

Abstract
Lytic polysaccharide monooxygenases have attracted vast attention due to their abilities to disrupt glycosidic bonds via oxidation instead of hydrolysis and to enhance enzymatic digestion of recalcitrant substrates including chitin and cellulose. We have determined high resolution X-ray crystal structures of an enzyme from Neurospora crassa in the resting state and of a copper(II)–dioxo intermediate complex formed in the absence of substrate. X-ray crystal structures also revealed “pre-bound” molecular oxygen adjacent to the active site. An examination of protonation states enabled by neutron crystallography and density functional theory calculations identified a role for a conserved histidine in promoting oxygen activation. These results provide a new structural description of oxygen activation by substrate free lytic polysaccharide monooxygenases and provide insights that can be extended to reactivity in the enzyme–substrate complex.
Keywords: density functional calculations, neutron diffraction, oxidoreductases, polysaccharide monooxygenases, reaction mechanisms
Graphical abstract
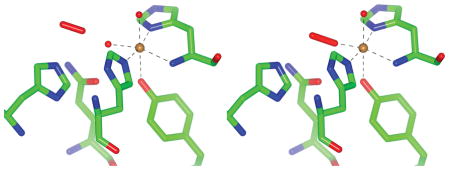
X-ray crystallography produces the first structure of an LPMO enzyme with an activated dioxo species bound in the plane of the histidine brace. A “pre-binding” site for molecular oxygen is also identified and characterized with neutron protein crystallography and DFT calculations.
Lytic polysaccharide monooxygenases (LPMOs) are copper-dependent metalloenzymes that activate molecular oxygen for the net insertion of one oxygen atom into the carbon–hydrogen bond of a glycosidic carbon in polymeric carbohydrates.[1] This oxygenation leads to spontaneous elimination of the glycosidic bond, polysaccharide chain cleavage and, in the case of crystalline substrates, local disruption of chain ordering.[2] Fungal LPMOs are commonly involved in the saprotrophic metabolism of cellulosic biomass, and this biological role has inspired industrial use of LPMOs to enhance the efficiency of cellulose deconstruction by hydrolytic enzymes that release soluble sugars for biofuels production.[3] Despite industrial use and rapidly growing scientific interest in LPMOs, the reaction mechanism, which overall requires the input of molecular O2, two electrons and two protons, remains poorly understood. It is generally accepted that the mechanism begins with one-electron reduction of the Cu(II) LPMO resting state to the Cu(I) state that has a high affinity for molecular O2. Oxygen binding to the Cu(I) state has been shown to yield rapid oxidation to Cu(II) with the presumed formation of a copper–superoxide complex.[4] Multiple mechanistic pathways exist from this intermediate complex. Superoxide, hydroperoxyl and oxyl, reflecting different redox and protonation states, have each been proposed as the reactive oxygen species responsible for abstracting hydrogen from a glycosidic carbon of the polysaccharide substrate.[3] Additionally, in the absence of substrate the superoxide species can dissociate from Cu(II) leading to the generation of hydrogen peroxide.[5] This communication presents new atomistic details of oxygen activation by a fungal LPMO in the absence of substrate and provides insights for understanding oxygen reactivity in both the productive cellulose disrupting and non-productive hydrogen peroxide generating reactions.
We have determined high resolution (1.20 Å) X-ray crystal structures of a heterologously expressed LPMO from Neurospora crassa, NcPMO-2 (NCU01050; UniProt Q1K8B6), which is a family AA9 LPMO (CAZy database).[6] As was the case for the previously reported structure of natively expressed NcPMO-2 (PDB 4EIR), crystallization occurred in the P21 space group with two protein molecules with non-crystallographic symmetry (NCS) per asymmetric unit.[7] Fig. 1 A–B shows the NcPMO-2 active site in the copper(II) oxidation resting state as determined from synchrotron X-ray diffraction data collected at 100 K (PDB 5TKG). (Dataset and model refinement statistics for all structures are listed in Table S1 and Table S2, respectively. Uncertainties were derived from diffraction precision index analysis of individual atomic coordinate precision; see SI Methods.) The single copper ion is coordinated by the amino (NH2) nitrogen and Nδ of the N-terminal His1 and Nε of His84 in a T-shaped “histidine brace” motif that is conserved in all known PMOs. The hydroxyl group of Tyr168 is oriented toward an axial copper coordination site though with an elongated copper–oxygen distance. The remaining axial and equatorial copper coordination sites are occupied by water molecules. The presence of the axial and equatorial waters in the copper coordination sphere (Cu–H2Oeq = 2.00 Å and 1.96 Å; Cu–H2Oax = 2.44 Å and 2.36 Å) is indicative of the Cu(II) oxidation state despite the readiness with which reduction to Cu(I) can occur during synchrotron X-ray exposure.[8] Copper–ligand distances and selected atomic displacement factors and occupancies for both NCS molecules are listed in Table S3.
Figure 1.

NcPMO-2 active site for both NCS molecules in the enzymatic resting state and after treatment with ascorbic acid: A) NCS molecule A resting state B) NCS molecule B resting state C) NCS molecule A ascorbate-treated D) NCS molecule B ascorbate-treated. Electron densities are shown as 2F0-Fc maps contoured at σ = 1.0.
In contrast with the resting state, treating crystals with an excess of ascorbic acid in the presence of atmospheric oxygen prior to flash freezing reduced the Cu(II) site to Cu(I) and permitted the formation of a Cu(II)–dioxo complex in NCS molecule A, shown in Fig. 1 C (PDB 5TKH). The ligand, present at 59% occupancy, coordinates to copper with η1 “end-on” geometry at the equatorial site with a Cu–O1 distance of 1.90 ± 0.05 Å and a Cu–O1–O2 angle of 141° (see Table S4). The equatorial oxygen ligand is modeled as a peroxo species (O22−) due to the observed O1–O2 bond length of 1.44 ± 0.06 Å; however, the precision of the bond length does not conclusively indicate “peroxo” as opposed to “superoxo” as the activated dioxo species observed. Omit maps for this species are shown in Fig. S1. The axial water molecule shows a reduced occupancy of 48%. Incomplete reduction of the Cu ion by ascorbate may account for the partial occupancies observed for the coordinating species.
The activated copper–dioxo complex observed in this structure has a geometry similar to that of a theoretical copper–superoxide active site model of Thermoascus aurantiacus LPMO9A; Kjaergaard et al. determined this model from density functional theory (DFT) calculations calibrated to solution-state extended X-ray absorption fine structure measurements of Ta-LPMO9A in the Cu(II) resting state.[4] Dioxo coordination in the available equatorial coordination site is also consistent with a classical associative displacement mechanism for hydroperoxo release by an axially coordinated water molecule in the absence of polysaccharide substrate.[9] The equatorial dioxo coordination observed is different from that modeled in the previous NcPMO-2 crystal structure (see Fig. S2) which shows superoxide instead occupying the available axial Cu coordination sites of both NCS molecules.[7] However, despite crystallographic evidence supporting these previous axial coordination models, the complexes exhibited Cu–O1 distances = 2.92–2.96 Å that are too distant for a copper–oxygen coordination interaction.[3, 10] Furthermore, the recently reported crystal structures of Lentinus similis (AA9)A in complex with either cellotriose or cellohexaose substrates (PDB 5ACJ and 5ACI, respectively) clearly show that the axial coordination site of this cellulose active LPMO is occluded by substrate binding while the equatorial site faces a cavity presumed to be suitable for oxygen binding.[11]
Electron density for the alternate NCS molecule in the ascorbate treated crystals, shown in Fig. 1 D, did not support the presence of a copper–dioxo complex but instead revealed the partial occupancy of ordered, “pre-bound” molecular O2 adjacent to the equatorial copper coordination site. The presence of molecular O2 at partial occupancies was also apparent in the electron density maps for both NCS molecules of the resting state crystals (Fig. 1 A–B). Residues His157 and Gln166, which are conserved among all AA9 LPMOs with known crystal structures (see Fig. S3.), along with Glu30 from the alternate NCS molecule (not shown) are immediately adjacent to the molecular O2 molecule. As such, this site lacks the hydrophobic character commonly observed for oxygen “pockets” identified by xenon binding studies and instead resembles the more polar oxygen binding sites that have been identified in chloride binding or high pressure O2 studies.[12]
We further characterized the NcPMO-2 active and molecular O2 sites by determining a room-temperature crystal structure jointly refined against 1.6 Å X-ray diffraction and 2.1 Å neutron diffraction data (PDB 5TKI). For joint X-ray/neutron refinement, heavy atoms are refined against the X-ray dataset while hydrogen atoms are simultaneously refined independently against the neutron diffraction data. The relatively large coherent neutron scattering lengths of hydrogen (and deuterium, 2H) atoms permit accurate modeling even at medium resolutions (dmin ≈ 2 Å).[13] In the NcPMO-2 structure, obtained at pH = 5.6 (pD = 6.0), neutron scattering length density is apparent in 2F0–Fc maps at sites occupied by 2H atoms introduced by vapor exchange (Fig. 2). For both NCS molecules His157 is modeled best as neutral and as the Nε protonated (2H) tautomer with torsion angle χ2 = −75.0° (Cα–Cβ–Cγ–Nδ1) (see Fig. S4). In this side chain conformation protonated Nε points directly toward the molecular oxygen pre-binding site observed in the higher resolution 100 K X-ray structures. It would be expected that at more acidic conditions (pH ≈ 5.0) where many cellulases show maximum activity, His157 would become doubly protonated and positively charged. The proximity of His157 to the O2 pre-binding site (Hε2–O distances ranging 1.80–2.30 Å) suggests that this doubly protonated histidine may promote O2 binding or activation.
Figure 2.
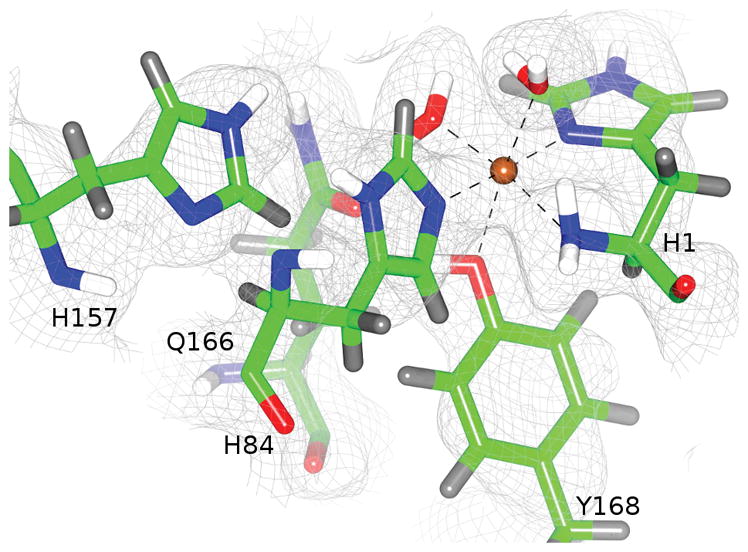
His157 conformation observed by room temperature neutron protein crystallography. Neutron scattering length densities are shown as 2F0-Fc maps contoured at σ = 1.0. NCS molecule A is shown. Covalently bonded hydrogen atoms are shown in gray, and hydrogens subject to 1H/2H exchange are shown in white.
The energetic consequences of the His157 sidechain conformation and Nε protonation that we observe from neutron diffraction have not been evaluated in prior DFT studies of LPMO oxygen activation.[4, 14] We performed DFT geometry optimizations of three active site models (ASMs) derived from our high-resolution resting state X-ray structure and calculated the free energy of molecular O2 “pre-binding” when His157 exhibits different side chain conformations and protonation states. (All atoms of the ASMs are shown in Fig. S5, and optimized atomic coordinates are given in Table S5.) Molecular O2 addition near neutral His157 in the observed conformation is approximately thermoneutral (ΔG = −1.1 kcal mol−1) which may explain the partial occupancy of O2 in each of the X-ray crystal structures. However, upon second protonation of His157 to form the positively charged sidechain, O2 addition becomes strongly thermodynamically favored (ΔG = −17.2 kcal mol−1). This suggests that under acidic conditions O2 may readily occupy the pre-binding site and be poised for activation immediately upon reduction of the resting state Cu(II) ion.
Overall, our results provide a new structural perspective on fungal LPMO reactivity with unique crystallographic and computational characterization of O2 binding and activation and of protein protonation states at and near the active site. The structures reported herein have greatest consequence for understanding the hydrogen peroxide generating reaction that occurs when LPMOs are exposed to O2 and reducing agent in the absence of substrate. It is also intriguing to consider the activated copper–dioxo complex in the context of substrate oxidation by superimposing its active site structure with those of the Ls-(AA9)–substrate complexes reported by Frandsen et al., as is shown in Fig. S6.[11] (Both Ls(AA9)-A and NcPMO-2 show regiospecificity for oxygenation at C4 of cellosaccharide glycosidic bonds.[15]) Considering the positions of O1 and O2 relative to C4 and C1′, C4 regiospecificity would result only from O1 acting as the hydrogen abstracting atom implying that the O1–O2 bond is broken beforehand. However, this structural comparison does not consider possible changes in atomic positions or oxygen species identity that could occur when a LPMO is bound to substrate and activated dioxygen simultaneously. Ultimately, combined X-ray and neutron crystallographic studies of an LPMO exhibiting, in crystallo, both O2 activation and carbohydrate substrate binding may reveal currently elusive details including the chemical identity of the copper–oxygen species responsible for lytic polysaccharide monooxygenation.
Experimental Section
Heterologous expression of NcPMO-2 from the host Pichia pastoris, protein purification and protein crystallization were performed according to the methods of O’Dell et al.[in review] Additional experimental and computational details are provided in the Supporting Information.
Supplementary Material
Table 1.
Calculated thermodynamics of molecular oxygen “pre-binding” as a function of His157 conformation and protonation.
| His157 | χ2 (°)[a] | protonation | ΔE (kcal mol−1) | ΔH (kcal mol−1) | ΔS (cal mol−1) | ΔG (kcal mol−1) |
|---|---|---|---|---|---|---|
| neutral | −70.2 | Nε | 0.7 | 0.7 | 5.6 | −1.1 |
| positive | −70.2 | Nε, Nδ | −22.8 | −22.8 | −18.5 | −17.2 |
| neutral, flipped | 106.2 | Nδ | 8.2 | 8.2 | −19.6 | 14.1 |
Reported torsion angles are those of the starting coordinates prior to geometry optimization.
Acknowledgments
Protein expression and purification experiments were conducted at the Center for Structural Molecular Biology, a DOE BER User Facility. Diffraction data were collected at SER-CAT 22-ID at the Advanced Photon Source and at CG-4D IMAGINE (NSF MRI 09229719) at the High Flux Isotope Reactor, DOE BES User Facilities. WBO acknowledges student support from NSF IGERT 1069091. FM acknowledges support from USDA NIFA Hatch 211001. PKA acknowledges support from NIH GM105978.
References
- 1.a) Harris PV, et al. Biochemistry. 2010;49:3305–3316. doi: 10.1021/bi100009p. [DOI] [PubMed] [Google Scholar]; b) Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sorlie M, Eijsink VG. Science. 2010;330:219–222. doi: 10.1126/science.1192231. [DOI] [PubMed] [Google Scholar]
- 2.a) Beeson WT, Phillips CM, Cate JHD, Marletta MA. J Am Chem Soc. 2012;134:890–892. doi: 10.1021/ja210657t. [DOI] [PubMed] [Google Scholar]; b) Eibinger M, Ganner T, Bubner P, Rosker S, Kracher D, Haltrich D, Ludwig R, Plank H, Nidetzky B. J Biol Chem. 2014;289:35929–35938. doi: 10.1074/jbc.M114.602227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beeson WT, Vu VV, Span EA, Phillips CM, Marletta MA. Annu Rev Biochem. 2015;84:923–946. doi: 10.1146/annurev-biochem-060614-034439. [DOI] [PubMed] [Google Scholar]
- 4.Kjaergaard CH, et al. Proc Natl Acad Sci U S A. 2014;111:8797–8802. doi: 10.1073/pnas.1408115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R. Biotechnol Biofuels. 2012:5. doi: 10.1186/1754-6834-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Scott BR, Huang HZ, Frickman J, Halvorsen R, Johansen KS. Biotechnol Lett. 2016;38:425–434. doi: 10.1007/s10529-015-1989-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Levasseur A, Drula E, Lombard V, Coutinho P, Henrissat B. Biotechnol Biofuels. 2013;6:41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Beeson WT, Phillips CM, Marletta MA, Cate JHD. Structure. 2012;20:1051–1061. doi: 10.1016/j.str.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudmundsson M, et al. J Biol Chem. 2014;289:18782–18792. doi: 10.1074/jbc.M114.563494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basolo F, Pearson RG. Mechanisms of Inorganic Reactions. 2. John Wiley and Sons, Inc; New York: 1967. [Google Scholar]
- 10.a) Persson I, Persson P, Sandstrom M, Ullstrom AS. J Chem Soc, Dalton Trans. 2002:1256–1265. [Google Scholar]; b) Frank P, Benfatto M, Qayyam M, Hedman B, Hodgson KO. J Chem Phys. 2015:142. doi: 10.1063/1.4908266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frandsen KE, et al. Nature Chem Biol. 2016;12:298–303. doi: 10.1038/nchembio.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Colloc’h N, et al. Biophys J. 2008;95:2415–2422. doi: 10.1529/biophysj.107.122184. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kommoju PR, Chen ZW, Bruckner RC, Mathews FS, Jorns MS. Biochemistry. 2011;50:5521–5534. doi: 10.1021/bi200388g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Afonine PV, Mustyakimov M, Grosse-Kunstleve RW, Moriarty NW, Langan P, Adams PD. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:1153–1163. doi: 10.1107/S0907444910026582. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) O’Dell WB, Bodenheimer AM, Meilleur F. Arch Biochem Biophys. 2016;602:48–60. doi: 10.1016/j.abb.2015.11.033. [DOI] [PubMed] [Google Scholar]; c) Meilleur F, Munshi P, Robertson L, Stoica AD, Crow L, Kovalevsky A, Koritsanszky T, Chakoumakos BC, Blessing R, Myles DA. Acta Crystallogr, Sect D: Biol Crystallogr. 2013;69:2157–2160. doi: 10.1107/S0907444913019604. [DOI] [PubMed] [Google Scholar]
- 14.Kim S, Stahlberg J, Sandgren M, Paton RS, Beckham GT. Proc Natl Acad Sci U S A. 2014;111:149–154. doi: 10.1073/pnas.1316609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillips CM, Beeson WT, Cate JH, Marletta MA. ACS Chem Biol. 2011;6:1399–1406. doi: 10.1021/cb200351y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.