

Abstract
The endocannabinoid system (ECS), and agonists acting on cannabinoid receptors (CBr), are known to regulate several physiological events in the brain, including modulatory actions on excitatory events probably through N-methyl-D-aspartate receptor (NMDAr) activity. Actually, CBr agonists can be neuroprotective. The synthetic CBr agonist WIN55,212-2 acts mainly on CB1 receptor. In turn, the mitochondrial toxin 3-nitropropionic acid (3-NP) produces striatal alterations in rats similar to those observed in the brain of Huntington’s disease patients. Herein, the effects of WIN55,212-2 were tested on different endpoints of the 3-NP-induced toxicity in rat brain synaptosomes and striatal tissue. Motor activity was also evaluated. The 3-NP (1 mM)-induced mitochondrial dysfunction and lipid peroxidation was attenuated by WIN55,212-2 (1 µM) in synaptosomal fractions. The intrastriatal bilateral injection of 3-NP (500 nmol/µL) to rats increased lipid peroxidation and locomotor activity, augmented the rate of cell damage, and decreased the striatal density of neuronal cells. These alterations were accompanied by transcriptional changes in the NMDA (NR1 subunit) content. The administration of WIN55212-2 (1 mg/kg, i.p.) to rats for six consecutive days, before the 3-NP injection, exerted preventive effects on all alterations elicited by the toxin. The prevention of the 3-NP-induced NR1 transcriptional alterations by the CBr agonist together with the increase of CB1 content suggest an early reduction of the excitotoxic process via CBr activation. Our results demonstrate a protective role of WIN55,212-2 on the 3-NP-induced striatal neurotoxicity that could be partially related to the ECS stimulation and induction of NMDAr hypofunction, representing an effective therapeutic strategy at the experimental level for further studies.
Keywords: 3-Nitropropionic acid, mitochondrial energy depletion, WIN55, 212-2, endocannabinoid system, neuroprotection, cannabinoid receptor agonists
Introduction
Since the first description of the endocannabinoid system (ECS) [1], evidence has been collected demonstrating its modulatory role in different physiological processes in mammals, with particular emphasis in the CNS. As integral part of the ECS, two types of G protein-coupled cannabinoid receptors (CBr) have been described in the brain: cannabinoid receptor 1 (CB1r) has been found in neuronal cells from cortex, hippocampus, striatum and cerebellum, whereas cannabinoid receptor 2 (CB2r) is mostly located in immune cells (microglia) throughout the brain, and in a minor density in other glial, endothelial and neuronal cells [2,3]. CB2r are likely to be more linked to the modulation of immunological responses and motor functions. Either through CB1r or CB2r stimulation, the regulation of the ECS can confer neuroprotective actions against excitotoxic insults [4]. To this regard, among the considerable number of synthetic cannabinoids available nowadays, WIN55,212-2 is a well-known agonist for CB1r that reproduces the in vivo effects elicited by delta-9-tetrahydrocannabinol (D9-THC) [5,6], representing a good candidate for studies aiming to stimulate the ECS in experimental models of chronic disorders in search for neuroprotective mechanisms.
Mitochondrial dysfunction and secondary excitotoxicity are well-known triggering factors leading to cell damage. Excitotoxicity refers to the overactivation of N-methyl-D-aspartate (NMDA) receptors (NMDAr), with the consequent increase in the intracellular levels of Ca2+. Altogether, these processes activate noxious signaling cascades, thus promoting neuronal cell death that can be related with the progression of neurodegenerative disorders [7,8]. The neurotoxic model produced by the mitochondrial toxin 3-nitropropionic acid (3-NP) in mammals mimics some important features of Huntington’s disease (HD) as it produces neurodegeneration similar to that observed in this disorder [9,10]. Among its toxic effects 3-NP is known to inhibit succinate dehydrogenase (SDH, mitochondrial complex II) activity by competing with succinate, thus blocking the electron transport chain and the Krebs cycle, depleting the levels of ATP and favoring the formation of reactive oxygen species (ROS) and secondary excitotoxicity, ultimately leading to mitochondrial dysfunction [11] and neuronal cell death [12,13]. Noteworthy, the protective role of cannabinoid receptor agonists has been poorly studied in this toxic model. Therefore, this toxic model constitutes a valuable tool to explore protective effects and mechanisms linked to the manipulation of the ECS as an experimental approach to identify new therapeutic alternatives for treatment of neurodegenerative disorders such as HD.
In this study we evaluated the preventive effect of WIN55,212-2 on behavioral, morphological and biochemical alterations induced by 3-NP after its intrastriatal infusion into the rat striatum. In addition, we evaluated the effect of WIN55,212-2 on biochemical markers of damage induced by 3-NP in rat brain synaptosomes. Our results demonstrate that rats pretreated with the synthetic cannabinoid receptor agonist reduced the toxic endpoints induced by 3-NP, probably involving mechanisms oriented to reduce excessive excitability in the brain. WIN55,212-2 also resulted protective against the 3-NP-induced mitochondrial dysfunction and oxidative damage in synaptosomes, confirming its protective properties.
The schematic representation of chemical structures of the agents tested in this study, WIN55,212-2 and 3-NP, is shown in Figure 1, where 3-NP is structurally compared with succinate, the SDH natural substrate.
Figure 1.
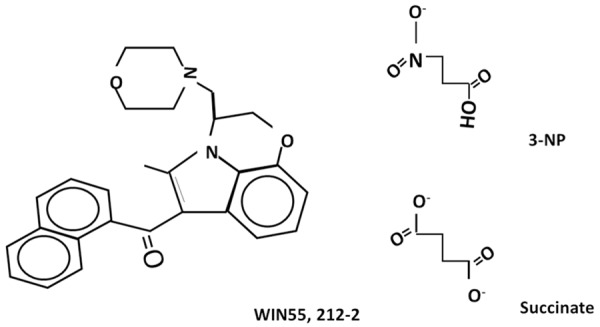
Schematic representation of the chemical structures of WIN55,212-2, 3-nitropropionic acid (3-NP) and succinate.
Materials and methods
Chemicals
3-NP, HEPES, dithiothreitol, protease and phosphatase inhibitor cocktail, WIN55,212-2, and the anti-NR1 antibody were all obtained from Sigma-Aldrich (St. Louis, MO). Other reagents were obtained from other commercial sources. Solutions were prepared using deionized water from a Milli-RQ system (Millipore, MA). Anti-CB1r (C-20), anti-NeuN and anti-actin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Donkey-anti rabbit IgG-horseradish peroxidase conjugate and donkey-anti mouse IgG-horseradish peroxidase conjugate secondary antibodies were obtained from Jackson Immunoresearch (Baltimore, MA, USA). Enhanced chemiluminescence (ECL) kit for Western blot was acquired from Amersham Life Sciences (Buckinghamshire, UK).
Animals
A total of 130 male Wistar rats (280-320 g) were used throughout the study. The animals were obtained from the animal houses of the Instituto Nacional de Neurología y Neurocirugía (INNN), the Universidad Autónoma Metropolitana-Iztapalapa (UAM-I) and the Instituto Nacional de Cardiología (INC), Mexico. Rats were housed five per cage, received a standard diet and water ad libitum, and synchronized with 12-h: 12-h light-dark cycles under standard conditions of temperature (25±1°C) and 50% relative humidity. All procedures were carried out strictly according to the local guidelines (Norma Official Mexicana NOM-062-ZOO-2001) for the use and care of laboratory animals, and the “Guidelines for the Use of Animals in Neuroscience Research” from the Society of Neuroscience. All the experiments were approved by the Ethics Committee of the INNN. In addition, all efforts were made to minimize animal suffering during the experiments.
In vitro experiments
Isolation and incubation of brain synaptosomes
Synaptosomal P2 fractions (also containing mitochondria) were isolated from rat brains, according to the method described by Lopachin et al. [14], with modifications [4]. The rat brains (without the cerebellum) were collected, weighted and homogenized in 10 volumes (g/ml) of sucrose (0.32 M). The obtained homogenates were centrifuged for 10 min at 1,073× g (4°C) and the supernatants recovered for re-centrifugation for 15 min at 17,000× g (4°C). Subsequently, the pellets were collected and resuspended in 40 volumes (ml) of HEPES buffer (0.1 M NaCl, 0.001 M NaH2PO4, 0.005 M NaHCO3, 0.001 M CaCl2, 0.006 M glucose, 0.01 M HEPES, pH 7.4). Final volumes were separated in aliquots of equal volumes. Total protein content was estimated through the Bradford’s method [15]. Freshly obtained synaptosomal fractions were incubated in the simultaneous presence of WIN55,212-2 (1 µM, 10 µL) and/or 3-NP (0.1-1 mM, variable volumes in µL) for 60 min at 37°C in a shaking water bath, to be further processed for biochemical analysis.
MTT assay
MTT assay represents an indirect index of the mitochondrial function [4]. Synaptosomal fractions were added with 8 µl of MTT (5 mg/ml) and re-incubated at 37°C for 60 min. Samples were then centrifuged at 15,300× g for 15 min and the corresponding pellets re-suspended in 1 ml of isopropanol. A second centrifugation step was carried out at 1,700× g for 3 min. Formazan was quantified in the supernatants in a Cytation 3 Imaging Reader (BioTek Instruments, Winooski, VT, USA) by measuring optical density at a wavelength of 570 nm. Results were expressed as percent of MTT reduction with respect to control (synaptosomal fractions not exposed to drug treatments).
Lipid peroxidation
In biological preparations from both in vitro and in vivo protocols, lipid peroxidation was estimated as the formation of thiobarbituric acid-reactive substances (TBARS), following specifications from a previous report [4]. Synaptosomal or homogenate aliquots (200 µL) were added to 500 µL of the TBA reagent (containing 0.75 g of TBA+15 g of trichloroacetic acid +2.54 mL of HCl) and immediately incubated at 100°C for 30 min. Samples were briefly ice-cooled and centrifuged at 3,000× g for 15 min. The optical density of supernatants was estimated at 532 nm in a Cytation 3 Imaging Reader (BioTek). The levels of TBARS -an index of malondialdehyde (MDA) formation- were calculated as nmoles of MDA per mg protein by interpolation of absorbance in a constructed standard curve of tetramethoxypropane, and expressed as percent vs. control.
In vivo experiments
Experimental design and drugs infusion
The study comprised the following four experimental groups (one group per condition, n=5-8 rats per group): Group A (Sham) received the corresponding vehicles (intrastriatal bilateral saline and intraperitoneal Tween + DMSO solution). Group B received the CBr agonist WIN55,212-2 (1 mg/kg, i.p.) [16] once a day for six consecutive days as pretreatment, and single intrastriatal bilateral injections of vehicle on day six (one h after the last WIN55,212-2 injection). WIN55,212-2 was prepared in vehicle containing 5% Tween 80 and 5% DMSO in 0.9% saline solution. Group C received intrastriatal bilateral injections of 3-NP (500 nmol/µL) after receiving vehicle during six days; 3-NP was dissolved in water. Group E received WIN55,212-2 for 6 days prior to the infusion of 3-NP (the last cannabinoid administration was given 1 h after 3-NP was injected). Intrastriatal injections were practiced at the following stereotaxic coordinates: 0.5 mm anterior to bregma, 2.6 mm lateral to bregma and 4.5 ventral to the dura.
Lipid peroxidation assay
Lipid peroxidation was estimated in tissue samples as described above, in the in vitro experiments’ section. This assay was carried out in striatal tissues 24 h after the intrastriatal infusion of 3-NP in order to confer this event a causative role of the brain damage registered six or seven days later.
Immunoblot assay
For this assay, the striata of animals were collected 24 h after the 3-NP or vehicle administration, assuming this time is optimum to find early (causal) protein expression regulation. The striata were homogenized in lysis buffer containing Triton (0.1%), PMFS (1 mM), aprotinin (1.7 mg/ml), EDTA (4 mM), and sodium azide (0.05%). Using Lowry’s method [17], equal amounts of protein were calculated and used for the assays. Proteins were separated in polyacrylamide gel (10%) and transferred to nitrocellulose membranes, which were blocked with PBS solution + milk powder (5%) for 2 h at room temperature and further incubated overnight at 4°C in the presence of the primary antibodies anti-CB1r (1:1,000), anti-NR1 (1:1,000) and anti-NeuN (1:1,000). Membranes were then washed trice in PBS-Triton and incubated with secondary mice (1:2,000) and goat (1:2,000) antibodies for 1 h at room temperature. The ImageJ software was used to perform the densitometric analysis of revealed membranes. An antibody against β-Actin (1:2,000) was used to normalize the protein content.
The open field test of motor activity assay
The open field test was carried out 6 days after the 3-NP injection, using an AccuScan device (AccusScan Instruments Inc., Columbus, OH) in a room with controlled conditions of light, temperature and noise, according to previous reports [18]. Briefly, rats were placed in the center of the device and their motor activity was recorded for 15 min in 5 min intervals. The estimated parameters of motor activity included the total number of horizontal and vertical movements, the time spent in the center of the device (sec) and the resting time (sec).
Histological assessment of striatal lesions
The brains of transcardially perfused rats were collected and paraffin-embedded at seven days post-lesion to obtain coronal sections (4 µm) with a Leica RM2255 microtome (Leica Geosystems AG, St. Gallen, Switzerland). The sections were fixed, dewaxed and dehydrated. Hematoxylin & Eosin staining was used for the histological analysis of the striatum. Processed in parallel, similar sections were obtained for histochemical purposes. Digital images were obtained in a Nikon ECLIPSE E200 microscope (DiaMedical USA, West Bloomfield, MI) using the Q-capture Pro 7 software.
Immunofluorescence assay for CB1r, NR1 and NeuN co-localization in the striatum
Immunofluorescence assays were carried out in slides from brain sections of paraffin blocks obtained from the histopathological study. The slides were treated with the primary antibodies to identify and co-localize CB1r, NR1 and NeuN. For this purpose, brain sections were previously dewaxed and the slides rehydrated through a decreasing ethanol series. Antigens were recovered with a citrate buffer (0.1 M monohydrated citric acid/0.1 M dihydrated trisodic citrate). Nonspecific antigens were blocked with universal blockade solution (Biogenex, Fremont, CA). Slides were incubated overnight with the corresponding antibodies (anti-NeuN, antiNMDAr and anti-CB1, all at a 1:200 dilution) to further undergo 3 washes of 10 min each with PBS-TWEEN (0.2%, pH7.2). Slides were then incubated with 0.6 M glycine for 20 min in order to remove autofluorescence of aldehydes, and further washed with 0.5% PBA (PBS albumin) three times. The anti-rabbit Alexa-488 (Thermo Fisher, Waltham, MA) and anti-mouse-Rodamine (Santa Cruz Biotechnology, Santa Cruz, CA) secondary antibodies were diluted (1:400) and incubated for 60 min at room temperature. Slides were washed trice with PBS-TWEEN (0.2%, pH 7.2). Nuclei were stained in blue with DAPI (Santa Cruz Biotechnology, Santa Cruz, CA) and sections were mounted with a fluorescent solution (DakoCytomation, Glostrup, Denmark). Images (20×) were obtained in a LSM-780 NLO Confocal Microscope (Carl Zeiss, Germany), using a lasser diode at 405 nm for DAPI, and an Ar/ML 458/488/514 nm lasser for Alexa 488 and Rodamine fluorophores. For the analysis of images, the ZEN 2010 6.0 software (Carl Zeiss, Germany) was used.
Statistical analysis
Data are shown as mean values ± one standard error (S.E.M.). The statistical analysis was performed employing the SPSS 21.0 software, using two-way ANOVA followed by Bonferroni’s test, with a minimum level of significance of P≤0.05.
Results
WIN55,212-2 attenuated the 3-NP-induced mitochondrial dysfunction and lipid peroxidation in rat brain synaptosomes
Figure 2 depicts the effects of 3-NP and/or WIN55,212-2 on MTT reduction and lipid peroxidation in rat brain synaptosomal/mitochondrial enriched fractions. A concentration-response effect of 3-NP on MTT reduction is presented in Figure 2A (upper left panel). 3-NP exhibited the maximum inhibitory effect at 0.5 mM concentration (51% below the control; P<0.05). The 0.1 and 1 mM concentrations exerted less intense effects (40 and 36% below the control, respectively). Despite 0.5 mM was more effective, the 1 mM concentration was chosen for further experiments as it also exhibited a prominent inhibitory effect.
Figure 2.
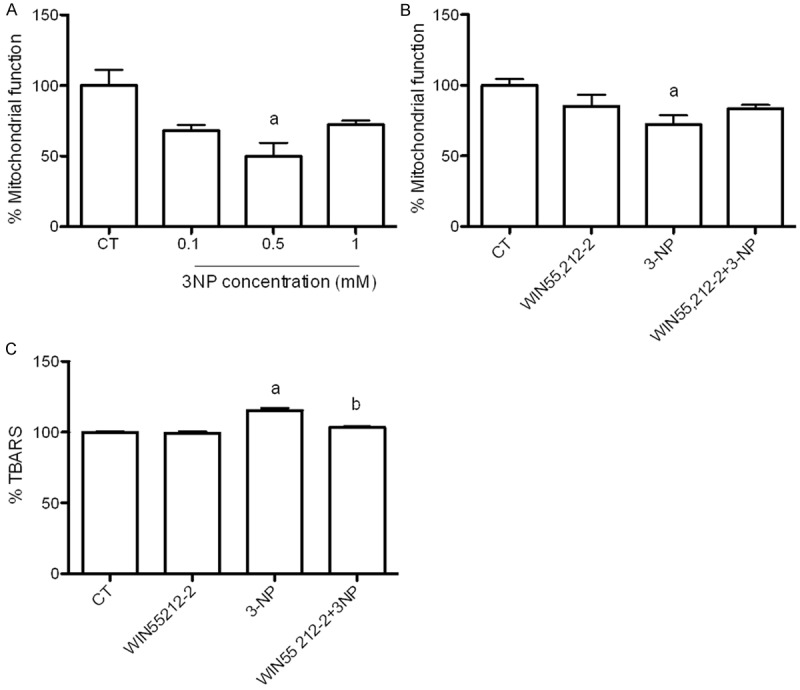
Effect of WIN55,212-2 (1 µM) on 3-NP (1 mM)-induced mitochondrial dysfunction (MTT reduction) and lipid peroxidation (TBARS levels) in rat brain synaptosomal/mitochondrial-enriched fractions. Synaptosomal fractions were pre-incubated with WIN55,212-2 and further incubated with 3-NP for 60 min. Data are expressed as mean values ± S.E.M. of n=6-8 experiments per group. Symbols denote statistical differences vs. control (aP<0.05) and against 3-NP (bP<0.05). Two-way ANOVA followed by Bonferroni’s test.
The effect of WIN55,212-2 (1 µM) on the 3NP-induced mitochondrial dysfunction is shown in Figure 2B (upper right panel). At 1 mM concentration, 3-NP decreased MTT reduction by 35% compared with the control (P<0.05). WIN55,212-2 ameliorated the 3-NP induced mitochondrial dysfunction by 20% (16% below the control). Per se, WIN55,212-2 had no effect on MTT reduction (10% below the control).
TBARS formation was measured as an index of lipid peroxidation (Figure 2C, bottom panel). 3-NP increased the peroxidative levels by 19% above the control (P<0.05). WIN55,212-2 significantly reduced the 3-NP-induced TBARS formation by 10% (P<0.05; 5% above the control). Once again, WIN55,212-2 per se did not affect the levels of lipid peroxidation (2% above the control).
The 3-NP-induced striatal lipid peroxidation was prevented by WIN55,212-2
The effect of WIN55,212-2 on the early (24 h) TBARS formation in striatal tissue of rats infused with 3-NP (500 nmol/µl) is shown in Figure 3. 3-NP increased the levels of lipid peroxidation by 158% above the control (P<0.05). In contrast, WIN55,212-2 significantly prevented this effect (77% below 3-NP; P<0.05) reaching levels even below the control (36%). WIN55,212-2 per se also decreased the TBARS formation below the control (52%).
Figure 3.
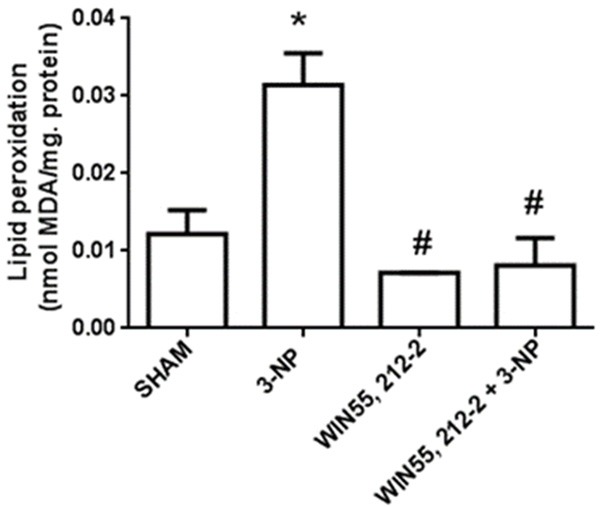
Effects of WIN55,212-2 on 3-NP-induced lipid peroxidation in the rat striatum. Animals received WIN55,212-2 (1 mg/kg, i.p. once a day ×6 days prior to 3-NP) or vehicle, plus 3-NP (500 nmol/µl, intrastriatal bilateral dose) or vehicle. The striatal levels of lipid peroxidation are shown. Mean values ± S.E.M of n=5 rats per group are depicted; *P<0.05, different of Sham; #P<0.05, different of 3-NP group. Two-way ANOVA followed by Bonferroni’s test.
WIN55,212-2 prevented the striatal changes in NR1 transcriptional regulation induced by 3-NP and augmented NR1 content
The transcriptional regulation of NMDAr (NR1) and CB1 receptor proteins induced by WIN55,212-2 and/or 3-NP at 24 h is shown in Figure 4, along with is densitometric representation. 3-NP augmented the NMDAr protein content (130% above Sham) and slightly decreased the CB1 content (15% below Sham; P<0.05). In animals receiving WIN55,212-2 as a pretreatment to 3-NP, the levels of NMDAr protein were decreased compared to 3-NP and similar to Sham (53% below 3-NP and 9% above Sham; P<0.01). In the same treatment, the CB1 content was increased compared to 3-NP and Sham (56% and 40% above 3-NP and Sham, respectively; P<0.01). Interestingly, WIN55,212-2 per se augmented the NMDAr and CB1 contents compared to Sham (97% and 179% above Sham, respectively; P<0.05).
Figure 4.
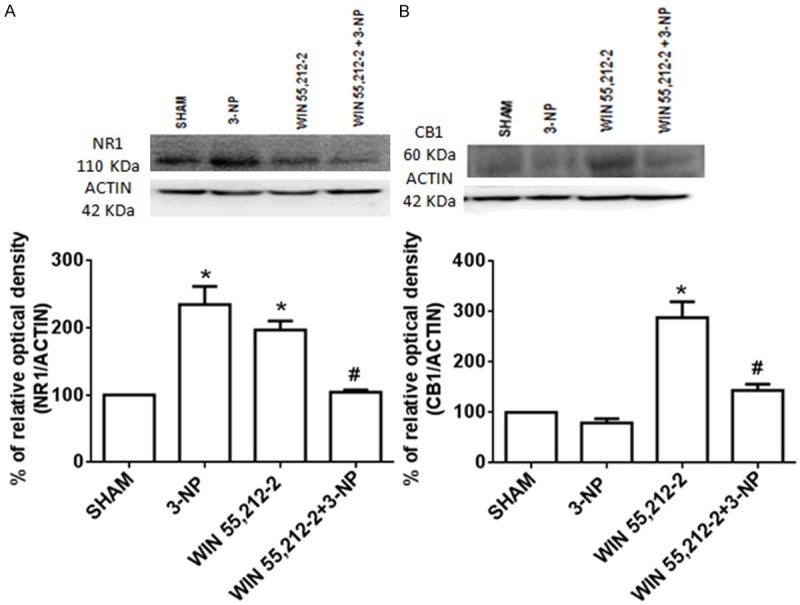
Effect of WIN55,212-2 on the 3-NP-induced early changes in the striatal protein content of the NR1 subunit of NMDA receptor and the CB1 receptor in rat. Animals received WIN55,212-2 (1 mg/kg, i.p. once a day × 6 days prior to 3-NP) or vehicle, plus 3-NP (500 nmol/µL, intrastriatal bilateral dose). Protein content was estimated 24 h after the animals were intrastriatally lesioned. Blotting bands and their represented densities are shown. Mean values ± S.E.M of n=5 rats per group are shown; *P<0.05 and **P<0.01, different of Sham; #P<0.05, different of 3-NP group. Two-way ANOVA followed by Bonferroni’s test.
WIN55,212-2 prevented the 3-NP-induced motor alterations in rats
Three parameters of motor activity altered by 3-NP in rats 6 days after intrastriatally lesioned are shown in Figure 5. 3-NP significantly increased the horizontal activity (Figure 5A) and the time spent in the center of the open field (Figure 5B) by 29% and 139% above Sham, respectively (P<0.05), suggesting a hyperactive pattern of motor activity coursing with disorientation. Consequently, 3-NP also decreased the resting time (Figure 5C) by 14% below Sham. When animals were treated with WIN55,212-2 before being lesioned with 3-NP, the number of horizontal movements remained close to Sham (2% above Sham and 27% below 3-NP; P<0.05). In addition, WIN55,212-2 partially reduced the time spent in the center (45% above Sham and 39% below 3-NP; P<0.05). WIN55,212-2 also prevented the 3-NP-induced decrease in resting time by 36% (4% above Sham; P<0.05). Despite WIN55,212-2 per se augmented the number of horizontal movements (27% above Sham; P<0.05), it did not significantly change the time spent in the center or the resting time of animals, suggesting that the motor activity stimulated in these rats did not comprise disorientation.
Figure 5.
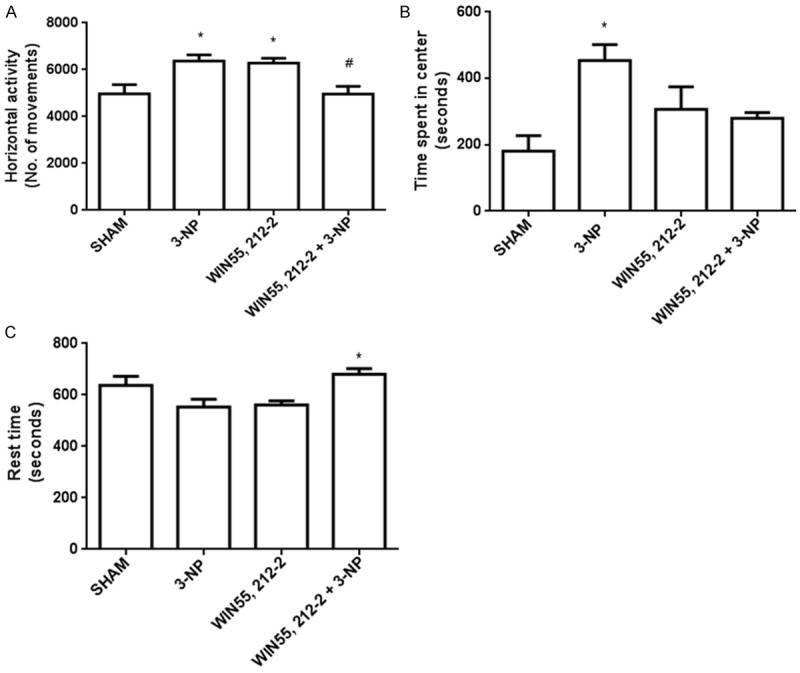
Effect of WIN55,212-2 on the 3-NP-induced alterations in locomotor activity in rats. Animals received WIN55,212-2 (1 mg/kg, i.p. once a day × 6 days prior to 3-NP) or vehicle, plus 3-NP (500 nmol/µL, intrastriatal bilateral dose) and challenged 6 days after intrastriatally lesioned in an open field device. In the upper left panel, the horizontal activity is presented; in the upper right panel, the time spent in the center of the device is shown; in the panel at the bottom, the resting time is depicted. Mean values ± S.E.M of n=5 rats per group are shown; *P<0.05, different of Sham; #P<0.05, different of 3-NP group. Two-way ANOVA followed by Bonferroni’s test.
WIN55,212-2 preserved the striatal cell integrity of rats exposed to 3-NP
Figure 6 shows two panels of photomicrographs (10× in left panel and 40× in right panel) and a graphic depicting the effect of WIN55,212-2 on 3-NP-induced striatal cell damage in rats at day seven after the toxin infusion. The panoramic view of the left panel (Figure 6A, 10×) demonstrates an extensive area of cell damage (necrotic area and massive cell infiltration) produced by the intrastriatal infusion of 3-NP, in contrast to the lesions produced by all other treatments. An intermediate level of damage was found in the WIN55,212-2+3-NP treatment, suggesting certain level of protection by the cannabinoid drug. These images help to demonstrate not only the needle trajectory, but also the extent of damage in the area surrounding the lesions.
Figure 6.

Effect of WIN55,212-2 on 3-NP-induced morphological alterations in the rat striatum. Animals received WIN55,212-2 (1 mg/kg, i.p. once a day ×6 days prior to 3-NP) or vehicle, plus 3-NP (500 nmol/µL, intrastriatal bilateral dose) and striata were histologically processed and stained with Hematoxylin & Eosin. In the left upper panel, the four micrographs of striatal tissue from rats of all treatments are shown at the 10× magnification. In the right upper panel, micrographs of the striatal from all treatments are presented at 40×. *Represent normal cells; +Denote pyknotic cells. In the bottom, counting of cell damage per field is depicted. Mean values ± S.E.M of n=5 rats per group are shown; *P<0.05, different of Sham; #P<0.05, different of 3-NP group. Two-way ANOVA followed by Bonferroni’s test.
In the right panel (Figure 6B), fields (40×) of striatal tissue from all treatments are shown. Representative images were taken all at the same distance from the lesion site (100 µm). In contrast to Sham (normal appearance), the striatal tissue from 3-NP treated rats depicts hyperchromatic/shrunken cells with pyknotic nuclei, vacuolation and damaged neuropil. The general integrity of the cyto-architecture has been compromised. While WIN55,212-2 per se produced no morphological changes, when pre-administered to 3-NP-treated rats, preserved the integrity of the majority of cells (partial protection), although neuropil remained altered by 3-NP. These tendencies were quantitatively confirmed through the counting of damaged cells per field (Figure 6C, graphic in the bottom), clearly showing that 3-NP was able to massively kill striatal cells in the vicinity of the lesion site (446% of cell damage above Sham; P<0.05), whereas WIN55,212-2 prevented this effect in 3-NP-treated animals (33% above Sham and 76% below 3-NP; P<0.05).
3-NP decreased the number of striatal neurons, but not the late regulation of CB1 or NMDAr: WIN55,212-2 changed the profiles of these proteins
In Figure 7, the co-localization of NMDAr (NR1) or CB1 proteins in striatal neuronal cells marked from rats exposed to all experimental treatments is shown. This experiment corresponds to day 7 after the animals were intrastriatally lesioned. The fluorescent mark for neuronal cells (NeuN, in green) revealed a decreased number and intensity in tissue from rats exposed to 3-NP (35% less cells than Sham; P<0.05). The number of neurons was preserved in 3-NP-lesioned rats pretreated with WIN55,212-2 (3% of cells below Sham, different of 3-NP at P<0.05). The cannabinoid agonist did not affect the number of cells when compared with Sham (3% below).
Figure 7.

Effect of WIN55,212-2 and/or 3-NP on the immunofluorescent co-localization of NMDAr and CB1 receptor in neuronal cells (NeuN) from the rat striatum. Animals received WIN55,212-2 (1 mg/kg, i.p. once a day × 6 days prior to 3-NP) or vehicle, plus 3-NP (500 nmol/µL, intrastriatal bilateral dose) and striata were histologically processed and stained with fluorescent antibodies against the mentioned proteins. In the upper panel, micrographs of striatal tissue showing the localization of NeuN (green), CB1 (red in the third, fourth and fifth columns), and NMDAr (red in the sixth, seventh and eight columns) from rats of all treatments are shown at a 20× magnification. In the bottom, counting of neuronal cells and fluorescence intensities is depicted. Mean values ± S.E.M of n=5 rats per group are shown; aP<0.05, different of Sham; bP<0.05, different of 3-NP group. Two-way ANOVA followed by Bonferroni’s test.
CB1 was moderately expressed in tissue from control rats (Sham). 3-NP augmented the level of expression of the protein by 35%; however, this effect was not significant since WIN55,212-2 was able to increase the expression levels of CB1 in a higher magnitude when pre-administered to 3-NP-injected rats (213% above Sham; P<0.05). WIN55,212-2 per se augmented de CB1 expression (138% above Sham; P<0.05). The merge revealed that CB1 was localized in some neuronal cells, but also in other cell types.
The basal expression of NMDAr was not affected by 3-NP treatment (6% below Sham). In contrast, WIN55,212-2 significantly stimulated the expression of this protein when administered to 3-NP-treated rats (167% above Sham; P<0.05) or per se (193% above Sham; P<0.05). The merge revealed that NMDAr was localized in NeuN-positive cells.
Discussion
In this report, we have evaluated for the first time the protective efficacy of an agent described as a modulator of the ECS on the toxic model produced by 3-NP in the CNS. We estimated the effect of a direct activation of CBr -mostly of CB1- by WIN55,212-2 on different toxic endpoints evoked by the mitochondrial toxin 3-NP. Particular emphasis was given to biochemical, behavioral, and morphological alterations. Under in vitro conditions, WIN55,212-2 was able to ameliorate the 3-NP-induced mitochondrial dysfunction and oxidative damage to lipids in rat brain synaptosomes. In the in vivo model of striatal damage induced by 3-NP in rats, WIN55,212-2 efficiently prevented the early induction of lipid peroxidation by 3-NP in the striatum, suggesting that the properties of this agent could comprise an antioxidant component. WIN55,212-2 also reduced the changes in transcriptional deregulation of NR1 in 3-NP-lesioned striata and augmented the CB1 content, supporting the concept that the pharmacological induction of reduced neuronal excitability accounts, at least in part, for the protective actions of this agent in excitotoxic models. Accompanying these early effects, WIN55,212-2 prevented the late (seven days post-lesion) behavioral and morphological endpoints of hyperactivity and cell damage, respectively, hence suggesting that the use of this agent readily constitutes a valuable therapeutic strategy at the experimental level to preserve the structural and functional integrity of the brain tissue. In addition, the prevention of the 3-NP-induced neuronal cell loss by WIN55,212-2 also at seven days evidenced by immunofluorescence assays-, as well as the compensatory effects that this agent exerted on CB1 and NMDAr regulation, serve to hypothesize that plasticity events could take place after the brain exposure to ECS modulators to compensate the early transcriptional modifications. Indeed, the fact that after WIN55,212-2 exposure, localization of CB1 was not remitted to NeuN positive cells suggest that other cells are involved in the ECS late compensatory changes.
An initial possible explanation to the protective actions of WIN55,212-2 in the toxic paradigm evoked by 3-NP is related with the ability of cannabinoid agents to activate CB1 to further trigger a mechanism that abducts NR1 for its degradation, therefore leading to NMDA hypofunction and the prevention of excitotoxicity [19]; however, other possible mechanisms that we will revise herein cannot be ruled out from the actions of this agent.
Although this is the first report describing the specific actions of WIN55,212-2 in the toxic paradigm produced by the systemic administration of 3-NP, the role of the ECS in this toxic model has been already explored and discussed. One of the first reports investigating this issue described the binding of the marked cannabinoid receptor agonist 3H[CP55,940 to cannabinoid receptors in the basal ganglia of rats [20]. The decreased binding in the striatum and other regions such as the globus pallidus revealed the participation of CBr in this model, despite no resemblance of the HD features was achieved. In the same toxic model, but injecting 3-NP bilaterally into the rat striatum, it was demonstrated that CB1 receptor binding and a CB1 receptor-mediated decrease in activation of GTP-binding proteins are mediated by the toxin in the basal ganglia [21]. Moreover, these changes were correlated with the hyperactive pattern elicited by 3-NP and decreased levels of AEA and 2-AG in the striatum (caudate-putamen), readily resembling HD features. This study supports the use of the 3-NP bilateral intrastriatal injection, the one we have used herein, as the most appropriate for modeling the general alterations seen in HD, and the specific changes in the ECS produced in early stages of the disorder. Congruent with this concept, the pharmacological manipulation of the ECS by agents with cannabinoid profiles -such as AM404, an inhibitor of the endocannabinoid reuptake and possible disturbances in early hyperkinetic stages produced by bilateral intrastriatal injections of 3-NP [22]. This agent also ameliorated neurochemical deficits (GABA depletion), supporting the idea that the stimulation of the ECS, either by endocannabinoids or by synthetic cannabinoids, can protect neuronal cells and prevent degenerative events similar to those of human neurological disorders.
The protective properties of other agents acting on the ECS and/or the endovanilloid system in the same model have been reported, hence supporting our present findings. However, as previously demonstrated [23], the role of CB1 receptors during the protection of the nervous system in the toxic model produced by 3-NP is, in most of the cases, only partial. Therefore, it seems that the involvement of the endovanilloid system and its receptor VR1 could play a major role in preventing striatal damage. Whether WIN55212-2 recruits in a direct or indirect manner the endovanilloid system to prevent striatal damage, remains to be tested in future studies. It is however, clear enough that CBr play a crucial role for the toxic model produced by 3-NP since loss of CB1 receptors occurs in the basal ganglia, at least during the akinetic stage of the disease model [24], prompting the development of strategies to target these receptors for therapeutic purposes.
The course of alterations of the ECS in the toxic model produced by 3-NP has been described and involves an early decrease of G-protein activation by CBr agonists occurring days before the onset of striatal degeneration, followed by severe alterations in CB1 receptors (decreased density, mRNA levels and coupling to G proteins), matching with the onset of degeneration [25]. It is therefore plausible to hypothesize that WIN55,212-2, when administered during several days before the toxic infusion of 3-NP -as we did herein-, could prevent all these stages from the very beginning, thus blocking the early signaling cascades that will be affected by 3-NP, as demonstrated by our evidence on the early prevention of oxidative damage to lipids, as well as the transcriptional regulation of CB1. Moreover, in our study, WIN55-212-2 acted as an antihyperkinetic agent in several locomotor endpoints, and this effect has been previously demonstrated with hybrid endocannabinoind and vanilloid compounds, such as Arvanil [26]. This effect is likely to be due to an increase in excitatory transmission at the globus pallidus, increasing the glutamate content in this region. Therefore, WIN55,212-2 and other synthetic cannabinoid receptor agonists require to be challenged through these mechanisms in order to know better how they are acting in toxic paradigms involving motor alterations.
Other inhibitors of anandamide uptake, such as UCM707, also exhibit anti-hyperkinetic activity in the toxic model of 3-NP, through mechanisms related with the amelioration of GABA and glutamate deficits in the globus pallidus and substantia nigra [27]. However, no protection against GABAergic cell loss was observed. This finding is relevant as reveals that not all the agents exhibiting a cannabinoid profile are adequate for tissue preservation. In contrast, the fact that a more natural molecule, cannabidiol (phytocannabinoid), is able to reduce striatal atrophy caused by 3-NP by mechanisms independent of the activation of CBr, vanilloid and adenosine A2A receptors, points out to specific profiles among cannabinoid agents [28]. Thereby, the fact that WIN55,212-2 was able to prevent both hyperkinetic activity and morphological alterations emphasizes the need to develop more detailed studies with this agent. Noteworthy, the protective actions of cannabidiol were attributed to its antioxidant properties. Although not a natural compound, since WIN55,212-2 was able to prevent the early oxidative damage to lipids produced by 3-NP, antioxidant properties of this molecule cannot be discarded and could be accounting for its protective actions.
Still, a major role of CB1 receptors cannot be ruled out of HD-like pathology. Knocking out CB1 worsened motor performances in N171-82Q transgenic mice, turning these animals more vulnerable to 3-NP toxicity, hence emphasizing a major role of CB1 for the physiopathological development of HD [29].
More recently, neuroprotective properties against the toxic actions of 3-NP have been attributed to cannabigenol [30], and its derivative, VCE-003.2. A novel finding in these studies is the action of this cannabinoid through the activation of the peroxisome proliferator-activated receptor-γ (PPARγ), a protein that activates neuroprotective signals. The fact that cannabinoid agents can exert beneficial actions through mechanisms independent of CBr or vanilloid receptors, recruiting anti-inflammatory and/or antioxidant pathways in the process, as well as neurotrophic and survival pathways, opens new avenues of research for agents like WIN55,212-2, exhibiting a wide range of protective properties.
Altogether, these studies, and the evidence collected in this report, demonstrate the potential of agents with cannabinoid profiles, such as WIN55,212-2, to counteract the deleterious actions of toxins like 3-NP in the brain, prompting the development of more detailed investigations in this field.
Conclusion
The use of CBr agonists such as WIN55,212-2 to induce protection in neurotoxic paradigms involving mitochondrial dysfunction, energy depletion, secondary excitotoxicity, oxidative damage and transcriptional deregulation, represents a valuable therapeutic tool at the experimental level and provides a relevant approach to characterize some major mechanisms underlying neuronal degeneration and brain dysfunction in experimental models of neurodegenerative disorders. The finding that WIN55,212-2 exhibited a protective profile on behavioral, morphological and biochemical endpoints of 3-NP-induced striatal toxicity emphasizes the concept that agents with a cannabinoid profile possess a considerable potential to design cannabinoid-based therapies aiming to ameliorate or control major behavioral and morphological alterations observed in disorders such as HD. Although the main mechanism suggested by which WIN55,212-2 produced preventive effects on the toxic events elicited by 3-NP in the rat striatum is likely to be related with its ability to stimulate CBr and reduce excessive excitation, in light of the evidence collected herein, we cannot discard other possible mechanisms of protection for this molecule, including a possible role as an antioxidant and/or energy modulator, or acting on other systems, such as PPAR-γ. A summary of the hypothesized mechanisms occurring in this model is shown in Figure 8.
Figure 8.
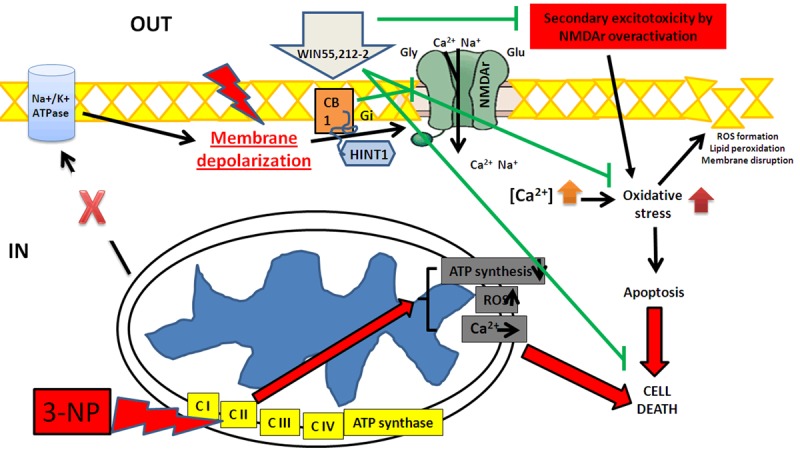
Schematic representation of the proposed mechanisms occurring in the toxic model induced by 3-NP and the preventive actions exerted by the synthetic cannabinoid agonist WIN55,212-2 in the rat striatum. 3-NP reaches and block the mitochondrial electron transfer chain Complex II (succinate dehydrogenase), producing a deregulation of electron transport and energy (ATP) depletion. Neuronal cells are gradually depolarized since their Na+/K+-ATPases are dysfunctional, thus over-activating voltage-gated NMDAr and prompting cells to secondary excitotoxicity. Increased toxic levels of intracellular Ca2+ then activate several inner metabolic pathways, increasing reactive oxygen species (ROS), oxidative damage, apoptosis, deadly cascades and cell death of striatal GABAergic neurons. Mitochondria also contributes to this toxic pattern by releasing ROS and increasing the intracellular levels of Ca2+ through a reversed potential. The mechanisms elicited by 3-NP are marked in red. The activation of pre- and post-synaptic CB1 receptors by WIN55,212-2 will modulate glutamatergic transmission, thereby reducing excessive excitatory events by limiting the NMDAr activation and/or the neurotransmitter release. In addition, WIN55,212-2 could also act on CB2 located in microglia, hence modulating immunological events and promoting protective responses or inhibiting toxic events, and preventing against 3-NP-induced striatal toxicity, leading cells to survival. The signals elicited by WIN55,212-2 are marked in green.
Acknowledgements
This work was supported by CONACyT-TUBITAK Grant 265991 (A.S.). Marisol Maya-López presents this article as the first author and as integral part of the experiments she performed for her Master thesis to obtain her Master degree in the Programa de Posgrado en Biología Experimental at the Universidad Autónoma-Metropolitana-Iztapalapa (UAM; Student No. 2143800919). During the process, she received a scholarship (No. 262025) from CONACyT-Mexico. Authors wish to express gratitude to Dr. Cecilia Zazueta and Gibrán Pedraza-Vázquez for their technical support.
Disclosure of conflict of interest
None.
References
- 1.Self DW. Anandamide: a candidate neurotransmitter heads for the big leagues. Nature Neurosci. 1999;2:303–304. doi: 10.1038/7210. [DOI] [PubMed] [Google Scholar]
- 2.Morales M, Bonci A. Getting to the core of addiction: Hooking CB2 receptor into drug abuse? Nat Med. 2012;18:504–505. doi: 10.1038/nm.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onaivi ES, Ishiguro H, Gu S, Liu QR. CNS effects of CB2 cannabinoid receptors: beyond neuro-immuno-cannabinoid activity. J Psychopharmacol. 2012;26:92–103. doi: 10.1177/0269881111400652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rangel-López E, Colín-González AL, Paz-Loyola AL, Pinzón E, Torres I, Serratos IN, Castellanos P, Wajner M, Souza DO, Santamaría A. Cannabinoid receptor agonists reduce the short-term mitochondrial dysfunction and oxidative stress linked to excitotoxicity in the rat brain. Neuroscience. 2015;285:97–106. doi: 10.1016/j.neuroscience.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 5.Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pertwee RG. Pharmacological actions of cannabinoids. Handb Exp Pharmacol. 2005;168:1–51. doi: 10.1007/3-540-26573-2_1. [DOI] [PubMed] [Google Scholar]
- 7.Sgambato-Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol. 2012;96:69–86. doi: 10.1016/j.pneurobio.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hariharan A, Shetty S, Shirole T, Jagtap AG. Potential of protease inhibitor in 3-nitropropionic acid induced Huntington’s disease like symptoms: mitochondrial dysfunction and neurodegeneration. Neurotoxicology. 2014;45:139–148. doi: 10.1016/j.neuro.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Túnez I, Tasset I, Pérez-De La Cruz V, Santamaria A. 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: past, present and future. Molecules. 2010;15:878–916. doi: 10.3390/molecules15020878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem. 2005;95:1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- 12.Burtscher J, Zangrandi L, Schwarzer C, Gnaiger E. Differences in mitochondrial function in homogenated samples from healthy and epileptic specific brain tissues revealed by high-resolution respirometry. Mitochondrion. 2015;25:104–112. doi: 10.1016/j.mito.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Chaturvedi RK, Beal MF. Mitochondrial disease of the brain. Free Radic Biol Med. 2013;63:1–29. doi: 10.1016/j.freeradbiomed.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Lopachin RM, Geohagen BC, Gavin T. Synaptosomal toxicity and nucleophilic targets of 4-hydroxy-2-nonenal. Toxicol Sci. 2009;107:171–181. doi: 10.1093/toxsci/kfn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 16.Balistreri E, Garcia-Gonzalez E, Selvi E, Akhmetshina A, Palumbo K, Lorenzini S, Maggio R, Lucattelli M, Galeazzi M, Distler JW. The cannabinoid WIN55,212-2 abrogates dermal fibrosis in scleroderma bleomycin model. Ann Rheum Dis. 2011;70:695–699. doi: 10.1136/ard.2010.137539. [DOI] [PubMed] [Google Scholar]
- 17.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 18.Tatem KS, Quinn JL, Phadke A, Yu Q, Gordish-Dressman H, Nagaraju K. Behavioral and locomotor measurements using an open field activity monitoring system for skeletal muscle diseases. J Vis Exp. 2014;29:51785. doi: 10.3791/51785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sánchez-Blázquez P, Rodríguez-Muñóz M, Garzón J. The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: implications in psychosis and schizophrenia. Front Pharmacol. 2014;4:169. doi: 10.3389/fphar.2013.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Page KJ, Besret L, Jain M, Monaghan EM, Dunnett SB, Everitt BJ. Effects of systemic 3-nitropropionic acid-induced lesions of the dorsal striatum on cannabinoid and mu-opioid receptor binding in the basal ganglia. Exp Brain Res. 2000;130:142–150. doi: 10.1007/s002210050016. [DOI] [PubMed] [Google Scholar]
- 21.Lastres-Becker I, Fezza F, Cebeira M, Bisogno T, Ramos JA, Milone A, Fernández-Ruiz J, Di Marzo V. Changes in endocannabinoid transmission in the basal ganglia in a rat model of Huntington’s disease. Neuroreport. 2001;12:2125–2129. doi: 10.1097/00001756-200107200-00017. [DOI] [PubMed] [Google Scholar]
- 22.Lastres-Becker I, Hansen HH, Berrendero F, De Miguel R, Pérez-Rosado A, Manzanares J, Ramos JA, Fernández-Ruiz J. Alleviation of motor hyperactivity and neurochemical deficits by endocannabinoid uptake inhibition in a rat model of Huntington’s disease. Synapse. 2002;44:23–35. doi: 10.1002/syn.10054. [DOI] [PubMed] [Google Scholar]
- 23.Lastres-Becker I, de Miguel R, De Petrocellis L, Makriyannis A, Di Marzo V, Fernández-Ruiz J. Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington’s disease. J Neurochem. 2003;84:1097–1109. doi: 10.1046/j.1471-4159.2003.01595.x. [DOI] [PubMed] [Google Scholar]
- 24.Lastres-Becker I, Gómez M, De Miguel R, Ramos JA, Fernández-Ruiz J. Loss of cannabinoid CB(1) receptors in the basal ganglia in the late akinetic phase of rats with experimental Huntington’s disease. Neurotox Res. 2002;4:601–608. doi: 10.1080/10298420290030514. [DOI] [PubMed] [Google Scholar]
- 25.Lastres-Becker I, Bizat N, Boyer F, Hantraye P, Fernández-Ruiz J, Brouillet E. Potential involvement of cannabinoid receptors in 3-nitropropionic acid toxicity in vivo. Neuroreport. 2004;15:2375–2379. doi: 10.1097/00001756-200410250-00015. [DOI] [PubMed] [Google Scholar]
- 26.de Lago E, Urbani P, Ramos JA, Di Marzo V, Fernández-Ruiz J. Arvanil, a hybrid endocannabinoid and vanilloid compound, behaves as an antihyperkinetic agent in a rat model of Huntington’s disease. Brain Res. 2005;1050:210–216. doi: 10.1016/j.brainres.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 27.de Lago E, Fernández-Ruiz J, Ortega-Gutiérrez S, Cabranes A, Pryce G, Baker D, López-Rodríguez M, Ramos JA. UCM707, an inhibitor of the anandamide uptake, behaves as a symptom control agent in models of Huntington’s disease and multiple sclerosis, but fails to delay/arrest the progression of different motor-related disorders. Eur Neuropsychopharmacol. 2006;16:7–18. doi: 10.1016/j.euroneuro.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Sagredo O, Ramos JA, Decio A, Mechoulam R, Fernández-Ruiz J. Cannabidiol reduced the striatal atrophy caused 3-nitropropionic acid in vivo by mechanisms independent of the activation of cannabinoid, vanilloid TRPV1 and adenosine A2A receptors. Eur J Neurosci. 2007;26:843–851. doi: 10.1111/j.1460-9568.2007.05717.x. [DOI] [PubMed] [Google Scholar]
- 29.Mievis S, Blum D, Ledent C. Worsening of Huntington disease phenotype in CB1 receptor knockout mice. Neurobiol Dis. 2011;42:524–529. doi: 10.1016/j.nbd.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Valdeolivas S, Navarrete C, Cantarero I, Bellido ML, Muñoz E, Sagredo O. Neuroprotective properties of cannabigerol in Huntington’s disease: studies in R6/2 mice and 3-nitropropionate-lesioned mice. Neurotherapeutics. 2015;12:185–199. doi: 10.1007/s13311-014-0304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]