

SUMMARY
How type I/II interferons prevent periodic reemergence of latent pathogens in tissues of diverse cell-types remains unknown. Using homogenous neuron cultures latently-infected with herpes simplex virus-1, we show that extrinsic type I or II interferon act directly on neurons to induce unique gene expression signatures and inhibit the reactivation-specific burst of viral genome-wide transcription called Phase I. Surprisingly, interferons suppressed reactivation only during a limited period early in Phase I preceding productive virus growth. Sensitivity to type II interferon was selectively lost if viral ICP0, which normally accumulates later in Phase I, was expressed before reactivation. Thus, interferons suppress reactivation by preventing initial expression of latent genomes but are ineffective once Phase I viral proteins accumulate, limiting interferon action. This demonstrates that inducible reactivation from latency is only transiently sensitive to interferon. Moreover, it illustrates how latent pathogens escape host immune control to periodically replicate by rapidly deploying an interferon-resistant state.
Graphical Abstract
INTRODUCTION
Besides combating acute infections, innate host immune defenses, such as type I / II interferons (IFN), play critical roles controlling persistent infections. Unlike viruses that persist in their host through continuous replication, herpesviruses establish their genomes in a non-replicating, epigenetically silenced state within nuclei called latency (Bloom et al., 2016; Wilson & Mohr, 2012). Periodically, in response to incompletely understood physiological cues or changes in immune status, latent viral episomes reactivate, reentering the productive replication cycle to produce infectious virus progeny. While the ability of IFNs to limit viral replication in productively infected cells by inducing interferon-stimulated genes (ISGs) is well documented (Schoggins, 2014), precisely how and when IFNs act to contain established latent herpesvirus reservoirs prone to episodic reactivation is poorly understood.
Infection with herpes simplex virus type 1 (HSV-1), an α-herpesvirus subfamily member, begins in epithelial cells at mucosal surfaces. While host defenses typically contain primary infections, some virus accesses the peripheral nervous system (PNS) through axon terminals that innervate epithelia and enter neuronal nuclei in ganglia to establish latency. The viral genome is epigenetically silenced, limiting transcription of the approximately 80 virus-encoded ORFs and preventing infectious virus production (reviewed in Roizman & Zhou, 2015; Bloom, 2016). Transcription is restricted to a single locus encoding LAT, the latency-associated transcript that is processed into non-coding, stable intronic-sequence RNAs and several microRNAs (Stevens et al., 1987; Umbach et al., 2008). To reactivate, this repressed state is reversed allowing expression of viral genes required for productive, lytic replication and infectious virus production (reviewed in Bloom et al., 2016; Roizman & Zhou, 2015). Using either isolated ganglia or a cultured neuron model that faithfully displays key hallmarks of latency defined in animal models, NGF-binding to the TrkA receptor tyrosine kinase (RTK) suppressed HSV-1 reactivation (Camarena et al., 2010; Du et al., 2011). Continuous NGF-signaling through phosphatidyl-inositol 3-kinase (PI3K), Akt, mTORC1 and its target the translational repressor 4E-BP1 maintains latency while transient interruption triggers reactivation (Kobayashi et al., 2012b).
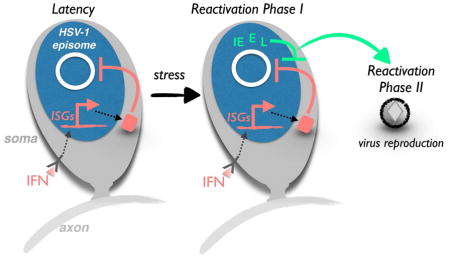
Unexpectedly, viral gene expression during reactivation from latency differed substantially from the temporally-ordered sequence characteristic of acute infection (Du et al., 2011; Kim et al., 2012). Two discrete waves of viral productive cycle gene transcription, termed Phases I and II, were identified in latently-infected cultured neurons following inducible reactivation (Kim et al., 2012). By phosphorylating histone H3 ser10 adjacent to repressive di- or trimethylated H3 lys9, the neuronal JNK stress pathway overrides epigenetic silencing to activate viral promoters in Phase I (Cliffe et al, 2015; Cliffe & Wilson, 2016). The resulting burst of transcription from viral genomes includes all HSV-1 genes irrespective of their kinetic class [immediate-early (IE), early, leaky-late and true-late], is independent of protein or DNA synthesis, and proceeds without infectious virus production. In contrast, Phase II depends upon synthesis and action of the viral transactivator VP16, resulting in DNA replication and infectious virus production. Normally expressed late during acute infection, VP16 is packaged into virions and delivered into newly infected cells where it activates viral IE promoters. The resulting temporally-controlled viral gene expression program proceeds through viral DNA replication and culminates in infectious virus production (Kristie, 2007). Although HSV-1 proteins including ICP0 and ICP27 also accumulate in Phase I (Kim et al., 2012), their contribution to one or both reactivation gene expression phases and ultimately infectious virus production has not been explored.
Besides intrinsic epigenetic silencing of HSV-1 genomes, varied host immune defenses, including infiltrating T-cells that patrol ganglia and cytokines like IFNs impact latency (Cantin et al., 1995; Chen et al., 2000; Khanna et al., 2003; Liu et al., 1996; Shimeld et al., 1995; St Leger & Hendricks, 2011; Leib & Enquist, 2016). While type I (α/β) IFN limits spread at initial infection sites and restricts replication to PNS neurons (Luker et al., 2003), type II (γ) IFN minimally impacts acute replication but helps control reactivation either alone or by synergizing with type I IFN (Cantin et al., 1999; Carr et al., 2009; Decman et al., 2005; Sainz & Halford, 2002). Indeed, IFNs are detected in sensory ganglia from latently-infected mice (Cantin et al., 1995; Chen et al., 2000; Halford et al., 1996, 1997; Shimeld et al., 1997) and promote latency establishment in dissociated ganglia cultures (De Regge et al., 2010; Wigdahl et al., 1982). As neurons reportedly lack an intrinsic response to HSV-1, they rely on and respond to extrinsic IFN sources (Liu et al., 2001; Low-Calle et al., 2014; Rosato & Leib, 2014; Van Opdenbosch et al., 2011). CD8+ T-cells within latently-infected ganglia suppress reactivation in part by producing IFNγ (Liu et al., 2001). In addition, the type I / II IFN signaling molecule STAT1 is essential to control HSV-1 infection in mice (Halford et al., 2006) and humans (Dupuis et al., 2003). Mice lacking STAT1 in neuroectoderm-derived cells, which includes neurons, glia, and astrocytes, could not contain viral replication and pathogenesis upon acute infection.
Paradoxically, while dispensable for establishing latency, fewer reactivation events were observed in ganglia explanted from these mice, perhaps suggesting IFN signaling prevents neuron loss within ganglia after initial infection (Rosato & Leib, 2015; Rosato et al., 2016). Whether STAT1 directly controls reactivation of latent HSV-1 within the neurons themselves, as opposed to other cells, was not addressed. Indeed, the complex tissue architecture of sensory and sympathetic ganglia, comprised of multiple types of neurons, peripheral glia, satellite cells and infiltrating leukocytes (Ernsberger, 2001; Le Pichon and Chesler, 2014; St Leger & Hendricks, 2011; Yang et al., 2000), has hampered efforts to define the mechanism of IFN action in latently-infected neurons. Although IFNs have been proposed to control latency and reactivation (Cantin et al., 1999; Carr et al., 2009; Carr et al., 2003; Decman et al., 2005; Ellison et al., 2000; Liu et al., 2001), precisely how IFNs influence the onset and progression of viral gene expression during reactivation remains unknown.
Here, we capitalize on a primary neuronal culture model of HSV-1 latency to demonstrate that extrinsic IFNs prevent inducible reactivation in a neuron-intrinsic manner. Besides stimulating complex networks of ISG expression, IFNs blocked reactivation downstream of the translation repressor 4E-BP1, a key regulatory point of the NGF-signaling pathway required for latency. Significantly, IFNβ and IFNγ each acted selectively to block Phase I viral genome transcription, preventing viral replication in Phase II. This block was lost when IFN was applied after the onset of Phase I allowing virus reproduction in Phase II to proceed, suggesting IFN functions at an exceptionally early stage of reactivation to contain the viral genome-wide Phase I transcription burst and thereby restrict stress-induced reactivation. While a common subset of host mRNAs accumulated in latently-infected neurons treated with IFNγ or IFNβ, unique IFNβ or γ-specific gene expression signatures were identified and constitute neuron-specific responses. Moreover, delivery of the HSV-1 IFN antagonist ICP0 prior to reactivation selectively suppressed the inhibitory response to IFNγ, but not IFNβ. This establishes that the capacity of IFNs to block HSV-1 reactivation in neurons is restricted to a discrete temporal window that closes prior to Phase II and shows that Phase I viral proteins function in part to overcome host antiviral defenses.
RESULTS
Exogenous IFNβ or IFNγ prevent HSV-1 reactivation in a neuron-intrinsic manner
To determine if IFNs control HSV-1 latency by direct action on neurons, independent of other cell types, we measured the impact of exogenous IFNs on inducible reactivation in primary sympathetic neurons latently-infected with a wild type HSV-1 reporter virus expressing enhanced green fluorescent protein (EGFP) fused to the true-late protein Us11 (Fig.1A). While background spontaneous reactivation levels were observed in control DMSO-treated cultures, application of the PI3K inhibitor LY294002 (LY) for 20 h induced HSV-1 reactivation in agreement with published studies (Fig. 1B,C). Besides enabling control over reactivation-inducing stimuli, this system allowed precise application of either type I or II IFN at defined temporal points. Significantly, inclusion of 100 U/ml IFNβ or IFNγ during and after LY-treatment reduced reactivation as measured by visible EGFP accumulation to 10% (p≤0.0001) or 33% (p≤0.0001) of wells, respectively. IFN similarly suppressed reactivation induced by Akt inhibitor VIII or the mTOR active-site inhibitor PP242 (Fig. S1A,B). The reduction in EGFP accumulation by IFN reflected a more than three orders of magnitude reduction in infectious virus production (Fig 1D). To determine if reactivation suppression required IFN signaling through cognate cell surface receptors, the impact of inhibiting JAK, a critical IFN receptor-interacting kinase, on inducible reactivation was evaluated. While IFNβ and γ each blocked reactivation, inducible reactivation in neurons treated with either IFN in the presence of the JAK inhibitor was not significantly reduced (Fig. 1E,F). While lower IFN concentrations less effectively suppressed reactivation, concentrations greater than 100 U/ml did not further decrease reactivation (Fig. S1C,D). Spontaneous reactivation was also reduced from 22% of wells by 100 U/ml IFNβ or IFNγ alone to 2% or 10% of wells, respectively (Fig 1B,C). Together, these data establish that responses to extrinsic type I or type II IFN are dependent upon JAK activity, which is consistent with the involvement of type I and II IFNs signaling through their respective cell surface receptors, and suppress HSV-1 reactivation in a neuron-intrinsic manner.
Figure 1. Suppression of HSV-1 reactivation by exogenous IFNβ or IFNγ in a neuron cell-intrinsic manner.

A) Protocol for establishing latency and reactivating HSV-1 using homogenous neuronal cultures isolated from rat SCGs. SCGs were isolated, dissociated and cultured for 6 d with mitotic inhibitors to yield a homogenous cultured neuron population (Camarena et al., 2010). Cultures latently-infected with HSV-1 EGFP were established using acyclovir (ACV) to suppress productive virus growth. ACV was removed after 6 d and reactivation induced by treating with the PI3-K inhibitor LY294002 (LY) +/− IFN. After 20 h, LY was removed and incubation continued +/− IFN. Reactivation was quantified by determining the percentage of EGFP-positive wells detected by fluorescence microscopy at 4 d post-treatment. (B) Neurons were treated with 20μM LY +/− 100 U/ml IFNβ for 20 h. At 20h, LY was removed, neurons were cultured +/− IFN, and reactivation measured as described in (A). (C) As in B except that 100 U/ml IFNγ was used. (D) As in B and C except that the amount of infectious virus produced was quantified by plaque assay using Vero cells. (E) As in B except neurons were treated +/− 10 μM JAK Inhibitor for 20 h. At 20h, media containing LY was removed and replaced with media containing IFN and / or JAK Inhibitor. (F) As in E except that 100 U/ml IFNγ was used.
IFN acts on targets downstream of the translation repressor 4E-BP1
Our prior studies demonstrated that continuous PI3K-Akt-mTORC1 signaling is necessary to maintain HSV-1 latency in neurons by inhibiting the translational repressor 4E-BP1, which differentially controls neuronal mRNA translation in response to physiological stress (Kobayashi et al., 2012b). Preventing 4E-BP1 hyperphosphorylation, either with chemical inhibitors of PI3K, Akt, or mTOR or by expressing a constitutively-active 4E-BP1 mutant unable to be phosphorylated by mTORC1, triggers viral reactivation presumably by inhibiting synthesis of proteins needed to maintain latency (Kobayashi et al., 2012b). Because IFN stimulates mTORC1 signaling under certain conditions (Kaur et al., 2007; Saleiro et al., 2015), the impact of IFN on 4E-BP1 phosphorylation was evaluated in latently-infected neuronal cultures induced to reactivate. Compared to the more slowly-migrating, hyperphosphorylated 4E-BP1 isofoms detected in DMSO-treated cultures (control), abundant faster-migrating, hypophosphorylated isoforms accumulated upon LY-induced reactivation (Fig 2A). Importantly, the distribution of 4E-BP1 isoforms in LY-induced cultures was indistinguishable from those treated concurrently with both LY and type I or II IFN. Thus, suppression of reactivation by type I or II IFN does not simply result from restored PI3K-Akt - mTORC1 signaling to preempt activation of the translational repressor 4E-BP1.
Figure 2. Inhibition of reactivation by IFN despite enforced interruption of PI3K-Akt-mTORC1 signaling.

(A) Latently-infected neurons were treated for 20 h with 20 μM LY alone or with 100 U/ml IFNβ or IFNγ. Total protein was isolated, fractionated by SDS-PAGE in a 17.5% gel and analyzed by immunoblotting. Slower-migrating, low abundance hyper-phosphorylated 4E-BP1 isoforms indicate active PI3K-Akt-mTORC1 signaling. Accumulation of faster-migrating, hypophosphorylated 4E-BP1 indicates that PI3-K-Akt-mTORC1 signaling is inhibited. Tubulin is a loading control. (B) Total protein isolated from latently-infected neurons treated as in A was separated by SDS-PAGE and analyzed by immunoblotting using STAT1 and DAXX antibodies. (C) Neurons transduced with a lentiviral vector expressing a doxycycline (dox)-inducible, constitutively-active 4E-BP1 (AA) mutant unresponsive to mTORC1 were treated with dox +/− 100 U/ml IFNβ or IFNγ. HSV-1 UL30 mRNA accumulation measured by qRT-PCR was plotted relative to untreated controls.
Because inhibiting PI3K-Akt-mTORC1 signaling differentially controls neuronal cap-dependent mRNA translation by activating the 4E-BP1 repressor, it could potentially influence ISG-encoded protein abundance (Kaur et al., 2007). To investigate this possibility, accumulation of representative proteins encoded by ISGs was examined. STAT1, one of the most highly induced ISGs in IFNβ and IFNγ-treated neuronal cultures (Yordy et al., 2012), and DAXX, which colocalizes with latent HSV viral episomes in neurons (Catez et al., 2012), were attractive candidates based on their potential to impact latency. STAT1 (Fig. 2B) and DAXX protein (Fig. 2B) and transcript (Fig S2A,B) levels were increased by IFNβ or IFNγ in untreated and LY-treated neurons. This indicates that inducing reactivation by interfering with PI3K-Akt-mTORC1 signaling did not preclude ISG product accumulation in latently-infected neurons.
To investigate the role of 4E-BP1 phosphorylation further without using chemical inhibitors to induce reactivation, a doxycycline (dox)-inducible 4E-BP1 mutant (4E-BP1-AA) that constitutively represses eIF4E and cap-dependent mRNA translation (Kobayashi et al., 2012b) was utilized. Compared to uninduced, latently-infected neurons, accumulation of the UL30 productive growth cycle transcript, an early mRNA encoding the DNA polymerase catalytic subunit, was greatly stimulated by dox-induced 4E-BP1-AA expression (Fig. 2C). Moreover, type I or II IFN reduced UL30 transcript abundance to levels below those observed in latently-infected control cultures, to 10% that of the control for IFNβ (p≤0.001) and 50% that of the control for IFNγ-treated cultures (p≤0.01). This argues that IFN does not directly reverse transmission of the reactivation signal itself, at least at the level of 4E-BP1 phosphorylation, but rather acts downstream to preclude the viral response(s) to the signal or via an independent pathway(s).
IFN restricts reactivation during a discrete window of opportunity within Phase I
Reactivation of HSV-1 from latency in cultured neurons occurs in two distinct phases of lytic gene transcription (Kim et al., 2012). Phase I or animation corresponds to a burst of viral genome-wide transcription without DNA replication or infectious virus production, which are delayed until a second wave of VP16-dependent viral transcription termed Phase II. To ask if these discrete reactivation phases might respond to IFN differently, we examined the consequences of applying IFN at varying times relative to LY, the reactivation inducer. First, latently-infected cultures were treated with DMSO or induced to reactivate with LY in the presence or absence of type I or II IFN. After 20 h, mRNA was isolated and representative productive cycle transcripts analyzed by RT qPCR. Two viral mRNAs were used as markers for Phase I, UL30 and ICP27 (Fig. 3A,B). Consistent with earlier results, both were approximately 8-fold more abundant in LY-treated samples compared to unreactivated, DMSO-treated controls. In contrast, inducing reactivation concurrently with IFNβ or IFNγ treatment reduced accumulation of these viral transcripts to baseline levels (Fig. 3A,B). Similar results were obtained with the HSV-1 KOS strain, showing that the effects of IFN on Phase I transcription are not strain specific (Fig. S3). This shows that both type I and II IFN impair productive cycle transcript accumulation during Phase I and thus counter reactivation at the earliest detectable stage of viral gene expression.
Figure 3. Suppression of reactivation by IFN is limited to a temporal window that closes before Phase II.

(A, B) Latently-infected neurons were treated with 20 μM LY alone or in the presence of 100 U/ml IFNβ or IFNγ. After 20 h, accumulation of HSV-1 lytic Phase I transcripts UL30 (A) or ICP27 (B) was analyzed by qRT-PCR. (C–E) Latently-infected neurons induced to reactivate with LY for 20 h (shaded arrow) were treated with 100 U/ml (D) IFNβ or (E) IFNγ for different periods: during and after the LY pulse (IFN for Phase I & II), only during the LY pulse (IFN for Phase I only), or only after the LY pulse when Phase I has already occurred (IFN for Phase II only). Reactivation was scored 4 d after LY application.
To determine if the responsiveness of reactivating neurons to IFN might be restricted to a precise temporal window, the capacity of IFN to suppress reactivation at various times after induction was evaluated (Fig. 3C–E). IFNβ or IFNγ treatment concurrent with LY-treatment (Phase I & II) or for the first 20 h (Phase I only) effectively suppressed reactivation (Fig. 3D,E). This supports our finding that type I and II IFN limited accumulation of productive growth cycle transcripts and antagonized the generalized burst of viral gene expression that defines Phase I. Surprisingly, inhibition of reactivation was not detected when type I or II IFN was added 20h after LY-application (Fig. 3D,E; see IFN for Phase II only). Indeed, the capacity of IFN to restrict reactivation was progressively lost when IFNβ or IFNγ was added between 5–10h after LY (Fig. S4A,B). Thus, the capacity of IFN to antagonize reactivation was diminished 5–10h after the onset of reactivation and lost entirely by 20h. This correlates with the beginning of Phase I and the transition to Phase II. Furthermore, it suggests that type I or II IFN cannot counter reactivation after Phase I has been fully engaged.
Discrete gene expression signatures in latently-infected neurons treated with IFNβ or IFNγ
While ISG induction by IFNs is well established, how gene expression is modified in neurons during HSV-1 reactivation, potentially shaping cell intrinsic neuronal responses, is not known (Leib & Enquist, 2016). To define the impact of IFNs on neuronal gene expression during HSV-1 reactivation, RNA was isolated from latently-infected neuron cultures treated with LY in the presence of IFNβ or IFNγ and analyzed by RNA-seq (Fig. 4). Using a fold change cut off of log2 > |0.5|, 266 and 437 responsive genes were respectively identified. The vast majority of these host genes were up-regulated in response to either IFNβ (Fig. 4A) or IFNγ (Fig. 4B). Clustered heat maps of all IFN responsive genes reveal distinct signatures unique to IFNβ or IFNγ (Fig. 4C). Among IFN-regulated transcripts, 176 responded similarly in both IFNβ and IFNγ-treated neurons (Fig. 4C,D; Table S1) and included many genes with known roles in anti-viral defenses and IFN signaling such as Mx1/2, STAT1/2, ISG15, IFIT2, IRF1/7/9, Daxx, PKR, TAP1, and USP18 (Schoggins, 2014). Nicotinamide phosphoribosyltransferase (Nampt) was induced by IFNβ and IFNγ (Table S1) and had been previously identified in a proteomic study of ISG products in uninfected axons (Song et al, 2016). By catalyzing the rate-limiting step in NAD+ biosynthesis, Nampt could conceivably counter the rapid decline of NAD+ associated with axonal injury in response to infection (Gerdts et al, 2016). In terms of unique expression signatures, transcripts for 90 genes were selectively altered by IFNβ-treatment, compared to 261 genes in response to IFNγ (Fig 4D; Table S2, S3). Genes specific to the IFNβ signature are involved in serotonin receptor signaling, complement function, agrin interactions at neuromuscular junctions, dopamine receptor signaling, natural killer cell signaling, and bacterial or viral pattern recognition receptor pathways (Fig. 4E, Table S4). In contrast, genes induced uniquely by IFNγ were those from the acute phase response to inflammation, death receptor function, apoptosis, IFN, TWEAK, TREM1, and TNFR1 signaling pathways (Fig 4F). This includes genes important for RIPK1-mediated regulated necrosis and TNFR induced NFKB signaling pathways (Tables S5). Significantly, we identified 36 genes enriched in neurons, 22 of which were not previously recognized as IFN inducible (Fig. 4G, Table S6) and might represent neuron-specific ISGs. Overall, this analysis shows that while IFNγ or IFNβ-treated neurons undergoing reactivation express a common set of genes, they also express unique IFNβ or γ-specific gene signatures that potentially modify the overall anti-viral response. It further raises the possibility that different virus-encoded effectors might preferentially antagonize discrete IFNβ or IFNγ-induced states.
Figure 4. Treatment with IFNβ and IFNγ elicit unique profiles of neuronal gene expression in reactivating neurons.

Volcano plots of genes (blue and orange points) differentially expressed following IFNβ (A) or IFNγ (B) treatment. Dotted line indicates y=0.01, vertical solid lines indicate the × = −0.5 and × = 0.5 thresholds. (C) Clustered heat maps of all differentially expressed genes showing log2 fold-change values in either condition. (D) Venn diagram depicting significant overlaps between genes whose expression changes in response to IFNβ or IFNγ. The significance of overlap was calculated using a hypergeomeric test. (E–F) The top 6 pathways enriched among genes induced uniquely by (E) IFNβ or (F) IFNγ. (G) Interferon induced genes were compared to a reference transcriptomic dataset of CNS cell types (Zhang et al, 2014). Heat map depicts the top 10 neuron-enriched genes and respective enrichment scores for each cell type.
IFNβ but not IFNγ can block HSV-1 reactivation in the presence of viral IE protein ICP0
If Phase I functions in part to counter IFN action, ectopic expression of HSV-1 proteins produced in Phase I might alter the responsiveness of latently-infected neurons to IFN. To test this, we asked if ectopic expression of a single viral factor, ICP0, would be sufficient to overcome the IFN-mediated block to Phase I. We chose ICP0 because it is a multifunctional protein with E3 ligase activity that stimulates viral gene expression and antagonizes host antiviral defenses, including IFN signaling (reviewed in Boutell & Everett, 2013). Moreover, ICP0 is required for efficient reactivation from latency (Halford & Schaffer, 2001; Thompson & Sawtell, 2006) and its ectopic expression is sufficient to trigger reactivation in mixed cultures prepared from latently-infected, murine trigeminal ganglia (Halford et al., 2001). Recently, it was found that ICP0 is present at low levels during latency where it helps to sustain the latent program (Raja et al., 2016). However, ICP0 synthesis is markedly upregulated during reactivation Phase I (Kim et al., 2012), raising the possibility that ICP0 might antagonize IFN action and contribute to the IFN insensitivity.
Cultured neurons latently-infected with HSV-1 were transduced with replication-defective adenovirus (Ad) vectors that allowed gene delivery to essentially all neurons in the culture. In agreement with in vitro studies using dissociated, latently-infected ganglia (Halford et al., 2001), an ICP0 expressing Ad (Ad-ICP0) was sufficient to induce reactivation in 84% of wells (Fig. 5A), obviating the need for LY-treatment. A control vector that does not express ICP0 was unable to stimulate reactivation (Fig. 5A). In the presence of Ad-ICP0, IFNγ did not detectably alter the number of EGFP-positive wells, indicating that when ICP0 was supplied either before or during Phase I, it was capable of negating IFNγ antiviral action (Fig 5A). Levels of ICP0 produced in transduced cultures were not significantly altered by IFN either in neurons alone (Fig. 5B) or in latently-infected neurons (Fig. 5C). Surprisingly, ICP0 was not able to overcome IFNβ action when provided in the same context and only 36% (p≤0.0001) of wells were EGFP-positive after 4 days (Fig 5A). In contrast, reactivation induced by Ad expressing VP16, a distinct HSV-1 protein made in Phase I, remained sensitive to either IFNβ or IFNγ similar to LY-induced reactivation (Fig. S5). This shows that ICP0 antagonizes the actions of IFNγ but not IFNβ in reactivating neurons and suggests that other viral functions produced in Phase I contribute to countering IFNβ. Moreover, expression of an HSV-1 protein prior to the onset of Phase I can alter the temporal interferon sensitivity profile of reactivating neurons. This is consistent with a biological role for Phase I proteins in countering host defenses and establishing a distinctive state that supports productive viral growth.
Figure 5. IFNβ but not IFNγ counters HSV-1 reactivation even in the presence of the viral protein ICP0.

ICP0 was ectopically expressed from a dox-inducible promoter using a recombinant adenovirus vector in neurons latently-infected with HSV-1. (A) The percentage of wells in which HSV-1 had reactivated −/+ 100 U/ml IFNβ or IFNγ was scored 4 d after inducing ICP0 expression. After 4 d of dox treatment, total protein was isolated from (B) neurons not latently-infected with HSV-1 or (C) latently-infected with HSV-1 and ICP0 accumulation was measured by immunoblotting. (D) Model depicting how a viral genome-wide expression burst or virion protein delivery represent different strategies to counter anti-viral defenses and promote productive viral growth during acute infection or reactivation. During acute infection, the tegument delivers a subset of viral proteins from different kinetic classes into newly infected cells that antagonize host cell-intrinsic immunity and foster reproductive replication. Unlike acute infection, which begins with virion entry and tegument deposition, reactivation begins with de novo expression of latent HSV-1 genomes organized in compacted chromatin within neuronal nuclei. Instead of delivering a preformed tegument loaded with viral proteins into the host, reactivation Phase I in neurons results in a genome-wide burst of viral gene expression allowing the simultaneous accumulation of viral proteins of all kinetic classes without viral DNA replication or infectious virus production. Both strategies achieve a similar function by providing a toolkit of viral proteins capable of countering cell-intrinsic host defenses, including responses to IFN and ISGs, and stimulating productive virus growth.
DISCUSSION
While control of HSV latency and reactivation by type I/II IFNs is well established (Cantin et al., 1999; Carr et al., 2009; Carr et al., 2003; Decman et al., 2005; Ellison et al., 2000; Liu et al., 2001), exactly where and how these cytokines act remains unclear. Sensory and autonomic ganglia harboring latent virus are complex, heterogenous tissues comprised of different neuron subtypes and an even greater number of non-neuronal cells, including support cells and lymphocytes (Ernsberger, 2001; Le Pichon and Chesler, 2014; St Leger and Hendricks, 2011; Yang et al., 2000). The involvement of numerous different cell types has compounded the difficulties in discerning precisely how IFNs antagonize reactivation within infected neurons and the relative contributions of non-neuronal cells. Indeed, evidence that tissue resident, HSV-1 specific CD8+ T-cells contain rather than prevent sub-clinical reactivation episodes underscores the importance of deciphering neuron-intrinsic control mechanisms (Mark et al. 2008; Zhu et al, 2013).
Using a latency model consisting of homogenous neuron cultures and wild type virus, we demonstrate that IFNβ and IFNγ act directly on neurons to induce unique, neuron-specific transcriptional responses that prevent Phase I, the initial viral genome-wide burst of transcription. By blocking Phase I, the earliest known response of HSV to reactivation stimuli, damage to otherwise irreplaceable neurons is likely averted. Unexpectedly, reactivation suppression by IFN was attenuated after the initiation of Phase I and subsequent IFN application did not restrict viral gene transcription. Furthermore, ectopic HSV-1 ICP0 expression prior to Phase I selectively protected reactivating neurons from IFNγ, but not IFNβ. The surprising finding that IFN acts within a limited window and is antagonized by a Phase I viral protein implicates the viral products of Phase I in the establishment of an IFN resistant state that allows reactivating virus to proceed into active replication during Phase II (Fig. 5D).
So how does IFN block Phase I transcription? Because reactivation induced by expressing a constitutively active 4E-BP1 translational repressor remained IFN sensitive, we conclude that IFN did not simply antagonize reactivation cues by stimulating PI3K-Akt-mTORC1 signaling. Thus, targets of IFN action are likely down-stream of 4E-BP1 and/or function in an alternate pathway(s). Unlike mitotic cells, DRG neurons reportedly limit HSV-1 lytic replication through IFN-induced autophagy (Yordy et al., 2012). While a role for IFN-induced autophagy cannot be discounted, we believe it unlikely as IFNs suppressed reactivation induced by LY, which inhibits PI3Ks required for autophagy (Blommaart et al., 1997). Notwithstanding mundane differences between these studies (mouse vs. rat or sympathetic vs. sensory ganglia), it potentially highlights different IFN-induced antiviral responses to acute HSV-1 infection compared to those that control reactivation.
Alternatively, IFN might maintain epigenetic silencing of viral lytic genes even in the presence of a reactivation stimulus. Recently Cliffe and colleagues showed that LY-treatment of latently-infected neurons activates c-Jun N-terminal kinase (JNK) resulting in phosphorylation of histone H3 ser10 adjacent to di- or tri-methylated lys9 on viral chromatin and reversing epigenetic repression (Cliffe et al. 2015). IFN signaling may directly antagonize this de-repression step to maintain HSV-1 genome silencing by blocking JNK or by clearing the phosphorylation mark. JNK phosphatases were not identified among IFNβ- or IFNγ-induced transcripts but there were increases in molecules, such as ISG15, known to manipulate a variety of signaling pathways. Likewise, ISGs could potentially modify or change the availability of JNK interacting proteins 3 (JIP3) and dual leucine zipper kinase (DLK), neuronal scaffold components required for JNK activation (Di et al, 2010; Holland et al, 2016). Lastly, IFN might activate additional mechanisms of transcriptional silencing that cannot be displaced by JNK-mediated phosphorylation, effectively bypassing the reactivation cue.
Given that Phase I results in IE, E and L viral mRNA accumulation, all viral productive cycle polypeptides are likely synthesized to some level. However, it is not known how many are correctly localized or available in sufficient abundance to be functional. Remarkably, overexpression of just a single viral protein, the SUMO dependent E3-ligase ICP0, prior to the application of IFN allowed the latent virus to reactivate in the presence of IFNγ, but interestingly, not with IFNβ. Surprisingly, delivery of VP16, another abundant Phase I protein, did not establish an effective IFN resistant state, even though it provided a functional reactivation stimulus. This seems counterintuitive because VP16 stimulates transcription of HSV-1 IE genes, including ICP0. The failure of VP16 to mimic direct delivery of ICP0 may be due to retention of VP16 in the cytoplasm during Phase I (Kim et al., 2012), resulting in reduced ICP0 accumulation. This might be compounded by miR138, an abundant neuronal microRNA that inhibits translation of ICP0 mRNAs produced from endogenous HSV-1 genomes (Pan et al. 2014).
Earlier studies reported that ICP0 countered IFN-induced antiviral responses in acutely-infected, non-neuronal cells (Boutell et al., 2011; Eidson et al., 2002; Harle et al., 2002; Mossman et al., 2000; Orzalli et al., 2012). Specificity with regard to type I and type II IFN-mediated responses has not been described and probably reflects the unique ISG expression signatures elicited by IFNβ and IFNγ in neurons as revealed by our RNA-Seq analysis. There is growing appreciation for the differences between IFN action in neurons and other cell types. For instance, restriction of HSV-1 replication in acutely-infected neurons by IFNβ occurs through separate mechanisms in the axonal and soma compartments (Rosato & Leib, 2015).
Exactly how ICP0 antagonizes IFNγ but not IFNβ action requires further study but likely involves host factors unique to the IFNγ profile. This category included genes associated with controlling programmed cell death and TNF superfamily signaling such as tumor necrosis factor receptor superfamily members [TNFSF] 6 (FAS/TNFSF6); TNFSF12A (TWEAK/TNFSF12). This was also true for receptor interacting protein kinase 1 (RIPK1), TNF receptor 1 (TNFR1), and caspase 8 and Fadd-like apoptosis regulator (CFLAR). ICP0 might antagonize these pathways directly, or by stimulating expression of other viral genes like ICP6 (Sze & Herman, 1992). ICP6 blocks caspase 8-dependent apoptosis in both rodent and human cells, and can suppress TNF-induced necroptosis in human cells by binding to RIPK1 (Dufour et al, 2011; Guo et al, 2015).
Although exposure to IFN is sufficient to trigger neuron-intrinsic, anti-viral responses that control reactivation, murine neurons produce very little IFN in response to acute HSV-1 infection and other cell types are the more likely sources of IFN (Rosato & Leib, 2014). HSV-specific CD8+ T-cells resident within ganglia secrete IFNγ (St Leger & Hendricks, 2011), which is thought to preferentially control reactivation (Cantin et al., 1999; Carr et al., 2009; Decman et al., 2005; Sainz & Halford, 2002). Other cell types, including those in peripheral tissues innervated by axons, are likely sources of IFNβ. The markedly different neuronal responses to IFNβ and IFNγ might engender different outcomes dependent upon the viral countermeasures deployed in Phase I.
Simultaneous expression of genes from all kinetic classes during Phase I equips the reactivating virus with a broad tool set to effectively defend against host antiviral responses triggered by the new viral activity. This creates an IFN-resistant state that precedes the onset of productive viral replication in reactivating neurons and is likely a critical step facilitating completion of the viral life-cycle, perhaps counteracting host neuron-intrinsic transcriptional responses to HSV-1 reactivation (Ma et al., 2014). Because ISG expression in response to infection and latent virus reactivation is stochastic (Rand et al., 2012; Teng et al., 2012), antagonism of IFNγ by ICP0 in Phase I might be a general strategy to increase the likelihood of progressing into Phase II, biasing the stochastic process in favor of reactivation. IE1 encoded by the β-herpesvirus HCMV is functionally analogous to ICP0, and might similarly help to establish an IFN-resistant state upon reactivation. The ability to establish IFN resistance at the onset of reactivation might also be a feature of other latent viruses such as HIV (Weinberger & Weinberger, 2013). During productive herpesvirus infection, a similar outcome favoring acute replication is achieved in part by selectively packaging numerous late proteins, including HSV-1 VP16, into the virion tegument for release into the cytoplasm at the beginning of infection, creating a permissive environment for viral productive replication (Kelly et al., 2009). Although tegument proteins are not present at the onset of reactivation, which begins with de novo viral gene expression from latent genomes (Du et al., 2011; Kim et al., 2012), the Phase I viral genome-wide expression burst solves this problem by allowing viral proteins of all kinetic classes to accumulate simultaneously without viral DNA replication or infectious virus production (Fig. 5D). Using two distinct routes to achieve a similar goal may have allowed HSV-1 to evolve a latency strategy requiring few if any viral proteins.
EXPERIMENTAL PROCEDURES
Cell Culture and Establishment of HSV-1 Latency
Superior cervical ganglia (SCG) neurons were isolated, cultured and latently-infected as described (Kim et al., 2014; Kobayashi et al., 2012a). Briefly, SCGs isolated from E21 Sprague-Dawley® rat pups were incubated at 37°C in trypsin (0.1%, Invitrogen) and collagenase (10 μg/ml, Sigma) for 20 min. After passage through 21G and 23G needles, dissociated cells were passed through a 70-μm cell strainer. 5000 cells/well were plated in 96-well plates coated with rat-tail collagen (0.66 mg/ml, Millipore) and laminin (2 mg/ml, Sigma). Cells were cultured in neurobasal medium (Invitrogen) supplemented with NGF (50 ng/ml, Harlan Laboratories) and with aphidicolin (5 mM, Calbiochem) and 5-fluorouracil (20 mM, Sigma) to eliminate proliferating cells. After 5–6 d in culture, acyclovir (ACV; 50 mM, Calbiochem) was added and the following day neuron cultures were infected with wild-type HSV-1 (Patton strain) expressing an EGFP-Us11 fusion protein (MOI=1 based on titer determined in Vero cells). The virus was removed after 2 h and the neurons maintained in culture with ACV until reactivation.
Induction of HSV-1 reactivation
After 12–14 d in vitro, (6–7 d after HSV-1 infection), ACV was removed and cultures were induced to reactivate by treatment with LY294002 (20 μM, Calbiochem), PP242 (2.5 μM, Sigma) or Akt Inhibitor VIII (5 μM, Millipore) for 20 h. IFNβ and IFNγ (100U/ml, PBL Interferon Source) and JAK Inhibitor (10 μM, Millipore) were added as indicated. For experiments measuring Phase I viral transcript accumulation, neurons were harvested after 20 h for qRT-PCR analysis. For assessing reactivation using EGFP fluorescence, neurons were re-fed with fresh media lacking inducer and wells visually scored over 4 d. Each data point represents 20 wells with data collected across a minimum of three separate experiments. Graphs and statistical calculations were made using Prism 5.0 software (GraphPad) with error bars indicating SEM.
RNA isolation, RNA-seq library construction and sequencing
Primary rat sympathetic neuron cultures latently infected with HSV-1 were induced to reactivate with LY in the presence or absence of IFNβ or IFNγ. After 20 h, total RNA was extracted using Trizol (Life Technologies) and precipitated with ethanol. Following treatment with DNase I (Promega), phenol-CHCl3 extraction, and ethanol precipitation, RNA yield and concentration was determined by absorption spectroscopy using a NanoDrop (NanoDrop Products). All samples were prepared in 3 biological replicates. RNA-seq libraries were generated following Illumina TruSeq protocols for rRNA removal using Ribo-Zero, fragmentation, and adaptor ligation. Multiplexed libraries were sequenced on HiSeq-2500 platform at the New York Genome Center, generating 125-bp paired end reads. RNAseq data has been deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) and is accessible through GEO Series accession number GSE90744.
Supplementary Material
Acknowledgments
We thank Jennifer Zalk for technical assistance, the late Priscilla Schaffer for generously providing adenovirus recombinants, and Wilson, Chao and Mohr lab members for helpful discussions. This work was supported by an individual NRSA fellowship to JAL (NS086333), HHMI support to RBD, and NIH grants to MVC (NS21072), ACW (AI103933, AI105896), RBD (NS34389, NS06943, NS081706) and IM (AI073898 and GM056927).
Footnotes
ACCESSION NUMBERS
The accession number for the RNA seq data reported in this paper is NCBI GEO: GSE90744.
AUTHOR CONTRIBUTIONS
J.A.L. and M.K. designed and conducted experiments and prepared figures. J.J.F. assisted MK with the RNA seq experiment, and V.R. performed the experiment in Fig S3. J.A.L., M.K., M.V.C., A.C.W, and I.M. designed experiments, evaluated and interpreted data, and wrote the manuscript. J.A.L., R.B.D., M.V.C., A.C.W, and I.M. acquired financial support and provide resources.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- Bloom DC. Alphaherpesvirus Latency: A Dynamic State of Transcription and Reactivation. Adv Virus Res. 2016;94:53–80. doi: 10.1016/bs.aivir.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011;7:e1002245. doi: 10.1371/journal.ppat.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C, Everett RD. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol. 2013;94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- Cai W, Schaffer PA. A cellular function can enhance gene expression and plating efficiency of a mutant defective in the gene for ICP0, a transactivating protein of herpes simplex virus type 1. J Virol. 1991;65:4078–4090. doi: 10.1128/jvi.65.8.4078-4090.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe. 2010;8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin E, Tanamachi B, Openshaw H. Role for gamma interferon in control of herpes simplex virus type 1 reactivation. J Virol. 1999;73:3418–3423. doi: 10.1128/jvi.73.4.3418-3423.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin EM, Hinton DR, Chen J, Openshaw H. Gamma interferon expression during acute and latent nervous system infection by herpes simplex virus type 1. J Virol. 1995;69:4898–4905. doi: 10.1128/jvi.69.8.4898-4905.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DJ, Austin BA, Halford WP, Stuart PM. Delivery of Interferon-gamma by an adenovirus vector blocks herpes simplex virus Type 1 reactivation in vitro and in vivo independent of RNase L and double-stranded RNA-dependent protein kinase pathways. J Neuroimmunol. 2009;206:39–43. doi: 10.1016/j.jneuroim.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DJJ, Al-khatib K, James CM, Silverman R. Interferon-β suppresses herpes simplex virus type 1 replication in trigeminal ganglion cells through an RNase L-dependent pathway. J Neuroimmunol. 2003;141:40–46. doi: 10.1016/s0165-5728(03)00216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catez F, Picard C, Held K, Gross S, Rousseau A, Theil D, Sawtell N, Labetoulle M, Lomonte P. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog. 2012;8:e1002852. doi: 10.1371/journal.ppat.1002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee AV, Lopez P, Pandolfi PP, Roizman B. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol. 2003;77:7101–7105. doi: 10.1128/JVI.77.12.7101-7105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Garber DA, Schaffer PA, Knipe DM, Coen DM. Persistent elevated expression of cytokine transcripts in ganglia latently infected with herpes simplex virus in the absence of ganglionic replication or reactivation. Virology. 2000;278:207–216. doi: 10.1006/viro.2000.0643. [DOI] [PubMed] [Google Scholar]
- Cliffe AR, Wilson AC. Restarting Lytic Gene Transcription at the Onset of Herpes Simplex Virus Reactivation. J Virol. 2016 doi: 10.1128/JVI.01419-16. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Regge N, Van Opdenbosch N, Nauwynck HJ, Efstathiou S, Favoreel HW. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS One. 2010;5:e13076. doi: 10.1371/journal.pone.0013076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decman V, Kinchington PR, Harvey SA, Hendricks RL. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J Virol. 2005;79:10339–10347. doi: 10.1128/JVI.79.16.10339-10347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di A, Kawamura T, Gao XP, Tang H, Berdyshev E, Vogel SM, Zhao YY, Sharma T, Bachmaier K, Xu J, Malik AB. A novel function of sphingosine kinase 1 suppression of JNK activity in preventing inflammation and injury. J Biol Chem. 2010;285:15848–15857. doi: 10.1074/jbc.M109.075549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Zhou G, Roizman B. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc Natl Acad Sci U S A. 2011;108:18820–18824. doi: 10.1073/pnas.1117203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour F, Sasseville AM, Chabaud S, Massie B, Siegel RM, Langelier Y. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFα- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis. 2011;16:256–271. doi: 10.1007/s10495-010-0560-2. [DOI] [PubMed] [Google Scholar]
- Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- Eidson KM, Hobbs WE, Manning BJ, Carlson P, DeLuca NA. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J Virol. 2002;76:2180–2191. doi: 10.1128/jvi.76.5.2180-2191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison AR, Yang L, Voytek C, Margolis TP. Establishment of latent herpes simplex virus type 1 infection in resistant, sensitive, and immunodeficient mouse strains. Virology. 2000;268:17–28. doi: 10.1006/viro.1999.0158. [DOI] [PubMed] [Google Scholar]
- Enquist LW, Leib DA. Intrinsic and innate defenses of neurons: Détente with the herpesviruses. J Virol. 2016 doi: 10.1128/JVI.01200-16. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernsberger U. The development of postganglionic sympathetic neurons: coordinating neuronal differentiation and diversification. Auton Neurosci. 2001;94:1–13. doi: 10.1016/S1566-0702(01)00336-8. [DOI] [PubMed] [Google Scholar]
- Everett RD, Parada C, Gripon P, Sirma H, Orr A. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol. 2008;82:2661–2672. doi: 10.1128/JVI.02308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol. 2006;80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, DiAntonio A. Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron. 2016;89:449–460. doi: 10.1016/j.neuron.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17:243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Gebhardt BM, Carr DJ. Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J Immunol. 1996;157:3542–3549. [PubMed] [Google Scholar]
- Halford WP, Gebhardt BM, Carr DJ. Acyclovir blocks cytokine gene expression in trigeminal ganglia latently infected with herpes simplex virus type 1. Virology. 1997;238:53–63. doi: 10.1006/viro.1997.8806. [DOI] [PubMed] [Google Scholar]
- Halford WP, Kemp CD, Isler JA, Davido DJ, Schaffer PA. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J Virol. 2001;75:6143–6153. doi: 10.1128/JVI.75.13.6143-6153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Schaffer PA. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J Virol. 2001;75:3240–3249. doi: 10.1128/JVI.75.7.3240-3249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Weisend C, Grace J, Soboleski M, Carr DJ, Balliet JW, Imai Y, Margolis TP, Gebhardt BM. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virol J. 2006;3:44. doi: 10.1186/1743-422X-3-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harle P, Sainz B, Jr, Carr DJ, Halford WP. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology. 2002;293:295–304. doi: 10.1006/viro.2001.1280. [DOI] [PubMed] [Google Scholar]
- Holland SM, Collura KM, Ketschek A, Noma K, Ferguson TA, Jin Y, Gallo G, Thomas GM. Palmitoylation controls DLK localization, interactions and activity to ensure effective axonal injury signaling. Proc Natl Acad Sci USA. 2016;113:763–768. doi: 10.1073/pnas.1514123113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014;10:e1004503. doi: 10.1371/journal.ppat.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Lal L, Sassano A, Majchrzak-Kita B, Srikanth M, Baker DP, Petroulakis E, Hay N, Sonenberg N, Fish EN, et al. Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J Biol Chem. 2007;282:1757–1768. doi: 10.1074/jbc.M607365200. [DOI] [PubMed] [Google Scholar]
- Kelly BJ, Fraefel C, Cunningham AL, Diefenbach RJ. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 2009;145:173–186. doi: 10.1016/j.virusres.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity. 2003;18:593–603. doi: 10.1016/s1074-7613(03)00112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 2012;8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Shiflett LA, Linderman JA, Mohr I, Wilson AC. Using homogeneous primary neuron cultures to study fundamental aspects of HSV-1 latency and reactivation. Methods Mol Biol. 2014;1144:167–179. doi: 10.1007/978-1-4939-0428-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Kim JY, Camarena V, Roehm PC, Chao MV, Wilson AC, Mohr I. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J Vis Exp. 2012a Apr 2;(62) doi: 10.3791/3823.. pii: 3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Wilson AC, Chao MV, Mohr I. Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev. 2012b;26:1527–1532. doi: 10.1101/gad.190157.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM. Early events pre-initiation of alphaherpes viral gene expression. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Chapter 8. Cambridge: 2007. [PubMed] [Google Scholar]
- Le Pichon CE, Chesler AT. The functional and anatomical dissection of somatosensory subpopulations using mouse genetics. Front Neuroanat. 2014;8:21. doi: 10.3389/fnana.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Khanna KM, Carriere BN, Hendricks RL. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J Virol. 2001;75:11178–11184. doi: 10.1128/JVI.75.22.11178-11184.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Tang Q, Hendricks RL. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J Virol. 1996;70:264–271. doi: 10.1128/jvi.70.1.264-271.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low-Calle AM, Prada-Arismendy J, Castellanos JE. Study of interferon-beta antiviral activity against Herpes simplex virus type 1 in neuron-enriched trigeminal ganglia cultures. Virus Res. 2014;180:49–58. doi: 10.1016/j.virusres.2013.12.022. [DOI] [PubMed] [Google Scholar]
- Luker GD, Prior JL, Song J, Pica CM, Leib DA. Biolumin-scence Imaging Reveals Systemic Dissemination of Herpes Simplex Virus Type 1 in the Absence of Interferon Receptors. J Virol. 2003;77:11082–11093. doi: 10.1128/JVI.77.20.11082-11093.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response. PLoS Pathog. 2014;10:e1004237. doi: 10.1371/journal.ppat.1004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark KE, Wald A, Magaret AS, Selke S, Olin L, Huang ML, Corey L. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis. 2008;198:1141–1149. doi: 10.1086/591913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman KL, Saffran HA, Smiley JR. Herpes Simplex Virus ICP0 Mutants Are Hypersensitive to Interferon. J Virol. 2000;74:2052–2056. doi: 10.1128/jvi.74.4.2052-2056.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negorev DG, Vladimirova OV, Ivanov A, Rauscher F, 3rd, Maul GG. Differential role of Sp100 isoforms in interferon-mediated repression of herpes simplex virus type 1 immediate-early protein expression. J Virol. 2006;80:8019–8029. doi: 10.1128/JVI.02164-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A. 2012;109:E3008–3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladino P, Mossman KL. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J Interfer Cytok Res. 2009;29:599–607. doi: 10.1089/jir.2009.0074. [DOI] [PubMed] [Google Scholar]
- Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, Rosato PC, Leib DA, Cullen BR, Coen DM. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe. 2014;15:446–456. doi: 10.1016/j.chom.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja P, Lee JS, Pan D, Pesola JM, Coen DM, Knipe DM. A Herpesviral Lytic Protein Regulates the Structure of Latent Viral Chromatin. MBio. 2016;7(3) doi: 10.1128/mBio.00633-16. pii: e00633–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand U, Rinas M, Schwerk J, Nöhren G, Linnes M, Kröger A, Flossdorf M, Kály-Kullai K, Hauser H, Höfer T, et al. Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol Syst Biol. 2012;8:584. doi: 10.1038/msb.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Zhou G. The 3 facets of regulation of herpes simplex virus gene expression: A critical inquiry. Virology. 2015;479–480:562–567. doi: 10.1016/j.virol.2015.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Leib DA. Intrinsic innate immunity fails to control herpes simplex virus and vesicular stomatitis virus replication in sensory neurons and fibroblasts. J Virol. 2014;88:9991–10001. doi: 10.1128/JVI.01462-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Leib DA. Neuronal Interferon Signaling Is Required for Protection against Herpes Simplex Virus Replication and Pathogenesis. PLoS Pathog. 2015;11:e1005028. doi: 10.1371/journal.ppat.1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Katzenell S, Pesola JM, North B, Coen DM, Leib DA. Neuronal IFN signaling is dispensable for the establishment of HSV-1 latency. Virology. 2016;497:323–327. doi: 10.1016/j.virol.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks WR, Schaffer PA. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J Virol. 1987;61:829–839. doi: 10.1128/jvi.61.3.829-839.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainz B, Halford WP. Alpha/Beta Interferon and Gamma Interferon Synergize To Inhibit the Replication of Herpes Simplex Virus Type 1. J Virol. 2002;76:11541–11550. doi: 10.1128/JVI.76.22.11541-11550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleiro D, Mehrotra S, Kroczynska B, Beauchamp EM, Lisowski P, Majchrzak-Kita B, Bhagat TD, Stein BL, McMahon B, Altman JK, et al. Central role of ULK1 in type I interferon signaling. Cell Rep. 2015;11:605–617. doi: 10.1016/j.celrep.2015.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol. 2014;6:40–46. doi: 10.1016/j.coviro.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimeld C, Whiteland JL, Nicholls SM, Grinfeld E, Easty DL, Gao H, Hill TJ. Immune cell infiltration and persistence in the mouse trigeminal ganglion after infection of the cornea with herpes simplex virus type 1. J Neuroimm. 1995;61:7–16. doi: 10.1016/0165-5728(95)00068-d. [DOI] [PubMed] [Google Scholar]
- Shimeld C, Whiteland JL, Williams NA, Easty DL, Hill TJ. Cytokine production in the nervous system of mice during acute and latent infection with herpes simplex virus type 1. J Gen Virol. 1997;78(Pt 12):3317–3325. doi: 10.1099/0022-1317-78-12-3317. [DOI] [PubMed] [Google Scholar]
- Song R, Koyuncu OO, Greco TM, Diner BA, Cristea IM, Enquist LW. Two Modes of the Axonal Interferon Response Limit Alphaherpesvirus Neuroinvasion. MBio. 2016;7(1):e02145–15. doi: 10.1128/mBio.02145-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Leger AJ, Hendricks RL. CD8+ T cells patrol HSV-1-infected trigeminal ganglia and prevent viral reactivation. J Neurovirol. 2011;17:528–534. doi: 10.1007/s13365-011-0062-1. [DOI] [PubMed] [Google Scholar]
- Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science. 1987;235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- Sze P, Herman RC. The herpes simplex virus type 1 ICP6 gene is regulated by a ‘leaky’ early promoter. Virus Res. 1992;26:141–152. doi: 10.1016/0168-1702(92)90153-z. [DOI] [PubMed] [Google Scholar]
- Teng MW, Bolovan-Fritts C, Dar RD, Womack A, Simpson ML, Shenk T, Weinberger LS. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell. 2012;151:1569–1580. doi: 10.1016/j.cell.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J Virol. 2006;80:10919–10930. doi: 10.1128/JVI.01253-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Opdenbosch N, De Regge N, Van Poucke M, Peelman L, Favoreel HW. Effects of interferon on immediate-early mRNA and protein levels in sensory neuronal cells infected with herpes simplex virus type 1 or pseudorabies virus. Vet Microbiol. 2011;152:401–406. doi: 10.1016/j.vetmic.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Weinberger AD, Weinberger LS. Stochastic fate selection in HIV-infected patients. Cell. 2013;155:497–499. doi: 10.1016/j.cell.2013.09.039. [DOI] [PubMed] [Google Scholar]
- Wigdahl BL, Scheck AC, De Clercq E, Rapp F. High efficiency latency and activation of herpes simplex virus in human cells. Science. 1982;217:1145–1146. doi: 10.1126/science.6180477. [DOI] [PubMed] [Google Scholar]
- Wilson AC, Mohr I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 2012;20:604–611. doi: 10.1016/j.tim.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Voytek CC, Margolis TP. Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J Virol. 2000;74:209–217. doi: 10.1128/jvi.74.1.209-217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe. 2012;12:334–345. doi: 10.1016/j.chom.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–119247. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, Klock A, Jin L, Diem K, Koelle DM, Wald A, et al. Immune surveillance by CD8αα+ skin-resident T cells in human herpes virus infection. Nature. 2013;497:494–497. doi: 10.1038/nature12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.