

Abstract
Memory deterioration and synapse damage with accumulation of β-amyloid and hyperphosphorylated tau are hallmark lesions of Alzheimer’s disease (AD). Methylglyoxal (MG), a key intermediate of glucose metabolism, is elevated in AD brains and modifies Aβ42, increasing misfolding and leading to the accumulation of senile plaques. Liraglutide, an analog of glucagon-like peptide-1 (GLP-1), is neurotrophic and neuroprotective. However, whether liraglutide can protect against AD-like memory-related deficits and tau hyperphosphorylation caused by MG in vivo is not known. Here, we report that MG induces tau hyperphosphorylation and causes ultrastructural hippocampal damage and cognitive impairment in C57BL/6J mice. Liraglutide reduced these effects via activation of the protein kinase B and glycogen synthase kinase-3β pathways. Our data reveal that liraglutide may alleviate AD-like cognitive impairment by decreasing the phosphorylation of tau.
Keywords: Alzheimer’s disease, methylglyoxal, glucagon-like peptide-1, glycogen synthase kinase-3β, tau protein
Introduction
Alzheimer’s disease (AD) is the leading cause of dementia, and is characterized by progressive cognitive decline, neuronal loss, memory impairment, and changes in behavior and personality [1]. It is becoming more prevalent in aging populations worldwide [2]. Accumulation of senile plaques composed of amyloid beta (Aβ) peptide and neurofibrillary tangles of hyperphosphorylated tau proteins, are pathological hallmarks of AD [3,4]. Aβ is a 40 or 42 amino acid peptide derived from the cleavage of amyloid precursor protein by γ-secretase [5]. Accumulation of Aβ can increase intracellular Ca2+ loading and induce oxidative stress and neurodegeneration [6]. Tau is a microtubule-associated protein involved in promoting and maintaining microtubule stability under normal physiological conditions [7]. In people with AD, tau is hyperphosphorylated and polymerized into straight and paired helical filaments, known as neurofibrillary tangles [8]. The loss of normal tau function leads to pathological cytoskeletal disturbances, which affect normal neuronal functions such as axonal transport and maintenance of appropriate morphology, leading to neurodegeneration and synaptic dysfunction [9].
The deregulation of glucose metabolism leads to the production of numerous reactive carbonyl compounds such as methylglyoxal (MG), which may be an important factor in the pathomechanism of cognitive impairment [10-12]. MG is a key intermediate of glucose metabolism, produced during glycolysis from triosephosphates or nonenzymatically by sugar fragmentation reactions [13,14]. It is the major precursor to advanced glycation endproducts (AGEs) [15]. Higher levels of baseline MG are associated with cognitive decline and neurodegeneration [16,17], and MG modifies Aβ42, increasing misfolding and leading to the accumulation of senile plaques [18]. In primary cultured hippocampal neurons, MG activates caspase-3 and decreases the ratio of Bcl-2/Bax, inducing apoptotic cell death [19]. Furthermore, MG increases tangle formation in vivo and this process might be accelerated by hyperphosphorylation [20]. MG rapidly modifies proteins and other substrates to generate AGEs, which can contribute to cross-linking of protein fibrils and to proinflammatory signaling, which all contribute to the pathological changes and dementia of AD [21].
Liraglutide, a glucagon-like peptide-1 analog, is a novel long-lasting incretin mimetic, which has been used to treat type 2 diabetes mellitus [22]. Emerging evidence indicates that GLP-1 is neurotrophic and neuroprotective, and that GLP-1 analogs protect neurons from glutamate toxicity in vitro [23], reduce the levels of Aβ in the brain [24], and decrease hyperphosphorylation of tau protein [25]. Furthermore, liraglutide has been shown to prevent synapse loss and deterioration of synaptic plasticity in the hippocampus in 7- and 14-month-old APP/PS1 mice [26,27]. The drug also enhances neuroblastoma cell viability, reduces cytotoxicity and apoptosis via the activation of the cell survival kinases Akt and MEK1/2 and the transcription factor p90RSK, and decreases pro-apoptotic Bax and Bik expression during MG-induced stress [28].
In the present study, we show that intracerebroventricular (i.c.v.) administrationof MG directly induced AD-like tau hyperphosphorylation via activation of glycogen synthase kinase-3β (GSK-3β). In addition, the effects of liraglutide on tau hyperphosphorylation and neuronal ultra-structure were evaluated.
Materials and methods
Reagents and animals
Male C57BL/6J mice (Department of Research Animal Center, Shanghai, China), were housed in groups of five in individually ventilated cages (Tecniplast, Buguggiate, Italy), in a pathogen-free, temperature- and humidity-controlled environment (23 ± 1°C; 50%-60% humidity) under a 12-h light/dark cycle at Fujian Medical University. Mice received a unique identity number (ear tag) and were randomized to individual experimental groups. All experiments were approved by the Fujian Animal Research Ethics committee. MG and Aβ1-42 (Sigma, St. Louis, MO, USA) were diluted with 0.9% (w/v) sterile physiological saline (NS). Liraglutide (Novo Nordisk) was dissolved in NS to a concentration of 2.5 nmol/ml as a 10× stock solution and diluted to 1× before use.
Drug administration
Body weight was monitored weekly throughout the experiment. Mice were 9 weeks old when i.c.v. administration began. At the beginning of the dark phase, mice were anesthetized with 10% (v/v) chloral hydrate (4 ml/kg) and placed in a stereotactic device (Narishige, Tokyo, Japan). A bolus injection was performed using a Hamilton syringe and an i.c.v. injection cannula (internal diameter, 0.11 mm). A micropump (KD Scientific, Holliston, MA, USA) was used to control the injection rate at 0.2 μl/min. MG (0.35, 0.7, or 1.4 µmol), Aβ1-42 (410 pmol) or NS, each in a volume of 3 µl, were injected i.c.v. according to the Allen brain atlas [29], using the following coordinates: anteroposterior: -1.0 mm; mediolateral: -0.4 mm; dorsoventral: -2.5 mm [30]. These mice were then tested in a Morris water maze (see 2.3, below), and the tissue from mice in the 0.7-µmol MG group was used for western blotting.
Another 40 mice were then randomly divided into four groups: control, liraglutide, MG, and MG + liraglutide. Mice received a 3-µl i.c.v. injection of 0.7 µmol of MG or NS, and subsequent once daily subcutaneous injections of liraglutide (25 nmol/kg body weight) or an equivalent volume of NS for 8 weeks. The mice were sacrificed and the left and right hippocampus was collected for transmission electron microscopy (TEM), enzyme-linked immunosorbent assay (ELISA) and western blot assay.
Morris water maze
The Morris water maze was used to evaluate learning and memory in the mice, using a circular pool (diameter, 120 cm; height, 30 cm) filled with water at 24 ± 1°C. A transparent escape platform (diameter, 10 cm; height, 23 cm) was submerged 1 cm beneath the water and placed at a fixed position at the center of one of the quadrants. The task was performed as described previously [31]. Briefly, in the acquisition phase (days 1-5), mice underwent four trials per day with a 10-min intertrial interval, for 5 consecutive training days. Four starting positions, equally spaced along the circumference of the pool, were randomized across the four trials each day. If a mouse did not reach the platform within 60 s, it was gently guided there and was allowed to remain on it for 15 s before being returned to its home cage. The time taken to search for the transparent platform was recorded as the escape latency. The escape latency and swimming speed were recorded by behavioral software (Ethovision 3.0, Noldus Information Technology, Wageningen, the Netherlands). On day 6, a probe trial was performed to assess spatial memory retention. During this trial, mice were allowed to swim freely for 60 s, but no platform was present. The time spent in the target quadrant where the escape platform had been located during the acquisition trial was calculated and the number of times mice crossed over the target quadrant was also recorded.
Glucose, insulin, glucagon, and GLP-1 receptor measurements
Blood glucose levels were measured in whole venous blood collected from the tail using a blood glucose meter (OneTouch UltraLink Meter, LifeScan, Shanghai, China). Insulin and glucagon plasma levels, and insulin and GLP-1 receptor levels in the right hippocampus, were measured using ELISA kits (Cloud-Clone Corp., Houston, TX, U.S.A.), according to the manufacturer’s instructions.
TEM
The mice were anesthetized with 10% chloral hydrate and perfused transcardially with physiological saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer solution (PBS), both at 4°C. The brain was removed and the CA1 subregion of the right hippocampus was dissected rapidly on ice. Samples were post-fixed by immersion in 2.5% buffered glutaraldehyde followed by 1% osmium tetroxide, dehydrated through an ascending ethanol gradient, and embedded in Epon 812. Images were acquired using a Philips EM 208 transmission electron microscope.
Western blot
Western blotting was performed as reported previously [32]. The protein concentration in the supernatant was detected using a bicinchoninic acid assay kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Proteins were separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 80 μg of protein lysate per line). The blots were incubated overnight at 4°C with the primary antibodies: β-actin, tau (both 1:1000; both from Cell Signaling Technology), phospho-tau Ser202/199 (1:3000), phospho-tau Ser396 (1:4000) (both from Abcam), Akt (1:200; BosterBio), phospho-Akt Ser473, GSK-3β, phospho-GSK-3β Ser9, procaspase-3, and cleaved caspase-3 (all 1:1000; all from Cell Signaling Technology). The membranes were then incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:3000; Cell Signaling Technology) for 1 h at room temperature and visualized by exposure to Kodak film after detection with a chemiluminescent reagent (Millipore). Image J analysis software was used to quantify the density of the bands.
Statistics
All data are expressed as the mean ± standard error. SPSS 18.0 and GraphPad Prism 5 were used for statistical analysis and diagrams. In the acquisition phase of the Morris water maze task, latency and swimming speed data were analyzed by two-way repeated measures analysis of variance (ANOVA); probe trial data were analyzed by one-way ANOVA. Other behavioral data and protein expression data were analyzed using one-way ANOVA with least significant difference (equal variances assumed) or Dunnett’s T3 (equal variances not assumed) post hoc tests and Bonferroni multiple comparisons correction. The statistical significance level was set at P<0.05.
Results
MG impaired spatial learning and memory
To explore the effect of MG on the acquisition and retrieval of spatial memory, we evaluated the performance of mice in the Morris water maze task after a single 3-µl i.c.v. injection of MG (0.35, 0.7 or 1.4 µmol). NS and Aβ were used as a negative and positive control, respectively. As expected, the mice in the NS group were quick to find the escape platform and spent a lot of time in the target quadrant. In comparison, mice in the Aβ group took longer to find the platform and spent less time in the target quadrant (P<0.05), indicating a marked deficit in cognitive ability. All concentrations of MG significantly impaired performance during the acquisition phase and in the probe trials (P<0.05; Figure 1). Mice in the MG group had longer escape latencies than those in the NS group (P<0.05; Figure 1A). However, no significant differences were found in swimming speeds among the five groups during the training days (P>0.05; Figure 1B). These results indicate that the increased latency in mice after MG was owing to their deficient spatial memory rather than any motor impairment. In the probe trials, the time spent in the target quadrant was significantly shorter in mice that received MG than in mice that received NS (P<0.05; Figure 1C) and mice in the MG groups crossed the position where the platform had been located significantly fewer times than those in the NS group (P<0.05; Figure 1D). However, there was no significant difference between the three doses of MG, indicating that the cognitive damage caused by MG does not occur in a concentration-dependent manner.
Figure 1.
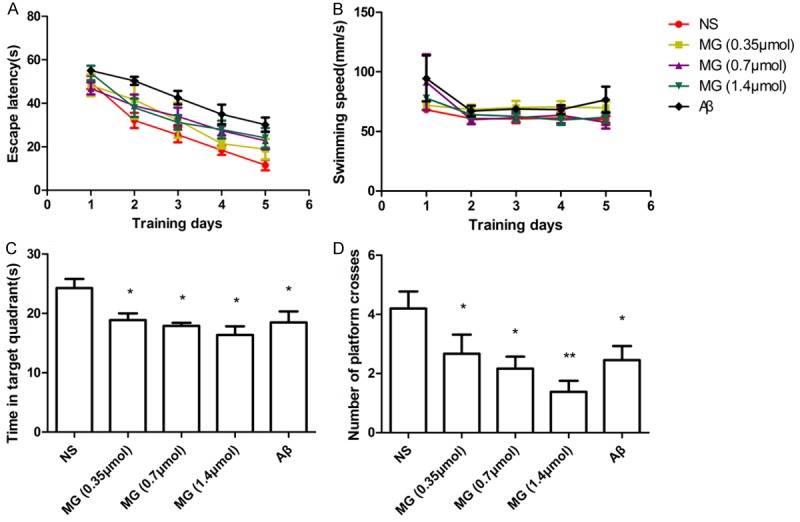
MG (i.c.v.) induced performance deficits (Morris water maze task). A. Mean escape latency decreased with increased training. Latency was significantly longer after MG (0.35, 0.7 or 1.4 µmol) than after NS (P<0.05). B. No differences in swimming speed were observed among the five groups. C. In the spatial probe trial, mice spent significantly less time in the target quadrant after MG (0.35, 0.7 or 1.4 µmol) than after NS (P<0.05). D. Mice made significantly fewer crossings of the target location after MG (0.35, 0.7 or 1.4 µmol) than after NS (P<0.05). The difference in effect between the three concentrations of MG was not significant (P>0.05). n = 7-10 mice per group. *P<0.05 vs. NS group; **P<0.01 vs. NS group. Aβ, amyloid beta (positive control); MG, methylglyoxal; NS, normal saline (negative control).
MG induced tau hyperphosphorylation through the GSK-3β pathway
To explore the molecular mechanisms underlying the memory deficit in mice that received 0.7 µmol of MG, the expression of phospho-tau Ser202/199 and phospho-tau Ser396 was measured by western blotting of hippocampal lysates. The expression of both was significantly greater in the MG group than in the NS group (P<0.05; Figure 2A). However, total tau protein was not significantly different between groups (P>0.05). These results indicate that MG may preferentially induce tau phosphorylation. To further investigate the molecular mechanism involved in MG-induced memory deficit, phospho-Akt and phospho-GSK-3β expression levels were also measured by western blot (Figure 2B). Expression of both was significantly lower in the MG group than in the NS group (P<0.05), whereas total levels of these proteins were unchanged (P>0.05). These results indicate that MG may promote tau hyperphosphorylation in miceby inhibiting Akt and activating GSK-3β.
Figure 2.
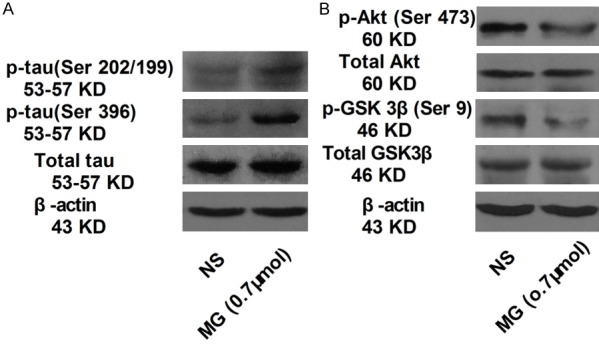
MG induced tau hyperphosphorylation via the GSK-3β pathway (Western blot). Phospho-tau (Ser396), phospho-tau (Ser202/199), tau, phospho-Akt (Ser473)/Akt and phospho-GSK-3β/GSK-3β protein in the hippocampus. Compared with the NS group, the 0.7-µmol MG group showed (A) higher phospho-tau (Ser202/199) and phospho-tau (Ser396) expression; (B) Lower phospho-Akt (Ser473) and phospho-GSK-3β protein expression. Total levels of these proteins were unchanged. The samples derived from the same experiment and blots were processed in parallel. β-actin was used as a loading control. n = 6 mice per group. GSK-3β, glycogen synthase kinase-3β; MG, methylglyoxal; NS, normal saline.
Liraglutide alone did not affect normal cognitive behavior but prevented MG-induced impairment of spatial learning and memory
To investigate the protective effect of liraglutide against cognitive impairment induced by 0.7 µmol of MG, the performance of mice that received NS, liraglutide, MG, or MG + liraglutide was examined in the Morris water maze. Liraglutide alone had no effect on escape latency or distance moved when searching for the hidden platform (P>0.05; Figure 3A). Importantly, liraglutide markedly protected against MG-induced spatial cognition deficits (P<0.05). In the acquisition phase, the escape latency in the MG + liraglutide group was significantly lower than that in the MG group (P<0.05). There was no difference between the four groups in the swimming speed on the training days (P>0.05; Figure 3B). In the probe trials, with the platform removed, liraglutide showed no effect on memory, with no significant difference being observed in the preference to swim in the target quadrant between this group and the control group (P>0.05; Figure 3C, 3D). However, 0.7 µmol of MG significantly decreased the percentage of time spent in the target quadrant (P<0.05) and the number of times the previous location of the platform was crossed (P<0.05). Furthermore, the time spent in the target quadrant and the number of target location crosses were both significantly greater in the MG + liraglutide group than in the MG group (P<0.05). Typical probe trial swimming tracks of mice in each group are shown in Figure 3E. These results indicate that liraglutide protected against MG-induced learning and memory impairments.
Figure 3.
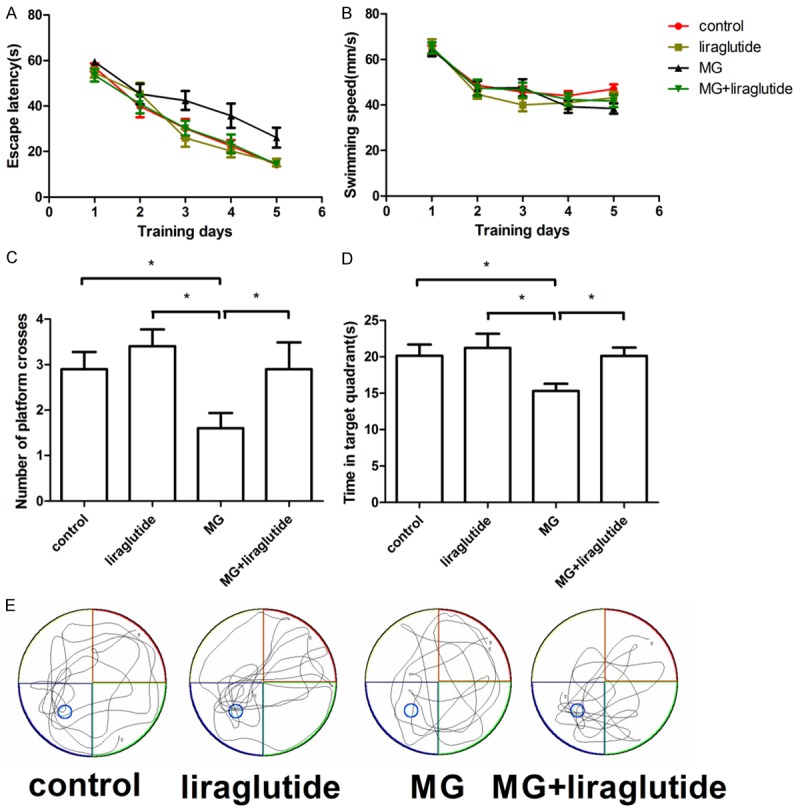
Liraglutide alleviated MG-induced performance deficits (Morris water maze). A. Escape latency in the MG group (0.7 µmol) was longer than in the NS (P<0.05) and liraglutide (P<0.05) groups, and the escape latency of mice in the MG + liraglutide group was shorter than that in the MG group (P<0.05). B. No difference in swimming speed was observed among the four groups (P>0.05). C. Mice in the NS, liraglutide, and MG + liraglutide groups spent significantly more time in the target quadrant than mice that received MG alone (P<0.05) in the spatial probe trial. D. NS, liraglutide, and MG + liraglutide groups crossed the area of the submerged platform significantly more often than mice in the MG group (P<0.05). E. Typical swimming tracks of mice during probe trials. n = 10 rats per group. *P<0.05. MG, methylglyoxal; NS, normal saline.
Liraglutide increased hippocampal GLP-1 receptor expression
After behavioral testing, plasma glucose (Figure 4A), plasma insulin (Figure 4B), plasma glucagon (Figure 4C), and hippocampal insulin (Figure 4D) were measured by ELISA. No differences were observed between the groups in any of these measures (P>0.05), indicating that liraglutide does not promote insulin secretion when plasma glucose is within the normal range. However, subcutaneous liraglutide increased hippocampal GLP-1 receptor expression compared with the NS group (P<0.05; Figure 4E).
Figure 4.
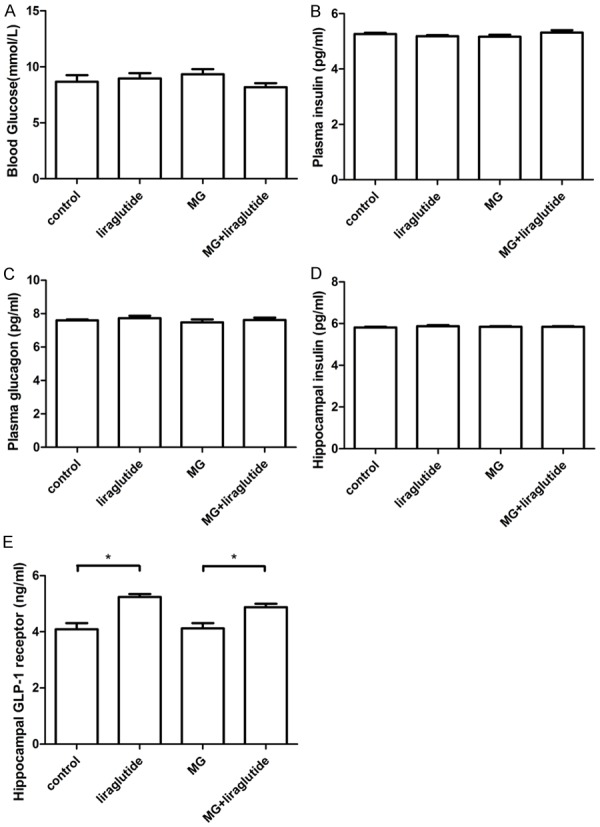
Liraglutide increased the expression of GLP-1 receptor in the hippocampus (ELISA). A: Blood glucose; B: Plasma insulin; C: Plasma glucagon; D: Hippocampal insulin; E: Hippocampal GLP-1 receptor. No differences were observed among the other groups (P>0.05); n = 6 per group *P<0.05. GLP-1, glucagon-like peptide 1; MG, methylglyoxal; NS, normal saline.
Liraglutide protects against MG-induced ultrastructural changes at the synaptic and cellular level
In mice in the NS and liraglutide groups, the typical structure of the chemical synapse [33] was observed in the hippocampal CA1 region using TEM (Figure 5A, 5B). The pre- and postsynaptic membranes were parallel to each other, and the synaptic cleft appeared as a narrow gap. Inside the presynaptic membrane, there were plenty of synaptic vesicles containing neurotransmitters. On the intracellular surface of the postsynaptic membrane was a centralized postsynaptic density. In mice that received 0.7 µmol of MG, synaptic structure differed from that of the control and liraglutide groups. The presynaptic region was deformed, there were fewer synaptic vesicles, and the areas of the postsynaptic region and postsynaptic density were smaller. The synaptic cleft was markedly expanded and the postsynaptic membrane appeared dilated (Figure 5C). However, in the MG + liraglutide group, the synaptic structure was more regular than in the MG group, with more vesicles evenly distributed in the anterior region, more regular presynaptic membranes, and a slightly thickened postsynaptic membrane (Figure 5D). These observations suggest that liraglutide protects the synaptic structure of the CA1 region against MG in mice.
Figure 5.
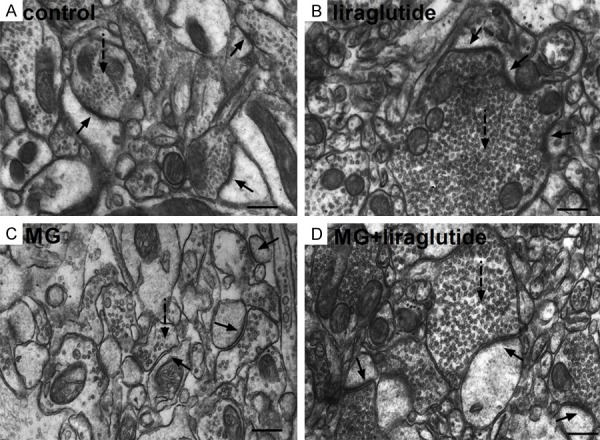
Liraglutide alleviates MG-induced synaptic structure degradation (TEM). (A, B) Chemical synapses in mice that received NS (A) or liraglutide (B) appeared normal: the postsynaptic density was centralized on the intracellular surface of the opposing postsynaptic membrane; synaptic vesicles were observed inside the presynaptic membrane; the synaptic cleft was narrow and with parallel membranes either side. (C) In mice that received MG, synaptic structure was destroyed, with fewer synaptic vesicles, a smaller postsynaptic region, deformed anterior region, and a smaller postsynaptic density; the synaptic cleft was markedly widened and the postsynaptic membrane was swollen. (D) In the MG + liraglutide group, vesicles were evenly distributed in the presynaptic area, and the postsynaptic membrane was slightly thickened. Compared with the MG group, the synaptic form was more regular and the cleft was recognizable. Solid arrow, synapse; dotted arrowheads, synaptic vesicles. n = 3 mice per group. Scale bar = 300 nm. TEM, transmission electron microscopy; MG, methylglyoxal.
The cellular ultrastructure (mitochondria, Golgi apparatus, and lysosomes) of the CA1 appeared normal in the NS and liraglutide groups (Figure 6A, 6B). In contrast, in the 0.7 µmol MG group, the structure of the organelles was irregular, mitochondria were swollen and showed fewer and fractured cristae, and aggregated lipofuscin was observed (Figure 6C). However, in the MG + liraglutide group, there was less organelle damage than in the MG group; organelles were more regularly shaped and larger numbers of mitochondrial cristae were observed, and there was less lipofuscin (Figure 6D). These results suggest that liraglutide alleviates the cell damage induced by MG.
Figure 6.
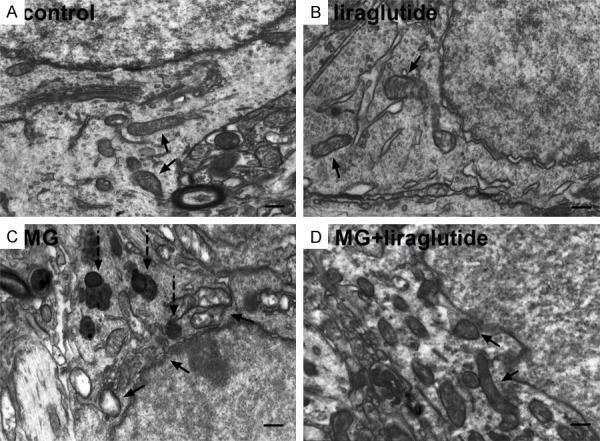
Liraglutide alleviates MG-induced cell structure degradation (TEM). (A, B) Cell ultrastructure appeared normal in mice that received NS (A) and liraglutide (B); cells contained many mitochondria, Golgi apparatus, and lysosomes, which all appeared structurally normal. (C) In the MG group, organelle structure was irregular, mitochondria were swollen, with few and fractured cristae; abundant lipofuscin was observed in the CA1 region. (D) In the MG + liraglutide group, damage to organelles was somewhat alleviated; observations included less lipofuscin, more regular organelle structure, and more mitochondrial cristae compared with the MG group. Solid arrow, mitochondria; dotted arrowheads, lipofuscin. n = 3 per group. Scale bar = 300 nm.
Liraglutide reduced MG-induced activated caspase-3 expression and tau hyperphosphorylation
Caspase-3 is an important biomolecule in intracellular apoptotic pathways and is activated in neurons and astrocytes in AD [34]. In the 0.7 µmol MG group, expression of cleaved caspase-3 was significantly greater than in the NS and liraglutide groups (P<0.05; Figure 7A, 7B). However, cleaved caspase-3 expression was significantly lower in the MG + liraglutide group than in the MG group (P<0.05). Total caspase-3 did not differ among the four groups (P>0.05).
Figure 7.
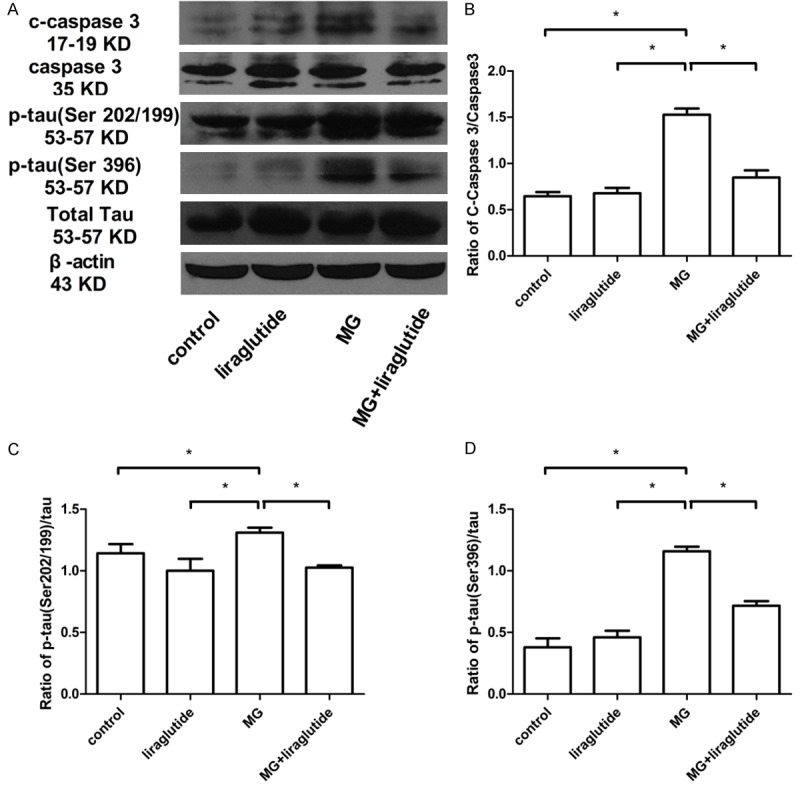
Liraglutide reduces activated (c)-caspase-3 and tau hyperphosphorylation induced by MG (Western blot). A: c-Caspase-3/caspase-3; B: Phospho-tau (Ser202/199)/phospho-tau (Ser396)/tau; C: Phospho-Akt (Ser473)/Akt; D: Phospho-GSK-3β/GSK-3β protein in the hippocampus (n = 6 mice per group). All samples shown derive from the same experiment and blots were processed in parallel. β-Actin was used as a loading control. *P<0.05.
Hyperphosphorylated tau protein is a key component of neurofibrillary tangles. Western blotting was carried out on hippocampal lysates to measure phospho-tau Ser202/199 and phospho-tau Ser396 expression. Compared with the NS group, the expression of both proteins was elevated in the MG group (Figure 7A-D). Furthermore, we observed that hyperphosphorylation of tau at Ser202/199 and Ser396 in the hippocampus was lower in the MG + liraglutide group than in the MG group. There was no significant difference in total tau protein expression among the groups. These results indicated that liraglutide reduced caspase-3 activation and tau hyperphosphorylation induced by MG.
Liraglutide activated the GSK-3β signaling pathway
To investigate whether liraglutide inhibited tau phosphorylation in mice with MG-induced AD-like features, we measured the expression of Akt and GSK-3β, two protein kinases important in modulating tau phosphorylation. GSK3β is a major tau kinase in the brain and phosphorylates tau at various sites including Ser199, Ser202, and Ser396 [35]. Activated phospho-Akt phosphorylates GSK-3β at Ser9 and inhibits its kinase activity [36]. In the present study, we found a significantly lower level of phosphorylation of both Akt and GSK-3β in mice that received MG than in those that received NS (P<0.05) whereas the total level of both kinases was unchanged (P>0.05) (Figure 8A, 8B). Liraglutide administration for 8 weeks resulted a recovery of phosphorylation of both Akt and GSK-3β (P<0.05) (Figure 8A-C). These results indicated that liraglutide delivery inhibits tau hyperphosphorylation by activating Akt and inhibiting GSK-3β activity in MG-treated mice.
Figure 8.
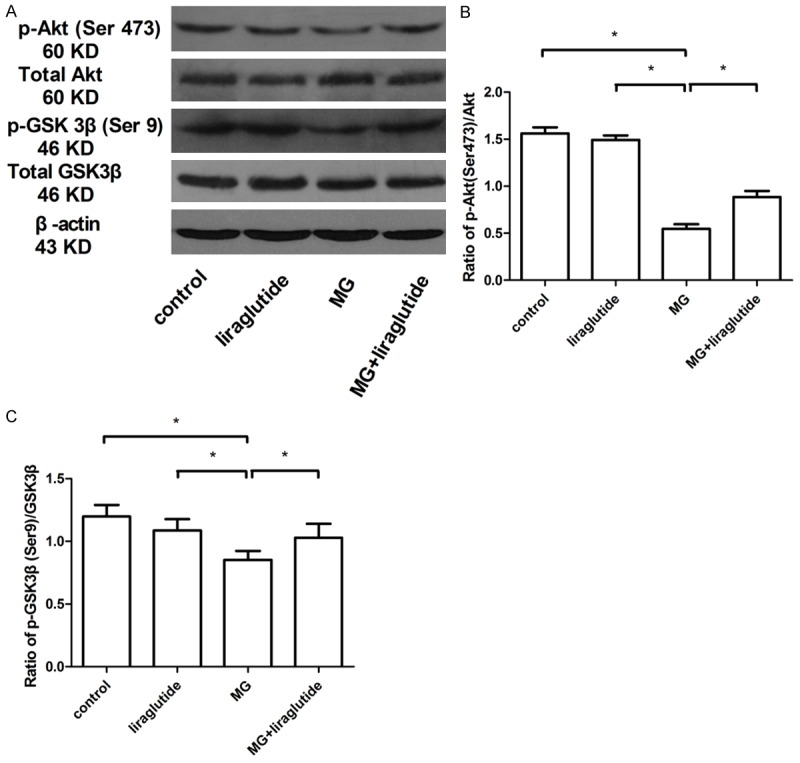
Liraglutide promotes activation of the p-Akt/GSK-3β signaling pathway (Western blot). Liraglutide increased the active (phosphorylated) form of Akt (A, B) and decreased the active form of GSK-3β (non-phosphorylated) (A, C). The samples derive from the same experiment and blots were processed in parallel (n = 6 mice per group). β-Actin was used as the loading control. *P<0.05.
Discussion
The present study has shown that i.c.v. administration of MG induces cognitive impairment accompanied by ultrastructural changes in pyramidal cells and synapses, and regulation of tau-associated proteins in the hippocampus. Importantly, we also found that liraglutide alleviated MG-induced cognitive impairment and activated the Akt/GSK3β signaling pathway in the hippocampus, which is known to decrease tau phosphorylation.
Tau hyperphosphorylation is a pathological hallmark and an early event in the development of AD [37,38]. Therefore, modulating tau phosphorylation may prevent the progression of AD pathology [39]. However, tau hyperphosphorylation remains incompletely understood owing to the contribution of many factors in the cellular environment. Previous studies have shown that MG induces tau hyperphosphorylation by promoting the formation of AGEs [40]. Furthermore, MG accelerates tangle formation in vivo, in terms of formation of tau dimers and higher molecular weight oligomers [20]. Therefore, we hypothesized that MG modulates tau phosphorylation. Serum MG concentration is associated with cognitive decline in elderly individuals [16] and MG levels are elevated in the cerebrospinal fluid of people with AD [41]. We used three concentrations of MG to explore its effect on cognitive ability in mice. The concentrations were based on previous reports of MG levels in the mouse brain [42-45]. We found that all three concentrations tested impaired learning and memory, but the difference between doses of MG was not significant owing to large intergroup variations. Importantly, we found that MG resulted in tau hyperphosphorylation at multiple AD-related sites and at all three doses tested.
Recent studies have shown that analogs of GLP-1, such as liraglutide, improve cognition via neurotrophic effects [46,47]. GLP-1(9-36) amide, a natural cleavage product of GLP-1, prevented impairments in long-term potentiation and enhanced long-term depression induced by exogenous Aβ1-42 [48]. Liraglutide significantly increased memory retention and numbers of hippocampal CA1 region pyramidal neurons in SAMP8 mice compared with age-matched vehicle-injected SAMP8 mice [49]. Furthermore, liraglutide increased the expression of the pro-survival signaling protein Mcl-1, activated the cell survival kinases Akt and MEK1/2 and the transcription factor p90RSK, increased the membrane potential, and caused an influx of calcium in human neuroblastoma SH-SY5Y cells exposed to MG stress [28]. Interestingly, learning and memory can be improved when GLP-1 receptors are overexpressed in the hippocampus, whereas long-term potentiation and spatial learning and memory decline when GLP-1 receptors are knocked out in the hippocampus [50,51]. Importantly, liraglutide can cross the blood-brain barrier and GLP-1 receptors are expressed in the brain, including the hippocampus, an area involved in memory formation [26,52]. Together, this indicated that subcutaneous injections of liraglutide might play a protective role against MG-induced impairments in learning and memory. Therefore, in the present study, we used subcutaneous injections of liraglutide to explore the possible protective effect on cognition, which was impaired by 0.7 µmol of MG. As expected, we found that liraglutide reversed MG-induced impairments in spatial learning and memory in the Morris water maze. Furthermore, morphological changes at the synapse corresponded to an overall decrease in synaptic function [53]. Our TEM images revealed that MG-induced neuronal and synaptic damage was reduced by liraglutide, supporting a previous study in which (Val8) GLP-1 reversed damage to the nucleolus and nucleus induced by i.c.v. streptozotocin [25].
In addition, peripheral liraglutide injections significantly decreased tau hyperphosphorylation at both the Ser199/202 and Ser396 sites, which are associated with AD [54,55]. Tau phosphorylation is regulated by protein kinases and phosphatases [56]. Several kinases phosphorylate tau in vitro, including GSK-3β [57,58], cyclin dependent protein kinase 5 [59,60], cAMP-dependent protein kinase, and stress-activated protein kinase [61,62]. Previous reports have shown that MG induces AD-like tau hyperphosphorylation via formation of AGEs, involving upregulation of the receptor for advanced glycation end products (RAGE), and activation of GSK-3β [40], In the brain of a rat model of type 2 diabetes, liraglutide was shown to decrease phosphorylation of Akt at Ser308 and GSK-3β at Ser9, which indicated inactivation of GSK-3β [36]. We therefore examined the effect of liraglutide on the Akt-GSK3β signaling pathway and, in agreement with previous studies, we found that it ameliorated the MG-induced increase in tau phosphorylation at Ser199/202 and Ser396, and that this effect appeared to be associated with the downregulation of brain GSK-3β activity.
Conclusions
Taken together, our results show that MG impairs cognitive function by inducing ultrastructural changes in synapses and pyramidal neurons, and increases tau phosphorylation. Liraglutide effectively reversed this cognitive impairment and prevented ultrastructural damage in AD-like model mice after MG. Furthermore, liraglutide reduced MG-induced hyperphosphorylation of tau in the brain at several phosphorylation sites by increasing Akt phosphorylation and decreasing GSK-3β activity. These findings provide evidence that targeting MG may be a promising therapeutic strategy to prevent AD-like tau hyperphosphorylation, and that liraglutide or similar stable GLP-1 analogs are a promising therapy for slowing or reversing degenerative processes observed in diseases such as AD.
Acknowledgements
This work was supported by grants from the National Clinical Key Specialty Discipline Construction Programs (grant number 2010306); Fujian Province’s Key Clinical Specialty Discipline Construction Programs (grant numbers 2010306 and 2015593); Fujian Medcial University Professor Fund (grant number FS15007) and the China International Medical Foundation (CIMF)-Novo Nordisk China Diabetes Young Scientific Talent Research Funding (grant number 2013-1).
Disclosure of conflict of interest
None.
References
- 1.Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006239. doi: 10.1101/cshperspect.a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mucke L. Alzheimer’s disease. Nature. 2009;461:895–897. doi: 10.1038/461895a. [DOI] [PubMed] [Google Scholar]
- 3.Goedert M. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science. 2015;349:1255555. doi: 10.1126/science.1255555. [DOI] [PubMed] [Google Scholar]
- 4.Shen C, Chen Y, Liu H, Zhang K, Zhang T, Lin A, Jing N. Hydrogen peroxide promotes Abeta production through JNK-dependent activation of gamma-secretase. J Biol Chem. 2008;283:17721–17730. doi: 10.1074/jbc.M800013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sierant M, Kubiak K, Kazmierczak-Baranska J, Paduszynska A, Kuwabara T, Warashina M, Nacmias B, Sorbi S, Nawrot B. RNA interference in silencing of genes of Alzheimer’s disease in cellular and rat brain models. Nucleic Acids Symp Ser (Oxf) 2008;52:41–42. doi: 10.1093/nass/nrn021. [DOI] [PubMed] [Google Scholar]
- 6.Qi JS, Qiao JT. Amyloid beta-protein fragment 31-35 forms ion channels in membrane patches excised from rat hippocampal neurons. Neuroscience. 2001;105:845–852. doi: 10.1016/s0306-4522(01)00244-5. [DOI] [PubMed] [Google Scholar]
- 7.Spillantini MG, Michel Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–433. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- 8.Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barage SH, Sonawane KD. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. 2015;52:1–18. doi: 10.1016/j.npep.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Gella A, Durany N. Oxidative stress in Alzheimer disease. Cell Adh Migr. 2009;3:88–93. doi: 10.4161/cam.3.1.7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matafome P, Sena C, Seica R. Methylglyoxal, obesity, and diabetes. Endocrine. 2013;43:472–484. doi: 10.1007/s12020-012-9795-8. [DOI] [PubMed] [Google Scholar]
- 12.Pratico D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci. 2008;29:609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Sousa Silva M, Gomes RA, Ferreira AE, Ponces Freire A, Cordeiro C. The glyoxalase pathway: the first hundred years. .. and beyond. Biochem J. 2013;453:1–15. doi: 10.1042/BJ20121743. [DOI] [PubMed] [Google Scholar]
- 14.Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. 1999;344:109–116. [PMC free article] [PubMed] [Google Scholar]
- 15.Vistoli G, De Maddis D, Cipak A, Zarkovic N, Carini M, Aldini G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radic Res. 2013;47(Suppl 1):3–27. doi: 10.3109/10715762.2013.815348. [DOI] [PubMed] [Google Scholar]
- 16.Beeri MS, Moshier E, Schmeidler J, Godbold J, Uribarri J, Reddy S, Sano M, Grossman HT, Cai W, Vlassara H, Silverman JM. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech Ageing Dev. 2011;132:583–587. doi: 10.1016/j.mad.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srikanth V, Westcott B, Forbes J, Phan TG, Beare R, Venn A, Pearson S, Greenaway T, Parameswaran V, Munch G. Methylglyoxal, cognitive function and cerebral atrophy in older people. J Gerontol A Biol Sci Med Sci. 2013;68:68–73. doi: 10.1093/gerona/gls100. [DOI] [PubMed] [Google Scholar]
- 18.Fawver JN, Schall HE, Petrofes Chapa, RD , Zhu X RD. Amyloid-beta metabolite sensing: biochemical linking of glycation modification and misfolding. J Alzheimers Dis. 2012;30:63–73. doi: 10.3233/JAD-2012-112114. [DOI] [PubMed] [Google Scholar]
- 19.Chen YJ, Huang XB, Li ZX, Yin LL, Chen WQ, Li L. Tenuigenin protects cultured hippocampal neurons against methylglyoxal-induced neurotoxicity. Eur J Pharmacol. 2010;645:1–8. doi: 10.1016/j.ejphar.2010.06.034. [DOI] [PubMed] [Google Scholar]
- 20.Kuhla B, Haase C, Flach K, Luth HJ, Arendt T, Munch G. Effect of pseudophosphorylation and cross-linking by lipid peroxidation and advanced glycation end product precursors on tau aggregation and filament formation. J Biol Chem. 2007;282:6984–6991. doi: 10.1074/jbc.M609521200. [DOI] [PubMed] [Google Scholar]
- 21.Munch G, Kuhla B, Luth HJ, Arendt T, Robinson SR. Anti-ageing defences against alzheimer’s disease. Biochem Soc Trans. 2003;31:1397–1399. doi: 10.1042/bst0311397. [DOI] [PubMed] [Google Scholar]
- 22.Drucker DJ, Dritselis A, Kirkpatrick P. Liraglutide. Nat Rev Drug Discov. 2010;9:267–268. doi: 10.1038/nrd3148. [DOI] [PubMed] [Google Scholar]
- 23.Gilman CP, Perry T, Furukawa K, Grieg NH, Egan JM, Mattson MP. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J Neurochem. 2003;87:1137–1144. doi: 10.1046/j.1471-4159.2003.02073.x. [DOI] [PubMed] [Google Scholar]
- 24.Gault VA, Holscher C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur J Pharmacol. 2008;587:112–117. doi: 10.1016/j.ejphar.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Zhang ZF, Holscher C, Gao C, Jiang YH, Liu YZ. (Val(8)) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. Eur J Pharmacol. 2012;674:280–286. doi: 10.1016/j.ejphar.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 26.McClean PL, Holscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014;76:57–67. doi: 10.1016/j.neuropharm.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 27.McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31:6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma MK, Jalewa J, Holscher C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J Neurochem. 2014;128:459–471. doi: 10.1111/jnc.12469. [DOI] [PubMed] [Google Scholar]
- 29.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 30.Lahmy V, Long R, Morin D, Villard V, Maurice T. Mitochondrial protection by the mixed muscarinic/sigma1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Abeta25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front Cell Neurosci. 2014;8:463. doi: 10.3389/fncel.2014.00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Zhang J, Zheng K, Shen H, Chen X. Long-term ginsenoside Rg1 supplementation improves age-related cognitive decline by promoting synaptic plasticity associated protein expression in C57BL/6J mice. J Gerontol A Biol Sci Med Sci. 2014;69:282–294. doi: 10.1093/gerona/glt091. [DOI] [PubMed] [Google Scholar]
- 33.Hernandez-Nicaise ML. The nervous system of ctenophores. III. Ultrastructure of synapses. J Neurocytol. 1973;2:249–263. doi: 10.1007/BF01104029. [DOI] [PubMed] [Google Scholar]
- 34.Bader Lange ML, St Clair D, Markesbery WR, Studzinski CM, Murphy MP, Butterfield DA. Age-related loss of phospholipid asymmetry in APP (NLh)/APP (NLh) x PS-1 (P264L)/PS-1 (P264L) human double mutant knock-in mice: relevance to Alzheimer disease. Neurobiol Dis. 2010;38:104–115. doi: 10.1016/j.nbd.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang JZ, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. 2008;85:148–175. doi: 10.1016/j.pneurobio.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Zhang J, Ma D, Zhang M, Hu S, Shao S, Gong CX. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J Alzheimers Dis. 2013;37:637–648. doi: 10.3233/JAD-130491. [DOI] [PubMed] [Google Scholar]
- 37.Chou JL, Shenoy DV, Thomas N, Choudhary PK, Laferla FM, Goodman SR, Breen GA. Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J Proteomics. 2011;74:466–479. doi: 10.1016/j.jprot.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Riederer BM, Mourton-Gilles C, Frey P, Delacourte A, Probst A. Differential phosphorylation of tau proteins during kitten brain development and Alzheimer’s disease. J Neurocytol. 2001;30:145–158. doi: 10.1023/a:1011991207942. [DOI] [PubMed] [Google Scholar]
- 39.Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Kayed R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. 2014;40(Suppl 1):S97–S111. doi: 10.3233/JAD-132477. [DOI] [PubMed] [Google Scholar]
- 40.Li XH, Xie JZ, Jiang X, Lv BL, Cheng XS, Du LL, Zhang JY, Wang JZ, Zhou XW. Methylglyoxal induces tau hyperphosphorylation via promoting AGEs formation. Neuromolecular Med. 2012;14:338–348. doi: 10.1007/s12017-012-8191-0. [DOI] [PubMed] [Google Scholar]
- 41.Kuhla B, Luth HJ, Haferburg D, Boeck K, Arendt T, Munch G. Methylglyoxal, glyoxal, and their detoxification in Alzheimer’s disease. Ann N Y Acad Sci. 2005;1043:211–216. doi: 10.1196/annals.1333.026. [DOI] [PubMed] [Google Scholar]
- 42.Hambsch B, Chen BG, Brenndorfer J, Meyer M, Avrabos C, Maccarrone G, Liu RH, Eder M, Turck CW, Landgraf R. Methylglyoxal-mediated anxiolysis involves increased protein modification and elevated expression of glyoxalase 1 in the brain. J Neurochem. 2010;113:1240–1251. doi: 10.1111/j.1471-4159.2010.06693.x. [DOI] [PubMed] [Google Scholar]
- 43.Kurz A, Rabbani N, Walter M, Bonin M, Thornalley P, Auburger G, Gispert S. Alpha-synuclein deficiency leads to increased glyoxalase I expression and glycation stress. Cell Mol Life Sci. 2011;68:721–733. doi: 10.1007/s00018-010-0483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Distler MG, Palmer AA. Role of Glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: recent advances and mechanistic insights. Front Genet. 2012;3:250. doi: 10.3389/fgene.2012.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jakubcakova V, Curzi ML, Flachskamm C, Hambsch B, Landgraf R, Kimura M. The glycolytic metabolite methylglyoxal induces changes in vigilance by generating low-amplitude non-REM sleep. J Psychopharmacol. 2013;27:1070–1075. doi: 10.1177/0269881113495596. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, An FM, Yin L, Liu AR, Yin DK, Yao WB, Gao XD. Glucagon-like peptide-1 protects hippocampal neurons against advanced glycation end product-induced tau hyperphosphorylation. Neuroscience. 2014;256:137–146. doi: 10.1016/j.neuroscience.2013.10.038. [DOI] [PubMed] [Google Scholar]
- 47.Porter WD, Flatt PR, Holscher C, Gault VA. Liraglutide improves hippocampal synaptic plasticity associated with increased expression of Mash1 in ob/ob mice. Int J Obes (Lond) 2013;37:678–684. doi: 10.1038/ijo.2012.91. [DOI] [PubMed] [Google Scholar]
- 48.Ma T, Du X, Pick JE, Sui G, Brownlee M, Klann E. Glucagon-like peptide-1 cleavage product GLP-1(9-36) amide rescues synaptic plasticity and memory deficits in Alzheimer’s disease model mice. J Neurosci. 2012;32:13701–13708. doi: 10.1523/JNEUROSCI.2107-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hansen HH, Fabricius K, Barkholt P, Niehoff ML, Morley JE, Jelsing J, Pyke C, Bjerre Knudsen L, Farr SA, Vrang N. The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of Alzheimer’s disease. J Alzheimers Dis. 2015;46:877–888. doi: 10.3233/JAD-143090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abbas T, Faivre E, Holscher C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: Interaction between type 2 diabetes and Alzheimer’s disease. Behav Brain Res. 2009;205:265–271. doi: 10.1016/j.bbr.2009.06.035. [DOI] [PubMed] [Google Scholar]
- 51.During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, Bland RJ, Klugmann M, Banks WA, Drucker DJ, Haile CN. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med. 2003;9:1173–1179. doi: 10.1038/nm919. [DOI] [PubMed] [Google Scholar]
- 52.Hamilton A, Holscher C. Receptors for the incretin glucagon-like peptide-1 are expressed on neurons in the central nervous system. Neuroreport. 2009;20:1161–1166. doi: 10.1097/WNR.0b013e32832fbf14. [DOI] [PubMed] [Google Scholar]
- 53.Adams MM, Shi L, Linville MC, Forbes ME, Long AB, Bennett C, Newton IG, Carter CS, Sonntag WE, Riddle DR, Brunso-Bechtold JK. Caloric restriction and age affect synaptic proteins in hippocampal CA3 and spatial learning ability. Exp Neurol. 2008;211:141–149. doi: 10.1016/j.expneurol.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu SJ, Zhang AH, Li HL, Wang Q, Deng HM, Netzer WJ, Xu H, Wang JZ. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem. 2003;87:1333–1344. doi: 10.1046/j.1471-4159.2003.02070.x. [DOI] [PubMed] [Google Scholar]
- 55.Zhou XW, Li X, Bjorkdahl C, Sjogren MJ, Alafuzoff I, Soininen H, Grundke-Iqbal I, Iqbal K, Winblad B, Pei JJ. Assessments of the accumulation severities of amyloid beta-protein and hyperphosphorylated tau in the medial temporal cortex of control and Alzheimer’s brains. Neurobiol Dis. 2006;22:657–668. doi: 10.1016/j.nbd.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 56.Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem. 2009;108:1480–1494. doi: 10.1111/j.1471-4159.2009.05882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu M, Waring JF, Gopalakrishnan M, Li J. Role of GSK-3beta activation and alpha7 nAChRs in Abeta(1-42)-induced tau phosphorylation in PC12 cells. J Neurochem. 2008;106:1371–1377. doi: 10.1111/j.1471-4159.2008.05483.x. [DOI] [PubMed] [Google Scholar]
- 58.Qi LQ, Ke LF, Liu XH, Liao LM, Ke SJ, Liu XY, Wang YP, Lin XW, Zhou Y, Wu LJ, Chen Z, Liu LB. Subcutaneous administration of liraglutide ameliorates learaning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3β pathway in an amyloid β protein induced Alzheimer disease mouse model. Eur J Pharmacol. 2016;783:23–32. doi: 10.1016/j.ejphar.2016.04.052. [DOI] [PubMed] [Google Scholar]
- 59.Hamdane M, Sambo AV, Delobel P, Begard S, Violleau A, Delacourte A, Bertrand P, Benavides J, Buee L. Mitotic-like tau phosphorylation by p25-Cdk5 kinase complex. J Biol Chem. 2003;278:34026–34034. doi: 10.1074/jbc.M302872200. [DOI] [PubMed] [Google Scholar]
- 60.Zhao H, Chang R, Che H, Wang J, Yang L, Fang W, Xia Y, Li N, Ma Q, Wang X. Hyperphosphorylation of tau protein by calpain regulation in retina of Alzheimer’s disease transgenic mouse. Neurosci Lett. 2013;551:12–16. doi: 10.1016/j.neulet.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 61.Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- 62.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]