

Abstract
This review is a summary of a talk presented at the 2015 American Epilepsy Society Annual Meeting. Its purposes are 1) to review developments in epilepsy genetics, 2) to discuss which groups of patients with epilepsy might benefit from genetic testing, and 3) to present a rational approach to genetic testing in epilepsy in the rapidly evolving era of genomic medicine.
In the past decade, we have experienced a genomic revolution in medicine. Now faced with single nucleotide variants and copy number abnormalities that might explain our patients' epilepsies, we must think carefully about which populations of patients with epilepsy might most benefit from genetic testing, what type of testing makes sense for them, and how best to educate patients and families about the possible outcomes and limitations of genetic testing. Finally, as genetic testing becomes more available, to most effectively manage the growing complexity of epilepsy genetics, we need to consider emphasizing genetic education for neurologists during training and partnering with geneticists and genetic counselors with expertise in epilepsy.
Epilepsy Genetics
Genetics has had a long-recognized role in epilepsy, particularly familial epilepsy and epilepsy of unknown cause that was formerly called “idiopathic epilepsy” (1). Since the sequencing of the human genome in 2001 (2, 3), we have experienced a paradigm shift in many fields of medicine, including epilepsy. The early discoveries of genes responsible for familial epilepsies—for example, SCN1A, CHRNA4, and LGI1—provided proof of principle that substantiated many decades of hypotheses that patients with familial epilepsy have a genetic etiology (4–6). The notion that some sporadic cases of epilepsy had a genetic basis was initially theoretical, until associations were established, for example, between SCN1A and severe myoclonic epilepsy of infancy (Dravet syndrome) (7). Over the past 15 years, the list of genes associated with epilepsy has expanded dramatically as modern sequencing technologies and analysis capacities have accelerated both research and clinical genetic analysis (8, 9). Though genome-wide association studies did not change the landscape of epilepsy genetics dramatically (10), a large number of studies implicating new dominant and recessive epilepsies—strengthened by genetic evidence and/or functional data—rapidly unfolded in the past decade (9). A key development is that de novo pathogenic variants in epilepsy-associated genes are now recognized to have a major role in the etiology of epileptic encephalopathies, in the form of single nucleotide variants (11–13) and copy number variants (14–18).
What Types of Genetic Variants Give Rise to Epilepsy?
A brief word on terminology: The term mutation refers to an event resulting in a change in the expected DNA sequence. In the medical literature, “mutation” has conventionally referred to such changes for which there was strong evidence for pathogenicity. More recently, the term “variant” has been suggested as a preferred term by the American College of Medical Genetics, with various levels of evidence for pathogenicity that can be attributed to each variant; these are based on the strengths of association between a given gene and a given phenotype (gene-level evidence) and based on amino acid conservation across species and gene paralogs, family segregation data, and presence or absence in clinical and research databases or population databases (variant-level evidence; e.g., Exome Aggregation Consortium, www.exac.broadinstitute.org) (19).
The primary types of genetic variants associated with epilepsy, as with other diseases, are single nucleotide variants (1 base pair), small insertions and deletions that may or may not result in a shift in the reading frame of the gene, and structural variation in the form of microdeletions/microduplications or chromosomal monosomy or trisomy. Chromosomal rearrangements are another category of genetic variation that may result in small or large losses or gains in copy number at their breakpoints.
Genetic Variants May Have Arisen Ancestrally or May Be De Novo
Mutational events give rise to DNA variation on a regular basis, sometimes leading to cancer and developmental disease and other times occurring without known direct consequence (20–24). Ancestral variants, arising several generations ago, may be tolerated in the heterozygous state such that the individual's cells all have one reference allele, or “normal” copy of the gene, and one alternate or variant allele, or “abnormal” copy of the gene. These variants may be passed down from generation to generation without causing symptoms or any discernable phenotype (e.g., overt seizures or EEG abnormality). Two copies of the same variant allele may be present in the same individual when the population allele frequency is sufficiently high that chance mating occurs between two carriers (e.g., the delta F508 allele associated with cystic fibrosis) or in the case of parental consanguinity, which occurs in many parts of the world. Two different variant alleles in the same gene may occur and result in a compound heterozygous condition. When an individual is found to have two alleles in “trans” configuration, having inherited one from each parent (or one inherited and the other de novo but affecting both alleles of the gene), compound heterozygous recessive disease results if both variant alleles affect normal protein function.
De novo variants are those that are present and detectable in an individual and apparently absent in his or her parents when DNA is assayed, traditionally from leukocytes. Both the inherited heterozygous variant allele in a gene associated with a recessive condition and the variant that is pathogenic in the heterozygous state (dominantly inherited or de novo) occurred at some point as a truly de novo, or new, mutation, likely during meiosis during the formation of a gamete (oocyte or spermatocyte) (25–27). In such a situation, the variant is present from the point when the individual was a zygote and onward and is thus detectable in blood.
Mutational events also occur after the stage of the zygote, during mitosis (21, 25, 28). These events have classically been associated with the development of cancer; when they occur early in the course of embryonic development, and do not result in the death of the involved daughter cells, they may present as a developmental lesion (20–24, 29). A role for postzygotic mutation, also referred to as somatic mutation, has been postulated for epilepsy (30). While not yet demonstrated for nonlesional focal epilepsy, this mechanism has been demonstrated to explain focal cortical dysplasia, hemimegalencephaly, and neurocutaneous syndromes with megalencephaly and prominent brain malformations (31–41). The extent to which focal malformations and, by extension, focal epilepsy can be explained by variants that arose in the postzygotic embryo remains to be determined.
To Whom Should We Offer Genetic Testing in Epilepsy?
Genetic discoveries in the research arena were soon followed by the availability of genetic testing in the clinical arena, initially with single gene sequencing and chromosomal microarray analysis (CMA) and later with gene panels for sequencing of a few genes at a time. Now, larger panels of genes can be assayed, with both sequencing that will identify single nucleotide variants and indels as well as deletion/duplication testing that can identify copy number variants at a resolution of ~20 kB. Exome sequencing is clinically available yet costly. In general, for epilepsy diagnoses, the more it costs the less likely it is to be covered by insurance, which presents a hurdle for many families and their physicians who are seeking a genetic diagnosis (42).
Who might benefit from genetic testing? And why should we embark on the process of seeking approval and coverage for genetic testing in the realm of epilepsy, when we are also managing medications, side effects, and developmental and behavioral concerns in ever more complex healthcare systems? Genetic testing can be time-consuming, particularly when done with proper pretest counseling about the range of effects that may ensue and their meaning (see below). Follow-up of results, parental testing in some cases, and return of positive results often demand additional visits and can be logistically challenging. Why then pursue genetic testing in the epilepsy clinic? The two main reasons for seeking a genetic diagnosis in epilepsy are 1) diagnostic certainty, which may aid in prognosis, and 2) potential impact on treatment (see examples in Table 1). So, it would make sense to focus testing at this time on those in whom there is a high likelihood of a diagnostic finding and on those whose refractory epilepsy may be influenced by a precise genetic diagnosis that can guide treatment. In most cases, we cannot answer the question of whether treatment will be affected until after we perform genetic testing and have the results. For patients with refractory epilepsy, particularly infants and young children, if there is any chance of that occurring, parents and physicians alike are naturally compelled to undertake all possible testing that could in any way ameliorate the clinical situation. This is the case regardless of the likelihood of the availability of a rational therapy based on the diagnosis. In this group, we seek an end to a “diagnostic odyssey” for families and a removal of a sense of blame that many of them report prior to a conclusive molecular diagnosis (42).
TABLE 1.
Genetic Epilepsies in Which Specific Etiology Influences Treatment
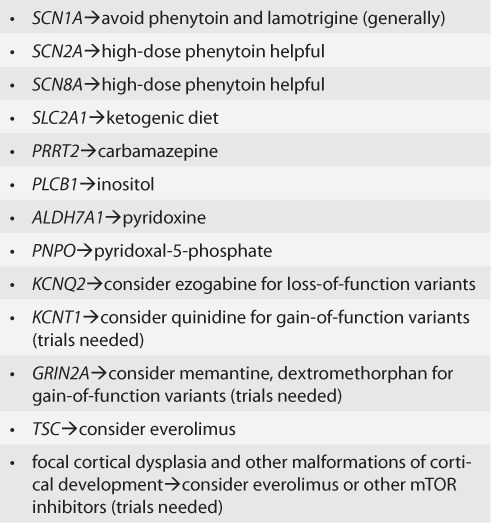
The highest yield group of patients for genetic testing to date can be summarized as “epilepsy plus,” consisting of epilepsy with accompanying dysmorphic features (though a specific genetic syndrome may not be evident early on), intellectual disability, autism, and cognitive regression (12, 13, 43–45). In these groups, the yield of CMA is about 5% (14) and sequencing via panel testing or exome sequencing, 20 to 50 percent (44, 46–48). This group is also highly likely to experience refractory epilepsy. There are some conditions, the inherited metabolic epilepsies, that are treatable and simply should not be missed; these include pyridoxine-dependent epilepsy caused by recessive variants in ALDH7A1 (49). There is a growing list of genes that suggest a modification in treatment (e.g., SCN1A—avoid phenytoin and lamotrigine, in general though not always; SCN2A and SCN8A—high-dose sodium channel-affecting agents such as phenytoin may be effective) (42). There are others for which observations across many centers may have a role in suggesting treatment (e.g., PCDH19 [50]) but for which prospective trials are still needed to know if there is a clear specific response to particular treatments. For some genes, there is a hope of “precision” medicine, or treatment based on the biology of the genetic dysfunction. This includes KCNT1 (51), GRIN2A (52), and DEPDC5 and other mTORopathies, including focal cortical dysplasia (53). Two major caveats should be mentioned here: 1) the presence of any variant in an epilepsy-associated gene does not automatically mean that the variant is pathogenic, nor does it inform us as to the type of functional change that may be present (e.g., gain vs loss of function), and 2) while case reports are helpful in establishing proof of principle and pointing to potential treatments, we need as a community to come together to conduct clinical trials with clear, uniform outcomes to study the effects of rational, target medications for each rare genetic epilepsy (54). Both careful vetting of variants and consultation with colleagues with expertise in specific genes are often needed to aid in the most accurate determination of whether or not to pursue gene-based treatment.
How to Test?
There is not a single algorithm for genetic testing in epilepsy, and there are pros and cons to each strategy. The first step is to consider the clinical diagnosis. Is the patient dysmorphic? If so, a CMA to assess for 20 kB or larger deletions and duplications is warranted, as such findings are likely to be missed with exome sequencing and possible gene-panel testing, depending on the panel used. Is there an epilepsy syndrome associated with a gene or list of genes, such as Dravet syndrome with SCN1A or Rett syndrome with MECP2? If so, complete sequencing with full coverage of every exon (not always a guarantee with exome sequencing) and deletion/duplication coverage of one or more genes should be a priority. If the CMA and initial panel suggested by a specific phenotype (e.g., infantile epilepsy panel, progressive myoclonic epilepsy panel, or Angelman-like panel) are not revealing of a genetic diagnosis, but a genetic cause is highly suspected based on the phenotype, exome sequencing is a logical next step in the evaluation. In some cases, where several phenotypes are present and waiting for results of successive panels would prove inefficient, exome sequencing (+/− CMA) may be pursued early on at some centers. The incremental yield of exome sequencing after a negative evaluation with CMA and panels is not well documented yet, but there are numerous case examples where exome sequencing has provided a diagnosis that was not made with CMA or panel testing. These include examples in which the relevant gene was not included on a panel because it had not yet been associated with epilepsy when the test was run and examples when mosaicism is present and exome sequencing analysis allows identification of variants where other techniques may not.
The Importance of Pre- and Post-testing Counseling
With all the new and evolving information about genes important for epilepsy, variant interpretation, functional characterization of epilepsy genes, and emerging clinical trials for gene-specified populations of patients, it is important for clinicians to inform their patients of what they are getting themselves into. Genetic test results may be positive—with unequivocal findings in genes strongly associated with epilepsy of variants that have been previously reported as associated with disease and/or functionally characterized. They may be negative, with no explanatory results discovered, and it should be noted that this does not rule out that the cause is genetic—it may be, and we in 2017 (or the current year) are not yet able to elucidate it with current knowledge and technologies. They may be equivocal with variants of uncertain significance in disease-relevant genes; this situation may require parental testing, particularly for severe cases in which the presence of a de novo variant would be most compelling, and may require an ongoing discussion that incorporates incrementally accrued knowledge over time. Families should be told what the next steps might be in each scenario—possible treatment considerations and referral to gene-based support networks if positive, or research enrollment if equivocal or negative (e.g., the Epilepsy Genetics Initiative supported by the National Institutes for Neurological Disease and Stroke and Citizens United for Research in Epilepsy).
Case Examples
Inherited Epilepsy
A 14-year-old young woman with juvenile myoclonic epilepsy (JME) and a family history of generalized epilepsy (mother) and febrile seizures (maternal aunt) comes for consultation to consider a possible genetic diagnosis.
If she had a mutation in a gene always associated with a benign course, we could provide reassurance that she has a nonprogressive epilepsy. Suppose, for example, we were faced with school decline in a patient like this, which can be multifactorial and needs to be addressed at many levels. In that case, confirmation of a nonprogressive condition may not be required but may be helpful. In a classic case of JME in an otherwise well patient, with normal EEG background, genetic testing may not add to the clinical picture in 2017. There may well be a role, however, for pharmacogenetics that will help with treatment choice and side effect minimization in the future. What can we tell her about the next generation—will she pass on a genetic trait such that some of her children will have epilepsy? As the genetics of the more common epilepsies is worked out, in part through large collaborative sequencing efforts (55), it may be possible in the future to help with risk stratification for siblings and for successive generations. Currently, data suggest a risk to family members of about 5% (56), but without specific genes to evaluate, this remains a generic prediction.
Highlights
Genetic variants take the form of inherited (dominant or recessive) and sporadic (de novo) changes in DNA. Some are associated with disease, and others are population variants that may not affect the function of the gene involved.
Genetic research in epilepsy has led to the advent of a range of clinically available testing, including CMA, gene-panel testing, and exome sequencing.
We should start with a clinical approach, defining a specific epilepsy syndrome when relevant, to guide the nature and sequence of genetic testing.
Pre- and posttesting counseling, whether provided by a physician or genetic counselor, is critical to maintaining clear communication with patients about the implications and limitations of their genetic testing.
Likely De Novo Epilepsy
A 6-month-old boy with new-onset infantile spasms with hypsarrhythmia and normal MRI is referred for genetic testing as part of his evaluation.
If he was found to have a pathogenic variant in an “epilepsy gene,” repeated testing for possible metabolic disease (with lumbar puncture, etc.) and repeated imaging, as often occurs in patients with infantile spasms who then go on to have focal seizures, could be stopped.
Would there be an impact on treatment? As discussed above, there is a growing list of genes associated with modifications in treatment as well as rational approaches to treatment that will hopefully coalesce into multi-center clinical trials in the coming years.
Conclusions
Genetics plays a major role in epilepsy, particularly in patients with refractory epilepsy. The well-informed neurologist can triage who most needs and could benefit from genetic testing, choose the appropriate testing, and explain the findings in the context of the ever-important clinical scenario. What is needed is a thoughtful genetic approach incorporated into the overall clinical evaluation of a patient with epilepsy. The ability to access and incorporate new information about types of testing, specific genes, and possible treatments can augment the success of this approach. Depending on the practice setting, a neurologist can address all of these issues and stay current by partnering with a colleague in genetics or by seeking epilepsy genetics expertise from physicians and genetic counselors in a specialized epilepsy genetics program.
Acknowledgments
Editor's Note: Authors have a Conflict of Interest disclosure which is posted under the Supplemental Materials (208.7KB, docx) link.
References
- 1. Thomas RH, Berkovic SF.. The hidden genetics of epilepsy—A clinically important new paradigm. Nat Rev Neurol 2014; 10: 283– 292. [DOI] [PubMed] [Google Scholar]
- 2. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigó R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X.. The sequence of the human genome. Science 2001; 291: 1304– 1351. [DOI] [PubMed] [Google Scholar]
- 3. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, Szustakowki J; International Human Genome Sequencing Consortium. . Initial sequencing and analysis of the human genome. Nature 2001; 409: 860– 921. [DOI] [PubMed] [Google Scholar]
- 4. Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF.. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 1995; 11: 201– 203. [DOI] [PubMed] [Google Scholar]
- 5. Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli Boneschi F, Choi C, Morozov P, Das K, Teplitskaya E, Yu A, Cayanis E, Penchaszadeh G, Kottmann AH, Pedley TA, Hauser WA, Ottman R, Gilliam TC.. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002; 30: 335– 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR, Lerman-Sagie T, Lev D, Mazarib A, Brand N, Ben-Zeev B, Goikhman I, Singh R, Kremmidiotis G, Gardner A, Sutherland GR, George AL Jr, Mulley JC, Berkovic SF.. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet 2001; 68: 859– 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P.. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001; 68: 1327– 1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poduri A, Lowenstein D.. Epilepsy genetics—Past, present, and future. Curr Opin Genet Dev 2011; 21: 325– 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Helbig I, Lowenstein DH.. Genetics of the epilepsies: Where are we and where are we going? Curr Opin Neurol 2013; 26: 179– 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. International League Against Epilepsy Consortium on Complex Epilepsies (www.epilepsy-austin@unimelb.edu.au). . Genetic determinants of common epilepsies: A meta-analysis of genome-wide association studies. Lancet Neurol 2014; 13: 893– 903. doi:10.1016/S1474-4422(14)70171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, Barth-Maron A, Greenberg ME, Stuhlmann T, Weinert S, Jentsch TJ, Pazzi M, Restifo LL, Talwar D, Erickson RP, Hammer MF.. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 2013; 54: 1270– 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli Nieh S, O'Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR.. De novo mutations in epileptic encephalopathies. Nature 2013; 501: 217– 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. . De Novo Mutations in Synaptic Transmission Genes Including DNM1 Cause Epileptic Encephalopathies. Am J Hum Genet 2014; 95: 360– 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Olson H, Shen Y, Avallone J, Sheidley BR, Pinsky R, Bergin AM, Berry GT, Duffy FH, Eksioglu Y, Harris DJ, Hisama FM, Ho E, Irons M, Jacobsen CM, James P, Kothare S, Khwaja O, Lipton J, Loddenkemper T, Markowitz J, Maski K, Megerian JT, Neilan E, Raffalli PC, Robbins M, Roberts A, Roe E, Rollins C, Sahin M, Sarco D, Schonwald A, Smith SE, Soul J, Stoler JM, Takeoka M, Tan WH, Torres AR, Tsai P, Urion DK, Weissman L, Wolff R, Wu BL, Miller DT, Poduri A.. Copy number variation plays an important role in clinical epilepsy. Ann Neurol 2014; 75: 943– 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mefford HC, Yendle SC, Hsu C, Cook J, Geraghty E, McMahon JM, Eeg-Olofsson O, Sadleir LG, Gill D, Ben-Zeev B, Lerman-Sagie T, Mackay M, Freeman JL, Andermann E, Pelakanos JT, Andrews I, Wallace G, Eichler EE, Berkovic SF, Scheffer IE.. Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol 2011; 70: 974– 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, Franke A, Malafosse A, Genton P, Thomas P, Gurnett CA, Schreiber S, Bassuk AG, Guipponi M, Stephani U, Helbig I, Eichler EE.. Genome-wide copy number variation in epilepsy: Novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet 2010; 6: e1000962 doi:10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, Kluck C, Muhle H, von Spiczak S, Ostertag P, Obermeier T, Kleefuss-Lie AA, Hallmann K, Steffens M, Gaus V, Klein KM, Hamer HM, Rosenow F, Brilstra EH, Trenité DK, Swinkels ME, Weber YG, Unterberger I, Zimprich F, Urak L, Feucht M, Fuchs K, Møller RS, Hjalgrim H, De Jonghe P, Suls A, Rückert IM, Wichmann HE, Franke A, Schreiber S, Nürnberg P, Elger CE, Lerche H, Stephani U, Koeleman BP, Lindhout D, Eichler EE, Sander T.. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 2010; 133: 23– 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heinzen EL, Radtke RA, Urban TJ, Cavalleri GL, Depondt C, Need AC, Walley NM, Nicoletti P, Ge D, Catarino CB, Duncan JS, Kasperaviciūte D, Tate SK, Caboclo LO, Sander JW, Clayton L, Linney KN, Shianna KV, Gumbs CE, Smith J, Cronin KD, Maia JM, Doherty CP, Pandolfo M, Leppert D, Middleton LT, Gibson RA, Johnson MR, Matthews PM, Hosford D, Kälviäinen R, Eriksson K, Kantanen AM, Dorn T, Hansen J, Krämer G, Steinhoff BJ, Wieser HG, Zumsteg D, Ortega M, Wood NW, Huxley-Jones J, Mikati M, Gallentine WB, Husain AM, Buckley PG, Stallings RL, Podgoreanu MV, Delanty N, Sisodiya SM, Goldstein DB.. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet 2010; 86: 707– 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. . Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405– 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EE, Verstegen MM, van der Laan LJ, de Jonge J, IJzermans JN, Vries RG, van de Wetering M, Stratton MR, Clevers H, Cuppen E, van Boxtel R.. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016; 538: 260– 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, Lee S, Chittenden TW, D'Gama AM, Cai X, Luquette LJ, Lee E, Park PJ, Walsh CA.. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015; 350: 94– 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A 2010; 107: 961– 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Erickson RP. Somatic gene mutation and human disease other than cancer: An update. Mutat Res 2010; 705: 96– 106. [DOI] [PubMed] [Google Scholar]
- 24. Evrony GD, Cai X, Lee E, Hills LB, Elhosary PC, Lehmann HS, Parker JJ, Atabay KD, Gilmore EC, Poduri A, Park PJ, Walsh CA.. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012; 151: 483– 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poduri A, Evrony GD, Cai X, Walsh CA.. Somatic mutation, genomic variation, and neurological disease. Science 2013; 341: 1237758 doi:10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu S, Zong C, Fan W, . et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 2012; 338: 1627– 1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang J, Fan HC, Behr B, Quake SR.. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 2012; 150: 402– 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, Martincorena I, Petljak M, Alexandrov LB, Gundem G, Tarpey PS, Roerink S, Blokker J, Maddison M, Mudie L, Robinson B, Nik-Zainal S, Campbell P, Goldman N, van de Wetering M, Cuppen E, Clevers H, Stratton MR.. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 2014; 513: 422– 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erickson RP. Somatic gene mutation and human disease other than cancer. Mutat Res 2003; 543: 125– 136. [DOI] [PubMed] [Google Scholar]
- 30. Lindhout D. Somatic mosaicism as a basic epileptogenic mechanism? Brain 2008; 131: 900– 901. [DOI] [PubMed] [Google Scholar]
- 31. Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BF, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA.. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 2012; 74: 41– 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG.. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012; 44: 941– 945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GM, Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, Uyanik G, Weksberg R, Zirn B, Beaulieu CL; Finding of Rare Disease Genes (FORGE) Canada Consortium, Majewski J, Bulman DE, O'Driscoll M, Shendure J, Graham JM Jr, Boycott KM, Dobyns WB.. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 2012; 44: 934– 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ, Hiatt JB, Roden WH, Gunter SA, Christian SL, Collins S, Adams C, Rivière JB, St-Onge J, Ojemann JG, Shendure J, Hevner RF, Dobyns WB.. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 2015; 138: 1613– 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mirzaa GM, Campbell CD, Solovieff N, Goold CP, Jansen LA, Menon S, Timms AE, Conti V, Biag JD, Olds C, Boyle EA, Collins S, Ishak G, Poliachik SL, Girisha KM, Yeung KS, Chung BH, Rahikkala E, Gunter SA, McDaniel SS, Macmurdo CF, Bernstein JA, Martin B, Leary RJ, Mahan S, Liu S, Weaver M, Dorschner MO, Jhangiani S, Muzny DM, Boerwinkle E, Gibbs RA, Lupski JR, Shendure J, Saneto RP, Novotny EJ, Wilson CJ, Sellers WR, Morrissey MP, Hevner RF, Ojemann JG, Guerrini R, Murphy LO, Winckler W, Dobyns WB.. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol 2016; 73: 836– 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. D'Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, Hossain A, Hatem NE, Barry BJ, Kwiatkowski DJ, Vinters HV, Barkovich AJ, Shendure J, Mathern GW, Walsh CA, Poduri A.. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol 2015; 77: 720– 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jamuar SS, Lam AT, Kircher M, D'Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, Servattalab S, Mehta BK, Topcu M, Amrom D, Andermann E, Dan B, Parrini E, Guerrini R, Scheffer IE, Berkovic SF, Leventer RJ, Shen Y, Wu BL, Barkovich AJ, Sahin M, Chang BS, Bamshad M, Nickerson DA, Shendure J, Poduri A, Yu TW, Walsh CA.. Somatic mutations in cerebral cortical malformations. N Engl J Med 2014; 371: 733– 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J.. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med 2013; 368: 1971– 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mirzaa GM, Conti V, Timms AE, Smyser CD, Ahmed S, Carter M, Barnett S, Hufnagel RB, Goldstein A, Narumi-Kishimoto Y, Olds C, Collins S, Johnston K, Deleuze JF, Nitschké P, Friend K, Harris C, Goetsch A, Martin B, Boyle EA, Parrini E, Mei D, Tattini L, Slavotinek A, Blair E, Barnett C, Shendure J, Chelly J, Dobyns WB, Guerrini R.. Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: A next-generation sequencing study. Lancet Neurol 2015; 14: 1182– 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries LS, Lev D, Kramer N, Hopkins E, Graham JM Jr, Dobyns WB.. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: Two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A 2012; 158A: 269– 291. [DOI] [PubMed] [Google Scholar]
- 41. Mirzaa GM, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, Logan CV, Roberts N, Johnson CA, Singh S, Kholmanskikh SS, Adams C, Hodge RD, Hevner RF, Bonthron DT, Braun KP, Faivre L, Rivière JB, St-Onge J, Gripp KW, Mancini GM, Pang K, Sweeney E, van Esch H, Verbeek N, Wieczorek D, Steinraths M, Majewski J; FORGE Canada Consortium, Boycott KM, Pilz DT, Ross ME, Dobyns WB, Sheridan EG.. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet 2014; 46: 510– 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poduri A, Sheidley BR, Shostak S, Ottman R.. Genetic testing in the epilepsies—Developments and dilemmas. Nat Rev Neurol 2014; 10: 293– 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE.. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012; 367: 1921– 1929. [DOI] [PubMed] [Google Scholar]
- 44. Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, Tang S, Helbig I.. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med 2016; 18: 898– 905. [DOI] [PubMed] [Google Scholar]
- 45. Moller RS, Dahl HA, Helbig I.. The contribution of next generation sequencing to epilepsy genetics. Expert Rev Mol Diagn 2015; 15: 1531– 1538. [DOI] [PubMed] [Google Scholar]
- 46. Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, Palmer R, Morrogh D, Boustred C, Hurst JA, Jenkins L, Kurian MA, Scott RH.. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet 2016; 53: 310– 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, Hahn CD, Kannu P, Kobayashi J, Minassian BA, Moharir M, Siriwardena K, Weiss SK, Weksberg R, Snead OC III. . Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 2015; 56: 707– 716. [DOI] [PubMed] [Google Scholar]
- 48. Mullen SA, Carvill GL, Bellows S, Bayly MA, Trucks H, Lal D, Sander T, Berkovic SF, Dibbens LM, Scheffer IE, Mefford HC.. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology 2013; 81: 1507– 1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. El Achkar CM, Olson HE, Poduri A, Pearl PL.. The genetics of the epilepsies. Curr Neurol Neurosci Rep 2015; 15: 39. [DOI] [PubMed] [Google Scholar]
- 50. Lotte J, Bast T, Borusiak P, Coppola A, Cross JH, Dimova P, Fogarasi A, Graneß I, Guerrini R, Hjalgrim H, Keimer R, Korff CM, Kurlemann G, Leiz S, Linder-Lucht M, Loddenkemper T, Makowski C, Mühe C, Nicolai J, Nikanorova M, Pellacani S, Philip S, Ruf S, Sánchez Fernández I, Schlachter K, Striano P, Sukhudyan B, Valcheva D, Vermeulen RJ, Weisbrod T, Wilken B, Wolf P, Kluger G.. Effectiveness of antiepileptic therapy in patients with PCDH19 mutations. Seizure 2016; 35: 106– 110. [DOI] [PubMed] [Google Scholar]
- 51. Milligan CJ, Li M, Gazina EV, Heron SE, Nair U, Trager C, Reid CA, Venkat A, Younkin DP, Dlugos DJ, Petrovski S, Goldstein DB, Dibbens LM, Scheffer IE, Berkovic SF, Petrou S.. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol 2014; 75: 581– 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pierson TM, Yuan H, Marsh ED, Fuentes-Fajardo K, Adams DR, Markello T, Golas G, Simeonov DR, Holloman C, Tankovic A, Karamchandani MM, Schreiber JM, Mullikin JC; PhD for the NISC Comparative Sequencing Program, Tifft CJ, Toro C, Boerkoel CF, Traynelis SF, Gahl WA.. GRIN2A Mutation and early-onset epileptic encephalopathy: Personalized therapy with memantine. Ann Clin Transl Neurol 2014; 1: 190– 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weckhuysen S, Marsan E, Lambrecq V, Marchal C, Morin-Brureau M, An-Gourfinkel I, Baulac M, Fohlen M, Kallay Zetchi C, Seeck M, de la Grange P, Dermaut B, Meurs A, Thomas P, Chassoux F, Leguern E, Picard F, Baulac S.. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 2016; 57: 994– 1003. [DOI] [PubMed] [Google Scholar]
- 54. EpiPM Consortium. . A roadmap for precision medicine in the epilepsies. Lancet Neurol 2015; 14: 1219– 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Epi4K Consortium. . Epi4K: Gene discovery in 4,000 genomes. Epilepsia 2012; 53: 1457– 1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peljto AL, Barker-Cummings C, Vasoli VM, Leibson CL, Hauser WA, Buchhalter JR, Ottman R.. Familial risk of epilepsy: A population-based study. Brain 2014; 137: 795– 805. [DOI] [PMC free article] [PubMed] [Google Scholar]