (Scheme 1).
Scheme 1.

Reagents and conditions: a) Aldehyde (2.0 equiv.), KF (5.0 equiv.), 9 (0.1 equiv., 10 mol%), CHCl3, 24 – 96 h, rt (d.r. = diastereomeric ratio); b) di-tert-butyl phosphite (1.5 equiv.), TMS-Cl (1.5 equiv.), TEA (2.0 equiv.), CH2Cl2, 3 – 6 h, rt; c) 20% aq. citric acid (w/v), MeOH, 16 h, rt; d) O-phenyl thiochloroformate (3 equiv.), DIEA (4 equiv.), DMAP (0.2 equiv.), CH2Cl2, rt, 3 – 16 h; e) tributyltin hydride (3.0 equiv.), azoisobutylnitrile (AIBN, 1.0 equiv.), toluene, reflux, 20 min; f) LiOH (3.0 equiv.), THF/H2O (3:1), 16 h; g) 1 atm H2, Pd/C (10% w/w, 0.2 equiv.), MeOH, 3 h, rt; h) Fmoc-OSu (1.5 equiv.), NaHCO3 (5.0 equiv.), THF/H2O (1:1), 16 h, rt; i) di-benzyl phosphite (1.5 equiv.), TMS-Cl (1.5 equiv.), TEA (2.0 equiv.), CH2Cl2, 3 h, rt; j) 20% TFA/CH2Cl2, 2 h, rt.
In agreement with previously reported results,24 we found that by reacting propionaldehyde with the racemic sulfone-containing substrate 8 (readily obtained on multigram scale; see Supporting Information) in the presence of catalyst 9 (10 mol%), we were able to produce the desired (3R)-methyl-containing aldehyde 10a on gram-scale with excellent diastereoselectivity (>10:1). A Pudovik reaction on 10a using di-tert-butyl phosphite gave 11a (83% yield), with subsequent Barton-McCombie deoxygenation provided the globally protected 12a (61% yield over two steps).21 Finally, saponification and exchange of the amine-protecting group gave the desired final product 13a on gram-scale in 8 steps and 22–25% overall yield (Scheme 1, upper panel).
A desirable feature of the 8-step procedure described above is that the initial anti-Mannich reaction is able to tolerate sterically hindered aldehydes.24 This allowed us to prepare the C3-substituted aldehyde intermediates 10b – 10e, which could be further transformed into the orthogonally protected Pmab variants having different 3R substituents (13b – 13e, Scheme 1, upper panel). We synthesized analogs 13b – 13e using reaction conditions similar to what we had employed to prepare 13a. However, in some instances the increased steric bulk of the 3-substituents as compared to the 3R-methyl group, required longer reaction times or resulted in slightly diminished yields.
Because the reported anti-Mannich protocol24 is compatible N-Boc protected imines, we were also able to prepare orthogonally protected Pmab analogs bearing di-benzyl protection of the phosphonic acid functionality (18, Scheme 1, lower panel). In certain situations, reagent 18 may have a significant advantage over 13a. This is exemplified by use in peptides that are cleaved from resins under acidic conditions, in which the benzyl groups can be retained, while tert-butyl phosphonic acid protection would be removed. This allows the possibility of performing further post-solid-phase modifications on these peptides, which otherwise would be contraindicated in the presence of a free phosphonic acid group.
Having developed facile access to new phosphonic acid-based mimetics of pThr that have a variety of 3R groups, we validated their biological usefulness using Plk1 interactions as a model. In order to compare the inhibitory potency of the Pmab-containing peptide 7 with a peptide containing the corresponding phosphonic acid-based mimetic of pSer, [phosphonoaminobutyric acid (Pab)], we prepared peptide 19 using the requisite orthogonally protected reagent (N-Fmoc-O, O-di-tert-butyl-Pab, 29: see Supporting Information, Scheme S1). We evaluated the ability of these peptides to inhibit the binding of full-length Plk1 to an immobilized pThr-containing peptide in a competitive ELISA format. Consistent with our previous results, the Pmab-containing peptide (7) showed approximately 5-fold greater inhibitory potency than the corresponding Pab-containing peptide (19), with IC50 values of 0.21 μM and 1.2 μM having been obtained, respectively (Table 1).13
Table 1.
Structure-activity relationships of the PBD-binding ligands using full-length Plk1.
| |||||
|---|---|---|---|---|---|
| Cpd. | X | R | R1 | R2 | IC50 (μM) [a] |
| 4 | O | Me | OH | H | ~475 |
| 5 | O | Me | OH | (CH2)8Ph | 0.17 ± 0.04 |
| 7 | CH2 | Me | OH | (CH2)8Ph | 0.21 ± 0.06 |
| 19 | CH2 | H | OH | (CH2)8Ph | 1.2 ± 0.3 |
| 20 | CH2 | Et | OH | (CH2)8Ph | 0.20 ± 0.06 |
| 21 | CH2 | i-Pr | OH | (CH2)8Ph | 0.06 ± 0.02 |
| 22 | CH2 | Bn | OH | (CH2)8Ph | 0.30 ± 0.08 |
| 23 | CH2 | (CH2)2Ph | OH | (CH2)8Ph | 0.07 ± 0.02 |
| 24 | CH2 | i-Pr | H | (CH2)8Ph | 11 ± 3 |
| 25 | CH2 | (CH2)2Ph | H | (CH2)8Ph | 70 ± 14 |
IC50 values = average ± SEM (n = 5).
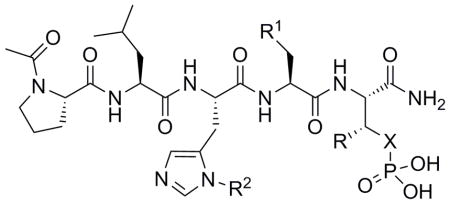
We hypothesized that more extensive structural variation at the C3 position could potentially have beneficial effects, either by providing conformational constraint (favorably restricting the χ1 angle14) or by introducing direct contacts with the PBD binding site. We utilized reagents 13b – 13e to synthesize peptides having Pmab analogs that were different at their C3 position (ethyl 20; isopropyl 21; benzyl 22 and phenylethyl 23). We found using our current full-length Plk1 PBD-competitive ELISA assay, that relative to the original Pmab-containing peptide (7, IC50 = 0.21 μM), the C3 variants either maintained or improved their IC50 values (Table 1). The most potent peptides (C3-substituted isopropyl 21, IC50 = 0.06 μM and phenylethyl 23, IC50 = 0.07 μM, respectively) were approximately 3-fold more potent than the parent pThr-containing peptide 5 and its corresponding Pmab-containing congener 7 (Table 1). Utilizing the X-ray co-crystal structure of 5 bound to the isolated PBD (PDB 3RQ715), we were able to visualize the likely binding orientations of compounds 7, 21, and 23 (Figure 2, Panels A–C). The PBD phosphate binding pocket is adjacent to the residues Leu 491, His538 and Arg557 and the C3-methyl group of pThr is directed toward these residues. The C3-isopropyl group of 21 likely improves potency by restricting conformational rotation of the side-chain phosphonate. The phenylethyl group of 23 may also serve to restrict rotation of the phosphonate, however modeling suggests that this group may also engage in a pi-cation interaction with Arg557. The Ser to Ala variants of 21 and 23 (24 and 25, respectively) displayed an approximate two orders-of-magnitude loss of potency. This provided evidence that the binding orientations of 21 and 23 are canonical in nature, since the Ser residue provides an important component of normal PBD-binding recognition.
Figure 2.

Visualization of the C-terminal C3-modified phosphonate residues of peptidomimetics 7 (A), 21 (B), and 23 (C). C3 substituents are shown as colored spheres and contacting protein residues are labeled. As described in the Supporting Information, structures were modeled based on the X-ray co-crystal structure of 5 bound to the isolated PBD (PDB: 3RQ7).
Next, we investigated the binding selectivity of certain analogs for the PBD of Plk1 relative to the PBDs of Plk2 and Plk3. We conducted these studies using fluorescence polarization assays, which measure the abilities of the inhibitors to compete with fluorescently labeled pThr-containing peptides for binding to isolated PBDs of Plks 1 – 3. For the labeled peptides, we used previously reported sequences that had been optimized for binding to each individual PBD isoform.26,27 We evaluated peptides 7, 19, 21, and 23, which contained 3-substituents consisting of –H, –Me, –i-Pr, and –(CH2)2Ph groups, respectively. We found that all analogs displayed high Plk1 PBD selectivity, with 50- to 800-fold decreases in potency against the PBDs of Plks 2 and 3 (Table 2).
Table 2.
Results of fluorescence polarzation assays measuring the inhibitory potencies of select analogs versus the PBDs of Plks 1 – 3.
| Cpd. | R | IC50 (nM) [a] | ||
|---|---|---|---|---|
| Plk1 | Plk2 | Plk3 | ||
| 7 | Me | 9 ± 1 | 1,400 ± 50 | 4,400 ± 100 |
| 19 | H | 11 ± 1 | 5,400 ± 600 | 8,800 ± 800 |
| 21 | i-Pr | 7 ± 1 | 360 ± 30 | 1,100 ± 200 |
| 23 | (CH2)2Ph | 4 ± 1 | 490 ± 50 | 1,200 ± 200 |
IC50 values = average ± SEM (n = 3).
One of the primary reasons for using phosphonic acid-based phosphate mimetics is to overcome biological inactivation by cytoplasmic phosphatases. We have previously demonstrated the phosphatase stability of phosphonic acid-based pTyr mimetics in Src homology 2 domain-binding systems.28 In like fashion, we determined the inhibitory potencies of the pThr and Pmab-containing constructs 5 and 7 following time-dependent incubation in concentrated cell lysates that lacked exogenous phosphatase inhibitors. Inhibitory potencies were measured using a competition ELISA assay, similar to what is shown in Table 1. However, this assay employed isolated Plk1 PBD, which lacks the N-terminal kinase domain (KD). We found that the inhibitory potency of 5 decreased dramatically following a two-hour incubation in the absence of phosphatase inhibitors, while the inhibitory potency of 7 remained unchanged under identical conditions (Table 3). It is worth noting that the IC50 values measured against the isolated PBD (t = 0 min) are ~10-fold more potent than those measured against full length Plk1. Consistent with previous observations of PBD-dependent binding to full length Plk1, this difference can be attributed to inhibitory interactions between the PBD and KD, which affect equilibrium binding to phosphopeptides.8 These results confirm the potential utility of using Pmab or other phosphonic acid-based analogs of pThr for maintaining efficacy in whole cell systems.
Table 3.
Efficacy of 5 and 7 in competitive ELISA assays vs Plk1 PBD following timed incubation in concentrated cell lysates lacking phosphatase inhibitors.
| Time (min) | 5, IC50 (nM) [a] | 7, IC50 (nM)[a] |
|---|---|---|
| 0 | 27 ± 10 | 14 ± 4 |
| 30 | 1,500 ± 500 | 11 ± 5 |
| 60 | >10,000 | 7 ± 2 |
| 120 | >10,000 | 14 ± 4 |
IC50 values = average ± SEM (n = 3).
To date, Pmab has been singular as a phosphonic acid-based pThr mimetic that retains excellent PBD-binding affinity. However, its routine use in ligand development has been limited by the inefficiency of its synthetic preparation. Additionally, very little had been reported on binding effects incurred by structural variation at the C3-position of either pThr or Pmab. Our current work addresses both of these issues by providing concise and high yielding access to orthogonally protected phosphatase-stable mimetics of pThr having different phosphonic acid protecting groups. This renders Pmab of practical use for inhibitor development, while at the same time achieving a systematic investigation of C3-substituents. One result of this work is our finding that several-fold enhancement of binding affinity can be achieved by suitable modification at the C3-position. This is potentially highly useful, since the starting parent 7 had been among the highest affinity PBD-binding peptides known, and significant improvements in its binding affinity can be obtained with simple modification to the synthetic route. Our demonstration that binding affinity is maintained for Pmab in the presence of active phosphatases and that peptides containing the new analogs retain good Plk1 PBD selectivity, are also noteworthy. Due to the dianionic nature of the phosphonate, cellular penetration of Pmab-containing peptidomimetics remains a significant challenge. We are currently focused on approaches to overcome this limitation and broaden the biological potential of these PBD-binding ligands. These new reagents may serve as useful tools in a variety of biological contexts, particularly when incorporated into compounds that are intended for cellular applications.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute, National Institutes of Health. We would like to thank Prof. Erich Nigg (Univ. of Basel, Switzerland) for generously providing plasmids encoding the PBDs of Plk 2 and 3.
Footnotes
Supporting Information for this article includes synthetic, biochemical and modeling experimental procedures as well as analytical data. This information can be accessed via a link at the end of the document.
Experimental Section
Assay binding curves, synthetic procedures, characterization data for all new compounds, and procedures for biological assays can be found in the Supporting Information.
References
- 1.Yaffe MB. Nat Rev Mol Cell Biol. 2002;3:177–186. doi: 10.1038/nrm759. [DOI] [PubMed] [Google Scholar]
- 2.Ladbury JE. Protein Rev. 2005;3:165–184. [Google Scholar]
- 3.Elia AEH, Yaffe MB. In: Modular Protein Domains. Cesare G, editor. Wiley-VCH; Weinheim: 2005. pp. 163–179. [Google Scholar]
- 4.Yun SM, Moulaei T, Lim D, Bang JK, Park JE, Shenoy SR, Liu F, Kang YH, Liao CZ, Soung NK, Lee S, Yoon DY, Lim Y, Lee DH, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR, Yaffe MB, Wlodawer A, Lee KS. Nat Struct Mol Bio. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burke TR, Lee K. Acc Chem Res. 2003;36:426–433. doi: 10.1021/ar020127o. [DOI] [PubMed] [Google Scholar]
- 6.Otaka A, Mitsuyama E, Kinoshita T, Tamamura H, Fujii N. J Org Chem. 2000;65:4888–4899. doi: 10.1021/jo000169v. [DOI] [PubMed] [Google Scholar]
- 7.Panigrahi K, Eggen M, Maeng JH, Shen Q, Berkowitz DB. Chem Biol. 2009;16:928–936. doi: 10.1016/j.chembiol.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elia AEH, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 9.Archambault V, Lepine G, Kachaner D. Oncogene. 2015;34:4799–4807. doi: 10.1038/onc.2014.451. [DOI] [PubMed] [Google Scholar]
- 10.Lee KS, Burke TR, Jr, Park JE, Bang JK, Lee E. Trends in Pharm Sci. 2015;36:858–877. doi: 10.1016/j.tips.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian W, Liu F, Burke TR., Jr J Org Chem. 2011;76:8885–8890. doi: 10.1021/jo201599c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elia AEH, Cantley LC, Yaffe MB. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 13.Qian W, Park JE, Liu F, Lee KS, Burke TR., Jr Biorg Med Chem. 2013;21:3996–4003. doi: 10.1016/j.bmc.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hruby VJ, Li G, Haskell-Luevano C, Shenderovich M. Biopol. 1997;43:219–266. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 15.Liu F, Park JE, Qian WJ, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR. Nat Chem Bio. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qian WJ, Park JE, Lim D, Lai CC, Kelley JA, Park SY, Lee KW, Yaffe MB, Lee KS, Burke TR., Jr Biopolymers. 2014;102:444–455. doi: 10.1002/bip.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ojea V, Ruiz M, Shapiro G, Pombovillar E. Tetrahedron Lett. 1994;35:3273–3276. [Google Scholar]
- 18.Ruiz M, Ojea V, Shapiro G, Weber HP, Pombovillar E. Tetrahedron Lett. 1994;35:4551–4554. [Google Scholar]
- 19.Ojea V, Ruiz M, Shapiro G, Pombo-Villar E. J Org Chem. 2000;65:1984–1995. doi: 10.1021/jo991402w. [DOI] [PubMed] [Google Scholar]
- 20.Ruiz M, Fernandez MC, Diaz A, Quintela JM, Ojea V. J Org Chem. 2003;68:7634–7645. doi: 10.1021/jo034707q. [DOI] [PubMed] [Google Scholar]
- 21.Liu F, Park JE, Lee KS, Burke TR. Tetrahedron. 2009;65:9673–9679. doi: 10.1016/j.tet.2009.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Notz W, Tanaka F, Barbas CF. Acc Chem Res. 2004;37:580–591. doi: 10.1021/ar0300468. [DOI] [PubMed] [Google Scholar]
- 23.Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]
- 24.Gianelli C, Sambri L, Carlone A, Bartoli G, Melchiorre P. Angew Chem Int Ed. 2008;47:8700–8702. doi: 10.1002/anie.200803819. [DOI] [PubMed] [Google Scholar]
- 25.Galzerano P, Agostino D, Bencivenni G, Sambri L, Bartoli G, Melchiorre P. Chem Eur J. 2010;16:6069–6076. doi: 10.1002/chem.200903217. [DOI] [PubMed] [Google Scholar]
- 26.Reindl W, Strebhardt K, Berg T. Anal Biochem. 2008;383:205–209. doi: 10.1016/j.ab.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 27.Reindl W, Graber M, Strebhardt K, Berg T. Anal Biochem. 2009;395:189–194. doi: 10.1016/j.ab.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 28.Burke TR, Jr, Smyth MS, Otaka A, Nomizu M, Roller PP, Wolf G, Case R, Shoelson SE. Biochemistry. 1994;33:6490–6494. doi: 10.1021/bi00187a015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.