

Abstract
Epidemiological studies have revealed the association between increased risk of bladder cancer and chronic arsenic exposure. Here, we explored biological effects of arsenic in T24. Microarray analysis was applied to analyze mRNA in T24 following 0, 2 or 5 μM sodium arsenite (As) exposure for 72 hours. Long term (up to 140 days) low-dose (200 nM) and high-dose (1,000 nM) As decreased E-cadherin protein level through different mechanisms because the mRNA levels of E-cadherin increased following low-dose As exposure but decreased following high-dose As exposure. Long term As increased the protein levels of N-cadherin, vimentin, β-catenin, and slug. Low-dose As exposure resulted in a change in the morphology of T24 cells from an epithelial to a mesenchymal-like appearance. Knockdown of E-cadherin increased the protein levels of N-cadherin, vimentin, β-catenin, and slug. Cell proliferation and growth of T24 with or without As exposure for 100 days were assayed using EdU and WST, respectively. Low-dose As exposure increased cell proliferation and growth while high-dose As exposure decreased both. Long term As activated p53 on account of increasing protein levels of p53, p-p53 (Ser15), and mRNA levels of p21. These demonstrate that arsenic exposure exerts multiple effects. Long term low- or high-dose arsenic induces epithelial-mesenchymal transition, likely via downregulation of E-cadherin, activates p53, and differently affects cell proliferation/growth.
Keywords: Bladder cancer, arsenic, E-cadherin, epithelial-mesenchymal transition, proliferation
Introduction
Arsenic, a chemical element classified as a metalloid, is widely distributed in nature, occurring in various compounds in the crust of the earth [1,2]. Arsenic toxicity in humans has been reported worldwide [1,3,4]. Significant arsenic exposure mostly occurs through the ingestion of arsenic contaminated water and food [1,5]. In countries, such as the USA and Chile, arsenic contaminated water has been consumed for more than 50 years [1,3,4]. Inhalation of airborne arsenic or arsenic-contaminated dust is the main occupational source of arsenic exposure [1].
In recent decades, epidemiological studies have revealed the association between arsenic and increased risk of various diseases, such as skin lesions (e.g., melanosis, leucomelanosis, and keratosis), neuropathy, obstetric problems, high blood pressure, diabetes mellitus, diseases of the respiratory system and of blood vessels, and cancers [1,3-5]. As a result, arsenic is considered one of the most significant environmental hazards [1,3-5].
Although arsenic elicits both acute and chronic toxic effects, chronic toxicity is most commonly observed and millions of people worldwide suffer from chronic arsenic poisoning [1,3,5]. Chronic arsenic exposure has been strongly associated with cancer of the bladder, prostate, lung, liver and skin [1,3-5]. The carcinogenicity of arsenic strongly depends on dose and duration of exposure [1,3-5]. The latency period of arsenic-related carcinogenesis in humans is considered to be 30-50 years [1]. Epidemiological studies have highlighted biological effects of long term arsenic exposure, and it has been shown that arsenic induces carcinogenesis through multiple mechanisms. However, the detailed biological effects of long term arsenic exposure remain unclear [1,2,6].
In this study, microarray technology was applied to identify differentially expressed mRNAs in the human transitional cell carcinoma cell line T24 following treatment with sodium arsenite (As). The biological effects of long term (up to 140 days) low-dose (200 nM) and high-dose (1,000 nM) As exposure in the T24 cell line were explored.
Materials and methods
Cell lines and cell culture
Human transitional cell carcinoma (bladder cancer) cell line, T24 and human embryonic kidney 293T were obtained from the Cell Bank, Chinese Academy of Science.
T24 was routinely grown and passaged as previously described [7]. In brief, cells were grown in McCoy’s 5A (Gibco, Grand Island, NY, USA, Cat: 16600108) supplemented with 100 ml/L fetal bovine serum (Gibco, Cat: 10438018), 100,000 IU/L penicillin, and 100 mg/L streptomycin (Gibco, Cat: 15140122) in the absence or presence of sodium arsenite (Sigma-Aldrich, St Louis, MO, USA, Cat: 38150) under a humidified atmosphere of 5% CO2 at 37°C.
293T was grown in DMEM (Gibco, Cat: 12100-046) supplemented with 100 ml/L fetal bovine serum (Gibco), 100,000 IU/L penicillin, and 100 mg/L streptomycin (Gibco) under a humidified atmosphere of 5% CO2 at 37°C [7].
Microarray analysis
T24 was cultured with or without sodium arsenite (2 μM, 5 μM) for 72 hours. Total RNA for microarray assay was extracted using TRIzol Reagent (Life Technologies, California, USA, Cat: 15596026) according to the manufacturer’s manual [8]. Gene Ontology (GO) analysis and Pathway analysis were performed by Gminix (Shanghai, PR China) [9].
GO analysis was applied to analyze the main function of the differential expression genes according to the GO which is the key functional classification of NCBI, which can organize genes into hierarchical categories and uncover the gene regulatory network on the basis of biological process and molecular function [9].
Specifically, two-side Fisher’s exact test and χ 2 test were used to classify the GO category, and the false discovery rate (FDR) was calculated to correct the P-value, the smaller the FDR, the small the error in judging the p-value. The FDR was defined as FDR = 1 - Nk/T (1), where Nk refers to the number of Fisher’s test P-values less than χ 2 test P-values. We computed P-values for the GOs of all the differential genes. Enrichment provides a measure of the significance of the function: as the enrichment increases, the corresponding function is more specific, which helps us to find those GOs with more concrete function description in the experiment. Within the significant category, the enrichment Re was given by: Re = (nf/n)/(Nf/N) (2), where “nf” is the number of flagged genes within the particular category, “n” is the total number of genes within the same category, “Nf” is the number of flagged genes in the entire microarray, and “N” is the total number of genes in the microarray [9].
Pathway analysis was used to find out the significant pathway of the differential genes according to KEGG, Biocarta and Reatome. Still, we turned to the Fisher’s exact test and χ 2 tested to select the significant pathway, and the threshold of significance was defined by P-value and FDR. The enrichment Re was calculated like the equation above [9].
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using TRIzol Reagent (Life Technologies) [8]. RNA was quantified through UV spectroscopy [8]. cDNA was synthesized using 2 μg of total RNA using the Fastquant RT Kit (Tiangen Biotech., Beijing, China, Cat: KR106-02) for mRNA analysis [8,10]. RT-PCR was performed with SuperReal PreMix (SYBR Green) (Tiangen Biotech., Cat: FP204-03) [8,10]. The primers used for RT-PCR are listed in Table 1. β-actin was used as an internal standard.
Table 1.
Primers for RT-PCR
| β-actin | Forward | CATGTACGTTGCTATCCAGGC |
| Reverse | CTCCTTAATGTCACGCACGAT | |
| ITGB3 | Forward | CTGTACCACGCGAGGTGTG |
| Reverse | CTCCTTCAGGTCACAGCGAG | |
| Notch1 | Forward | CGACGCACAAGGTGTCTTC |
| Reverse | AGGATCAGTGGCGTCGTG | |
| PRKCA | Forward | TTGTTACTTTTTCTTGTCCGGG |
| Reverse | GACCCACAGTGATCGCAGA | |
| NFATC2 | Forward | ACGAGCTTGACTTCTCCATCC |
| Reverse | GAGGCTGGTTCGAGGTGAC | |
| ZNF333 | Forward | AAACCCGAAGAGTTGCCTTC |
| Reverse | TCTTCTATGGAGGGCACCAG | |
| CDH1 | Forward | TCCTACACTGCCCAGGAGC |
| Reverse | AGTGGAAATGGCACCAGTGT | |
| p21 | Forward | TGGAGACTCTCAGGGTCGAA |
| Reverse | GGATTAGGGCTTCCTCTTGG | |
| ING4 | Forward | GAAGAAAGCTGCTCGTGCTC |
| Reverse | CCACACAGGCAAAATGGAA |
Lentiviral delivery of shRNA
E-cadherin was knocked down through the use of lentiviral vector-mediated shRNA interference using The RNAi Consortium System (Open Biosystems, Inc., Huntsville, AL, USA) according to the manual [7,11]. Sense sequence of shRNA targeting E-cadherin is AAGATAGGAGTTCTCTGATGC [7,11]. Control shRNA (Scrambled) is targeted against GFP and the sense sequence of shRNA is TACAACAGCCACAACGTCTAT. E-cadherin-targeting shRNA-pLKO.1 vector or a control shRNA-pLKO.1 vector with pCMV-Dr8.91 (the packaging plasmid) and pCMV-VSV-G (the enveloping plasmid) were co-transfected into 293T cells with Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA, Cat: 11668-030) according to the manual [7,11]. Virus-containing media was collected at 24, 48 and 72 hours posttransfection, and was filtered through microfilters (Millipore, Bedford, MA, USA; 0.22 μm, Cat: SLGP033RB). T24 cells were infected with lentivirus encoding shRNA targeting E-cadherin or control lentivirus, respectively. Then, cells were selected using puromycin (Sigma-Aldrich, St Louis, MO, USA, Cat: P8833). Knockdown efficiency was confirmed by western blot.
Western blot analysis
Cells were lysed in RIPA Buffer (50 mM Tris base, 150 mM NaCl, 1% Nonidet P-40, 0.25% Na-deoxycholate, 1 mM EDTA) with protease inhibitors and phosphatase inhibitors (1 mM PMSF, 5 μg/ml leupeptin, 2 μg/ml pepstatin, 4 μg/ml aprotinin, 10 mM NaF, 1 mM Na3VO4, 10 mM β-Glycerophosphate disodium salt pentahydrate) by incubating for 30 minutes on ice. Following centrifugation (26,000 g, 4°C, 16 minutes), the supernatant was collected as total cell protein [7,11,12].
Protein was resolved by SDS/PAGE and blotted on Nitrocellulose Membranes (Bio-Rad, Richmond, California, USA, Cat: 162-0115) as previously described [12]. Nitrocellulose membranes were incubated with specific primary antibodies at 4°C overnight. After incubating with secondary antibodies for 1 hour at room temperature, immunoreactive proteins were visualized by the Enhanced Chemiluminescnet Substrate (Thermo Scientific, Pittsburgh, PA, USA, Cat: 34094) [12].
E-cadherin antibody was from Abcam Inc. (Cambridge, MA, USA, Cat: ab1416, 1:1,000). β-catenin antibody (Cat: 9582, 1:1,000), vimentin (Cat: 9782, 1:1,000), slug (Cat: 9782, 1:1,000), TCF8/ZEB1 (Cat: 9782, 1:1,000), snail (Cat: , 1:1,000), ZO-1 (Cat: 9782, 1:1,000), N-cadherin (Cat: 9782, 1:1,000), p53 (Cat: 2524, 1:1,000), phospho-p53 (Ser15) (p-p53) (Cat: 9947, 1:1,000), HRP-linked secondary antibody were from Cell Signaling Technology. c-myc (Cat: sc-40, 1:500) and c/EBP β(Cat: sc-7962, 1:500)were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). GAPDH (Cat: KC-5G4, 1:500) was from Kangchen Biotech. (China).
Cell proliferation assay
Cell proliferation was analyzed with the Cell-Light™ EdU Apollo®567 In Vitro Imaging Kit (RiboBio Co. Ltd., Guangzhou, China, Cat: c10316-1) according to the manufacturer’s manual [13]. The proliferative cells were stained with EDU (red) and the nuclei were stained with Hoechst 33342 (blue). In brief, T24 cells with or without As (200 nM or 1,000 nM) treatment for 100 days were inoculated in 96-well plates at a density of 2000 cells/well. 24 hours later, cells were incubated with 100 μL 50 μM EDU for 2 hours. Then, cells were fixed with 4% formaldehyde in PBS, incubated with 2 mg/ml glycin for 5 minutes to counteract formaldehyde, permeabilized with 0.5% Triton X-100 for 10 minutes, incubated with Apollo reaction buffer for 30 minutes, permeabilized with 0.5% Triton X-100 for 10 minutes twice, incubated with Hoechst 33342 for 30 minutes. Cells were observed using the fluorescence microscopy (Olympus, Tokyo, Japan).
Cell growth assay
Cell growth was determined by water-soluble tetrazolium salt-based assay (WST assay) using the cell counting kit 8 (Dojindo Laboratories, Kumamoto, Japan, Cat: CK04-11) according to the manual as previously described [7,11]. In brief, T24 cells with or without As (200 nM or 1,000 nM) treatment for 100 days were inoculated in 96-well plates at a density of 2000 cells/well. The plates were incubated for 4 hours to let cells attach the bottom. 0, 24, 36, 48 hours after cell attachment, 10 μL of WST solution were added to each well and the plates were incubated for 30 minutes. Absorbance was measured at 450 nm using a microplate reader with reference wavelength of 650 nm. The absorbance (OD) values were normalized to time point 0, respectively.
Statistical analyses
The data shown represent the mean ± standard error. Statistical differences between groups were analyzed by one-way ANOVA. P<0.05 was considered statistically significant.
Results
Arsenic treatment affects genes expression
In order to elucidate the effects of arsenic exposure in bladder cancer, the human transitional cell carcinoma cell line, T24, was treated with 0, 2 or 5 μM As for 72 hours. Microarray analysis was applied to analyze mRNA. The GO results indicated that cell adhesion, including cell-cell adhesion and cell-substrate adhesion, was significantly affected. The data imply that multiple mechanisms were involved because junction assembly, junction organization, cytoskeleton organization were affected (Figure 1A, 1B). Cell proliferation was also significantly affected, and both positive and negative regulation of cell proliferation were involved (Figure 1A, 1B).
Figure 1.

Microarray analysis of gene expression in T24 cells following As exposure (0, 2 or 5 μM) for 72 hours. Significantly enriched GOs (A) and enrichment of GOs (B) show As exposure (2 μM and 5 μM) exerted multiple biological effects in T24 cells, including on cell adhesion and proliferation. (C) Pathways corresponding to regulated transcripts, pathways related to cell adhesion and cancer were significantly affected.
Pathway analysis data showed that pathways related to cell adhesion, such as cell adhesion molecules, focal adhesion, were significantly affected (Figure 1C). The prominent effects were related to cancer. Transcriptional dysregulation implicated in cancer, proteoglycans known to play a role in cancer, and several prominent pathways involved in carcinogenesis, such as the Hippo signaling pathway [14] and MAPK signaling pathway [15], were found to be significantly affected (Figure 1A, 1B). The data indicate that short term (72 hours) As (2 μM and 5 μM) exposure exerts multiple effects in the T24 cell line.
The latency period of arsenic-related carcinogenesis in humans is considered to be 30-50 years; however, the mechanisms involved remain elusive [1]. Therefore, effects of long term arsenic exposure were explored. T24 cells were treated with low-dose (200 nM) or high-dose (1,000 nM) As for gradient times (0, 3, 14, 35, 86 or 140 days). mRNA levels of genes were measured using qRT-PCR (Figure 2A-E). ITGB3 (a cell adhesion molecule strongly associated with cancer [16]), Notch1 (a single-pass transmembrane receptor strongly associated with carcinogenesis [17]), PRKCA (a serine- and threonine-specific protein kinase strongly associated with carcinogenesis [18]), NFATC2 (a nuclear factor strongly associated with cancer [19]) and ZNF333 (a member of the zinc finger gene complex, which regulated transcription [20]) were all found to be regulated by As exposure. Protein levels of cells treated with low-dose As at gradient times (0, 3, 14, 35, 86 or 140 days) was measured using western blot (Figure 2F). c-Myc and c/EBP β (two transcription factors strongly associated with carcinogenesis [21,22]) were affected by long term low-dose As exposure. These findings demonstrate that long term arsenic exposure exerts multiple carcinogenic effects in the T24 cell line.
Figure 2.
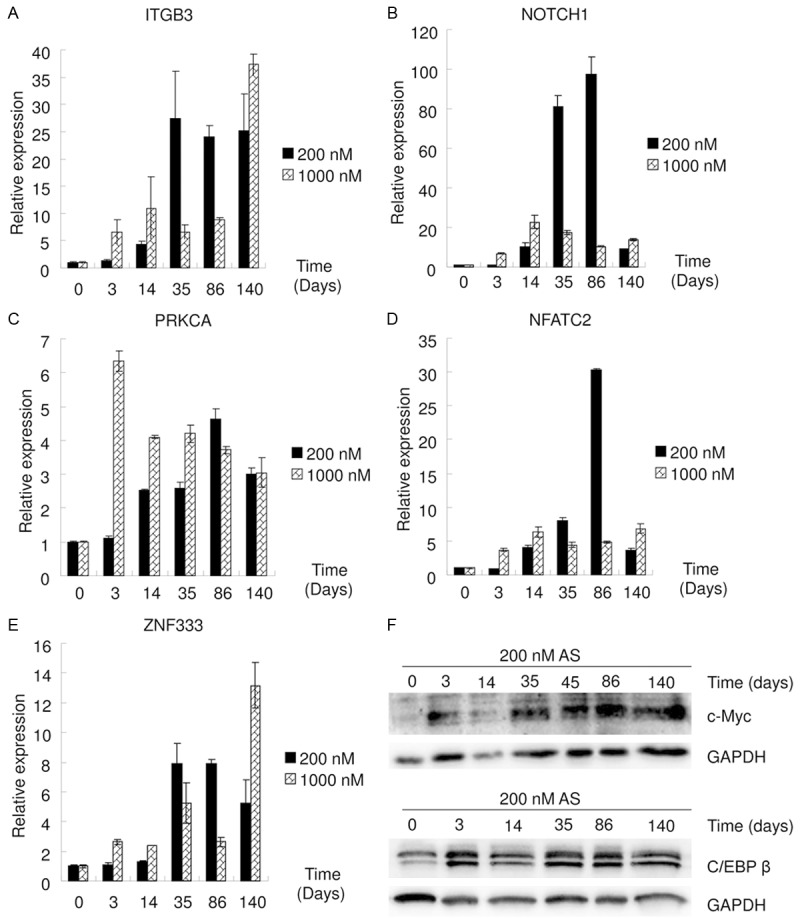
Multiple effects of long term As exposure. A-E. qRT-PCR data show that low-dose (200 nM) or high-dose (1,000 nM) As exposure at gradient times affected mRNA levels of ITGB3, Notch1, PRKCA, NFATC2 and ZNF333. F. Western blot data show that low-dose As exposure at gradient times affected the protein levels of c-Myc and c/EBP β.
Long term low- and high-dose arsenic exposure induces epithelial-mesenchymal transition
In the present study, microarray data showed that As exposure significantly affected cell adhesion molecules and cell-cell adhesion (Figure 1). E-cadherin, one of the most important cell adhesion molecules, plays a key role in cell-cell adhesion [7,11]. E-cadherin mRNAs levels in the T24 cells treated with As for 0, 3, 14, 35, 86, or 140 days were measured using qRT-PCR (Figure 3A). E-cadherin mRNA level increased following long term low-dose (200 nM) As exposure, but decreased following long term high-dose (1,000 nM) As exposure. E-cadherin protein levels in the T24 cells treated with As for 0, 3 or 140 days were assayed using western blot (Figure 3B). E-cadherin protein levels were found to decrease following As exposure, regardless of whether the dose was low or high. These findings demonstrate that long term low- and high-dose arsenic exposure decreases E-cadherin protein levels, but through different mechanisms.
Figure 3.
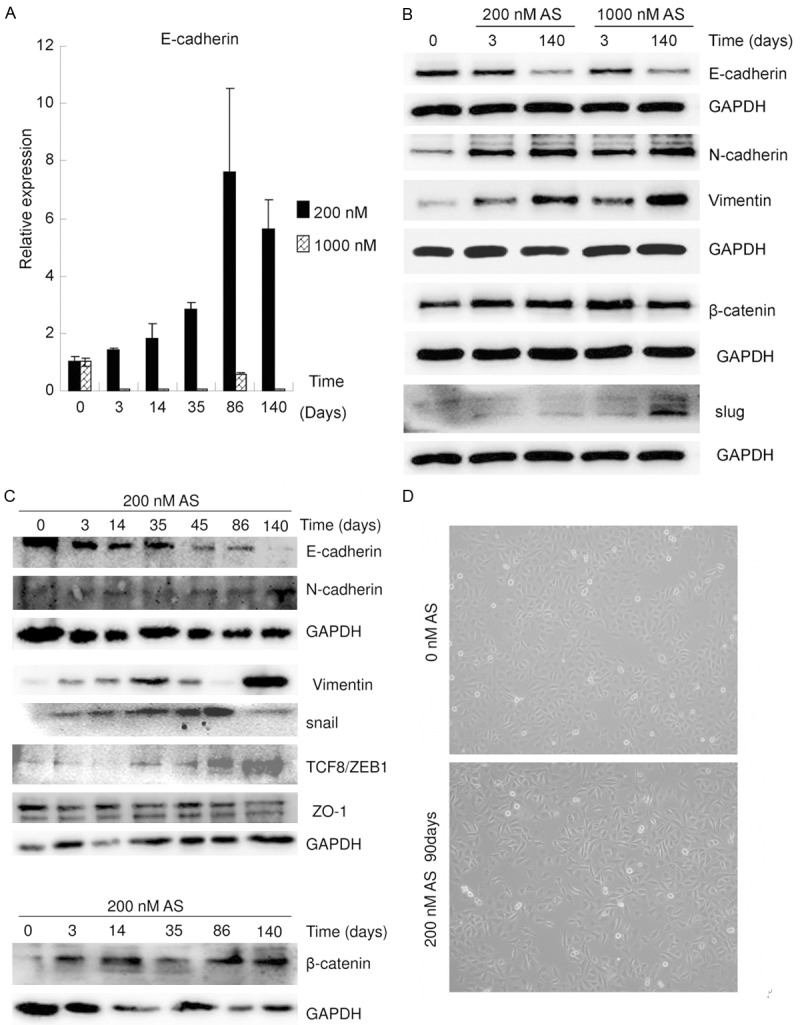
Long term low- and high-dose As exposure induced EMT. A. qRT-PCR data show that the mRNA levels of E-cadherin increased following long term low-dose As exposure but decreased following high-dose As exposure. B. Western blot data show that long term As exposure led to the loss of epithelial identity (decrease in E-cadherin expression) and gain of mesenchymal identity (increased N-cadherin, vimentin, β-catenin, and slug expression) in a time dependent manner. C. Western blot data showed that low-dose As exposure decreased the levels of E-cadherin and increased those of N-cadherin, vimentin, β-catenin, and slug in a time dependent manner. D. The morphology of T24 cells, with or without exposure to 200 nM As for 90 days, was examined.
E-cadherin is a key marker of epithelial-mesenchymal transition (EMT) [23]. A decline in the expression of E-cadherin implies that EMT might be intrinsically orchestrated [23]. Therefore, the protein levels of other EMT markers were assayed in the T24 cells treated with As for 0, 3 or 140 days, using western blot (Figure 3B). The levels of N-cadherin, vimentin, β-catenin and slug were all increased in a time dependent manner following As exposure, regardless of whether the dose was low or high. These data indicate that long term low- and high-dose arsenic exposure induces EMT. Protein levels of EMT markers in T24 cells treated with low-dose As for 0, 3, 14, 35, 86 or 140 days were assayed using western blot (Figure 3C). E-cadherin levels were found to decrease in a time dependent manner, while those of N-cadherin, vimentin, β-catenin and slug were all increased in a time dependent manner. The morphology of T24 cells, with or without exposure to 200 nM As for 90 days, was examined (Figure 3D). Exposure to 200 nM As for 90 days resulted in the loss of cell-cell adhesion, acquisition of a fusiform shape, and an increase in the number of heteromorphic cells. These results demonstrate that long term low- or high-dose arsenic exposure induces EMT in a time dependent manner.
E-cadherin plays a key role in EMT in specific cell types [23]; therefore, the role of E-cadherin in the mechanism underlying EMT in T24 cells was explored. E-cadherin was knocked down by lentiviral delivery of shRNA and knockdown efficiency was confirmed using western blot (Figure 4) [7,11]. Delivery of scrambled shRNA did not elicit a detectable change in the protein levels of E-cadherin, N-cadherin, vimentin, β-catenin, or slug. The delivery of shRNA targeting E-cadherin efficiently decreased E-cadherin levels, and knockdown of E-cadherin increased the levels of N-cadherin, vimentin, β-catenin, and slug (Figure 4). These findings demonstrate that E-cadherin plays a key role in EMT in T24 cells and suggest that arsenic exposure may induce EMT via downregulation of E-cadherin.
Figure 4.
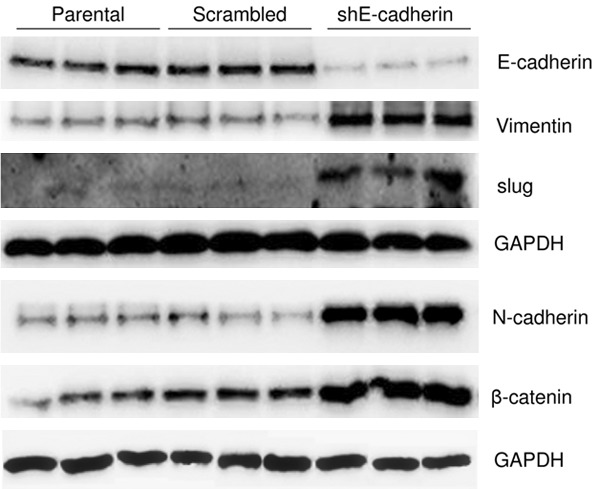
Knockdown of E-cadherin induced EMT, as indicated by western blot data. Lentiviral delivery of shRNA targeting E-cadherin (shE-cadherin) efficiently decreased E-cadherin levels. Knockdown of E-cadherin resulted in increased N-cadherin, vimentin, β-catenin, and slug levels, while the delivery of scrambled shRNA did not elicit a detectable change in the levels of them.
Long term low- and high-dose arsenic exposure affects cell proliferation differently. According to the GO results, both positive and negative regulation of cell proliferation were significantly affected by As exposure (Figure 1A, 1B). The cumulative result of arsenic exposure on cell proliferation was explored. Cell proliferation and growth of T24 cells with or without As (200 nM or 1,000 nM) exposure for 100 days were assayed (Figure 5). Cell proliferation was assayed using EDU. The ratio of proliferative cells (EDU positive) to total cells treated with low-dose As was higher than in the control while that of cells treated with high-dose As was lower than in the control (Figure 5A). This finding indicates that cell proliferation is increased following long term low-dose arsenic exposure but decreased following long term high-dose arsenic exposure. WST assay was employed to measure cell growth (Figure 5B). Long term low-dose As exposure increased cell growth while long term high-dose As decreased cell growth. The OD value of control cells at 48 hours was 6.43±0.01 while that of long term low-dose As treated cells at 48 hours was 7.44±0.17 (vs control: P<0.05). Furthermore, the OD value of long term high-dose As treated cells at 48 hours was 5.28±0.22 (vs control: P<0.05). Together, these findings indicate that long term low-dose arsenic exposure increases cell proliferation and growth in T24 cells, while long term high-dose arsenic exposure decreases both.
Figure 5.
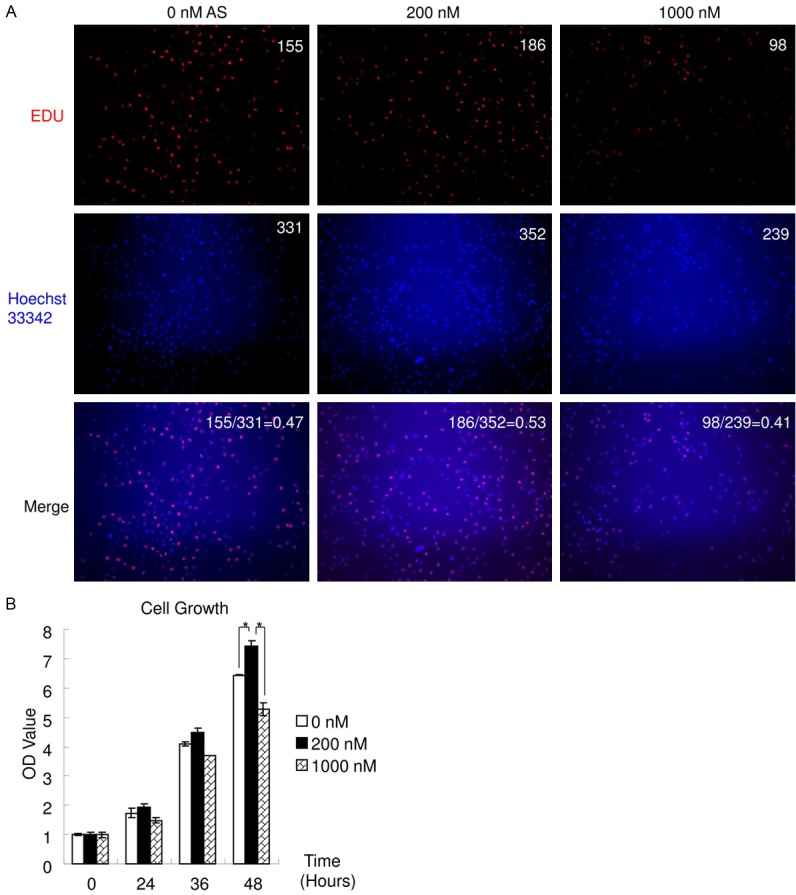
Long term low- and high-dose As affected cell proliferation and growth. Cell proliferation and growth of T24 cells, following As exposure (0, 200 or 1,000 nM) for 100 days, were assayed. A. The number of EDU positive cells (red) or of the nuclei (blue) and the ratio of EDU positive cells/all cells were labeled on the up-right, respectively. Cell proliferation increased following As exposure at 200 nM, but decreased following exposure at 1,000 nM. B. Cell growth was examined using WST assay. Cell growth increased following As exposure at 200 nM, but decreased following exposure at 1,000 nM. (*P<0.05).
Arsenic exposure activates p53. GO and Pathway analysis data indicated that As exposure was strongly associated with cancer (Figure 1). Arsenic is a carcinogen and induces DNA damage [2,6]. p53 plays a key role in the DNA damage response and is strongly related with cancer, especially with carcinogenesis [24-26]. Thus, p53 protein levels in T24 cells treated with As for 0, 3 or 140 days were assayed using western blot. Both low- and high-dose As exposure increased p53 protein levels (Figure 6A). The DNA damage response involves the phosphorylation of p53 at Ser15, which promotes both the accumulation and activation of p53 [25]. As exposure increased p-p53 (Ser15) protein levels (Figure 6A). The expression of p21, a target gene of p53 [26,27], in T24 cells treated with As for 0, 3, 14, 35, 86 or 140 days was assayed using qRT-PCR (Figure 6B). As exposure increased the mRNA levels of p21. Collectively, these findings demonstrate that p53 is activated by arsenic exposure.
Figure 6.
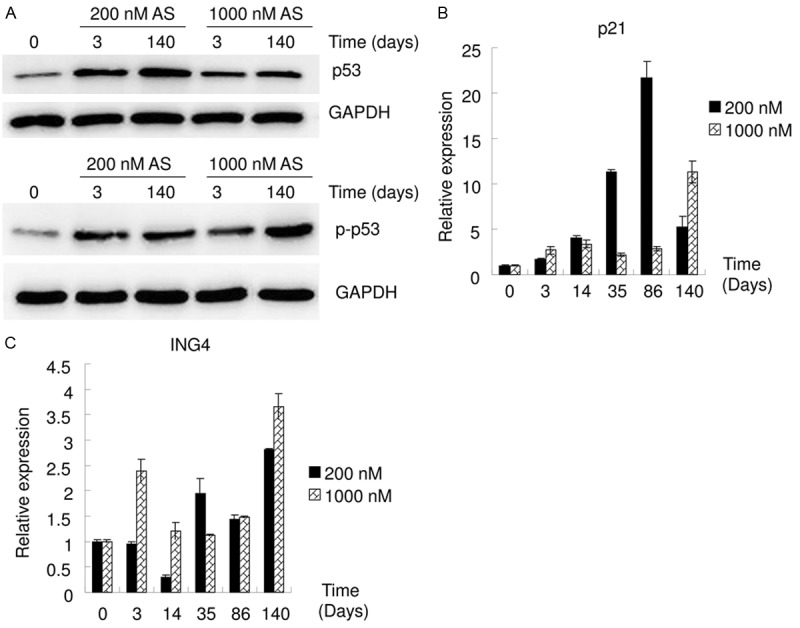
Long term As exposure activated p53. A. Western blot data show that As exposure increased the protein levels of p53 and p-p53 (Ser15). B, C. qRT-PCR data show that As exposure at low (200 nM) or high dose (1,000 nM) at gradient times affected the mRNA levels of p21 and ING4.
ING4 is known to enhance p53 transcriptional activity, partly by inducing acetylation of p53, and thereby promoting downstream p21 expression [28]. The ING4 mRNA levels in T24 cells treated with As for 0, 3, 14, 35, 86, or 140 days were assayed using qRT-PCR (Figure 6C). ING4 mRNA levels were affected by As exposure; however, these effects were not consistent with that of As affecting p53 or p21 (Figure 6). This suggests that ING4 might not play a dominant role in the mechanism of arsenic affecting p53 or p21.
Discussion
Chronic arsenic exposure is strongly associated with cancer. The evidence for the association between arsenic exposure and bladder cancer in humans is very robust, however, detailed biological effects and the mechanism remain unclear [1,4,5]. The investigation of mRNA expression profile helps in understanding these. In the present study, the expression of numerous genes and signaling pathways in the T24 cells were affected following arsenic exposure (Figures 1, 2, 3 and 6). Long term As affected lots of genes mRNAs levels, not always in a time dependent manner (Figures 1, 2, 3 and 6). Furthermore, long term low- and high-dose As exposure affected the expression of genes, such as E-cadherin, in a different manner. These findings indicate that the mechanisms underlying the association between arsenic exposure and cancer are complex.
E-cadherin protein levels were found to decrease following long term low- and high-dose As exposure; this effect was considered to involve varying mechanisms, as low-dose As exposure elicited an increase in E-cadherin mRNA levels while high-dose As exposure resulted in a decrease (Figure 3A, 3B). A decline in E-cadherin protein expression implies that EMT might be intrinsically orchestrated [23,29]. The findings of this study are consistent with this conclusion. Long term low- and high-dose As exposure led to the loss of epithelial identity (decreasing E-cadherin protein level) and gain of mesenchymal identity (increasing protein levels of N-cadherin, vimentin, β-catenin and slug) (Figure 3B, 3C). Exposure to 200 nM As for 90 days resulted in a change in the morphology of T24 cells from an epithelial to a mesenchymal-like appearance; this involved the loss of cell-cell adhesion, acquisition of a fusiform shape, and an increase in the number of heteromorphic cells (Figure 3D). These findings unanimously indicate long term arsenic exposure induces EMT in T24 cells.
Cell adhesion is important for the maintenance of epithelial phenotype, and disruption of cell adhesion results in EMT [29,30]. Cell adhesion is maintained by several types of junctional complexes, such as adherens junctions (AJs), tight junctions, desmosomes/semidesmosomes [7,11,29]. Numerous proteins are involved in regulation of junctional complexes through multiple mechanisms and occasionally, vice verse [11,29,31]. For example, AJs are established mainly via the cadherins or the nectins. Cadherins establish adhesive contacts through either heterophilic or homophilic binding of the extracellular domains. The cytoplasmic domains of the cadherins in the AJs recruit the catenins, which in turn connect the AJs to the actin cytoskeleton [23,29,31]. Linking catenins with the cytoskeleton may be mediated by the clustering of cadherin/catenin complexes to recruit high levels of α-catenin or by other cytoplasmic factors [23,29,31]. Therefore, a decline in the expression of E-cadherin leads to disruption of cell adhesion [11,29], consequentially results in EMT [23,29]. In this study, knockdown of E-cadherin was observed to induce EMT (Figure 4). Because long term As exposure was found to decrease E-cadherin protein levels and induce EMT (Figure 3), it is reasonable to conclude that long term As exposure may induce EMT via E-cadherin. But, in turn, cell-cell adhesion increases E-cadherin protein expression level [11,29,31]. In other words, disruption of cell adhesion decreases E-cadherin [11,29,31]. The present microarray data indicated that cell adhesion molecules, cytoskeleton, junction assembly and junction organization were affected by As treatment (Figure 1). Therefore, the relationship between the decline in E-cadherin expression and EMT remains uncertain, as it is not clear whether the former induces the latter or vice versa.
During EMT, differentiated epithelial cells undergo a series of dramatic changes in their morphology characterized by the loss of polarization and acquisition of a fusiform shape. In addition, loss of cell to cell contact and matrix remodeling into less differentiated and invasive mesenchymal cells is observed [29,30,32]. The EMT program simultaneously endows tumor cells with the capacity to self-renew, lose contact inhibition, survive under different circumstance (such as in the circulation, under chemotherapy or radiotherapy), invade and migrate through tissues, and form colonies in distant organs [29,30,32]. Therefore, EMT plays a key role in carcinogenesis, cancer progression, cancer invasion and metastasis, chemoresistance, and radioresistance [29,30,32]. Epidemiological studies have revealed that arsenic induces human transitional cell carcinoma in a dose- and time-dependent manner [3,4]. A significantly higher risk of cancers of the urinary organs (including bladder) and related mortality were observed among individuals who had consumed arsenic contaminated water for decades [3,4]. The induction of EMT in cells following arsenic exposure should, at least partly, explain the results of these epidemiological studies.
EMT is strongly associated with cell proliferation and/or growth [33,34]. The present GO results indicated that both positive and negative regulation of cell proliferation were significantly affected by As exposure (Figure 1A, 1B). The cumulative result of long term low-dose As exposure was an increase in cell growth and cell proliferation while that of high-dose As exposure was a decrease in both (Figure 5). However, both low- and high-dose long term As exposure induced EMT, implying that the change in cell proliferation/growth following As exposure did not result from the induction of EMT. p21, a potent cyclin-dependent kinase inhibitor, functions as an important regulator of cell proliferation and is a central player in various oncogenic signaling pathways [26,27,35]. The expression of P21 is tightly controlled by the tumor suppressor protein p53 [26,27,35]. Phosphorylation of p53 at Ser15 promotes both the accumulation and activation of p53 and is a marker of DNA damage response [25,26]. As exposure significantly increased p53 and p-p53 (Ser15) (Figure 6A). The mRNA levels of p21, a target gene of p53 [26,27], were also found to be increased following As exposure (Figure 6B), suggesting that arsenic exposure activates p53. Differences in the levels of p53, p-p53 (Ser15) and p21 between T24 cells treated with low- and high-dose As were observed; however, in each case, an increase was found to occur. The findings indicate that, while p53 and p21 might be involved in the low- and high-dose arsenic exposure-induced changes in cell proliferation/growth, they may not play a dominant role in mediating these effects.
In summary, the present microarray analysis showed that arsenic exposure exerts multiple effects in T24 cells. Long term low-dose (200 nM) and high-dose (1,000 nM) As exposure induced EMT, likely by decreasing the protein level of E-cadherin; however, the precise underlying mechanisms are not clearly understood. Long term low- and high-dose As exposure activated p53 and affected cell proliferation/growth in different ways. The present findings serve to elucidate the association between arsenic exposure and the elevated risk of various diseases, typically bladder cancer, as reported by epidemiological studies. Future studies should focus on investigating the precise mechanisms underlying these effects.
Acknowledgements
This work was supported by National Natural Science Foundation of China grant (21107140 and 81502765) and Chongqing Science & Technology Commission grant CSTC2011BB5041.
Disclosure of conflict of interest
None.
References
- 1.Tapio S, Grosche B. Arsenic in the aetiology of cancer. Mutat Res. 2006;612:215–246. doi: 10.1016/j.mrrev.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Sun J, Yu M, Lu Y, Thakur C, Chen B, Qiu P, Zhao H, Chen F. Carcinogenic metalloid arsenic induces expression of mdig oncogene through JNK and STAT3 activation. Cancer Lett. 2014;346:257–263. doi: 10.1016/j.canlet.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall G, Ferreccio C, Yuan Y, Bates MN, Steinmaus C, Selvin S, Liaw J, Smith AH. Fifty-year study of lung and bladder cancer mortality in Chile related to arsenic in drinking water. J Natl Cancer Inst. 2007;99:920–928. doi: 10.1093/jnci/djm004. [DOI] [PubMed] [Google Scholar]
- 4.Chiou HY, Chiou ST, Hsu YH, Chou YL, Tseng CH, Wei ML, Chen CJ. Incidence of transitional cell carcinoma and arsenic in drinking water: a follow-up study of 8,102 residents in an arseniasis-endemic area in northeastern Taiwan. Am J Epidemiol. 2001;153:411–418. doi: 10.1093/aje/153.5.411. [DOI] [PubMed] [Google Scholar]
- 5.Rahman MM, Ng JC, Naidu R. Chronic exposure of arsenic via drinking water and its adverse health impacts on humans. Environ Geochem Health. 2009;31(Suppl 1):189–200. doi: 10.1007/s10653-008-9235-0. [DOI] [PubMed] [Google Scholar]
- 6.Bach J, Peremarti J, Annangi B, Marcos R, Hernandez A. Oxidative DNA damage enhances the carcinogenic potential of in vitro chronic arsenic exposures. Arch Toxicol. 2016;90:1893–1905. doi: 10.1007/s00204-015-1605-7. [DOI] [PubMed] [Google Scholar]
- 7.Liang X, Xu X, Wang F, Chen X, Li N, Wang C, He J. E-cadherin knockdown increases beta-catenin reducing colorectal cancer chemosensitivity only in three-dimensional cultures. Int J Oncol. 2015;47:1517–1527. doi: 10.3892/ijo.2015.3137. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, He J, Zhong D, Li J, Liang H. High-mobility group box 1 protein activating nuclear factor-kappaB to upregulate vascular endothelial growth factor C is involved in lymphangiogenesis and lymphatic node metastasis in colon cancer. J Int Med Res. 2015;43:494–505. doi: 10.1177/0300060515581671. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Zhao LJ, Tan YX, Ren H, Qi ZT. MiR-138 induces cell cycle arrest by targeting cyclin D3 in hepatocellular carcinoma. Carcinogenesis. 2012;33:1113–1120. doi: 10.1093/carcin/bgs113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Y, He Y, Guan Q, Wu Q. A tetracycline expression system in combination with Sox9 for cartilage tissue engineering. Biomaterials. 2014;35:1898–1906. doi: 10.1016/j.biomaterials.2013.11.043. [DOI] [PubMed] [Google Scholar]
- 11.Liang X, Xu X, Wang F, Li N, He J. E-cadherin increasing multidrug resistance protein 1 via hypoxia-inducible factor-1alpha contributes to multicellular resistance in colorectal cancer. Tumour Biol. 2016;37:425–435. doi: 10.1007/s13277-015-3811-6. [DOI] [PubMed] [Google Scholar]
- 12.He J, Shin H, Wei X, Kadegowda AK, Chen R, Xie SK. NPC1L1 knockout protects against colitis-associated tumorigenesis in mice. BMC Cancer. 2015;15:189. doi: 10.1186/s12885-015-1230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi DB, Wang YW, Xing AY, Gao JW, Zhang H, Guo XY, Gao P. C/EBPalpha-induced miR-100 expression suppresses tumor metastasis and growth by targeting ZBTB7A in gastric cancer. Cancer Lett. 2015;369:376–385. doi: 10.1016/j.canlet.2015.08.029. [DOI] [PubMed] [Google Scholar]
- 14.Crose LE, Galindo KA, Kephart JG, Chen C, Fitamant J, Bardeesy N, Bentley RC, Galindo RL, Chi JT, Linardic CM. Alveolar rhabdomyosarcoma-associated PAX3-FOXO1 promotes tumorigenesis via Hippo pathway suppression. J Clin Invest. 2014;124:285–296. doi: 10.1172/JCI67087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okada T, Sinha S, Esposito I, Schiavon G, Lopez-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M, Okada M, Viale A, Heguy A, Socci ND, Sapino A, Seshan VE, Long S, Inghirami G, Rosen N, Giancotti FG. The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat Cell Biol. 2015;17:81–94. doi: 10.1038/ncb3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao B, Han H, Chen J, Zhang Z, Li S, Fang F, Zheng Q, Ma Y, Zhang J, Wu N, Yang Y. MicroRNA let-7c inhibits migration and invasion of human non-small cell lung cancer by targeting ITGB3 and MAP4K3. Cancer Lett. 2014;342:43–51. doi: 10.1016/j.canlet.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 17.Thomas MM, Zhang Y, Mathew E, Kane KT, Maillard I, Pasca di Magliano M. Epithelial Notch signaling is a limiting step for pancreatic carcinogenesis. BMC Cancer. 2014;14:862. doi: 10.1186/1471-2407-14-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang XH, Tu SP, Cui JT, Lin MC, Xia HH, Wong WM, Chan AO, Yuen MF, Jiang SH, Lam SK, Kung HF, Soh JW, Weinstein IB, Wong BC. Antisense targeting protein kinase C alpha and beta1 inhibits gastric carcinogenesis. Cancer Res. 2004;64:5787–5794. doi: 10.1158/0008-5472.CAN-03-1172. [DOI] [PubMed] [Google Scholar]
- 19.Gerlach K, Daniel C, Lehr HA, Nikolaev A, Gerlach T, Atreya R, Rose-John S, Neurath MF, Weigmann B. Transcription factor NFATc2 controls the emergence of colon cancer associated with IL-6-dependent colitis. Cancer Res. 2012;72:4340–4350. doi: 10.1158/0008-5472.CAN-11-4155. [DOI] [PubMed] [Google Scholar]
- 20.Jing Z, Liu Y, Dong M, Hu S, Huang S. Identification of the DNA binding element of the human ZNF333 protein. J Biochem Mol Biol. 2004;37:663–670. doi: 10.5483/bmbrep.2004.37.6.663. [DOI] [PubMed] [Google Scholar]
- 21.Ho C, Wang C, Mattu S, Destefanis G, Ladu S, Delogu S, Armbruster J, Fan L, Lee SA, Jiang L, Dombrowski F, Evert M, Chen X, Calvisi DF. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55:833–845. doi: 10.1002/hep.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sterneck E, Zhu S, Ramirez A, Jorcano JL, Smart RC. Conditional ablation of C/EBP beta demonstrates its keratinocyte-specific requirement for cell survival and mouse skin tumorigenesis. Oncogene. 2006;25:1272–1276. doi: 10.1038/sj.onc.1209144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auersperg N, Pan J, Grove BD, Peterson T, Fisher J, Maines-Bandiera S, Somasiri A, Roskelley CD. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc Natl Acad Sci U S A. 1999;96:6249–6254. doi: 10.1073/pnas.96.11.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, Karnezis AN, Attardi LD. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 26.Rong JJ, Hu R, Song XM, Ha J, Lu N, Qi Q, Tao L, You QD, Guo QL. Gambogic acid triggers DNA damage signaling that induces p53/p21(Waf1/CIP1) activation through the ATR-Chk1 pathway. Cancer Lett. 2010;296:55–64. doi: 10.1016/j.canlet.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Udden SM, Morita-Fujimura Y, Satake M, Ikawa S. c-ABL tyrosine kinase modulates p53-dependent p21 induction and ensuing cell fate decision in response to DNA damage. Cell Signal. 2014;26:444–452. doi: 10.1016/j.cellsig.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Shiseki M, Nagashima M, Pedeux RM, Kitahama-Shiseki M, Miura K, Okamura S, Onogi H, Higashimoto Y, Appella E, Yokota J, Harris CC. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003;63:2373–2378. [PubMed] [Google Scholar]
- 29.Le Bras GF, Taubenslag KJ, Andl CD. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh Migr. 2012;6:365–373. doi: 10.4161/cam.21326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Z, Hu W, Xu J, Kaufmann AM, Albers AE. MicroRNA-34a regulates epithelial-mesenchymal transition and cancer stem cell phenotype of head and neck squamous cell carcinoma in vitro. Int J Oncol. 2015;47:1339–1350. doi: 10.3892/ijo.2015.3142. [DOI] [PubMed] [Google Scholar]
- 31.Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, Ben-Ze’ev A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol. 2003;163:847–857. doi: 10.1083/jcb.200308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Celia-Terrassa T, Meca-Cortes O, Mateo F, Martinez de Paz A, Rubio N, Arnal-Estape A, Ell BJ, Bermudo R, Diaz A, Guerra-Rebollo M, Lozano JJ, Estaras C, Ulloa C, Rholvarez-Simon D, Mila J, Vilella R, Paciucci R, Martinez-Balbas M, Garcia de Herreros A, Gomis RR, Kang Y, Blanco J, Fernandez PL, Thomson TM. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122:1849–1868. doi: 10.1172/JCI59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Z, Wang J, Zheng T, Liang Y, Yin D, Song R, Pei T, Pan S, Jiang H, Liu L. FTY720 inhibits proliferation and epithelial-mesenchymal transition in cholangiocarcinoma by inactivating STAT3 signaling. BMC Cancer. 2014;14:783. doi: 10.1186/1471-2407-14-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo S, Xu X, Tang Y, Zhang C, Li J, Ouyang Y, Ju J, Bie P, Wang H. miR-15a inhibits cell proliferation and epithelial to mesenchymal transition in pancreatic ductal adenocarcinoma by down-regulating Bmi-1 expression. Cancer Lett. 2014;344:40–46. doi: 10.1016/j.canlet.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 35.Liu T, Qin W, Hou L, Huang Y. MicroRNA-17 promotes normal ovarian cancer cells to cancer stem cells development via suppression of the LKB1-p53-p21/WAF1 pathway. Tumour Biol. 2015;36:1881–1893. doi: 10.1007/s13277-014-2790-3. [DOI] [PubMed] [Google Scholar]