

Abstract
Background
The adipokine hormone, leptin, is a major component of body weight homeostasis. Numerous studies have been performed administering recombinant mouse leptin as an experimental reagent; however, the half life of circulating leptin following exogenous administration of recombinant mouse leptin has not been carefully evaluated.
Methods
Exogenous leptin was administered (3 mg leptin/kg body weight) to ten week old fasted non-obese male mice and plasma was serially collected at seven time points; plasma leptin concentration was measured by ELISA at each time point to estimate the circulating half life of mouse leptin.
Results
Under the physiological circumstances tested, the half life of mouse leptin was 40.2 (+/− 2.2) minutes. Circulating leptin concentrations up to one hour following exogenous leptin administration were 170-fold higher than endogenous levels at fasting.
Conclusions
The half life of mouse leptin was determined to be 40.2 minutes. These results should be useful in planning and interpreting experiments employing exogenous leptin. The unphysiological elevations in circulating leptin resulting from widely used dosing regimens for exogenous leptin are likely to confound inferences regarding some aspects of the hormone’s clinical biology.
Keywords: Leptin, half life, obesity
Introduction
Leptin (Lep) is a peptide hormone produced in adipocytes that signals via isoforms of the leptin receptor (LepR) (1). A major function of leptin is to signal the levels of peripheral energy stores in adipose tissue to the central nervous system (2). Mice and humans hypomorphic for LEP or LEPR develop severe, hyperphagic obesity (3–6). Circulating leptin concentrations are closely correlated with adipose tissue mass in both humans and mice (7–9).
The major site of leptin synthesis is the adipocyte; leptin synthesis is regulated by nutritional status, insulin, glucocorticoids, catecholamines, cold exposure, and growth hormone, amongst other signals (10). Long term nutritional status regulates leptin at the transcriptional level; leptin mRNA levels are elevated in obesity and decreased by calorie restriction (11). Circulating signals such as insulin regulate leptin posttranscriptionally; ex vivo insulin stimulation of adipose tissue explants increases the association of leptin mRNA with translationally active polysomes (12). Leptin is secreted from adipocytes via small intracellular vesicles in both constituitive and regulated pathways (13). Leptin secretion in humans is diurnal, with a nighttime zenith and morning nadir (14). In the circulation, leptin exists in both bound and free forms and is mainly cleared by the kidneys (14).
Leptin signals through its receptor, LepRb, in the arcuate nucleus of the hypothalamus primarily through JAK/STAT signaling, but also through PI3K, SHP2/ERK, and p38/MAPK signaling pathways (15). Hypothalamic levels of phosphorylated STAT3 following recombinant leptin administration in mice are commonly used as a read out of leptin activity at this receptor isoform (16). The response of other downstream signaling in the leptin-melanocortin pathway is also studied in response to leptin administration (17). Despite a multitude of studies utilizing the experimental paradigm of exogenous leptin stimulation, there have been limited studies investigating the pharmacokinetic properties of leptin. While the circulating half life of leptin has been determined in adult human subjects and female rats, the circulating half life of leptin in mice, the most commonly used model in obesity-related research, has not been rigorously calculated (18, 19). Here, we report the circulating half life of recombinant leptin in mice and compare the blood levels of commonly used concentrations of injected recombinant leptin to endogenous levels during fasting and refeeding in mice.
Materials and Methods
Mouse anthropometric, blood glucose, and food intake measurements
All animal studies were conducted in accordance with IACUC and CUMC standards and were IACUC approved under protocol AC-AAAH1203. Ten week old male C57BL/6J mice from Jackson Laboratories were used (stock #000664). Body weights, fat mass, and lean mass were measured at 8 and 10 weeks of age. Fat mass and lean mass was measured using the Bruker Minispec TD NMR. Mice were fed Research Diets PicoLab Rodent Diet 20 (#5053) with a fat content of 4.5% of calories. Blood was collected by submandibular bleeding and used for both blood glucose and plasma leptin measurements. Blood glucose was measured following an overnight (16 hours) fast and following 4 hours of ad lib re-feeding using a FreeStyle Lite blood glucose meter and strips (accurate range 30–372 mg/dL). Food intake during the 4-hour refeeding period was measured on a per-cage basis. Using an Acculab Vicon VIC212 scale; five similarly sized chow pellets were weighed and placed in the cage for the feeding period; and were weighed again at its completion; the difference in these numbers is the cumulative cage food intake for the 4 hour period. Statistical analysis was done using Prism 7 from GraphPad Software, Inc.
Measurement of circulating leptin at fasting and refeeding
Plasma leptin concentrations were measured from plasma following an overnight fast (16 hours), after 4 hours of re-feeding, and subsequent to injections of recombinant mouse leptin in fasted animals. Blood was collected in heparinized tubes and immediately placed on ice then centrifuged at 2,000 RCF for 15 minutes at 4 degrees Celsius. The R&D systems Mouse/Rat Leptin Quantikine ELISA Kit (SMOB00) was used to measure plasma leptin concentrations. Statistical analysis was performed using Prism 7 from GraphPad Software, Inc. Variance is reported as the standard error of the mean.
Measurement and calculation of circulating leptin half life
Mouse recombinant leptin was purchased from the National Hormone and Peptide Program (www.humc.edu); lyophilized leptin was dissolved in 1X PBS, pH 8.0 (1X PBS = 137 mM NaCl, 2.7 mM KCl; 10 mM Phosphate buffer). (Note that mouse recombinant leptin must be dissolved into PBS with a slightly basic pH, i.e. pH 8.0). Forty-five mice were arbitrarily separated into nine groups containing five mice each. Seven groups received intraperitoneal leptin injected at 3 mg/kg body weight at 7:45 AM following an overnight (16 hours) fast. Of the seven groups receiving recombinant leptin, circulating leptin concentrations were measured at the following time points post-injection: 0.25 hours, 0.5 hours, 1 hour, 2 hours, 4 hours, 6 hours, and 8 hours (naming convention as follows: 0.25 Hr L, 0.5 Hr L, 1 Hr L or 1HL, 2 Hr L, 4 Hr L or 4HL, 6 Hr L or 6HL, and 8 Hr L, respectively). All mice remained fasting for the duration of the experiment. Two groups of mice were injected with vehicle (saline) only, and circulating leptin concentrations were measured at 0.5 hours and 4 hours post-injection (naming convention: 0.5 Hr S, 4 Hr S, respectively). Blood collection and measurement of plasma leptin concentrations were carried out as described above. The half life of leptin was estimated by modeling the data with a single exponential decay process. Modeling the half life of leptin using a double exponential decay process did not significantly improve curve fitting. Half life modeling was carried out in Origin8 software from OriginLab.
Results
Modeling the plasma concentrations post-injection over time with a single exponential decay model yielded an estimate of the half life of circulating leptin of 40.2 minutes (+/− 2.2 minutes) (Figure 1). We also modeled the half life of circulating leptin using a double exponential decay process which did not improve the curve fit; thus we used the single exponential decay model. This estimate is similar to that reported by others for injected leptin in human subjects (24.9 +/− 4.4 minutes) and for ad lib fed female rats (71 minutes) (18, 19).
Figure 1. The half life of mouse recombinant leptin is (40.2 +/−2.2) minutes.
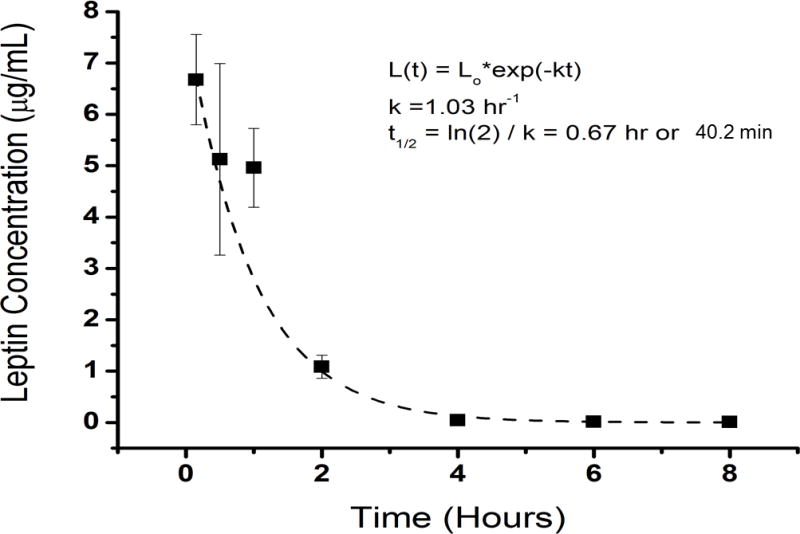
The half life of leptin was estimated by modeling the data with a single exponential decay process. (n=5 WT, 10 week old male mice/group, error bars are SEM; fasted mice were administered recombinant leptin at 3 mg/kg body weight).
We then compared plasma concentrations of leptin following 3 mg/kg injection to those of mice administered saline. Plasma leptin concentrations of fasted mice administered saline were not different from fasted mice that were not injected. At 1 hour following leptin administration, the concentration of circulating leptin was 170-fold higher than endogenous fasted leptin levels, and 13-fold higher than endogenous fed leptin concentrations in mice that were not injected with exogenous leptin (Figure 2). Four hours following injection, circulating leptin levels were 76-fold higher than endogenous leptin concentrations in fasted animals and 4.75-fold higher than endogenous leptin concentrations in fed animals. Six hours following the intraperitoneal injection of exogenous leptin into fasted mice, the concentrations of leptin were not different from the endogenous concentrations of leptin in non-fasted mice. There were no significant differences in body weights, fat or lean mass, food intake, or blood glucose or leptin concentrations among the groups of animals (Figure 3A–G). All fasted and fed blood glucose concentrations were in the normal physiological range (Figure 3 C, F). Blood glucose concentrations increased for all groups from fasting to re-feeding (Figure 3C, F). Circulating leptin concentrations also increased for all groups from fasting to re-feeding and were similar among the designated groups (Figure 3 D, G).
Figure 2. Commonly used recombinant leptin injection concentrations are supraphysiological.
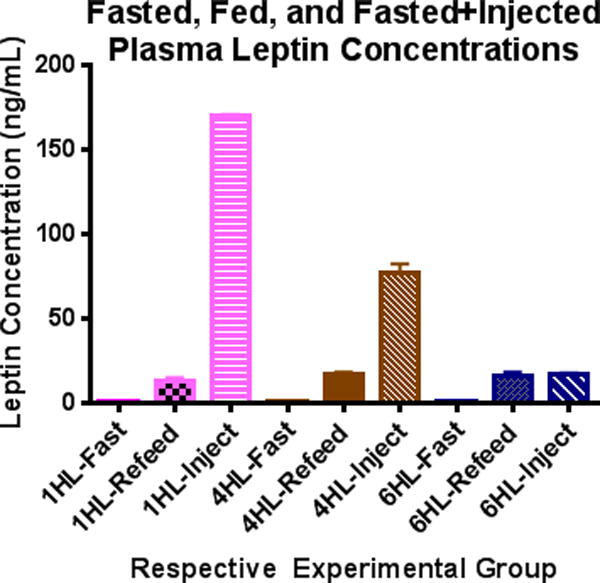
Plasma leptin concentrations were measured in the same three groups of wild type (WT) mice at fasting, refeeding (5 hours), and following recombinant mouse leptin injection into fasted animals. 1HL, 4HL, and 6HL refer to three separate groups of mice in which circulating leptin concentrations were measured 1, 4, or 6 hours following leptin injection. One hour following injection of recombinant mouse leptin to fasted mice (dose = 3 mg/kg body weight) plasma leptin concentrations were 170-fold higher than those at fasting and 13-fold higher than those at refeeding. By six hours following leptin injection, plasma leptin concentrations were not different from those at refeeding (n=5 WT, 10 week old male mice/group, error bars are SEM).
Figure 3. Body weight/compositions, blood glucose, food intake, and plasma leptin concentrations of wild type mice.
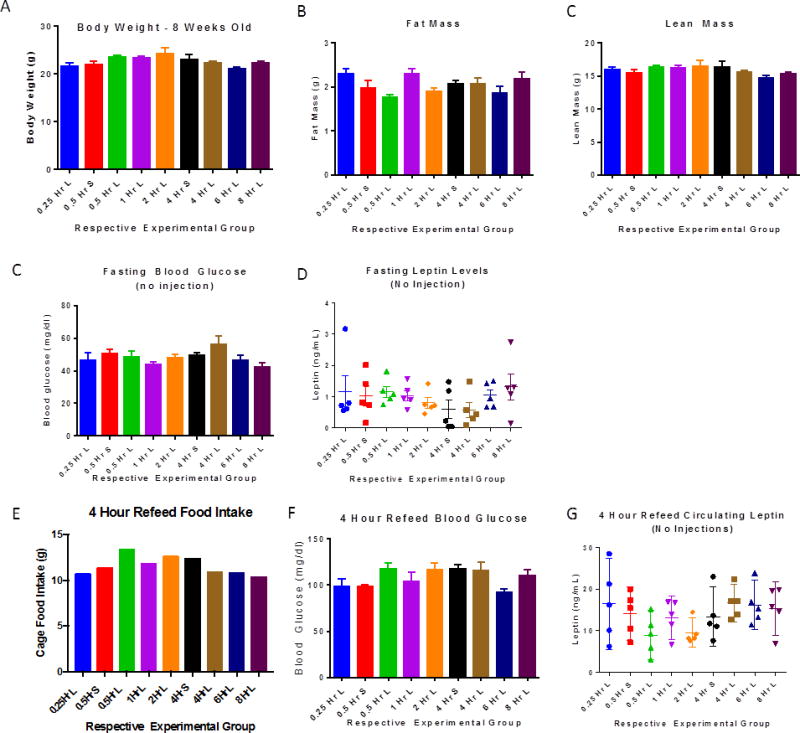
There is no difference in body mass or composition measures, blood glucose levels, or leptin values for animals used in the study. Naming convention follows that described in Figure 2 and Methods, S=saline, L=leptin (n=5 WT 10 week old male mice/group, error bars are SEM; food intake measurement in panel E is of each cage containing 5 mice).
Discussion
We measured plasma leptin concentrations in fasted, non-obese, male mice at successive time points following injection of mouse recombinant leptin. From these data we estimated that the half life of circulating mouse leptin is 40.2 (+/− 2.2) minutes (Figure 1). The half life was calculated by fitting the serial plasma leptin concentrations to a single exponential decay process. Modeling the data with a double exponential decay process did not significantly improve the fit. The half life for mice is consistent with those reported for humans (24.9 minutes) and rats (71 minutes) (18, 19). These data are also consistent with those reported by Ahima et al., with regard to the decay of circulating leptin concentrations following administration of recombinant mouse leptin to fasted mice at 1 mg/kg body weight (20).
Leptin is secreted from the adipocyte in small intracellular vesicles; ~60% of cellular leptin is co-secreted bound to soluble leptin receptor (13, 21). Soluble leptin receptor is the main carrier protein for leptin; however, c-reactive protein and other proteins can also bind circulating leptin (22, 23). The main sites of soluble leptin receptor production are adipose tissue and liver; fasting upregulates hepatic soluble leptin receptor expression (24, 25). Mouse soluble leptin receptor can be produced from alternative splicing of the LepR gene to LepRe; other splice forms can also be posttranslationally modified to soluble leptin receptor by ectodomain shedding (26, 27). In humans, LEPR transcript is not spliced to LepRe; the soluble leptin receptor is produced exclusively by ectodomain shedding (26, 27). Free circulating leptin is the bioactive form of leptin and bound circulating leptin has greater stability (longer circulating half life) than that of free circulating leptin (28, 29). The KD of the leptin-leptin receptor complex has been reported to be 0.23+/−0.08 nM, suggesting high affinity ligand binding (30). In fasted, lean animals the majority of circulating leptin is bound and this correlates with a minimal pSTAT3 signal in arcuate LepRb neurons at fasting (31). The proportion of free to bound leptin decreases in the fed state, thus resulting in higher concentrations of circulating free leptin, which likely results in a shorter half life for total circulating leptin in the fed state than in the fasted state. In states of chronic hyperleptinemia, such as mice in which leptin is continuously perfused by minipumps or transgenic mice overexpressing leptin, the half life of circulating leptin may differ from what is reported here circumstances (32, 33). Although it is unclear what fraction of leptin is free or bound in these circumstances, it is likely that the proportion of free leptin is increased thus resulting in a decreased half life of circulating leptin as compared non-hyperleptinemic adiposity-matched animals (32, 33).
Plasma leptin accesses the cerebrospinal fluid by active transport mediated at least partially by LepRa, LepRc, and LRP2 expression in median eminence tanycytes and ependymal cells of the choroid plexus (34–36). Upon leptin binding to LepRb or LepRa homodimers, leptin is internalized by clathrin coated vesicles (37). The percentage of leptin receptors at the cell surface at any given time is small; however in the presence of leptin at the cell surface, leptin receptors may cluster, resulting in enhanced ligand-receptor complexing and endocytosis of leptin (38–41). Phosphorylated STAT3 in response to intraperitoneal leptin injection (1–6 mg leptin/kg body weight) in non-obese, fasted mice can be observed within 30 minutes following injection (42–45). We found that 30 minutes following injection of recombinant mouse leptin (3mg/kg body weight) plasma leptin concentrations were 4,976-fold higher than endogenous fasted plasma leptin concentrations and 391-fold higher than endogenous re-fed plasma leptin concentrations (Figures 1, 2). The highest plasma leptin concentrations were measured at the first time point of blood collection, 15 minutes following injection, at which point plasma leptin concentrations of injected mice were 6,481-fold higher than endogenous fasted plasma leptin concentrations and 509-fold higher than endogenous fed plasma leptin concentrations (Figures 1, 2). Reduced leptin signaling through LepRb as well as decreased plasma concnetrations of soluble leptin receptor in obesity, may play roles in determining both the physical and biological half life of leptin in the obese state, which may differ from those of fasted, lean mice receiving exogenous leptin.
The results reported here have important implications for data obtained in the many studies intended to understand the systemic and neuro-molecular physiology of leptin. As we show, the widely applied doses of 2–4 mg/kg in mice clearly produce un-physiological elevations in circulating leptin, with consequences for leptin response pathways that could confound inferences regarding the biology of leptin in body weight homeostasis, by altering leptin sensitivity.
For example, among the most pressing clinical questions regarding regulation of body weight are those relating to whether the apparent “set point” for body fat can be increased by environmentally-induced chronic weight gain (46); and whether the compensatory behavioral and metabolic responses to maintenance of reduced body fat (hence reduced circulating leptin) can be mitigated by the administration of “replacement” exogenous leptin (47–49). With regard to the former, experimental parsing of the relative contributions of elevated dietary fat - and the elevations of circulating leptin consequent to secondary gains in body fat – requires careful titration of administered leptin to levels consistent with those resulting from increments in fat mass (32, 50). For the latter, use of high doses of leptin in the induction of weight loss, or in the maintenance of reduced body weight, can change central leptin responses in ways that will confound interpretation of the efficacy of the interventions (51). In short, many of the relevant studies conducted to date have used doses of leptin that might obscure important aspects of clinical biology.
Acknowledgments
The authors thank Yiying Zhang, Jayne Martin Carli, and Yann Ravussin for helpful discussions and Benjamin J. Burnett for assistance in half life modeling. We thank the Foundation for Prader-Willi Research, the Russell Berrie Foundation, and RO1 DK52431 for funding.
Footnotes
Conflict of Interest: The authors have no conflicts of interest.
References
- 1.Myers MG, Leibel RL. Lessons from Rodent Models of Obesity. In: De Groot LJB-PP, Chrousos G, et al., editors. Endotext. South Dartmouth: MDText.com, Inc.; 2015. [Google Scholar]
- 2.Rosenbaum M, Leibel RL. 20 years of leptin: role of leptin in energy homeostasis in humans. J Endocrinol. 2014;223(1):T83–96. doi: 10.1530/JOE-14-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 4.Clement K, Vaisse C, Lahlou N, Cabrolk S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homolog. Nature. 1994;372:425–32. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 6.Chua SC, Chung WK, Wu-Peng XS, Zhang Y, Liu S-M, Tartaglia L, et al. Phenotypes of Mouse diabetes and Rat fatty Due to Mutations in the OB (Leptin) Receptor. Science. 1996;271:994–6. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 7.Maskari MYA, Alnaqdy AA. Correlation between Serum Leptin Levels, Body Mass Index and Obesity in Omanis. Sultan Qaboos Univ Med J. 2006;6(2):27–31. [PMC free article] [PubMed] [Google Scholar]
- 8.Lönnqvist F, Nordfors L, Jansson Mr, Thörne A, Schalling M, Arner P. Leptin Secretion from Adipose Tissue in Women Relationship to Plasma Levels and Gene Expression. The Journal of Clinical Investigation. 1997;99(10):2398–404. doi: 10.1172/JCI119422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravussin Y, LeDuc CA, Watanabe K, Leibel RL. Effects of ambient temperature on adaptive thermogenesis during maintenance of reduced body weight in mice. Am J Physiol Regul Integr Comp Physiol. 2012;303(4):R438–48. doi: 10.1152/ajpregu.00092.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahima’ RS, Flier JS. Adipose Tissue as an Endocrine Organ. Trends in Endocrinology and Metabolism. 2000;11(8):327–32. doi: 10.1016/s1043-2760(00)00301-5. [DOI] [PubMed] [Google Scholar]
- 11.Lee MJ, Fried SK. Integration of hormonal and nutrient signals that regulate leptin synthesis and secretion. Am J Physiol Endocrinol Metab. 2009;296(6):E1230–8. doi: 10.1152/ajpendo.90927.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee M-J, Yang R-Z, Gong D-W, Fried SK. Feeding and Insulin Increase Leptin Translation. The Journal of Biological Chemistry. 2007;282(1):77–80. doi: 10.1074/jbc.M609518200. [DOI] [PubMed] [Google Scholar]
- 13.Cammisotto PG, Bukowiecki LJ, Deshaies Y, Bendayan M. Leptin biosynthetic pathway in white adipocytes. Biochem Cell Biol. 2006;84(2):207–14. doi: 10.1139/o06-032. [DOI] [PubMed] [Google Scholar]
- 14.Ahima RS, Flier JS. Adipose tissue as an endocrine organ. Trends Endocrinol Metab. 2000;11(8):327–32. doi: 10.1016/s1043-2760(00)00301-5. [DOI] [PubMed] [Google Scholar]
- 15.Fruhbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393(Pt 1):7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaisse C, Halaas J, Horvath C, Darnell J, Stoffel M, Friedman J. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nature Genetics. 1996;14(1):95–7. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, et al. Leptin Increases Hypothalamic Pro-opiomelanocortin mRNA Expression in the Rostral Arcuate Nucleus. Diabetes. 1997;46:2119–23. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 18.Hill R, Margetic S, Pegg G, Gazzola C. Leptin: its pharmacokinetics and tissue distribution. International Journal of Obesity. 1998;22:765–70. doi: 10.1038/sj.ijo.0800656. [DOI] [PubMed] [Google Scholar]
- 19.Klein S, Coppack SW, Mohamed-Ali V, Landt M. Adipose Tissue Leptin Production and Plasma Leptin Kinetics in Humans. Diabetes. 1996;45:984–7. doi: 10.2337/diab.45.7.984. [DOI] [PubMed] [Google Scholar]
- 20.Ahima RS, Prabakaran D, Mantzoros CS, Qu D, Lowell BB, Maratos-Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–2. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 21.Brabant G, Nave H, Mayr B, Behrend M, Harmelen VV, Arber P. Secretion of Free and Protein-Bound Leptin from Subcutaneous Adipose Tissue of Lean and Obese Women. The Journal of Clinical Endocrinology & Metabolism. 2002;87(8):3966–70. doi: 10.1210/jcem.87.8.8758. [DOI] [PubMed] [Google Scholar]
- 22.Lammert A, Kiess W, Bottner A, Glasow A, Kratzsch J. Soluble leptin receptor represents the main leptin binding activity in human blood. Biochem Biophys Res Commun. 2001;283(4):982–8. doi: 10.1006/bbrc.2001.4885. [DOI] [PubMed] [Google Scholar]
- 23.Chen K, Li F, Li J, Cai H, Strom S, Bisello A, et al. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat Med. 2006;12(4):425–32. doi: 10.1038/nm1372. [DOI] [PubMed] [Google Scholar]
- 24.Bornstein S, Abu-Asab M, Glasow A, Päth G, Hauner H, Tsokos M, et al. Immunohistochemical and ultrastructural localization of leptin and leptin receptor in human white adipose tissue and differentiating human adipose cells in primary culture. Diabetes. 2000;49(4):532–8. doi: 10.2337/diabetes.49.4.532. [DOI] [PubMed] [Google Scholar]
- 25.Cohen P, Yang G, Yu X, Soukas AA, Wolfish CS, Friedman JM, et al. Induction of leptin receptor expression in the liver by leptin and food deprivation. J Biol Chem. 2005;280(11):10034–9. doi: 10.1074/jbc.M413684200. [DOI] [PubMed] [Google Scholar]
- 26.Chua SC, Koutras IK, Han L, Liu S-M, Kay J, Young SJ, et al. Fine Structure of the Murine Leptin Receptor Gene: Splice Site Suppression is Required to Form Two Alternatively Spliced Transcripts. Genomics. 1997;45:264–70. doi: 10.1006/geno.1997.4962. [DOI] [PubMed] [Google Scholar]
- 27.Maamra M, Bidlingmaier M, Postel-Vinay M, Wu Z, Strasburger C, Ross R. Generation of human soluble leptin receptor by proteolytic cleavage of membrane-anchored receptors. Endocrinology. 2001;142(10):4389–93. doi: 10.1210/endo.142.10.8442. [DOI] [PubMed] [Google Scholar]
- 28.Huang L, Wang Z, Li C. Modulation of circulating leptin levels by its soluble receptor. J Biol Chem. 2001;276(9):6343–9. doi: 10.1074/jbc.M009795200. [DOI] [PubMed] [Google Scholar]
- 29.Yang G, Ge H, Boucher A, Yu X, Li C. Modulation of direct leptin signaling by soluble leptin receptor. Molecular Endocrinology. 2004;18(6):1354–62. doi: 10.1210/me.2004-0027. [DOI] [PubMed] [Google Scholar]
- 30.Mistrík P, Moreau F, Allen J. BiaCore analysis of leptin-leptin receptor interaction: evidence for 1:1 stoichiometry. Analytical Biochemistry. 2004;327(2):271–7. doi: 10.1016/j.ab.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 31.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AWK, Wang Y, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–9. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 32.Ravussin Y, LeDuc CA, Watanabe K, Mueller BR, Skowronski A, Rosenbaum M, et al. Effects of chronic leptin infusion on subsequent body weight and composition in mice: Can body weight set point be reset? Molecular Metabolism. 2014;3(4):432–40. doi: 10.1016/j.molmet.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu J, Ogus S, Lu R, Chehab F. Transgenic mice overexpressing leptin accumulate adipose mass at an older, but not younger, age. Endocrinology. 2001;142(1):342–58. doi: 10.1210/endo.142.1.7909. [DOI] [PubMed] [Google Scholar]
- 34.Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metabolism. 2014;19(2):293–301. doi: 10.1016/j.cmet.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodríguez E, Blázquez J, Guerra M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: the former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides. 2010;4(757–776):757. doi: 10.1016/j.peptides.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Roujeau C, Jockers R, Dam J. New pharmacological perspectives for the leptin receptor in the treatment of obesity. Front Endocrinol. 2014;5:167. doi: 10.3389/fendo.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uotani S, Bjørbæk C, Tornøe J, Flier JS. Functional Properties of Leptin Receptor Isoforms Internalization and Degradation of Leptin and Ligand-Induced Receptor Downregulation. Diabetes. 1999;48:279–86. doi: 10.2337/diabetes.48.2.279. [DOI] [PubMed] [Google Scholar]
- 38.Stratigopoulos G, LeDuc CA, Cremona ML, Chung WK, Leibel RL. Cut-like Homeobox 1 (CUX1) Regulates Expression of the Fat Mass and Obesity-associated and Retinitis Pigmentosa GTPase Regulator-interacting Protein-1-like (RPGRIP1L) Genes and Coordinates Leptin Receptor Signaling. The Journal of Biological Chemistry. 2010;286:2155–70. doi: 10.1074/jbc.M110.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan L, Guo K, Cremona M, McGraw T, Leibel R, Zhang Y. TNF-α up-regulates protein level and cell surface expression of the leptin receptor by stimulating its export via a PKC-dependent mechanism. Endocrinology. 2012;153(12):5821–33. doi: 10.1210/en.2012-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seo S, Guo D-F, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Human Molecular Genetics. 2009;18(7):1323–31. doi: 10.1093/hmg/ddp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byun Kyunghee, G SY, Namkoong Churl, Youn Byung-Soo, Huang Hu, Shin Mi-Seon, Kang Gil Myoung, Kim Hyun-Kyong, Lee Bonghee, Kim Young-Bum, Kim Min-Seon. Clusterin/ApoJ enhances central leptin signaling through Lrp2-mediated endocytosis. EMBO Reports. 2014;15:801–8. doi: 10.15252/embr.201338317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Björnholm M, Münzberg H, Leshan R, Villanueva E, Bates S, Louis G, et al. Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. Journal of Clinical Investigation. 2007;117(5):1354–60. doi: 10.1172/JCI30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Enriori PJ, Sinnayah P, Simonds SE, Rudaz CG, Cowley MA. Leptin Action in the Dorsomedial Hypothalamus Increases Sympathetic Tone to Brown Adipose Tissue in Spite of Systemic Leptin Resistance. Neurobiology of Disease. 2011;31(34):12189–97. doi: 10.1523/JNEUROSCI.2336-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ernst MB, Wunderlich CM, Hess S, Paehler M, Mesaros A, Koralov SB, et al. Enhanced Stat3 Activation in POMC Neurons Provokes Negative Feedback Inhibition of Leptin and InsulinSignaling in Obesity. Neurobiology of Disease. 2009;29(37):11582–93. doi: 10.1523/JNEUROSCI.5712-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stratigopoulos G, Carli JFM, O’Day DR, Wang L, LeDuc CA, Lanzano P, et al. Hypomorphism for RPGRIP1L, a Ciliary Gene Vicinal to the FTO Locus, Causes Increased Adiposity in Mice. Cell Metabolism. 2014;19:767–79. doi: 10.1016/j.cmet.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ravussin Y, Gutman R, Diano S, Shanabrough M, Borok E, Sarman B, et al. Effects of chronic weight perturbation on energy homeostasis and brain structure in mice. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2011;300(6):R1352–R62. doi: 10.1152/ajpregu.00429.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenbaum M, Goldsmith R, Bloomfield D, Magnano A, Weimer L, Heymsfield S, et al. Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J Clin Invest. 2005;115(12):3579–86. doi: 10.1172/JCI25977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galgani J, Greenway F, Caglayan S, Wong M, Licinio J, Ravussin E. Leptin replacement prevents weight loss-induced metabolic adaptation in congenital leptin-deficient patients. Journal of Clinical Endocrinology & Metabolism. 2010;95(2):851–5. doi: 10.1210/jc.2009-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hambly C, Duncan J, Archer Z, Moar K, Mercer J, Speakman J. Repletion of TNFα or leptin in calorically restricted mice suppresses post-restriction hyperphagia. Disease Models and Mechanisms. 2012;5(1):83–94. doi: 10.1242/dmm.007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knight Z, Hannan K, Greenberg M, Friedman J. Hyperleptinemia is required for the development of leptin resistance. PLoS One. 2010;5(6):e11376. doi: 10.1371/journal.pone.0011376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montez J, Soukas A, Asilmaz E, Fayzikhodjaeva G, Fantuzzi G, Friedman J. Acute leptin deficiency, leptin resistance, and the physiologic response to leptin withdrawal. Proceedings of the National Academy of Sciences. 2005;102(7):2537–42. doi: 10.1073/pnas.0409530102. [DOI] [PMC free article] [PubMed] [Google Scholar]