

Abstract
Brain injury secondary to birth asphyxia is the major cause of death and long-term disability in newborns. Intranasal drug administration enables agents to bypass the blood-brain barrier (BBB) and enter the brain directly. In this study, we determined whether intranasal basic fibroblast growth factor (bFGF) could exert neuroprotective effects in neonatal rats after hypoxic-ischaemic (HI) brain injury and assessed whether attenuation of endoplasmic reticulum (ER) stress was associated with these neuroprotective effects. Rats were subjected to HI brain injury via unilateral carotid artery ligation followed by 2.5 h of hypoxia and then treated with intranasal bFGF or vehicle immediately after HI injury. We found that the unfolded protein response (UPR) was strongly activated after HI injury and that bFGF significantly reduced the levels of the ER stress signalling proteins GRP78 and PDI. bFGF also decreased brain infarction volumes and conferred long-term neuroprotective effects against brain atrophy and neuron loss after HI brain injury. Taken together, our results suggest that intranasal bFGF provides neuroprotection function partly by inhibiting HI injury-induced ER stress. bFGF may have potential as a therapy for human neonates after birth asphyxia.
Keywords: bFGF, hypoxic-ischaemia brain injury, ER stress, neuroprotection
Introduction
An alarming rise in neonates suffer from birth asphyxia has created a substantial health and economic burden worldwide [1]. An estimated 4 to 9 million new-borns are diagnosed as asphyxia each year, along with approximately 1.2 million deaths and the same number of infants with severe disabilities [2,3]. Hypoxic-ischemia (HI) encephalopathy is one of the leading causes of neurological disabilities. Several long-term neurological or cognitive disabilities related to HI encephalopathy include cerebral palsy, epilepsy and mental retardation [4-6]. However, an effective therapeutic approach mitigating HI encephalopathy, especially in the developing countries, has not been established.
The endoplasmic reticulum (ER) is an intracellular organelle, characterized a network of membrane-enclosed sacs and tubules, found in eukaryotic cells. Both physiological and pathological functions of ER have been studied extensively. While ER is responsible for production of proteins and lipids, ER stress induces activation of the unfolded protein response (UPR), which resets ER homeostasis by enhancing protein folding, reducing protein translation and increasing ER associated degradation [7]. Several physiological triggers, such as HI [8], depletion of ER Ca2+ stores [9], free radicals [10], and elevated unfolded or misfolded protein levels [11], have been identified to activate the UPR. It is important to understand that the UPR is triggered to allow the cells to restore homeostasis by regulating cell growth, differentiation and apoptosis. Previous studies have demonstrated that ER stress-induced injury is associated with activation of glucose-regulated protein 78 (GRP78), protein disulfide isomerase (PDI), EBP homologous transcription factor (CHOP), X-box binding protein 1 (XBP-1), and transcription factor-6 (ATF-6) [12-15]. If the ER stress is chronically prolonged and the adaptive mechanism fails, tissue injury manifested by cellular dysfunction and cell death often occurs during HI [16,17]. Thus, it has been hypothesized that the ER stress initiates extensive neuronal cell death and malfunction, and this pathological mechanism may contribute to the neonatal HI injury.
Basic fibroblast growth factor (bFGF or FGF-2) is a member belongs to the fibroblast growth factor family, which is implicated in a diverse biological function, including cellular proliferation, differentiation and survival during development [18-20]. Several lines of evidence had highlighted that bFGF played an essential role in neurotropic activities, including neural stem cell maintenance and proliferation, in the subventricular zone of neonatal rat brains [21]. Furthermore, neural stem/progenitor cell transplantation improved brain function after cerebral ischaemic injury, and such protective effect was further enhanced by bFGF, in which increased NSCs/neural progenitor cell proliferation and functional recovery were observed in transplanted rats [22]. Although the beneficial effect has been well documented, the molecular mechanisms underlying the therapeutic effects of bFGF in neonatal HI recovery are still undefined. In this study, we sought to examine the molecular mechanisms by which bFGF mitigated ER stress-induced brain injury in neonatal rats.
Materials and methods
Reagents and antibodies
Dulbecco’s modified Eagle medium (DMEM) and foetal bovine serum (FBS) were purchased from Invitrogen (Invitrogen, Carlsbad, CA). Recombinant human basic fibroblast growth factor (bFGF) was purchased from Key Laboratory of Biotechnology and Pharmaceutical Engineering, Zhejiang, China. Hyaluronidase and 2, 3, 5-triphenyltetrazolium chloride (TTC) (dissolved in PBS) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies against ATF6, GRP78, PDI, XBP-1, ATF4, caspase12 and caspase3 were purchased from Abcam (Cambridge, MA, USA). Anti-actin, CHOP, microtubuli associated protein 2 (MAP-2) and myelin basic protein (MBP) were acquired from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The appropriate secondary antibodies were obtained from Santa Cruz Biotechnology or Abcam.
Animals and surgical procedures
Sprague-Dawley (SD) rats were purchased from the Animal Centre of the Chinese Academy of Sciences. Adult rats were crossed to give new-borns for subsequent studies. All animal work was performed under the animal use and care protocol conformed to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and Wenzhou Medical University. The day of delivery was considered day 0 for the pups. Neonatal rats were maintained in an environment of constant temperature and regular 12-h day, 12-h night cycles with access to food and water. The model used in this study was adapted from the study by Vannucci R.C. et al [23]. On postnatal day 7 (P7), the pups underwent unilateral ligation of the left common carotid artery via a midline neck incision after anaesthesia with diethyl ether. After ligation, the wound was sutured, and they were allowed to recover for 2 h before exposed to humidified nitrogen-oxygen mixture (92% and 8%, respectively, delivered at 3-4 L/min) for 2.5 h. The chamber was partially submerged in a 37°C water bath to maintain a constant thermal environment. Sham animals received anaesthesia, and their common carotid arteries were exposed but were not subjected to ligation. After the neonatal HI model was established, pups were administered bFGF or saline and returned to their dams for subsequent experiments.
Intranasal administration
Neonatal rats were randomly assigned to the treatment group receiving intranasal bFGF or the sham group receiving saline. bFGF was prepared by dissolving in 0.9% saline solution (100 μg/ml) and stored at -20°C for future use. bFGF or saline was administered as drops to the nostrils (100 ng/g) immediately after HI injury. Before bFGF administration, each neonatal rat was given hyaluronidase (Sigma-Aldrich) to increase the permeability of bFGF to the CNS. Each rat was held ventral-side up, and a small-modified 27-French catheter was inserted into either nare. bFGF or saline was slowly administered, and the rats were held for 1-2 min to ensure absorption. Then, bFGF or saline was administered twice a day until postnatal day 14 (P14).
Cell culture
PC12 cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. Cells were grown in DMEM supplemented with 10% FBS and 1% antibiotics and then incubated in a humidified atmosphere containing 5% CO2 at 37°C. PC12 cells were pretreated with bFGF (50 μg/ml) for 2 h. All experiments were performed in triplicate.
Immunohistochemistry and antibodies
Freshly cut, free-floating brain sections (5 μm thick) from P14 rats were prepared. Sections were incubated with 3% H2O2 in methanol for 10 min, followed by blocking with 5% BSA in PBS at 37°C for 30 min [24,25] and incubating with primary antibodies (1:200 for MAP-2 or 1:200 for MBP) at 4°C overnight. Sections were then washed with PBS and incubated with an appropriate secondary antibody donkey anti-rabbit (1:1000; Abcam) or donkey anti-goat (1:300; Santa Cruz, CA, USA) at 37°C for 1 h [26]. Nuclei were counterstained with 4’, 6-diamidino-2-pheny-lindole (DAPI) [27]. The images of MAP-2 and MBP were visualized using a Nikon ECLIPSE Ti microscope (Nikon, Tokyo, Japan). The extent of tissue damage was determined by calculating the amount of surviving tissue in each section. Briefly, brain damage was analyzed using the ImageJ software (http://imagej.nih.gov/ij/) by outlining both hemispheres on full section images. The ipsilateral MAP-2 area loss was measured as [1-(area ipsilateral MAP-2 staining/area contralateral MAP-2 staining)] ×100. The MBP area was determined as same as the calculation method of the MAP-2 area loss.
Measurement of infarct ratio
TTC staining was used to measure infarct volumes as previously described [28]. In brief, animals were perfused with saline under deep anaesthesia at 24 h after HI brain injury. Brains were isolated, sectioned into 2 mm slices, and then immersed in 1% TTC solution at 37°C for 15-20 min, followed by 4% paraformaldehyde. Images were captured, and infarct volumes were analyzed using the ImageJ software (http://imagej.nih.gov/ij/) by outlining both hemispheres on full section images.
Haematoxylin-eosin and Nissl staining
For HE and Nissl staining, brain sections from the P14 rats were hydrated using a Haematoxylin-eosin (HE) Staining Kit (Beyotime) or Nissl staining solution (Beyotime) for 10 min at room temperature. After being rinsed with double distilled water, sections were dehydrated and mounted with Mowiol. Images of the cortex and CA1 area of the hippocampus were captured accordingly. Quantitation was performed by counting the number of normal neurons in five randomly chosen fields within each slide at 400× magnification using a Nikon ECLIPSE Ti microscope. The average cell density was calculated as the ratio of the overall normal neurons number to the total area of the chosen field.
Apoptosis assay
For in vivo studies, DNA fragmentation was detected using an In Situ Cell Death Detection Kit (Roche, South San Francisco, CA, USA). According to the standard protocol, brain sections from the P14 rats were deparaffinized and rehydrated, which followed by an incubation with proteinase K (20 μg/ml) at 37°C for 15 min. For in vitro studies, cells were incubated in 3% H2O2 for 2 min with subsequent washes with PBS for three times. After washing, sections were incubated with TUNEL reaction mixture with terminal deoxynucleotidyl transferase (TdT) at 37°C for 60 min under humidified condition, and nuclei were stained with DAPI. Negative controls were obtained by omitting the TdT enzyme. Images were captured by using a Nikon ECLIPSE Ti microscope and were analysed by using ImageJ software. Apoptotic cells were characterized by green fluorescence of the nucleus and nuclear membrane according to the manufacturer’s protocol. Quantitation was performed by counting the number of positive cells in five randomly chosen fields within each slide at 400× magnification. The index of apoptosis was calculated as the ratio of the overall number of apoptotic cells to the total number of cells.
Immunofluorescence staining
Brain sections obtained at 24 h after HI were incubated with 5% BSA at 37°C for 30 min in PBS. Sections were then incubated with the appropriate primary antibody overnight at 4°C in the same buffer. Nuclei were stained with DAPI for 5 min at room temperature and mounted with Mowiol [24,25]. For ER stress detection, brain sections were prepared as previously described, and primary antibodies against GRP78 (1:500, Abcam) and PDI (1:500, Abcam) were used. After primary antibody incubation, sections were washed and then incubated for 1 h with the appropriate secondary antibody (1:1000; Invitrogen Corporation, Carlsbad, CA, USA) [26]. Images were captured by using a Nikon ECLIPSE Ti microscope at 600× magnification and were analysed by using ImageJ software.
Western blot analysis
PC12 cells were lysed in protein extraction reagent with protease and phosphatase inhibitors. For in vivo studies, animals were euthanized at 24 h after hypoxia and were subjected to perfusion with ice-cold 0.9% NaCl after decapitation. Total proteins were isolated from the hippocampus using mammalian tissue extraction reagent according to the manufacturer’s protocol. An equal amount of protein was fractionated by 12% SDS-PAGE and transferred onto a PVDF membrane (Bio-Rad, Hercules, CA). Membranes were blocked with 5% skim milk for 1.5 h at room temperature and were subsequently incubated with the primary antibodies, including GRP78 (1:1000), ATF6 (1:1000), PDI (1:1000), ATF4 (1:1000), XBP-1 (1:1000), CHOP (1:300), caspase12 (1:1000) and caspase3 (1:1000) at 4°C overnight. In the following day, membranes were washed with TBST for 3 times and incubated with appropriate HRP-conjugated secondary antibodies at room temperature for 1 h [29,30]. Proteins were detected by the ChemiDocTM XRS+ Imaging System (BioRad) and signal intensities were densitometrically quantified by Image J software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are expressed as the mean ± SEM from three independent experiments. Statistical significance was determined using Student’s t-test if comparing only two experimental groups or one-way ANOVA followed by Dunnett’s post hoc tests if analysing more than two groups. Differences were considered to be statistically significant when P<0.05.
Results
bFGF-induced brain recovery is associated with increased neuronal survival after HI brain injury in rats
We first examined whether treatment of bFGF protected against neonatal HI-induced brain injury in vivo. bFGF was administered immediately after hypoxia, and the severity of brain injury was determined at 24 h after HI by evaluating the infarct volume via TTC staining. Treatment of bFGF significantly attenuated post-HI brain infarction, reducing the infarct volume by 13% in bFGF-treated HI animals as compared to their vehicle-treated HI littermates (P<0.05, Figure 1A, 1B). While extensive tissue damages in the ipsilateral cerebral cortex and hippocampus were noticed in the HI group 7 days after brain injury, no observable tissue injury was found in contralateral cerebral tissue. When compared with the HI group, treatment of bFGF significantly reduced necrosis, nuclear pyknosis and fragmentation, and infiltration of polymorphonuclear leukocytes and macrophages, which mediated a neuroprotective effect against brain injury in neonatal rats (Figure 1C).
Figure 1.
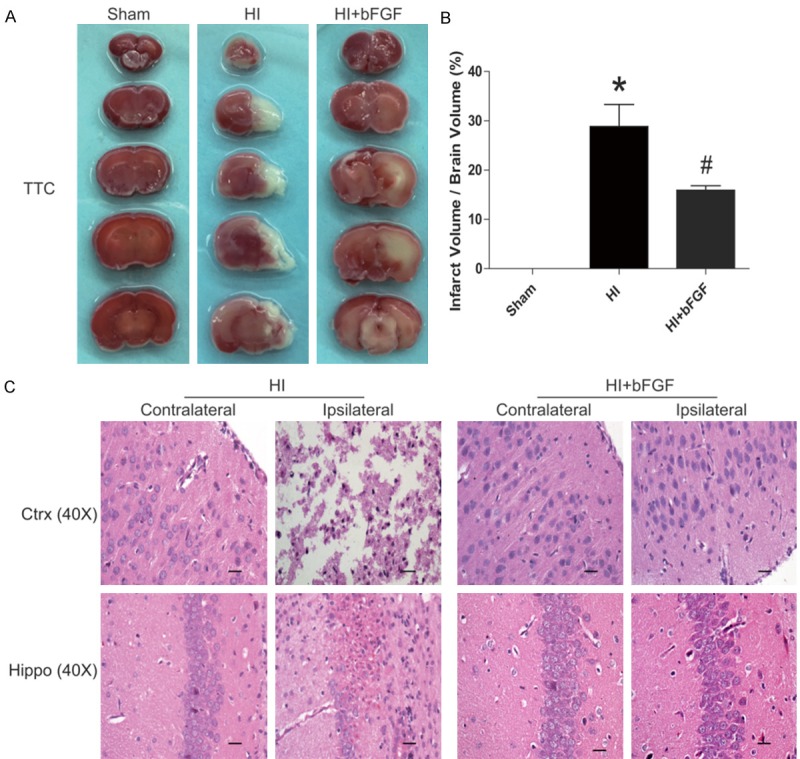
bFGF protects against brain infarction and improves histological recovery after neonatal HI injury. A. TTC staining of the sham rats, HI rats and HI rats treated with bFGF. B. Quantification of the TTC content of the brain (%). C. HE staining of the cortex and hippocampus on the lesioned (ipsilateral) and unlesioned (contralateral) sides in HI rats and HI rats treated with bFGF at 1 week after HI. *represents P<0.05 versus the sham group, and #represents P<0.01 versus the HI group. All data represent the mean value ± SEM, n=4.
Previous studies had shown that HI-induced brain injury was associated with an extensive neuronal death occured in different areas of the brain, such as the cortex and hippocampus [31,32]. We showed that bFGF attenuated brain infarction after HI injury, thus we postulated that such protective effect was attributed to reduced neuronal loss. Nissl staining was used to study morphology and pathology changes of neuronal tissue. It showed that healthy neurons which were characterized by round, pale-stained nuclei with scattered Nissl-stained bodies were throughout the cerebral cortex and hippocampus in the sham group. However, HI injury induced pyramidal neuron death in the cerebral cortex and hippocampus CA1 regions 7 d after injury, which was significantly reduced by bFGF treatment (Figure 2A). These findings suggest that bFGF administration protects neuronal survival and mediates tissue recovery in the brain after HI-induced injury.
Figure 2.
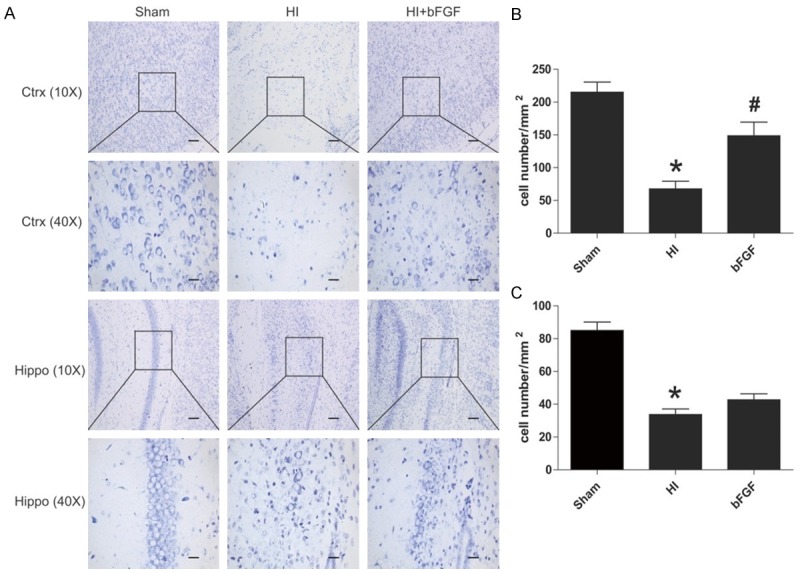
bFGF inhibits neuronal cell death after HI brain injury. Coronal brain sections were obtained from the sham group, HI group and bFGF-treated group at 1 week after HI. A. Nissl staining pictures of the cortex and hippocampus of the lesioned (ipsilateral) side are shown. B. Analysis of neuron numbers in the boxes of the cortex in different groups. *represents P<0.01 versus the sham group, and #represents P<0.05 versus the HI group. C. Analysis of the neuron numbers in the sections of the hippocampus in different groups. *represents P<0.01 versus the sham group. All data represent the mean value ± SEM, n=4.
Treatment of bFGF attenuates neuronal cell death after HI-induced brain injury in rats
Neuronal cell apoptosis was quantified by TUNEL staining. As shown in Figure 3A, increased TUNEL-positive neuronal cell were detected in the ipsilateral cerebral cortex at 7 d post-HI brain injury in neonatal rats as compared with the sham group (P<0.05). bFGF administration significantly decreased TUNEL-positive cells in the brain sections of the bFGF-treated HI group (P<0.05). Increased neuronal apoptosis was further corroborated by western blotting analyses and showed that the protein level of cleaved caspase3 was decreased significantly in the bFGF-treated group as compared with the HI group treated with vehicle (Figure 3B). Our data support that bFGF administration reduces neuronal apoptosis and decreases the activation of caspase 3 protein.
Figure 3.
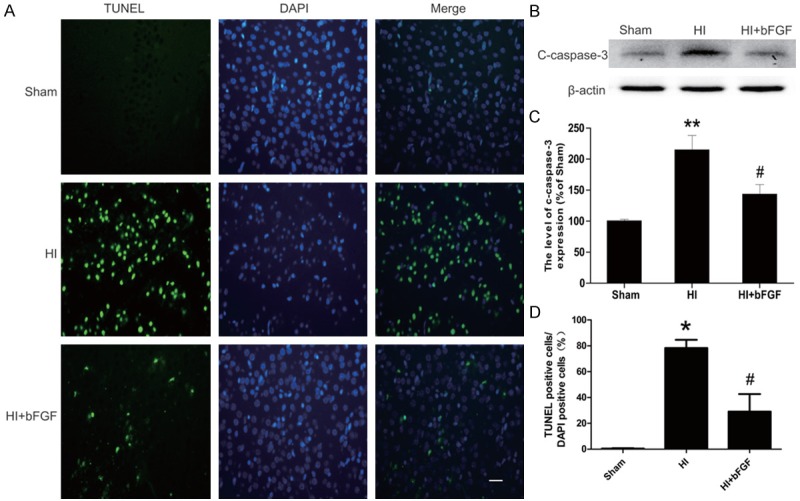
bFGF attenuates neuronal apoptosis and the caspase cascade in the injury areas of rats after HI. A. TUNEL immunofluorescence (green) and DAPI (blue) staining of sections from the cortical area of the lesioned side in each group. B. Protein expression levels of cleaved caspase3 in the brains of the sham rats, HI rats and HI rats treated with bFGF. C. Optical density analysis of cleaved caspase3 protein. *represents P<0.01 versus the sham group, and #represents P<0.05 versus the HI group. D. Quantitative analysis of TUNEL staining data from A. The percentage of TUNEL-positive cells was expressed as the number of TUNEL-stained nuclei divided by the total number of DAPI-stained nuclei. *represents P<0.01 versus the sham group, and #represents P<0.05 versus the HI group. All data represent the mean value ± SEM, n=4.
Treatment of bFGF inhibits ER stress-related protein expression in neonatal rats after HI-induced brain injury
Based on the results obtained from aforementioned results and previous studies, we hypothesized that the neuroprotective effects of bFGF were mediated by inhibition of ER stress. We measured ER stress proteins by western blotting and immunofluorescence staining. Our results indicated that protein levels of ATF-6, GRP-78, XBP-1, ATF-4, CHOP and cleaved caspase12 were significantly up-regulated in the brains of HI rats. However, bFGF treatment markedly decreased expression of these proteins suggesting the ER stress was down-regulated in the HI rat brains (Figure 4C). These findings were consistent with immunofluorescence results which demonstrated that abundant GRP78-positive and PDI-positive cells were observed in the cortex of vehicle-treated HI rats, while significantly reduced GRP78 and PDI staining was found in the same regions of the brain sections from bFGF-treated rats (Figures 4A, 5A). These results demonstrate that bFGF effectively attenuates the ER stress in neonatal rats after HI injury.
Figure 4.
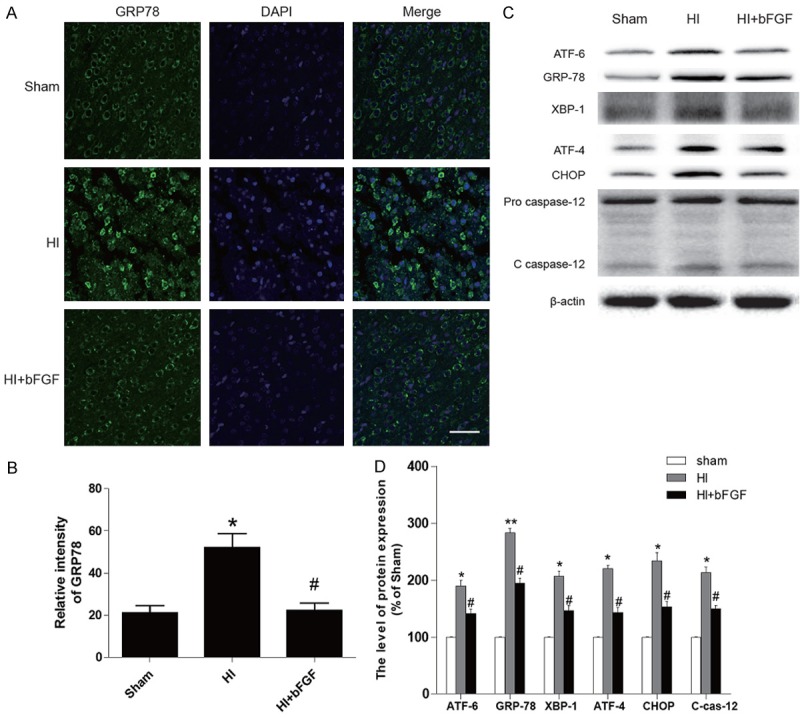
The effects of bFGF on ER stress-related proteins in the brains of rats after HI injury. A. Immunofluorescence staining of GRP78 in the cortical region in the sham group, HI group and bFGF-treated group at 1 week after HI. The nuclei were labelled by DAPI. B. Optical density analysis of GRP78 in each group. *represents P<0.01 versus the sham group, and #represents P<0.05 versus the HI group. C. Protein expression levels of the ER stress markers ATF-6, GRP78, XBP-1, ATF-4, CHOP and caspase12 in the sham rats, HI rats and HI rats treated with bFGF. D. Quantification of western blot data from C. *represents P< 0.05 versus the sham group, and #represents P<0.05 versus the HI group. All data represent the mean value ± SEM, n=4.
Figure 5.
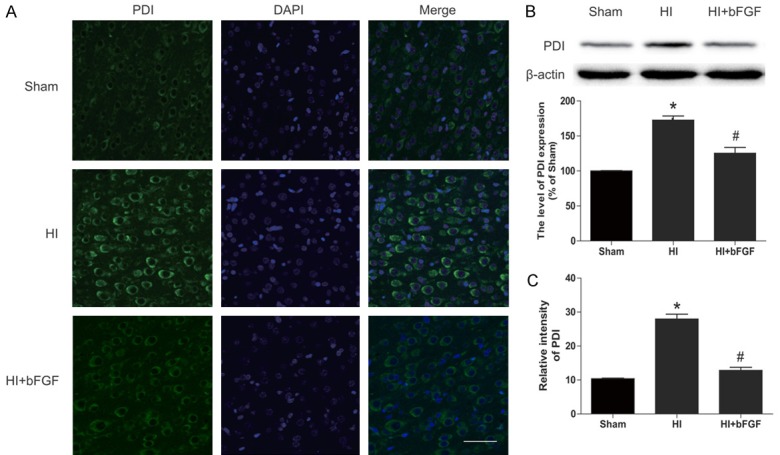
The effects of bFGF on the ER stress-related protein PDI in the brains of rats after HI. A. Representative micrographs showing immunofluorescence with PDI. Nuclei were labelled with DAPI (blue) in each group. B. Representative western blot and quantification data of the ER stress marker PDI in the sham group, HI group and HI bFGF-treated group. *represents P<0.05 versus the sham group, and #represents P<0.05 versus the HI group. C. Optical density analysis of PDI from A. *represents P<0.01 versus the sham group, and #represents P<0.01 versus the HI group. All data represent the mean value ± SEM, n=4.
Intranasal bFGF treatment preserves the brain structure in neonatal rats after HI-induced brain injury
We showed that bFGF provided short-term (1 day) protection against HI brain injury in neonatal rats. To examine whether bFGF treatment improve long-term outcomes in neonatal rats after HI-induced brain injury, the brain structure was studied at 7 d post-HI injury. Severe degree of brain atrophy and subsequent loss of brain mass were occurred in the left cerebral hemisphere in neonatal rats at 7 d after HI (Figure 6A). Interestingly, intranasal administration of bFGF twice a day for 7 consecutive days significantly reversed these injuries. Our immunohistochemical staining also revealed that MAP-2, a biomarker of neuronal damage or grey matter injury, and MBP, a measure of oligodendrocyte loss or white matter injury, were rarely found in the brain of HI group, but intranasal administration of bFGF significantly reduced neuronal loss (vehicle group 53±10.7 vs. bFGF group 26±11.1, P<0.05) and oligodendrocyte loss (vehicle group 50±9.7 vs. bFGF group 27±10.8, P<0.05) at 7 d after HI. Taken together, these results demonstrate potent neuroprotective effects of bFGF and it could be used to treat HI injury in rats.
Figure 6.
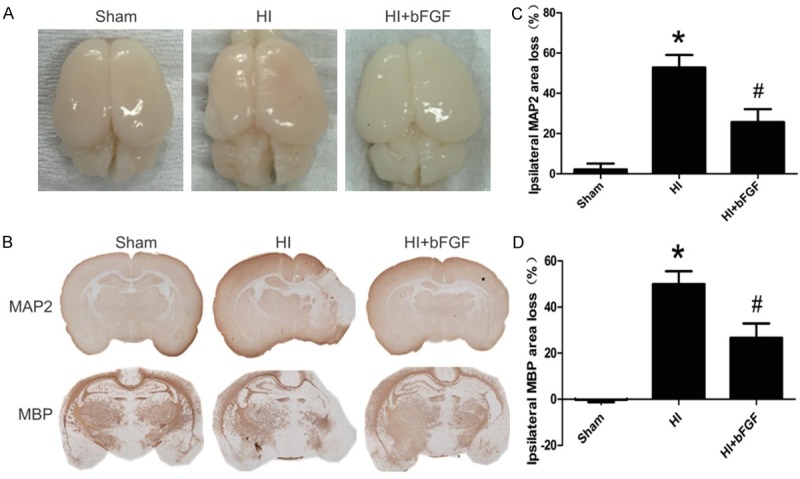
bFGF exerts long-term protective effects on brain structures after neonatal HI injury. Rats were treated with bFGF twice a day for 7 d after HI, and their brains were obtained at 1 week post-HI. A. Representative brain images of the sham rats, HI rats and HI rats treated with bFGF. B. Representative images of immunohistochemical staining for MAP-2 and MBP. C. Quantification of ipsilateral MAP-2 area loss. *represents P<0.01 versus the sham group, and #represents P<0.05 versus the HI group. D. Quantification of ipsilateral MBP area loss. *represents P<0.01 versus the sham group, and #represents P<0.05 versus the HI group. Area loss was measured as 1-(ipsi-/contralateral MAP-2-positive or MBP-positive area). All data represent the mean value ± SEM, n=4.
Treatment of bFGF reduces oxygen-glucose deprivation (OGD)-induced cell death and ER stress in PC12 cells
TUNEL assay was used to characterize cell death in OGD-treated PC12 cells. As shown in Figure 7A, there were few apoptosis-positive cells in the sham group, but the number of apoptotic cells was markedly increased after 24 h of the OGD challenge. Less TUNEL-positive PC12 cells in the OGD+bFGF group suggested that bFGF reduced cell death in vitro. Furthermore, protein level of cleaved caspase3 decreased significantly in the bFGF-treated group when compared with the OGD group (Figure 7B). To further investigate whether bFGF mitigated the ER stress in PC12 cells, the ER stress proteins were measured via western blot analyses in PC12 cells at 24 h after OGD stimulation. As expected, protein levels of GRP78, CHOP, ATF-4 and PDI were significantly higher in the OGD group as compared to the control group. Moreover, treatment of bFGF can effectively down-regulated these ER stress-related protein levels (Figure 8). Taken together, our data suggest that the protective effects of bFGF may involve inhibition of ER stress-related proteins.
Figure 7.
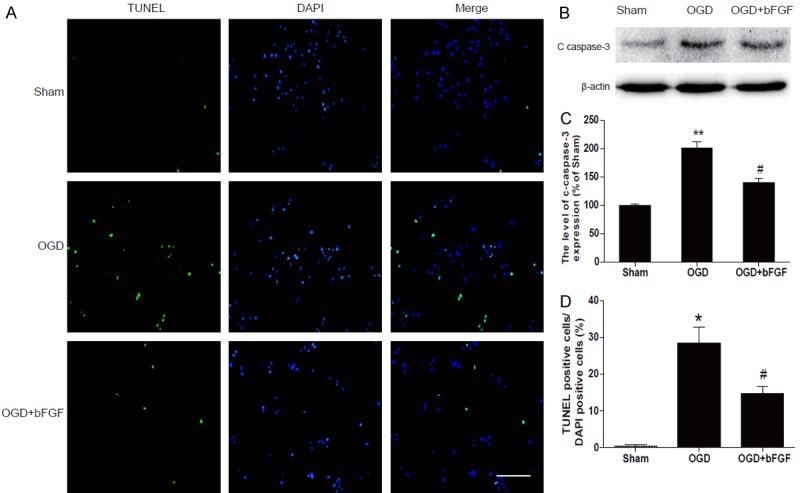
bFGF inhibits apoptosis induced by OGD in PC12 cells. A. Detection of apoptotic cells by TUNEL (green) and DAPI (blue) staining assay. Bright green dots were deemed apoptosis-positive cells. B. Protein expression of cleaved caspase3 in the sham, OGD and bFGF-treated group. C. Optical density analysis of cleaved caspase3 protein. **represents P<0.01 versus the sham group, and #represents P<0.05 versus the OGD group. D. Analysis of apoptosis in the sham, OGD and bFGF-treated group. *represents P<0.01 versus the sham group, and #represents P<0.01 versus the OGD group. All data represent the mean value ± SEM, n=4.
Figure 8.
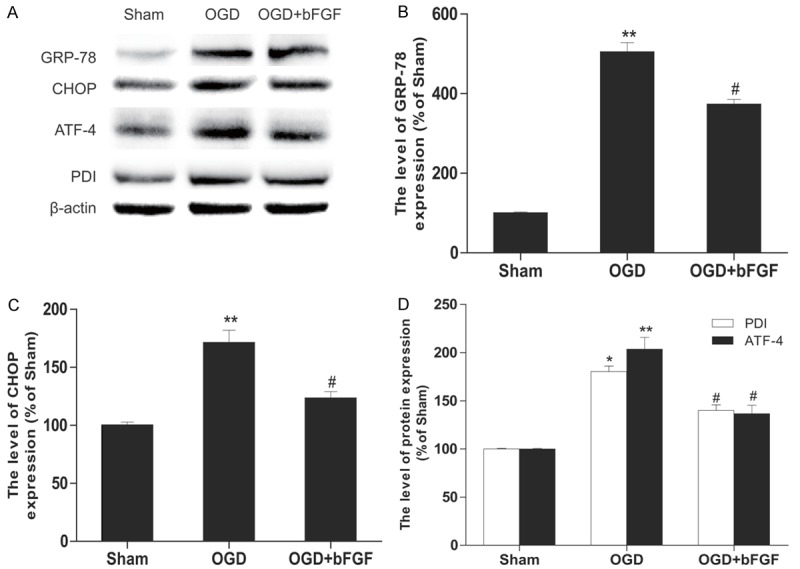
bFGF inhibits OGD-induced ER stress in PC12 cells. A. The western blotting results for GRP78, CHOP, ATF-4, and PDI. B. Optical density analysis of GRP78. C. Optical density analysis of CHOP. D. Optical density analysis of PDI and ATF-4. **represents P<0.01 versus the sham group, and #represents P<0.05 versus the OGD group. All data represent the mean value ± SEM, n=4.
Discussion
Neonatal HI brain injury is a leading cause of morbidity and mortality. Current therapeutic options for this disease are rather limited, as many drugs have disappointed us in human trials although they were shown to be effective in preclinical animal studies. In this study, we sought to demonstrate that intranasal bFGF is a promising therapeutic strategy to protect against HI-induced brain injury in neonatal rats, which may provide further insight in developing an effective therapeutic approach treating neonatal HI brain injury in humans. One of the biggest hurdles is that most systemic medications do not cross the blood-brain barrier (BBB) to the CNS. Nevertheless, intranasal administration has emerged as a non-invasive alternative route for drug delivery into the CNS [34-36], and clinical trials incorporating this method have shown encouraging results [37]. Specifically, intranasal drug delivery allows rapid distribution of molecules to the CNS via bulk flow through the olfactory and trigeminal perivascular channels and slower delivery via olfactory bulb axonal transport. Olfactory neurons are the only first-order receptor cells with specialized processes that are directly exposed in the upper nasal passage, while the other parts of the neurons project to the olfactory bulb [38]. Moreover, drug molecules are transported into olfactory neurons via endocytosis and are further delivered to the olfactory epithelium via extracellular flow through intercellular clefts. Drug molecules reached the olfactory epithelium effectively diffuse into the subarachnoid space [33]. In the present study, we clearly demonstrate, for the first time, that intranasal treatment of bFGF attenuates neonatal HI brain damage, which leads to improved neurological outcomes in neonatal rats.
HI is one of the major pathological factors causing neonatal brain injury, which leads to neurodegeneration and neuronal cell death as a result. In developing brain, the cerebral cortex and the hippocampal CA1 and CA3 subregions in particular, is vulnerable to insults mediated by HI [39,40]. Previous studies have shown that extensive neuronal cell apoptosis is found in the brain of newborn with HI brain injury, suggesting that cell apoptosis may be the mechanism underlying the onset of neurological disabilities associated with HI [41,42]. Several lines of evidence reinforces the finding of neuronal cell apoptosis triggers subsequent brain dysfunctions in the early phase of HI brain injury and its detrimental effects could last for days or even weeks [43]. Nevertheless, the extent of neuronal cell death in neonatal death associated with HI brain injury remains unclear [44-46]. In this study, our results clearly indicated that treatment of bFGF significantly decreases the number of TUNEL-positive cells induced by HI, suggesting that bFGF improves neuronal cell death in the cortical and hippocampal subregions of the neonatal rats after HI brain injury.
The ER stress pathway, also known as the UPR, is a defensive cellular response characterized by the accumulation of unfolded proteins in the ER to preserve organelle function. The UPR pathway can be activated by various cellular stress processes and triggers programmed cell death [47]. Previous studies have shown that activation of the UPR pathway is commonly observed in neurodegenerative diseases related to ageing, such as Alzheimer’s disease, Parkinson’s disease, or diabetes and cardiovascular diseases. Furthermore, the UPR pathway also plays a major role in the acute symptoms associated with ischemia or exposure to hypoxic conditions [48]. In our study, we examined whether treatment of bFGF mitigates the ER stress, which in turn inhibits the UPR pathway in neonatal rats after HI brain injury. GRP78 is an ER chaperone representing an indicator of ER stress [49]. PDI presents within the lumen of the ER, is known to catalyse the post-translational protein modification. In the quiescent state, a scarce amount of neurons are expressing GRP78 and PDI. However, when exposed to the HI insult, up-regulation of GRP78 and PDI is observed in injured neurons [50]. In our study, we found that GRP78 and PDI expression was significantly increased in the cortical and hippocampal regions of the brain in neonatal rats at 24 h after HI injury. Intranasal treatment of bFGF decreased the levels of ER stress-related proteins and inhibited neuronal cell apoptosis. These results indicated that the protective role of bFGF in neonatal HI injury was related to suppression of ER stress. Further study was needed to determine whether inhibition of ER stress-induced apoptosis by bFGF also leads to reversal of the UPR pathway and subsequently reactivates innate immunity, metabolism and cell differentiation. Our results were consistent with previous studies suggesting that bFGF attenuated ER stress in different animal models of HI injury. For instance, treatment of bFGF inhibited ER stress induced by ischaemic oxidative injury via activation of the PI3K/Akt and ERK1/2 pathways [49]. Similar findings were also observed in others studies investigating myocardial ischaemia/reperfusion injury [51] and spinal cord injury [52]. Thus, it is conceivable that bFGF may play an important role in ER stress induced by neonatal HI injury.
bFGF is highly expressed in the nervous system where it has multiple roles and exogenous bFGF has been shown to have neuroprotective function in brain injury. However, it is no doubt that the limitations of bFGF in HI injury therapy still need further investigations and improvements. For example, it is increasingly clear that exploiting a single pathophysiological pathway might not be sufficient to combat neonatal HI brain injury. Promising neuroprotective drugs should be studied in combination with hypothermia, because temperature change can greatly alter the pharmacokinetics and pharmacodynamics of various drugs. Moreover, bFGF has a short biological half-life due to the presence of degrading enzymes in vivo. Therefore, future work focuses on developing a delivery system that increases bFGF stability may bolster its therapeutic function for treating infants with HI brain injury.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NO. 81302775, 81472165, 81560375), The Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents (to J.X.), and the Wenzhou Science and Technology Foundation (Y20140681). Science and technology project of Zhejiang Province (LY17H090017 to Z.W.).
Disclosure of conflict of interest
None.
References
- 1.Walt G. WHO’s World Health Report 2003. BMJ. 2004;328:6. doi: 10.1136/bmj.328.7430.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bang AT, Bang RA, Baitule SB, Reddy HM, Deshmukh MD. Management of birth asphyxia in home deliveries in rural Gadchiroli: the effect of two types of birth attendants and of resuscitating with mouth-to-mouth, tube-mask or bag-mask. J Perinatol. 2005;25(Suppl 1):S82–91. doi: 10.1038/sj.jp.7211275. [DOI] [PubMed] [Google Scholar]
- 3.Minino AM, Heron MP, Murphy SL, Kochanek KD. Centers for Disease C and Prevention National Center for Health Statistics National Vital Statistics S. Deaths: final data for 2004. Natl Vital Stat Rep. 2007;55:1–119. [PubMed] [Google Scholar]
- 4.Lynch JK, Nelson KB. Epidemiology of perinatal stroke. Curr Opin Pediatr. 2001;13:499–505. doi: 10.1097/00008480-200112000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Sran SK, Baumann RJ. Outcome of neonatal strokes. Am J Dis Child. 1988;142:1086–1088. doi: 10.1001/archpedi.1988.02150100080031. [DOI] [PubMed] [Google Scholar]
- 6.Sreenan C, Bhargava R, Robertson CM. Cerebral infarction in the term newborn: clinical presentation and long-term outcome. J Pediatr. 2000;137:351–355. doi: 10.1067/mpd.2000.107845. [DOI] [PubMed] [Google Scholar]
- 7.Jing G, Wang JJ, Zhang SX. ER stress and apoptosis: a new mechanism for retinal cell death. Exp Diabetes Res. 2012;2012:589589. doi: 10.1155/2012/589589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badiola N, Penas C, Minano-Molina A, Barneda-Zahonero B, Fado R, Sanchez-Opazo G, Comella JX, Sabria J, Zhu C, Blomgren K, Casas C, Rodriguez-Alvarez J. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149. doi: 10.1038/cddis.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zha BS, Zhou H. ER stress and lipid metabolism in adipocytes. Biochem Res Int. 2012;2012:312943. doi: 10.1155/2012/312943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding W, Yang L, Zhang M, Gu Y. Reactive oxygen species-mediated endoplasmic reticulum stress contributes to aldosterone-induced apoptosis in tubular epithelial cells. Biochem Biophys Res Commun. 2012;418:451–456. doi: 10.1016/j.bbrc.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 11.Liu XD, Ko S, Xu Y, Fattah EA, Xiang Q, Jagannath C, Ishii T, Komatsu M, Eissa NT. Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. J Biol Chem. 2012;287:19687–19698. doi: 10.1074/jbc.M112.350934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akerfeldt MC, Howes J, Chan JY, Stevens VA, Boubenna N, McGuire HM, King C, Biden TJ, Laybutt DR. Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes. 2008;57:3034–3044. doi: 10.2337/db07-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- 14.Wu CT, Sheu ML, Tsai KS, Weng TI, Chiang CK, Liu SH. The role of endoplasmic reticulum stress-related unfolded protein response in the radiocontrast medium-induced renal tubular cell injury. Toxicol Sci. 2010;114:295–301. doi: 10.1093/toxsci/kfq006. [DOI] [PubMed] [Google Scholar]
- 15.Xiao J, Chu Y, Hu K, Wan J, Huang Y, Jiang C, Liang G, Li X. Synthesis and biological analysis of a new curcumin analogue for enhanced anti-tumor activity in HepG 2 cells. Oncol Rep. 2010;23:1435–1441. doi: 10.3892/or_00000781. [DOI] [PubMed] [Google Scholar]
- 16.Galano A, Tan DX, Reiter RJ. Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res. 2011;51:1–16. doi: 10.1111/j.1600-079X.2011.00916.x. [DOI] [PubMed] [Google Scholar]
- 17.Laliena A, San Miguel B, Crespo I, Alvarez M, Gonzalez-Gallego J, Tunon MJ. Melatonin attenuates inflammation and promotes regeneration in rabbits with fulminant hepatitis of viral origin. J Pineal Res. 2012;53:270–278. doi: 10.1111/j.1600-079X.2012.00995.x. [DOI] [PubMed] [Google Scholar]
- 18.Carloni S, Albertini MC, Galluzzi L, Buonocore G, Proietti F, Balduini W. Melatonin reduces endoplasmic reticulum stress and preserves sirtuin 1 expression in neuronal cells of newborn rats after hypoxia-ischemia. J Pineal Res. 2014;57:192–199. doi: 10.1111/jpi.12156. [DOI] [PubMed] [Google Scholar]
- 19.Wang ZG, Wang Y, Huang Y, Lu Q, Zheng L, Hu D, Feng WK, Liu YL, Ji KT, Zhang HY, Fu XB, Li XK, Chu MP, Xiao J. bFGF regulates autophagy and ubiquitinated protein accumulation induced by myocardial ischemia/reperfusion via the activation of the PI3K/Akt/mTOR pathway. Sci Rep. 2015;5:9287. doi: 10.1038/srep09287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang HY, Wang ZG, Wu FZ, Kong XX, Yang J, Lin BB, Zhu SP, Lin L, Gan CS, Fu XB, Li XK, Xu HZ, Xiao J. Regulation of autophagy and ubiquitinated protein accumulation by bFGF promotes functional recovery and neural protection in a rat model of spinal cord injury. Mol Neurobiol. 2013;48:452–464. doi: 10.1007/s12035-013-8432-8. [DOI] [PubMed] [Google Scholar]
- 21.Jin-Qiao S, Bin S, Wen-Hao Z, Yi Y. Basic fibroblast growth factor stimulates the proliferation and differentiation of neural stem cells in neonatal rats after ischemic brain injury. Brain Dev. 2009;31:331–340. doi: 10.1016/j.braindev.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Matsuse D, Kitada M, Ogura F, Wakao S, Kohama M, Kira J, Tabata Y, Dezawa M. Combined transplantation of bone marrow stromal cell-derived neural progenitor cells with a collagen sponge and basic fibroblast growth factor releasing microspheres enhances recovery after cerebral ischemia in rats. Tissue Eng Part A. 2011;17:1993–2004. doi: 10.1089/ten.TEA.2010.0585. [DOI] [PubMed] [Google Scholar]
- 23.Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, Vannucci SJ. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res. 1999;55:158–163. doi: 10.1002/(SICI)1097-4547(19990115)55:2<158::AID-JNR3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 24.Feng ZC, Popell A, Li J, Silverstein J, Oakie A, Yee SP, Wang R. c-Kit receptor signaling regulates islet vasculature, beta-cell survival, and function in Vivo. Diabetes. 2015;64:3852–3866. doi: 10.2337/db15-0054. [DOI] [PubMed] [Google Scholar]
- 25.Feng ZC, Li J, Turco BA, Riopel M, Yee SP, Wang R. Critical role of c-Kit in beta cell function: increased insulin secretion and protection against diabetes in a mouse model. Diabetologia. 2012;55:2214–2225. doi: 10.1007/s00125-012-2566-5. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Feng ZC, Yeung FS, Wong MR, Oakie A, Fellows GF, Goodyer CG, Hess DA, Wang R. Aldehyde dehydrogenase 1 activity in the developing human pancreas modulates retinoic acid signalling in mediating islet differentiation and survival. Diabetologia. 2014;57:754–764. doi: 10.1007/s00125-013-3147-y. [DOI] [PubMed] [Google Scholar]
- 27.Choi J, Diao H, Feng ZC, Lau A, Wang R, Jevnikar AM, Ma S. A fusion protein derived from plants holds promising potential as a new oral therapy for type 2 diabetes. Plant Biotechnol J. 2014;12:425–435. doi: 10.1111/pbi.12149. [DOI] [PubMed] [Google Scholar]
- 28.Yin D, Zhou C, Kusaka I, Calvert JW, Parent AD, Nanda A, Zhang JH. Inhibition of apoptosis by hyperbaric oxygen in a rat focal cerebral ischemic model. J Cereb Blood Flow Metab. 2003;23:855–864. doi: 10.1097/01.WCB.0000073946.29308.55. [DOI] [PubMed] [Google Scholar]
- 29.Feng ZC, Riopel M, Li J, Donnelly L, Wang R. Downregulation of Fas activity rescues early onset of diabetes in c-Kit(Wv/+) mice. Am J Physiol Endocrinol Metab. 2013;304:E557–565. doi: 10.1152/ajpendo.00453.2012. [DOI] [PubMed] [Google Scholar]
- 30.Feng ZC, Donnelly L, Li J, Krishnamurthy M, Riopel M, Wang R. Inhibition of Gsk3beta activity improves beta-cell function in c-KitWv/+ male mice. Lab Invest. 2012;92:543–555. doi: 10.1038/labinvest.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 32.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 33.Topkoru BC, Altay O, Duris K, Krafft PR, Yan J, Zhang JH. Nasal administration of recombinant osteopontin attenuates early brain injury after subarachnoid hemorrhage. Stroke. 2013;44:3189–3194. doi: 10.1161/STROKEAHA.113.001574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doyle KP, Yang T, Lessov NS, Ciesielski TM, Stevens SL, Simon RP, King JS, Stenzel-Poore MP. Nasal administration of osteopontin peptide mimetics confers neuroprotection in stroke. J Cereb Blood Flow Metab. 2008;28:1235–1248. doi: 10.1038/jcbfm.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorne RG, Emory CR, Ala TA, Frey WH 2nd. Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res. 1995;692:278–282. doi: 10.1016/0006-8993(95)00637-6. [DOI] [PubMed] [Google Scholar]
- 36.Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH 2nd, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain. 2008;131:3311–3334. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- 37.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dhuria SV, Hanson LR, Frey WH 2nd. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99:1654–1673. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- 39.Delivoria-Papadopoulos M, Mishra OP. Mechanisms of perinatal cerebral injury in fetus and newborn. Ann N Y Acad Sci. 2000;900:159–168. doi: 10.1111/j.1749-6632.2000.tb06226.x. [DOI] [PubMed] [Google Scholar]
- 40.Puka-Sundvall M, Sandberg M, Hagberg H. Brain injury after hypoxia-ischemia in newborn rats: relationship to extracellular levels of excitatory amino acids and cysteine. Brain Res. 1997;750:325–328. doi: 10.1016/s0006-8993(97)00024-3. [DOI] [PubMed] [Google Scholar]
- 41.Ma H, Sinha B, Pandya RS, Lin N, Popp AJ, Li J, Yao J, Wang X. Therapeutic hypothermia as a neuroprotective strategy in neonatal hypoxic-ischemic brain injury and traumatic brain injury. Curr Mol Med. 2012;12:1282–1296. doi: 10.2174/156652412803833517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hossain MA. Molecular mediators of hypoxic-ischemic injury and implications for epilepsy in the developing brain. Epilepsy Behav. 2005;7:204–213. doi: 10.1016/j.yebeh.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 43.Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol. 2011;69:743–758. doi: 10.1002/ana.22419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 45.Graham SH, Chen J. Programmed cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:99–109. doi: 10.1097/00004647-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 46.MacManus JP, Buchan AM. Apoptosis after experimental stroke: fact or fashion? J Neurotrauma. 2000;17:899–914. doi: 10.1089/neu.2000.17.899. [DOI] [PubMed] [Google Scholar]
- 47.Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012;22:274–282. doi: 10.1016/j.tcb.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Nakajima A, Tsuji M, Inagaki M, Tamura Y, Kato M, Niiya A, Usui Y, Oguchi K. Neuroprotective effects of propofol on ER stress-mediated apoptosis in neuroblastoma SH-SY5Y cells. Eur J Pharmacol. 2014;725:47–54. doi: 10.1016/j.ejphar.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 49.Wang Z, Wang Y, Ye J, Lu X, Cheng Y, Xiang L, Chen L, Feng W, Shi H, Yu X, Lin L, Zhang H, Xiao J, Li X. bFGF attenuates endoplasmic reticulum stress and mitochondrial injury on myocardial ischaemia/reperfusion via activation of PI3K/Akt/ERK1/2 pathway. J Cell Mol Med. 2015;19:595–607. doi: 10.1111/jcmm.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka S, Uehara T, Nomura Y. Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J Biol Chem. 2000;275:10388–10393. doi: 10.1074/jbc.275.14.10388. [DOI] [PubMed] [Google Scholar]
- 51.Wang Z, Zhang H, Xu X, Shi H, Yu X, Wang X, Yan Y, Fu X, Hu H, Li X, Xiao J. bFGF inhibits ER stress induced by ischemic oxidative injury via activation of the PI3K/Akt and ERK1/2 pathways. Toxicol Lett. 2012;212:137–146. doi: 10.1016/j.toxlet.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Zhang HY, Zhang X, Wang ZG, Shi HX, Wu FZ, Lin BB, Xu XL, Wang XJ, Fu XB, Li ZY, Shen CJ, Li XK, Xiao J. Exogenous basic fibroblast growth factor inhibits ER stress-induced apoptosis and improves recovery from spinal cord injury. CNS Neurosci Ther. 2013;19:20–29. doi: 10.1111/cns.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]