

Abstract
Hepatocellular carcinoma (HCC) is one of the most lethal malignancies of cancers and its prognosis remains dismal due to the paucity of effective therapeutic targets. Up-regulation of glutathione-s-transferase A 4 (GSTA4) is associated with poor prognosis of HCC, but its functional mechanism in HCC remains unclear. In this study, we investigated the roles of GSTA4 in tumor growth and metastasis of HCC and found that GSTA4 was frequently up-regulated in HCC tissues. Through gain- and loss-of-function studies, GSTA4 was demonstrated to significantly regulate cell proliferation, migration, and invasion in vitro. Furthermore, GSTA4 overexpressing significantly promoted the tumorigenicity and metastasis of HCC cells in nude mice models bearing human HCC, whereas silencing endogenous GSTA4 caused an opposite outcome. Moreover, we demonstrated that GSTA4 enhanced HCC aggressiveness by activating protein kinase B (AKT) signaling. In multivariate analysis, our results GSTA4 overexpression promotes the progression of hepatocellular carcinoma and might represent a novel therapeutic target for its treatment.
Keywords: GSTA4, hepatocellular carcinoma, metastasis, AKT
Introduction
Glutathione S-transferases (GSTs) are a broadly expressed family of phase II isoenzymes that protect normal cells against damage induced by hepatitis virus or aflatoxin-related hepatocarcinogen [1]. Numerous reports have shown that these glutathione-s-transferases dysfunction increase the risk of cancer and is involved in the development and progression of various human cancers, including liver cancer, breast cancer, prostate cancer and colorectal carcinoma [2]. In murine chemically-induced carcinogenesis models and human cancer, GST genes are frequently over-expressed. GSTA1 hypermethylation is an early event during prostate cancer formation, occurring in atrophic hyperplasia and prostatic intraepithelial neoplasia [3]. Functional loss of GSTA1 has been proposed to promote the development of genomic instability and prostate cancer [4]. Studies have shown that up-regulated GSTA1 activity is a hallmark of a malignant bladder cancer phenotype and is involved in the maintenance of the prooxidant-antioxidant balance towards a more reduced state during tumor progression [5]. GST1 has a high level of expression in the bladder and plays a central role in the inactivation of toxic and carcinogenic compounds. Recently, polymorphisms in these genes have been shown to produce significant alterations in the metabolism of many substrates, including carcinogens and chemotherapeutic agents. The metabolizing enzymes for detoxification or activation of these carcinogens to lower toxicity, potentially affect the individual cancer risk and progression of several cancer [6]. Moreover, emerging data show that some of GST have also been suggested to affect the survival status of cancer patients and are crucial for tumorigenicity, proliferation, apoptosis, angiogenesis, invasion, and metastasis of hepatocellular carcinoma (HCC).
Hepatocellular carcinoma (HCC) is the fifth most common malignancy worldwide and the second leading cause of cancer death in Asia [7]. Although the survival of patients with HCC has improved due to advances in surgical techniques, long-term survival after surgical resection remains low. Metastasis is the major reason for the high mortality of HCC patients after surgical resection [8]. Nonetheless, the molecular mechanisms underlying HCC metastasis remain largely unclear. Thus, it is critical to discover the mechanism of HCC metastasis. GSTA4 has been shown to exhibit oncogenic properties in glioma, lung cancer, hepatocellular carcinoma and cholangiocarcinoma [9]. Numerous studies have shown that GSTA4 promotes cell proliferation in nasopharyngeal carcinoma, breast cancer, and liver cancer cells [10]. Interestingly, GSTA4 was highly expressed in lymph node metastatic tumors compared with primary tumors and enhanced lung cancer cell migration and invasion. To date, experimental data suggest that GSTA4 directly regulates several target genes, including activator protein 1 (AP1), MTDH, CDK6, CCNE1, CCNE2, CCND2, EZH2, PTEN, RB1, MAP3K2, and GSK-3β [11]. However, the potential mechanisms by which GSTA4 affects the growth and metastasis of HCC cells remain largely unknown.
In this study, we investigated the potential function of GSTA4 in the growth and metastasis of HCC. We found that GSTA4 expression was up-regulated in the majority of HCC tissues and its over-expression was significantly associated with tumor metastasis in HCC patients. Through gain- and loss-of-function studies, GSTA4 was demonstrated to regulated hepatocellular carcinoma cells proliferation clone formation ability in vitro. In addition knock-down GSTA4 suppressed in vivo tumor growth in nude mice models bearing human HCC. Importantly, by silencing and over-expression of GSTA4, we confirmed the role of GSTA4 in the migration and invasion of HCC cells in vitro. Moreover, reduction and increasing of GSTA4 expression in HCC cells significantly impaired and promoted their metastasis in vivo. Further investigation demonstrated that AKT signaling pathway is involved in GSTA4-promoted hepatocellular carcinoma metastasis. To conclude, GSTA4 could promote tumor growth and metastasis of HCC through AKT signaling and is a novel therapeutic target for HCC.
Materials and methods
Oncomine analysis
The expression level of GSTA4 gene in the selected cancers was analyzed using Oncomine. For this, we compared clinical specimens of cancer vs. normal patient datasets. In order to reduce our false discovery rate, we selected P < 0.01 as a threshold. We analyzed the results for their p-values, fold change, and cancer subtype. In many instances we found several significant correlations in different tumor types, but we showed only one to three of them [12].
Cell lines and culture
Human hepatocellular cell lines HepG2, SMMC-7402, SMMC-7721, MHCC97, HCCLM3 and normal human liver cell line LO2 were purchased from Cell Resource Center, Shanghai Institutes for Biological Sciences and preserved in our laboratory. Cells were cultured in DMEM or 1640 with 10% fetal calf serum (FCS), 1% Penicilin/Streptomycin mix and maintained at 37°C with 5% CO2.
Transfection of siRNA and GSTA4 plasmid
MHCC97-H cells were transfected with siRNA and Lipofectamine-2000 according to the manufacturer’s instructions. Briefly, MHCC97-H cells were seeded in a 24-well-plate at a density of 0.5 × 105 cells/well with antibiotics-free medium 12 hours before the transfection. The GSTA4 siRNA (Santa Cruz Biotechnology, sc-105424) were mixed with Lipofectamine-2000 in serum-free RPMI-1640 medium and were incubated for 25 minutes to form a complex. After washing cells with PBS, the transfection mixtures were added to each well. Twenty-four hours after the transfection, the medium was replaced with fresh RPMI-1640 medium containing 10% FBS. GSTA4 expression plasmid was generated by PCR sub-cloning human GSTA4 coding sequence into retroviral transfer plasmid pQCXIP (Clontech) to generate plasmid pQCXIP-GSTA4. Retroviral production and infection were performed as we previously described, and stable HepG2 cell lines were selected by treatment with 0.5 μg/ml puromycin for 10 days, beginning at 24 h after infection [13].
Soft agar assay
Colony formation ability was examined by anchorage-independent soft agar assay on MHCC97-H or HepG2 cells. Briefly, 1.5 ml FBS supplemented medium containing 1% agarose were added in 35-mm cell culture dishes and allowed to solidify (base agar). Next, 1 × 104 GSTA4 siRNA transfected MHCC97-H cells or 1 × 104 HepG2 transfected with GSTA4 plasmid were mixed with 1.5 ml FBS-supplemented medium containing 0.1% agarose and added to the top of base agar. The cells were then cultured for 14 days at 37°C in 5% carbon dioxide. The dishes were stained with 0.1% crystal violet, and the colonies were examined with microscope and digital camera [14].
Wound healing assay
1 × 105 GSTA4 siRNA transfected MHCC97-H cells or 1 × 105 HepG2 transfected with GSTA4 plasmid were seeded in 6-well plates. Once the cells reached 90% confluency, a wound area was carefully created by scraping the cell monolayer with a sterile 10 μl pipette tip. The cells were then washed once with PBS to remove detached cells. Subsequently, the cells were incubated at 37°C in 5% carbon dioxide. The width of the wound area was monitored with an inverted microscope at various time points. The normalized wound area (wound area 48 hours/wound area 0 hours) was calculated using the software SPSS [15].
Transwell invasion assay
We evaluated the effect of GSTA4 on invasiveness properties of breast cancer cells using Transwell invasion assay. 24 hours after the transfection GSTA4 siRNA or GSTA4 plasmid, cells (1 × 105) were plated in the top chamber of the Transwell with a coated polycarbonate membrane (6.5 mm diameter insert, 8.0 μm pore size; Corning Incorporated). Medium with 10% FBS was added to the lower chamber as a chemo-attractant. After incubation for 24 hours, cells on the lower surface of the membrane were fixed with 10% formalin and stained with 0.1% crystal violet. The images of invasive cells were acquired by an inverted microscope. The number of invaded cells was counted from five randomly selected fields in a blind way [16].
Cell proliferation assay
The effect of siRNA or GSTA4 plasmid on cell proliferation was measured using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay according to the manufacturer’s protocol. After transfected with GSTA4 siRNA or GSTA4 plasmid, cells (5,000 cells/well) were seeded in 96-well plates. Subsequently at 1, 2, 3 and 4 days, 20 μl MTT (Sigma) was added to each well and incubated for 4 h. The culture medium was removed, and 200 μl dimethyl sulfoxide (DMSO) (Sigma) was added to each well. The plates were then shaken for 30 min, and the optical density (OD) at 490 nm was measured using a microplate reader. Each experiment was performed in triplicates [17].
Western blotting
The cultured cells were washed with PBS and lysed on ice in RIPA lysis buffer containing protease and phosphatase inhibitors. An equal amount of cell lysates (30 μg) was loaded and separated by 10% SDS-PAGE. The protein was transferred to PVDF membranes, blocked and incubated with monoclonal antibody against GSTA4 (1:1000, Santa Cruz Biotechnology), GSK-3β (1:500, Signalway Antibody) and phosphor-GSK3β Ser-9 (1:10000, Epitomics), AKT and phosphor-AKT Ser473 (1:1000, Cell Signaling Technology), mTOR and phospho-mTOR Ser2448 (1:1000, Cell Signaling Technology), FAK and phospho-FAK Tyr925 (1:1000, Cell Signaling Technology), Src and phospho-Src Ser17 (1:1000, Cell Signaling Technology), GPADH (1:5000, Bioworld Technology) followed by incubation with horseradish peroxidase-conjugated IgGs (1:10000, Bioworld Biotechnology). The protein was then visualized using ECL system (Millipore) and visualized with the ChemiDoc XRS system (Bio-Rad) [18].
Immunofluorescence analysis
The effects of GSTA4 silencing and over-expression on the expression of GSTA4 in cells were examined using an immunocytochemical method. Cells on glass coverslips were transfected with indicated agents, they were fixed by pre-cold acetone, then rinsed three times with PBS. The cells were permeabilized in 0.1% Triton X-100 and incubated with 1% BSA/PBS on ice. Subsequently, the cells were immunostained by incubating with anti-GSTA4 antibody (1:500, Santa Cruz Biotechnology) overnight at 4°C. After being washed with PBS, cells were incubated with FITC-conjugated goat anti-rabbit secondary antibody (1:60, Boster Biotechnology). Nuclei were counterstained with DAPI (Biotime Biotech). Images were taken and analyzed using the ZEN 2011 imaging software on a Zeiss invert microscope (CarlZeiss) under 200-fold magnification [19].
In vivo tumorigenesis assay and experimental metastasis assay
Animal protocols were approved by the Ethical Committee of Centre-south University. BALB/c nude mice (Shanghai Slack Laboratory Animal Co., LTD, China) were injected subcutaneously with 5 × 107 indicated MHCC97-H or HepG2 cells, which were transfected with a GSTA4 siRNA or GSTA4 plasmid, in 0.1 ml of PBS in the right flank. Tumor volumes were monitored with calipers and calculated by the formula: 0.5 × length × width2. Mice were sacrificed after 6 weeks. BALB/c nude mice (15-20 g) were purchased from Shanghai Slack laboratory animal co., LTD. Mice were maintained at dark/light cycles of 12 h duration with food and water available. For experimental metastasis analysis, the mice were injected at the lateral tail vein with (5 × 105) cells carrying GSTA4 siRNA or GSTA4 plasmid. Mice were sacrificed 2 weeks after inoculation and all organs were examined for the presence of macroscopic metastases. Lung and liver metastatic nodules were determined under a dissecting microscope. The data were presented as mean ± SE. Differences in the results of different treatment groups were evaluated using either two-tailed Student’s t test or one-way ANOVA followed by post hoc Dunnett’s test [20].
Statistical analysis
Statistical analysis was performed with SPSS. Values are expressed as the mean ± SD. The difference between groups was analyzed using a Student t test when comparing only two groups or one-way analysis of variance when comparing more than two groups. P < 0.05 was considered statistically significant.
Results
Up-regulation of GSTA4 is associated with metastasis of HCC
We extracted data on transcript expression for GSTA4 from the database Oncomine for lung and bronchus, prostate, breast, colon and rectum, ovarian and pancreatic cancers, focusing on clinical specimens of cancer vs. normal patient datasets, and separated by subtype when possible. The expression level of GSTA4 was significantly up-regulated in HCC tissues versus adjacent nontumor liver tissues (P < 0.01) (Figure 1A). Oncomine analysis of neoplastic vs. normal tissue showed that GSTA4 were significantly overexpressed in different types of lung cancer in different datasets (Supplementary Figure 1). Furthermore, in comparison to non-metastatic HCC tissues, GSTA4 levels were significantly higher in metastatic HCC tissues (Figure 1B). To further evaluate the association of GSTA4 with HCC metastasis, we analyzed GSTA4 levels in a panel of human HCC cell lines with different metastatic potentials. The GSTA4 level was significantly increased in four established HCC cell lines relative to the nontransformed hepatic cell line L02 (Figure 1C). Moreover, the expression levels of GSTA4 in the highly metastatic HCC cell lines MHCC97 and HCC-LM3 were much lower than those in the HCC cell lines that have low metastatic potential, including HepG2, SMMC-7402 and SMMC-7721 (Figure 1C). These results indicate that significant up-regulation of GSTA4 expression occurs in HCC and correlates with HCC metastasis.
Figure 1.

The expression levels of GSTA4 in HCC tissues and HCC cell lines. A. This graphic compared the number of datasets that had significant mRNA over-expression (left column, red) and under-expression (right column, blue) of the specified gene GSTA4 in cancer versus normal tissue. The datasets were obtained with the following parameters: P-value threshold of 0.01. B. Comparing differences in the expression levels of GSTA4 between metastatic and non-metastatic HCC tissues. C. Expression levels of GSTA4 in HCC cell lines with different metastatic potentials and the non-transformed hepatic cell line L02 were determined by western blotting assay. The experiment was repeated three times and GAPDH used as a loading control.
The effects of GSTA4 on HCC cells proliferation
To further explore the biological significance of GSTA4 in HCC, we transfected GSTA4 siRNA plasmid or GSTA4 vector into human HCC cells. Expression of GSTA4 was verified by western blotting (Figure 2A) and cellular immunofluorescence assays (Figure 2B). Down-regulation of GSTA4 in MHCC97 cells, which has high metastasis potential, resulted in significant suppression of cell proliferation. Transfection of GSTA4 for 24, 48, 72, or 96 hours inhibited proliferation of MHCC97 cells (Figure 2C). Metastatic malignant cells possess resistance to detachment induced cell death, or anoikis resistance, which ensures anchorage independent growth and survival during their dissemination. Cells proliferate rapidly and form sizable colonies from a single cell in semi-solid agar, whereas GSTA4 siRNA treatment decreased the number of sizable colonies and reduced colony size in MHCC97 cells (Figure 2D). In contrast, GSTA4 over-expression using GSTA4 vector in HepG2 cells (Figure 2A and 2B), which have low metastatic potential, significantly increased cell proliferation (Figure 2E) and anchorage independent growth (Figure 2F) compared with the negative control. These results demonstrated that GSTA4 regulated proliferation of HCC cells in vitro.
Figure 2.
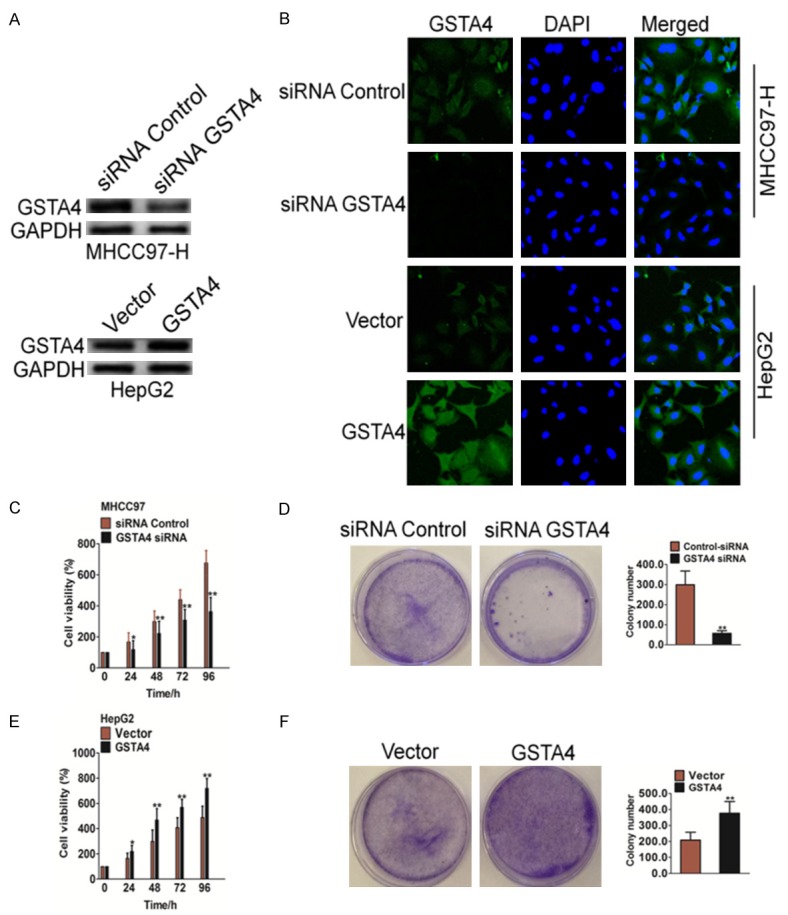
The effect of GSTA4 on the proliferation of hepatocellular carcinoma cells in vitro. A. siRNA against GSTA4 were transfected into MHCC97 cells or GSTA4 vector were transfected into HepG2 cells. After 24 h post transfections, western blot analysis of MHCC97 protein expression in the MHCC97 cell lines. B. MHCC97 cells were transfected with control siRNA or siRNA against GSTA4 and HepG2 cells were transfected with control vector or GSTA4 vector, then cells were fixed and incubated with primary antibodies against GSTA4. Cells were immunostained with anti-rabbit FITC-conjugated secondary antibody and then stained with DAPI. The specimens were visualized and photographed using a fluorescence microscope. C. MTT assay analysis of cell proliferation in MHCC97-siGSTA4 and control cells. Down-regulation of GSTA4 significantly inhibited MHCC97 cell proliferation compared with controls. The data are presented as mean ± SD. For indicated comparisons, *P < 0.05, **P < 0.01. D. A soft agar assay was conducted to examine the colony formation ability of MHCC97 cells. Twenty-four hours after the siRNA transfection, MHCC97 cells were seeded in 0.1% agarose in RPMI-1640-supplemented 10% FBS at a density of 1 × 104 per 25 mm2 culture dish and allowed to grow for 14 days. Results were representative picture of colonies of three independent experiments done in triplicate. The data are presented as mean ± SD. **P < 0.01 vs. negative control siRNA. E. MTT assay analysis of cell proliferation in GSTA4-overexpression HepG2 cells and control cells. Up-regulation of GSTA4 significantly increased MHCC97 cell proliferation compared with controls. The data are presented as mean ± SD. For indicated comparisons, *P < 0.05, **P < 0.01. F. The colony formation rate in HepG2 cells transfected with GSTA4 vector was significantly increased compared to the control cells. Results were representative picture of colonies of three independent experiments done in triplicate. The data are presented as mean ± SD. **P < 0.01 vs. negative control vector cells.
GSTA4 reduces migration and invasion of HCC cells
Decreased clonogenic potential is usually associated with the loss of migration and invasion capabilities in tumor cells, which are critical properties for the spreading of cancer cells and metastases. The cell metastasis potential of hepatocellular carcinoma cell was therefore tested using a classic wound healing assay and Transwell Matrigel invasion analysis. Decreased clonogenic mobility of MHCC97 cells in wound healing assay was significantly decreased after transfection of GSTA4. Compared with cells transfected with scrambled siRNA, the cells treated with GSTA4 siRNA showed a wider wound area 48 hours after wound generation, and took a longer time to fill the wound area, indicating a defect in migration (Figure 3A). In contrast, over-expression GSTA4 in HepG2 cells increased wound healing (Figure 3C). Additionally, Matrigel-coated Transwell chambers were used to access the invasive capacities of MHCC97 cancer cells. Consistent with the finding in migration assay, cells treated with GSTA4 siRNA demonstrated significant reduction in cell invasion ability by 70% in MHCC97 cells in comparison with control siRNA-treated cells (Figure 3B). Conversely, in Matrigel invasion assay, up-regulation of GSTA4 markedly increased invasion of HepG2 cells (Figure 3D). Taken together, these results indicate that silencing of GSTA4 decreased the invasive properties of human hepatoma cells.
Figure 3.
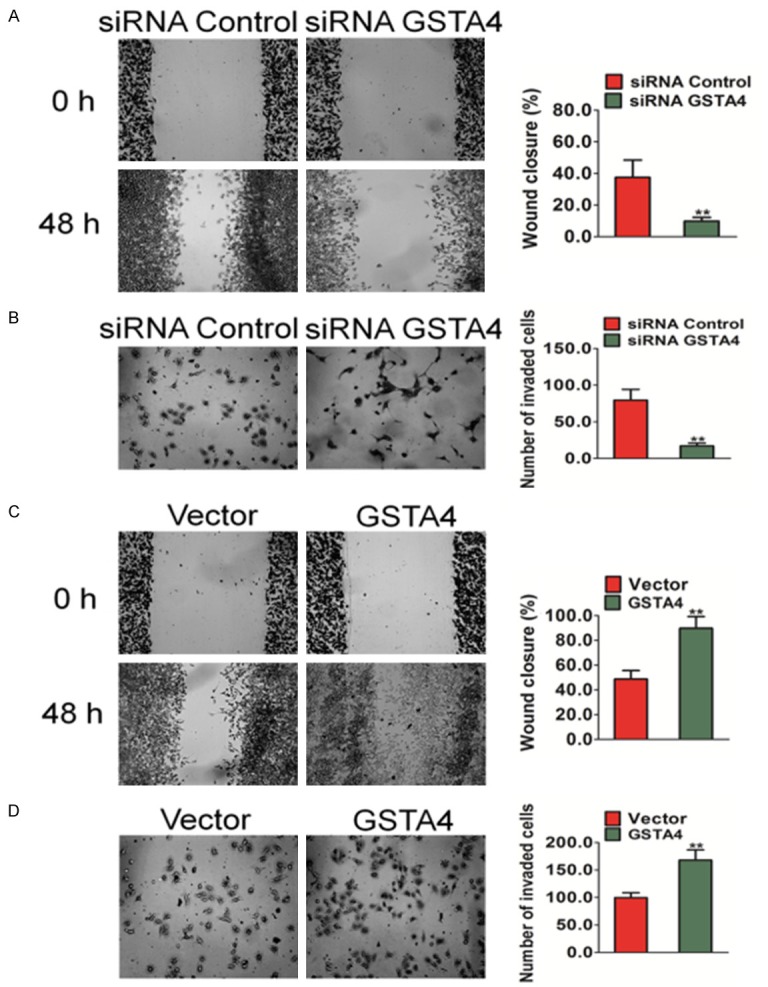
The effects of GSTA4 on migration and invasion of HCC cells in vitro. A. A wound healing assay was used to evaluate the migration of MHCC97 cells after silencing GSTA4. 24 hours after the transfection of siRNA, cells were wounded and monitored with a microscope every 48 hours. The migration was determined by the rate of cells filling the scratched area. B. Cell invasion was assayed in Transwell coated with Matrigel. MHCC97 cells crossed the Matrigel-coated filter were fixed, stained and counted. Representative pictures of the bottom surface were shown. Five random microscopic fields were counted for each group. The results presented were an average of five random microscopic fields from three independent experiments. The data are presented as mean ± SD. For indicated comparison, **P < 0.01 vs. control cells. C. Representative pictures of (left panel) and quantification (right panel) of migration in HepG2 cells transfected with GSTA4 were analyzed using a wound healing assay. 24 hours after the transfection of GSTA4 plasmid, cells were wounded and monitored with a microscope every 48 hours. The migration was determined by the rate of cells filling the scratched area. D. Significant induction of invasion was observed after transfected over-expression GSTA4 in HepG2 cells. The data were presented as mean ± SD. For indicated comparisons, **P < 0.01 vs. control cells.
Effects of GSTA4 on tumor growth of HCC in vivo
We subcutaneously injected MHCC97-siGSTA4 cells in nude mice to induce xenograft tumors and the tumors gradually enlarged over time (Figure 4A). Our results indicated that the growth rates of tumors in nude mice that received transplants of MHCC97-siGSTA4 cells were significantly lower than those in mice transplanted with control cells (Figure 4B). The average tumor volume of MHCC97 cells transfected with GSTA4 siRNA was 2.29 ± 0. 2 cm3, which was significantly lower than tumors in the control group (1.37 ± 0.14 cm3; P < 0.01). Western blot analysis of protein extracted from the tumor tissues showed that the GSTA4 knockdown was maintained in the transplanted tumors in the nude mice (Figure 4C). HepG2 cells stably expressing GSTA4 or control vector were injected subcutaneously into nude mice. Palpable tumors formed within 1 week. Tumor volume was measured each three days, and mice were sacrificed 6 weeks after tumor cell implantation (Figure 4D). The average tumor volume of HepG2 cells stably transfected with GSTA4 was 2.86 ± 0.21 cm3, which was significantly greater than tumors in the control group (1.69 ± 0.15 cm3; P < 0.01) (Figure 4E). Western blot analysis of protein extracted from the tumor tissues showed that the GSTA4 knock-in was maintained in the transplanted tumors in the nude mice (Figure 4F). Our results indicate that the absence of GSTA4 was responsible for the inhibition of transplanted tumor growth in nude mice.
Figure 4.
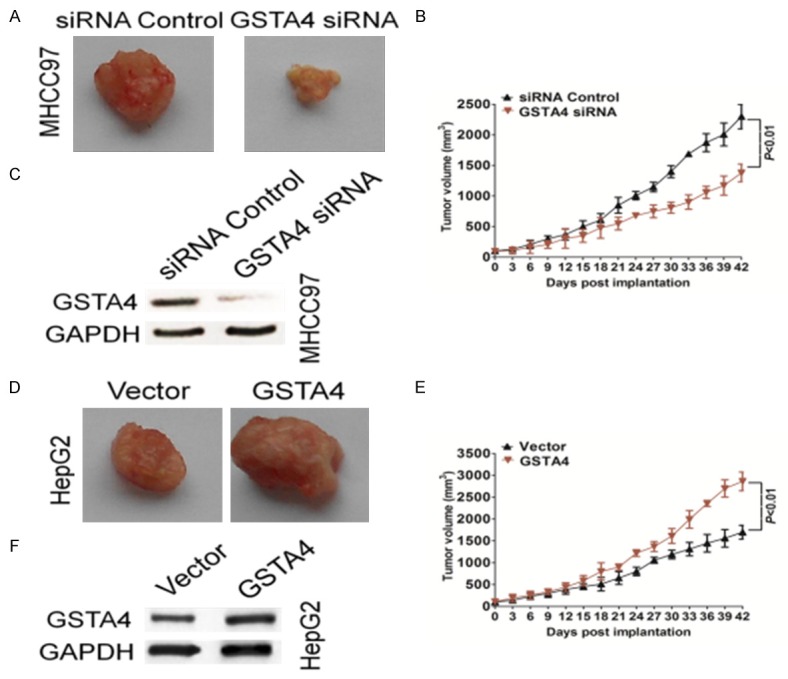
The effects of GSTA4 on in vivo tumor growth of HCC. A. Representative images of tumor in each group. GSTA4 down-expression suppressed tumor growth in subcutaneous implantation mouse models of MHCC97 cells. B. Tumor volumes and tumor growth curves of subcutaneous implantation models of HCC were shown. Data were presented as means ± SD **P < 0.01 indicated significant difference comparing GSTA4 siRNA and control. C. Confirmation that GSTA4 knockdown level was maintained in vivo. Protein samples were subjected from indicated tumor tissues to western blot for measuring GSTA4 and GAPDH. D. Nude mice were subcutaneously injected with HepG2-GSTA4 cells, and the tumors were excised after 6 weeks. Representative image of tumor tissue from nude mice. E. The tumor volumes were recorded every three days for 6 weeks. Data were presented as means ± SD **P < 0.01 indicated GSTA4 vs. vector transfected HepG2 cells (control). F. Confirmation that GSTA4 knock-in level was maintained in vivo. Protein samples were subjected from indicated tumor tissues to western blot for measuring GSTA4 and GAPDH.
Effects of GSTA4 on tumor metastasis of HCC in vivo
To further investigate the role of GSTA4 in the metastasis of hepatoma carcinoma cells in vivo, an experimental metastasis assay was performed. Control and GSTA4 knock down cells were injected into the lateral tail vein of nude mice. Two weeks post inoculation, animals were sacrificed and all the major organs were checked for the generation of tumor metastasis. We found that injection of MHCC97 wild type cells resulted in the formation of numerous lung colonies whereas silencing of GSTA4 significantly suppressed pulmonary metastasis (Figure 5A). To further confirm the role of GSTA4 in the metastasis of melanoma cells in vivo, another experimental metastasis assay was performed. Control and GSTA4 over-expression cells were injected into the lateral tail vein of nude mice. Two weeks post inoculation, animals were sacrificed and we found that injection of HepG2 over-expression cells resulted in the formation of more lung colonies than control cells (Figure 5B). These results implied that GSTA4 indeed perturbed the metastasis of melanoma cells not only in vitro but also in vivo.
Figure 5.
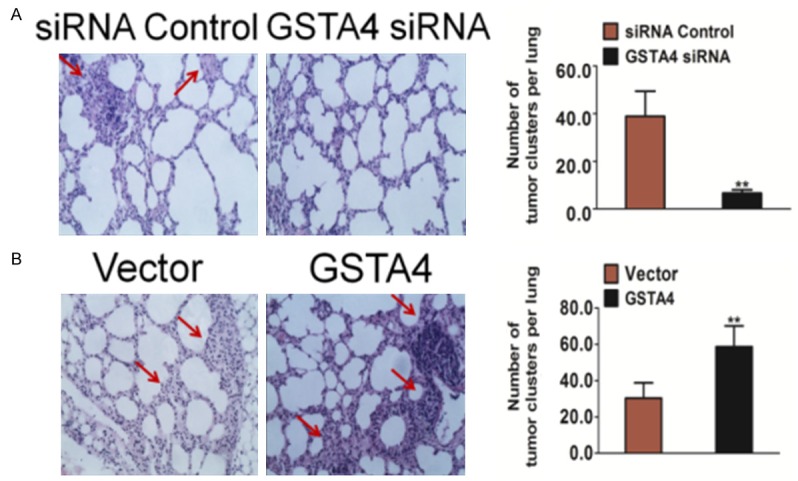
The metastasis of HCC cells is impaired by GSTA4 silencing in vivo. A. Control siRNA and GSTA4 siRNA transfected cells were injected into the lateral vein of mice respectively. Metastatic lesions in the lungs of the metastasis models were shown at two weeks after implantation. The total numbers of lung metastatic lesions in the GSTA4 transfection groups were much less than those in controls. Data were from three independent experiments and were average ± SD. n = 6, **P < 0.01, compared to mice injected with control siRNA cells. B. Control vector transfected and GSTA4 over-expression cells were injected into the lateral vein of nude mice respectively. Metastatic lesions in the lungs from mice injection with control cells or with GSTA4 over-expression cells were shown. The total numbers of lung metastatic lesions in the GSTA4 transfection groups were much less than those in controls. Data were from three independent experiments and were average ± SD. n = 6, **P < 0.01, compared to mice injected with transfected vector cells.
GSTA4 Inhibits the AKT signaling pathway
To determine the signaling pathways which are involved in GSTA4 mediated melanoma cell migration, multiple potential signaling pathways related to migration and invasion of cancer cells were screened. As shown in Figure 6A, only the basal level of AKT activation was found to be significantly up-regulated in HepG2 cells over-expressing GSTA4. In contrast, no obvious difference could be observed for many other signaling pathways, such as Focal Adhesion Kinase (FAK), Src, GSK-3β and mTOR. Consistently, when GSTA4 was silenced by siRNA in melanoma cells, AKT activation was also down regulated in MHCC97 cells (Figure 6B). To confirm the role of AKT in GSTA4-mediated cell migration, constitutively active form of AKT (D2AKT) was introduced into MHCC97 silenced cells. The expression of D2AKT was confirmed by western blot (Figure 6C) and immunofluorescence assay with anti-AKT antibody (Figure 6D). The metastasis of cells was then examined by wound healing assay and Transwell invasion analysis. As expected, active AKT largely restored the impaired migration and invasion in GSTA4-silenced MHCC97 cells (Figure 6E and 6F). Furthermore, GSK690693, an AKT specific inhibitor was also employed to dissect the role of AKT signaling in cell migration. As shown in Figure 7A and 7B, GSTA4 mediated phosphorylation of AKT was completely blocked by GSK690693 in HepG2 cells. As a result, GSTA4 promoted migration and invasion in HepG2 cells were also abolished by GSK690693, as shown by the wound healing migration assay (Figure 7C) and Transwell invasion analysis (Figure 7D). To conclude, these data indicate that AKT signaling was involved in GSTA4 promoted metastasis of HCC cells.
Figure 6.
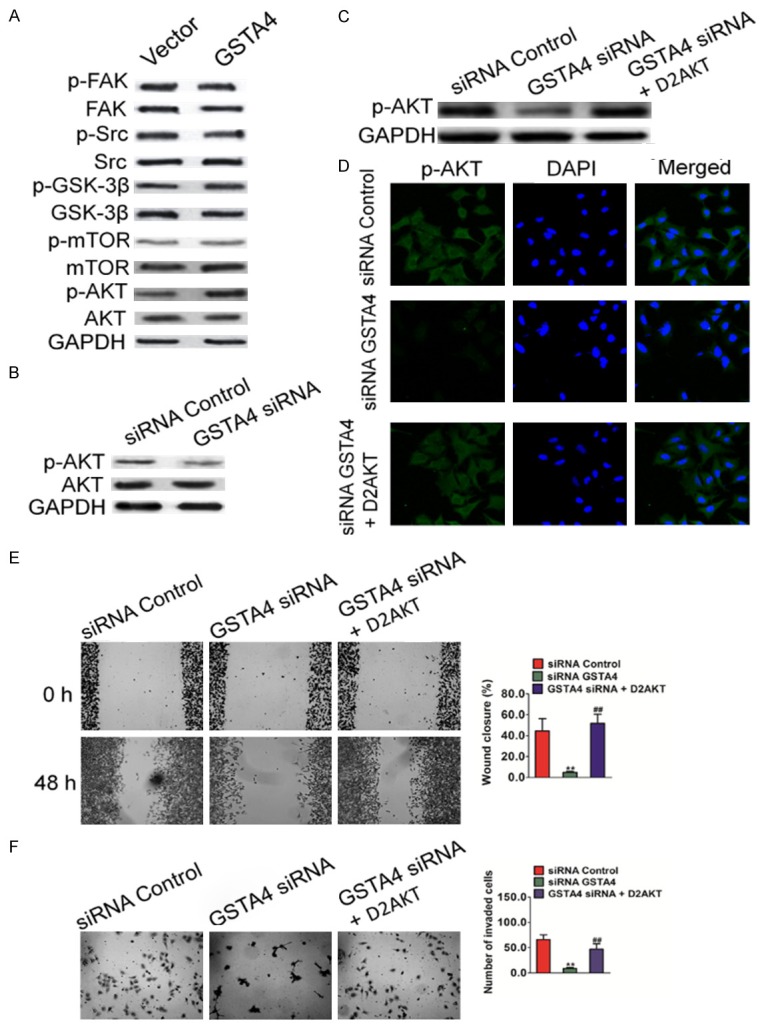
GSTA4 regulates AKT pathway in HCC cells. A. Western blot shown that the phosphorylation of AKT was elevated in MHCC97 cells transfected with GSTA4. B. The phosphorylation of AKT was perturbed in cells transfected with GSTA4 siRNA plasmid. C. Expression of D2AKT was confirmed by western blot with antibody against Flag-tag and AKT, and GAPDH was used as loading control. D. Expression of D2AKT was confirmed by Immunofluorescence assay with antibody against AKT. E. Wound healing assay was performed to determine the motility of cells co-transfected with GSTA4 silencing plasmid and D2AKT plasmid. Columns were data collected from three independent experiments. Representative pictures were taken after staining with crystal violet. Data were from three independent experiments and are average ± SD. **P < 0.01, compared to control siRNA cells; ##P < 0.01, compared to GSTA4 siRNA cells. F. Representative pictures of (left panel) and quantification (right panel) of invasion in MHCC97 cells transfected with GSTA4 siRNA or D2AKT plasmid were analyzed using a Transwell invasion assay. Data were from three independent experiments and are average ± SD. **P < 0.01, compared to control siRNA cells; ##P < 0.01, compared to GSTA4 siRNA cells.
Figure 7.

GSTA4 regulates HCC cells metastasis through AKT pathway. A. In the presence of GSK690693, HepG2 cells were incubated for 1 h, protein extracts were analyzed by western blot with antibodies against phosphorylated AKT (S473) or AKT. B. Expression of AKT was confirmed by Immunofluorescence assay with antibody against AKT. C. In the presence of GSK690693, wound healing migration assay was conducted to evaluate the cell motility after transfection. Data were from three independent experiments and were average ± SD. **P < 0.01, compared to transfected vector cells; ##P < 0.01, compared to transfected GSTA4 cells. D. In the presence of GSK690693, Transwell assay was conducted to evaluate the cell invasion after transfection. Data were from three independent experiments and were average ± SD. **P < 0.01, compared to transfected vector cells; ##P < 0.01, compared to transfected GSTA4 cells.
Discussion
Currently, late diagnosis and high recurrence are the major causes of the poor overall survival of patients with HCC [21]. Therefore, it is necessary to elucidate the molecular mechanisms responsible for HCC progression and to identify efficient therapeutic targets. In previous studies, GSTA4 has been reported to regulate hepatocyte proliferation during liver regeneration, block G1/S transition, and suppress in vivo tumor growth [22]. The molecular mechanisms by which GSTA4 inhibits HCC remain unclear. In the present study, we found that GSTA4 expression was markedly increased in HCC tissues versus adjacent non-tumor liver tissues. For the first time, we illuminate the role of GSTA4 up-regulation in tumor growth and metastasis using in vitro and in vivo assays. We found that GSTA4 down-expression suppressed in vitro cell proliferation and invasion, inhibited cell clonality, and restrained in vivo tumor growth and metastasis. Conversely, up-regulation of GSTA4 promoted proliferation, invasion, and tumor growth as well as metastasis in vivo. These results indicate that GSTA4 might be a novel cancer-promoting gene that plays important roles in the regulation of tumor growth and metastasis of HCC.
GSTs belong to a super-family of detoxification enzymes, which play a role in resisting a large variety of chemical carcinogens and environmental toxicants [23]. In murine chemically-induced carcinogenesis models and human cancer, GST genes are frequently over-expressed [24]. GSTA4, which metabolizes a reactive lipid peroxidation by-product associated with tumor promotion, was increased in the epidermis of TPA-treated C57BL/6 mice relative to DBA/2 mice, suggesting that polymorphisms that affect GSTA4 transcript abundance may modify susceptibility to skin tumor promotion [25]. The data from the mouse skin carcinogenesis model indicate that GSTA4 modifies susceptibility to skin tumor promotion. Although the precise role of GSTA4 in hepatocellular carcinoma oncogenesis is unknown, previous studies have shown that in the liver, GSTA4 has been proposed to protect against hepatitis B virus-related injury, which is partly manifested as extensive oxidative DNA damage [26]. Herein, we demonstrated that GSTA4 expression was significantly increased in HCC tissues compared with normal tissues. In addition, expression level of GSTA4 was positively associated metastasis in HCC and was over-expressed in highly metastatic HCC cell lines. In vitro assay, hepatocellular carcinoma cells MHCC97 transfected with GSTA4 siRNA showed dramatically low colony formation ability in soft agar compared with cells transfected with the control siRNA. Consistent with these observations, we found that silencing of GSTA4 with siRNA led to significant reduction in proliferation in MHCC97 cells. Meanwhile, we conducted experiment including the would-healing assay and invasion assay to assess the effect of GSTA4 siRNA on invasiveness properties of MHCC97 cells. The invasiveness properties were significantly inhibited in cells treated with the GSTA4 siRNA in comparison with cells treated with the scrambled siRNA. In agreement with these findings, we observed a significant anti-growth effect of GSTA4 siRNA in hepatocellular carcinoma cells in vivo. Overexpression of GSTA4 augmented the anchorage-independent growth and the invasive abilities of hepatocellular carcinoma cells HepG2, provoked their ability to growth and metastasis in vivo.
AKT promotes tumor growth and invasion in many types of human cancer, and high AKT levels are associated with the development of HCC [27]. AKT stimulated mTOR phosphorylation and promoted cell survival. Further, inhibition of AKT signaling has been shown to anti-cancer activity in human liver cancer cells. Activation of AKT induces transcription of target genes, including Bcl-2, Mcl-1, cyclin D1, and MMPs In contrast; blocking AKT activity significantly suppresses MMPs expression, tumor invasion, and brain and lung metastasis [28]. MMPs are a family of enzymes that proteolytically degrade various components of the extracellular matrix. High levels of certain MMPs are closely correlated with the invasive and metastatic potential of tumors [29]. In this study, we confirmed that the activation of AKT was reduced in MHCC97 cells in which GSTA4 was knocked down whereas up-regulation of GSTA4 significantly increased AKT levels in HCC HepG2 cells. More importantly, the effects of GSTA4 modulation on cell metastasis of HCC cells were accompanied by changes in AKT activities. Therefore, down-regulation of AKT is one important mechanism by which GSTA4 promotes HCC invasion and metastasis.
The fundamental role of GSTA4 in tumor cell proliferation, invasion, and metastasis suggests its potential application as a potential cancer therapeutic target. In conclusion, our results show that GSTA4 is up-regulated in HCC, and related to tumor metastasis. Using in vitro and in vivo studies, GSTA4 was confirmed as a novel inhibitor of tumor growth, invasion, and metastasis of HCC. The multiple tumor-promote functions of GSTA4 were mediated by the AKT pathway. These data suggest that GSTA4 up-regulation may play an important role in tumor growth and metastasis and may be a novel metastasis marker and potential therapeutic target in HCC.
Acknowledgements
Key research project of hunan province department of education (2014A092).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Baltruskeviciene E, Kazbariene B, Badaras R, Bagdonaite L, Krikstaponiene A, Zdanavicius L, Aleknavicius E, Didziapetriene J. Glutathione and glutathione S-transferase levels in patients with liver metastases of colorectal cancer and other hepatic disorders. Turk J Gastroenterol. 2016;27:336–341. doi: 10.5152/tjg.2016.15457. [DOI] [PubMed] [Google Scholar]
- 2.Zmorzynski S, Swiderska-Kolacz G, Koczkodaj D, Filip AA. Significance of polymorphisms and expression of enzyme-encoding genes related to glutathione in hematopoietic cancers and solid tumors. Biomed Res Int. 2015;2015:853573. doi: 10.1155/2015/853573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng Q, He B, Pan Y, Sun H, Liu X, Chen J, Ying H, Lin K, Peng H, Wang S. Polymorphisms of GSTA1 contribute to elevated cancer risk: evidence from 15 studies. J BUON. 2015;20:287–295. [PubMed] [Google Scholar]
- 4.Parsons JK, Nelson CP, Gage WR, Nelson WG, Kensler TW, De Marzo AM. GSTA1 expression in normal, preneoplastic, and neoplastic human prostate tissue. Prostate. 2001;49:30–37. doi: 10.1002/pros.1115. [DOI] [PubMed] [Google Scholar]
- 5.Savic-Radojevic A, Djukic T, Simic T, Pljesa-Ercegovac M, Dragicevic D, Pekmezovic T, Cekerevac M, Santric V, Matic M. GSTM1-null and GSTA1-low activity genotypes are associated with enhanced oxidative damage in bladder cancer. Redox Rep. 2013;18:1–7. doi: 10.1179/1351000212Y.0000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kassogue Y, Quachouh M, Dehbi H, Quessar A, Benchekroun S, Nadifi S. Effect of interaction of glutathione S-transferases (T1 and M1) on the hematologic and cytogenetic responses in chronic myeloid leukemia patients treated with imatinib. Med Oncol. 2014;31:47. doi: 10.1007/s12032-014-0047-z. [DOI] [PubMed] [Google Scholar]
- 7.Chow AK, Yau TC, Ng L, Chu AC, Law WL, Poon RT, Pang RW. A preclinical study on the combination therapy of everolimus and transarterial chemoembolization in hepatocellular carcinoma. Am J Cancer Res. 2015;5:2376–2386. [PMC free article] [PubMed] [Google Scholar]
- 8.Qu K, Liu SS, Wang ZX, Huang ZC, Liu SN, Chang HL, Xu XS, Lin T, Dong YF, Liu C. Polymorphisms of glutathione S-transferase genes and survival of resected hepatocellular carcinoma patients. World J Gastroenterol. 2015;21:4310–4322. doi: 10.3748/wjg.v21.i14.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li CG, Zhao ZM, Hu MG, Liu R. Predictive role of glutathione-S-transferase gene polymorphisms in risk and prognosis of hepatocellular carcinoma. Asian Pac J Cancer Prev. 2012;13:3247–3252. doi: 10.7314/apjcp.2012.13.7.3247. [DOI] [PubMed] [Google Scholar]
- 10.Ates NA, Tamer L, Ates C, Ercan B, Elipek T, Ocal K, Camdeviren H. Glutathione S-transferase M1, T1, P1 genotypes and risk for development of colorectal cancer. Biochem Genet. 2005;43:149–163. doi: 10.1007/s10528-005-1508-z. [DOI] [PubMed] [Google Scholar]
- 11.Song K, Yi J, Shen X, Cai Y. Genetic polymorphisms of glutathione S-transferase genes GSTM1, GSTT1 and risk of hepatocellular carcinoma. PLoS One. 2012;7:e48924. doi: 10.1371/journal.pone.0048924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ortega CE, Seidner Y, Dominguez I. Mining CK2 in cancer. PLoS One. 2014;9:e115609. doi: 10.1371/journal.pone.0115609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Lan H, Li J, Su Y, Xu L. Muc1 promotes migration and lung metastasis of melanoma cells. Am J Cancer Res. 2015;5:2590–2604. [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JK, Kim JY, Kim HJ, Park KG, Harris RA, Cho WJ, Lee JT, Lee IK. Scoparone exerts anti-tumor activity against DU145 prostate cancer cells via inhibition of STAT3 activity. PLoS One. 2013;8:e80391. doi: 10.1371/journal.pone.0080391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin B, Cheng K. Silencing of the IKKepsilon gene by siRNA inhibits invasiveness and growth of breast cancer cells. Breast Cancer Res. 2010;12:R74. doi: 10.1186/bcr2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan G, Yan S, Xue H, Zhang P, Sun J, Li G. JSI-124 suppresses invasion and angiogenesis of glioblastoma cells in vitro. PLoS One. 2015;10:e0118894. doi: 10.1371/journal.pone.0118894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pass HI, Lavilla C, Canino C, Goparaju C, Preiss J, Noreen S, Blandino G, Cioce M. Inhibition of the colony-stimulating-factor-1 receptor affects the resistance of lung cancer cells to cisplatin. Oncotarget. 2016;7:56408–56421. doi: 10.18632/oncotarget.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramani R, Lopez-Valdez R, Arumugam A, Nandy S, Boopalan T, Lakshmanaswamy R. Targeting insulin-like growth factor 1 receptor inhibits pancreatic cancer growth and metastasis. PLoS One. 2014;9:e97016. doi: 10.1371/journal.pone.0097016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang G, Tao L, Shen S, Chen L. Hypoxia induced CCL28 promotes angiogenesis in lung adenocarcinoma by targeting CCR3 on endothelial cells. Sci Rep. 2016;6:27152. doi: 10.1038/srep27152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, Liang L, Zhang XF, Jia HL, Qin Y, Zhu XC, Gao XM, Qiao P, Zheng Y, Sheng YY, Wei JW, Zhou HJ, Ren N, Ye QH, Dong QZ, Qin LX. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology. 2013;58:158–170. doi: 10.1002/hep.26305. [DOI] [PubMed] [Google Scholar]
- 21.Arslanoglu A, Seyal AR, Sodagari F, Sahin A, Miller FH, Salem R, Yaghmai V. Current guidelines for the diagnosis and management of hepatocellular carcinoma: a comparative review. AJR Am J Roentgenol. 2016:W1–W11. doi: 10.2214/AJR.15.15490. [DOI] [PubMed] [Google Scholar]
- 22.Mavis CK, Morey Kinney SR, Foster BA, Karpf AR. Expression level and DNA methylation status of glutathione-S-transferase genes in normal murine prostate and TRAMP tumors. Prostate. 2009;69:1312–1324. doi: 10.1002/pros.20976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raza H, John A. Implications of altered glutathione metabolism in aspirin-induced oxidative stress and mitochondrial dysfunction in HepG2 cells. PLoS One. 2012;7:e36325. doi: 10.1371/journal.pone.0036325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Townsend D, Tew K. Cancer drugs, genetic variation and the glutathione-S-transferase gene family. Am J Pharmacogenomics. 2003;3:157–172. doi: 10.2165/00129785-200303030-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riggs PK, Angel JM, Abel EL, DiGiovanni J. Differential gene expression in epidermis of mice sensitive and resistant to phorbol ester skin tumor promotion. Mol Carcinog. 2005;44:122–136. doi: 10.1002/mc.20127. [DOI] [PubMed] [Google Scholar]
- 26.Smith MW, Yue ZN, Geiss GK, Sadovnikova NY, Carter VS, Boix L, Lazaro CA, Rosenberg GB, Bumgarner RE, Fausto N, Bruix J, Katze MG. Identification of novel tumor markers in hepatitis C virus-associated hepatocellular carcinoma. Cancer Res. 2003;63:859–864. [PubMed] [Google Scholar]
- 27.Cidado J, Park BH. Targeting the PI3K/Akt/mTOR pathway for breast cancer therapy. J Mammary Gland Biol Neoplasia. 2012;17:205–216. doi: 10.1007/s10911-012-9264-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Y, Wang H, Mills GB. Targeting PI3K-AKT pathway for cancer therapy. Rev Clin Exp Hematol. 2003;7:205–228. [PubMed] [Google Scholar]
- 29.Davidson B, Reich R, Risberg B, Nesland JM. The biological role and regulation of matrix metalloproteinases (MMP) in cancer. Arkh Patol. 2002;64:47–53. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.