

Abstract
Angiogenesis is an essential process for tissue growth and embryo development. However, inflammation, abnormal wound healing, vascular diseases, and tumor development and progression can result from inappropriate angiogenesis. Lipopolysaccharide (LPS) can activate various cells and alter endothelium function and angiogenesis. This study investigated the underlying molecular events involved in LPS-induced angiogenesis and revealed a novel strategy for controlling abnormal angiogenesis. LPS treatment promoted wound healing and tube formation in human umbilical vein endothelial cell (HUVEC) cultures and induced their expression of miR-146a. miR-146a was previously shown to regulate angiogenesis in HUVECs. Knockdown of miR-146a expression antagonized LPS-induced angiogenesis in vitro. Moreover, bioinformatic analyses predicted TGF-β1 as a target gene for miR-146a, which was confirmed by aluciferase reporter assay. Expression of miR-146a in HUVECs resulted in downregulation of TGF-β1 in HUVECs, whereas a miR-146a inhibitor upregulated the expression of TGF-β1 and TGF-β1 downstream proteins, such as phosphoraylation-Smad2 and plasminogen activator inhibitor type 1 (PAI-1). Furthermore, the TGF-β1 signaling inhibitor SB431542 impaired the ability of miR-146a knockdown to suppress LPS-induced angiogenesis. Thus, LPS-induced angiogenesis of HUVECs functions through miR-146a upregulation and TGF-β1 inhibition. This study suggests that knockdown of miR-146a could activate TGF-β1 signaling to inhibit angiogenesis as a potential therapy for angiogenesis-related diseases.
Keywords: Angiogenesis, HUVECs, miR-146a, LPS, TGF-β1
Introduction
Angiogenesis is a pivotal process in embryo development, tissue growth, and wound healing [1]. During angiogenesis, the endothelial cells that line the inner wall of the vasculature respond to growth signals, such as basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), cytokines, or nitric oxide, to proliferate and migrate to form new vessels [2,3]. Pathologically, angiogenesis can promote inflammation, abnormal wound healing, vascular diseases, and tumor development and progression [4]. Thus, angiogenesis may be a therapeutic target for the treatment of various human diseases. For example, anti-angiogenesis treatments could be used in tumor therapy, whereas angiogenesis promotion could be used to treat cardiovascular heart diseases [5,6].
Many studies have shown that infectious disease-induced inflammation can promote cancer development and progression [7-11]. Gram-negative bacterial endotoxin lipopolysaccharides (LPS) can induce inflammation-related cells to secrete a variety of pro-inflammatory and pro-coagulant cytokines, nitric oxide, and eicosanoids that induce tissue inflammation and alter endothelium functions and angiogenesis [12,13]. A previous study showed that LPS induces migration and tube formation of HUVECs [13]. Thus, LPS is a useful agent for triggering angiogenesis in vitro in order to assess angiogenesis and the underlying molecular events. Here, we used an in vitro angiogenesis model using LPS-stimulated human umbilical vein endothelial cells (HUVECs) to examine the underlying molecular events of angiogenesis.
Transforming growth factor-beta (TGF-β) is a multifunctional cytokine that is involved in the regulation of proliferation, differentiation, migration, and survival of many different cell types [14], including blood endothelium. The role of TGF-β as a regulator of blood vessel endothelium is complicated and controversial, and the mechanisms by which TGF-β decreases angiogenesis in vitro are not well understood [15].
MicroRNAs (miRNAs), a class of small, naturally occurring, noncoding RNAs, function to posttranscriptionally suppress expression of specific genes coding for target proteins. miRNAs bind to the 3’-untranslated region (UTR) of their target mRNAs via complete or partial base-pair complementarity, resulting in degradation or translational repression of these mRNA molecules and subsequent regulation of various biological processes [16-20]. miRNAs can be used to therapeutically alter gene expression. A previous study showed that LPS induces the upregulation of miR-146a in HUVECs [13].
In this study, we utilized an in vitro model of HUVEC angiogenesis to investigate the underlying molecular mechanisms of LPS-induced angiogenesis. We confirmed that LPS induces angiogenesis and upregulates expression of miR-146a in HUVECs. Bioinformatic analysis identified TGF-β1 as a target gene of miR-146a. Our study shows that LPS-induced angiogenesis occurs through miRNA-146a inhibition of TGF-β1 and that in vitro angiogenesis can be altered by inducing or inhibiting expression of miR-146a or TGF-β1. Thus, targeting miR-146a could be a therapeutic strategy for treating human cancers and other diseases involving angiogenesis.
Materials and methods
Cell lines and knockdown of miR-146a
Human umbilical vein endothelial cells (HUVECs) were obtained from Clonetics (San Diego, CA, USA) and cultured according to a previous report [13]. miR-146a expression was knocked down by treating HUVECs with locked nucleic acid (LNA)-anti-miR-146a [13]. Briefly, the LNA-anti-miR-146a molecules were synthesized to specifically knock down hsa-miR-146a. An LNA control was designed with mismatched oligonucleotides in the seed region of known miRNAs to control for any interference during silencing. The LNA-anti-miR-146a and the control were synthesized using the following sequences: LNA-anti-miR-146a: 5’-UGAGAACUGAAUUCCAUGGGUU-3’ and control: 5’-CAGUACUUUUGUGUAGUACAA-3’. Synthetic miR-146a and RNA controls were purchased from Shanghai Genechem (Shanghai, China). HUVECs were seeded at a density of 1 × 106 cells in 6-cm plates and treated with 1-25 nM LNA-anti-miR-146a or LNA-control for 24 h. miR-146a levels were measured using qRT-PCR to determine the most effective concentrations.
RNA isolation and quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA isolation and synthesis were performed according to commercial standard methods. Specially, for miR-146a qRT-PCR, total RNA was reverse transcribed using a miScript Reverse Transcription Kit (Qiagen, Hilden, Germany) with a miRNA-specific primer and qPCR was performed in an ABI PRISM 7500 Real-Time PCR System (Applied Biosciences, Benicia, CA, USA) with miRNA-specific primers [13]. U6 was used as a normalization control. For qPCR, we used TGF-β1 specific primers (5’-GGCCAGATCCTGTCCAAGC-3’, 5’-GTGGGTTTCCACCATTAGCAC-3’); PAI-1 specific primers (5’-CCTGGGCACTTACAGGAAGG-3’, 5’-GGTCCGATTCGTCGTCAAATAAC-3’); and used GAPDH as an internal control (5’-TCACCAGGGCTGCTTTTAAC-3’ and 5’-GACAAGCTTCCCGTTCTCAG-3’). Relative expression levels were calculated using the comparative Ct method.
Wound healing assay
HUVECs were grown and transfected with the LNA-anti-miR-146a or LNA-control forfor 24 h and then treated with 1 μg/ml LPS (L3755; Sigma, St. Louis, MO, USA) for 24 h with or without pretreatment for 1 h with aTGF-β type I receptor inhibitor SB431542 (0.5 mM; Tocris, Bristol, UK) according to a methoddescribed in a previous study [21] to inhibit TGF-β family signaling. When HUVEC monolayer cultures reached approximately 95-100% confluency, three scratches were made using a 200-μl pipette tip across the plates. After plates were washed with endothelium growth medium, HUVECs were grown for 24 h and photographs were taken at different time points. Data are summarized as means ± SD.
In vitro matrigel tube formation assay
HUVECs were grown and transfected with the LNA-anti-miR-146a or LNA-control forfor 24 h and then treated with 1 μg/ml LPS for 24 h with or without SB431542 pretreatment for 1 h. HUVECs (3 × 104 cells per well) were seeded onto matrigel plates (containing 200 μl matrigel; BD Biosciences, San Jose, CA, USA) and cultured for 18 h at 37°C in 5% CO2. Capillary-like structures were evident and counted using a phase-contrast microscope and the networks formed by HUVECs were quantified with VIDEOMET software (Videojet Technologies Inc., Chicago, IL, USA). Data are summarized as means ± SD.
ELISA and western blot
A human TGF-β1 ELISA kit (eBioscience Inc., San Diego, CA, USA) was used to measure the level of supernatant TGF-β1 in HUVECs according to the manufacturer’s instructions. Briefly, after miR-146a transfection and treatment of HUVECs, cells were collected and homogenized in a lysis buffer containing protease and phosphatase inhibitors (Sigma). Protein concentrations were determined using BCA protein assay kits (Pierce, Waltham, MA, USA) and then subjected to ELISA analysis of TGF-β1 levels. For each reaction in a 96-well plate, 100 µg of protein samples was used and ELISAs were performed according to the manufacturer’s protocol (eBioscience Inc) with inclusion of a standard curve in each experiment.
Antibodies used for western blot analyses included anti-phosphoraylation-Smad2 (p-Smad2, phospho S250; ab184557, Abcam, Cambridge, MA, USA, 1:1000), anti-PAI-1 (ab125687, Abcam, 1:1000) and anti-β-actin (ab6276, Abcam, 1:2000), antibodies. Band intensities were analyzed using ImageJ software.
Bioinformatic analysis
miRDB (http://mirdb.org) was used to predict miR-146a targets. Gene ontology analysis and pathway enrichment analyses were performed using the DAVID website (http://david.abcc.ncifcrf.gov). Expression profiling of mRNAs was downloaded from correlative normalized GEO dataset (GSM1224744). The dataset was also used to compare the mRNA expression between LNA-antimir-146a LPS-treated HUVECs and LNA-control LPS-treated HUVECs.
Plasmid construction and luciferase assays
The 3’-UTR of TGF-β1 was amplified from human genomic DNA and cloned into a modified pGL3 luciferase vector (Promega, Madison, WI, USA) using the following primers: 5’-GTCTGTGTGAGGGAGGGCT-3’ and 5’-GGAAATGTCTGTGCTGT-3’. PCR products with the appropriate primers to generate point substitutions in miRNA-complementarity binding sites were also inserted into the pGL3 vector (pGl3-TGF-β1-UTR-mut) and used as a mutant control. After amplification and DNA sequencing verification, the constructedDNAwere used for luciferase reporter assays. HUVECs were grown and transfected with 1 μg of the pRL construct containing Renillareniformis luciferase gene (a normalization control), TGF-β1-luciferase-reporter construct (pGL3-TGF-β1 UTR), or constructs containing miR-146a binding motifs, miR-146a mimics (miR-146a) or negative controls (control) for 24 h using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Relative luciferase activity was assessed using a dual luciferase assay system (Promega). Each transfection was performed in triplicate and repeated three times.
Statistical analyses
All statistical analyses were conducted by using PRISM software, version 4 (Graph Pad Software, La Jolla, CA, USA). All experiments were performed in triplicate and repeated at least three times. Data are expressed as means ± SD and were statistically analyzed using two-tailed Student’s t tests or analysis of variance (ANOVA) tests. P < 0.05 (*), P < 0.01 (**), or P < 0.001 (***) were considered statistically significant.
Results
LPS induction of HUVEC migration, tube formation, and miR-146a expression
We confirmed that LPS acts as an inflammatory mediator to induce angiogenesis in HUVECs in vitro as previously reported for several chronic inflammatory diseases [22,23]. Specifically, the wound healing assay showed that LPS promoted HUVEC migration, and tube formation assay indicated that LPS induced HUVECs to form microvessels in vitro (Figure 1A-D). LPS induced the upregulation of miR-146a expression (Figure 1E), which is consistent with a previous study [13].
Figure 1.
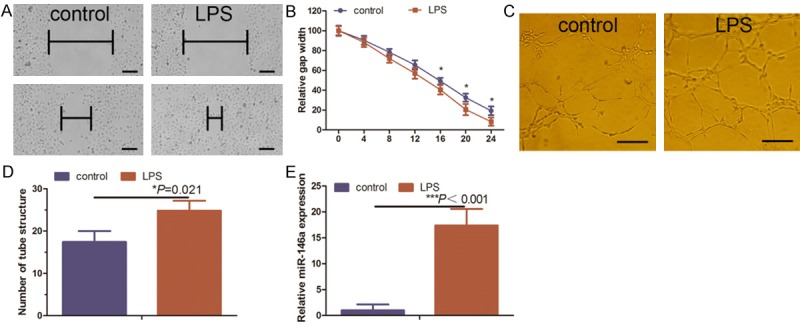
Effects of LPS on angiogenesis and miR-146a expression in HUVECs. A. HUVEC cultures were treated with 1 μg/ml LPS for 24 h and then subjected to wound healing assay at selected time points (every 4 h over the course of 24 h). Scale bar = 100 μm. B. Quantified data from A. C. HUVEC cultures were treated with 1 μg/ml LPS for 24 h and then subjected to in vitro tube formation assay. Scale bar = 50 μm. D. Quantified data from C. E. Duplicate HUVEC cultures were subjected to RNA isolation and qRT-PCR analysis of miR-146a expression. *Comparison between LPS-treated and control HUVECs.
Effects of miR-146a knockdown on LPS-induced angiogenesis
To better understand the pathways affected by miR-146a knockdown in HUVECs, we mimicked the HUVEC miR-146a-knockdown model developed in a previous report (Figure 2A) [13]. To investigate gene expression differences between miR-146a-knockdownLPS-treated HUVECs and miR-control LPS-treated HUVECs, we analyzed the expression profiles of a normalized Gene Expression Omnibus (GEO) dataset. We selected upregulated genes with at least a 2.0-fold increase in the miR-146a knockdown dataset compared with the control dataset. Pathway analysis of these differentially expressed genes revealed significant enrichment of genes involved in several important cellular processes, including intracellular signaling cascades, cytokine-mediated signaling pathways, negative regulation of endothelial cell migration, and blood vessel maturation (Figure 2B). Moreover, miR-146a inhibitor suppressed LPS-induced HUVEC migration and, also, inhibited the capacity of LPS-induced HUVEC tube formation in vitro (Figure 2C-F).
Figure 2.
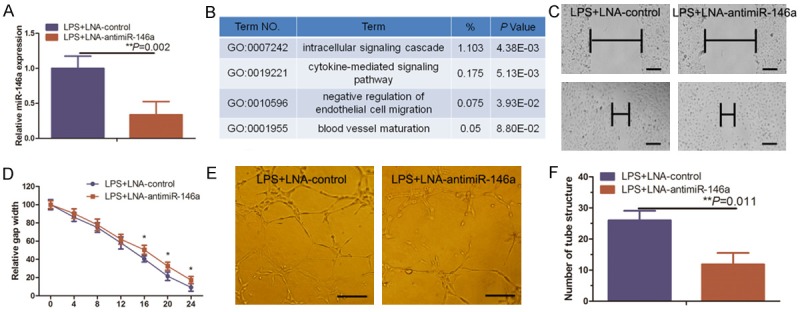
Knockdown of miR-146a suppressed LPS-induced angiogenesis. A. HUVEC cultures were transfected with miR-146a inhibitor (LNA-anti-miR-146a) and then treated with 1 µg/ml LPS for 24 h before being subjected to qRT-PCR analysis of miR-146a expression. B. Gene ontology classification of genes that were upregulated 2.0-fold or more in GSM1224744. C. HUVEC cultures were transfected with LNA-anti-miR-146a and treated with 1 µg/ml LPS for 24 h before being subjected to the wound healing assay. Scale bar = 100 μm. D. Quantified data from C. E. Duplicate HUVEC cultures were subjected to the in vitro tube formation assay. Representative images were taken at 0 h and 24 h. Scale bar = 50 μm. F. Quantified data from E.
Effects of miR-146a knockdown on LPS-induced TGF-β signaling
Previous studies indicated that overexpression of miR-146a modulates activation of hepatic stellate cells during TGF-β1 induction [19]; however, the mechanism of this effect has not been well characterized. Knockdown of miR-146a expression enhanced the expression of TGF-β1 and plasminogen activator inhibitor type 1 (PAI-1) mRNA (Figure 3A), and further confirmed the microarray data (Figure 3A). Knockdown of miR-146a expression also enhanced TGF-β1, PAI-1, and p-Smad2 protein levels in HUVECs (Figure 3B, 3C). These findings suggest thatmiR-146a knockdown may negatively regulate TGF-β signaling.
Figure 3.
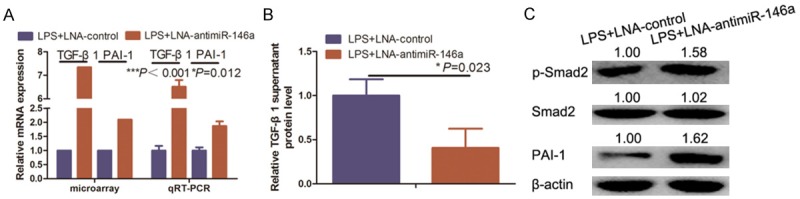
Knockdown of miR-146a-suppressed LPS-induced TGF-β signaling. A. HUVEC cultures were simultaneously subjected to microarray and qRT-PCR analyses of TGF-β1 and PAI-1 expression. B. HUVEC cultures were also subjected to ELISA analysis of TGF-β1 expression. C. HUVEC cultures were further subjected to western blot analysis of p-Smad2, Smad2, and PAI-1 expression.
TGF-β1 is a target gene of miR-146a
miRNAs function to posttranscriptionally inhibit the expression of genes, coding for their target protein, by binding to the 3’-UTR of their respective mRNA. Thus, we used qRT-PCR and ELISAs to compare TGF-β1 expression in HUVECs transfected with miR-146a or control (Figure 4A). TGF-β1 RNA and protein expression were significantly reduced after overexpression of miR-146a in HUVECs (Figure 4B, 4C). To obtain further direct evidence that miR-146a alters TGF-β1 expression via posttranscriptional effects on the 3’-UTR of the gene, we constructed a luciferase reporter plasmid containing the 3’-UTR of TGF-β1 (pGl3-TGF-β1-UTR) (Figure 4D). As shown in Figure 4E, the luciferase activity in pGl3-TGF-β1-UTR-transfected cells was significantly reduced compared with luciferase activity in cells transfected with miR-146a target site mutant TGF-β1 3’-UTR and with that negative control cells.
Figure 4.
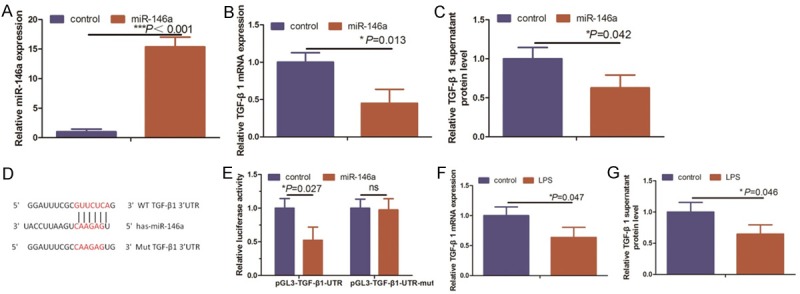
miR-146a directly targets TGF-β1. A and B. HUVECs were transfected with miR-146a and subjected to qRT-PCR analysis of miR-146a and TGF-β1 expression, respectively. C. Supernatants from miR-146a-transfected HUVECs were subjected to ELISA analysis of TGF-β1 protein expression. D. The predicted miR-146a target sequence in the 3’-UTR of TGF-β1 and the mutantcontaining eight mutated nucleotides (TGF-β1-mut). E. HUVEC cells were transfected with a reporter vector containing the WT (wild-type) or MUT (mutant) TGF-β 3’-UTR along with a control mimic (control) or miR-146a mimics (miR-146a). HUVECs were co-transfected with miR-146a and luciferase reporters containing either the predicted miRNA target site in the TGF-β1 3’-UTR or its corresponding mutant 48 hours after transfection. Levels were normalized to Renilla luciferase activity. F and G. TGF-β1 mRNA and protein levels were assessed by qRT-PCR and ELISA, respectively, in LPS-induced HUVECs.
Because LPS-treated HUVECs show enhanced miR-146a expression, we hypothesized that LPS would induce angiogenesis by upregulating miR-146a expression that subsequently would reduce TGF-β1 expression. To test this hypothesis, we analyzed the expression of TGF-β1 in LPS induced HUVECs. Using qRT-PCR, we observed that TGF-β1 mRNA levels were reduced in LPS induced HUVECs compared to control group (Figure 4F). Furthermore, ELISA analyses showed a negative correlation between miR-146a levels and secreted TGF-β1 protein levels in LPS induced HUVECs (Figure 4G). Our results show that miR-146a directly binds and negatively regulates TGF-β1 mRNA stability.
Effects of the TGF-β1 signaling inhibitor SB431542 on miR-146a inhibitor-mediated suppression of LPS-induced angiogenesis and gene expression in HUVECs
Thus far, we demonstrated that LPS-induced HUVEC angiogenesis occurs through miR-146 upregulation resulting in TGF-β1 inhibition. To further confirm this finding, we utilized the TGF-β1 signaling inhibitor SB431542 to antagonize the effect of LNA-anti-miR-146a on suppression of LPS-induced HUVEC angiogenesis in vitro. HUVECs were transfected with the miR-146a inhibitor, treated with 0.5 mM SB431542 for 1 h, and further treated with 1 µm/ml of LPS for 24 h. Wound healing and tube formation assays show that SB431542 treatment inhibited HUVEC migration and tube formation, respectively (Figure 5A-D). Our data further indicate that, as compared with the data shown in Figure 2B-E, SB431542 significantly impaired the ability of miR-146a inhibitor to suppress LPS-induced HUVEC angiogenesis. However, HUVEC angiogenesis following treatment with SB431542 was similar to that of LPS-treated HUVECs (Figure 1A-D). Moreover, the TGF-β1 pharmacological inhibitor downregulated the expression of p-Smad2 and PAI-1 (Figures 5E, 4F). Thus, our data indicate that LPS-induced angiogenesis occurs via upregulation of miR-146a and subsequent inhibition of TGF-β1 expression.
Figure 5.
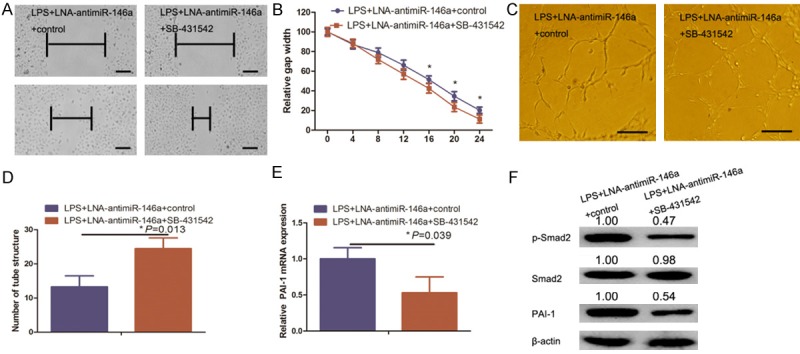
A. TGF-β1 inhibitor blocked miR-146a inhibitor-mediated suppression of LPS-induced angiogenesis and TGF-β signaling in HUVECs. A. HUVEC cultures were transfected with a miR-146a inhibitor (LNA-anti-miR-146a), pretreated with the TGF-β1 signaling inhibitor SB431542 for 1 h, and treated with 1 µg/ml LPS for 24 h before being subjected to the in vitro wound healing assay. Representative images were taken at 0 h and 24 h. Scale bar = 100 μm. B. Quantified data from A. C. Duplicate HUVEC cultures were subjected to the in vitro tube formation assay. Scale bar = 50 μm. D. Quantified data from C. E and F. HUVEC cultures were also subjected to qRT-PCR analysis of PAI-1 expression and western blot analysis of Smad2, p-Smad2, and PAI-1 expression, respectively.
Discussion
In this study, we explored the underlying mechanisms of angiogenesis using an in vitro model of HUVEC angiogenesis following LPS treatment. Our results suggest a novel strategy for manipulating tissue angiogenesis, which can help in clinically treating tumor angiogenesis and other angiogenesis-related diseases. Our current data indicate that i) LPS treatment promotes HUVEC angiogenesis in vitro and induces miR-146a expression, ii) knockdown of miR-146a expression antagonizes LPS-induced angiogenesis, iii) TGF-β1 is a target gene of miR-146a, and iv) TGF-β1 signaling inhibitor SB431542 impairs the ability of miR-146a knockdown to suppress LPS-induced angiogenesis. Therefore, the mechanism of LPS-induced angiogenesis involves the upregulation of miR-146a, which subsequently inhibits TGF-β1 expression. Future studies will investigate the utility of miR-146a inhibitors for the treatment of human cancers and miR-146a mimics for the treatment of cardiovascular diseases such as myocardial infarction.
LPS is a well characterized immune modulator that can induce tissue inflammation and promote angiogenesis [13,24] and human cancer development and progression [25]. Furthermore, LPS can be used as a stimulator of HUVEC angiogenesis to examine the molecular mechanisms of angiogenesis [13].
Profiling of miRNA regulatory genes can further the understanding of gene regulation and molecular signaling in normal and disease processes. Emerging evidence accumulated during the past decades implicate miRNAs in the pathogenesis of some human cancers [26]. Altered miRNA expression can contribute to cell transformation, tumorigenesis [27], and angiogenesis [28]. miR-146a is reported to exert a tumor-suppressive role in many types of cancer [29], but other studies have shown that miR-146a expression is upregulated in myeloma and prostate cancer and may, therefore, function as an oncogene [30]. Our previous study showed that miR-146a regulates HUVEC angiogenesis via the NF-κB/CARD10 axis [13]. Therefore, we investigated whether miR-146a is involved in LPS-mediated angiogenesis.
Here, microarray data (GEO dataset: GSM1224744), GO analysis, and our qRT-PCR data indicate that knockdown of miR-146a results in increased expression of TGF-β1 and other downstream genes. LPS induced miR-146a expression in HUVECs, which is consistent with data published previously in similar contexts [13,31]. Upregulation of miR-146a expression is pro-angiogenic in LPS-induced HUVECs [13]. In contrast, blockage of miR-146a expression with locked nucleic acids in LPS-treated HUVECs impairs angiogenesis [13,31].
miR-146a appears to play conflicting roles in different disease settings. A previous study showed that miR-146a plays a positive role in promoting the angiogenic activity of hepatocellular carcinoma-associated endothelial cells [32]. However, other studies have reported that overexpression of miR-146a in castration-resistant prostate cancer cell lines suppresses tumor cell growth, colony formation, and migration in vitro, and reduces tumorigenicity and angiogenesis in vivo [31-33]. Furthermore, miR-146a can function as an inhibitor of angiogenesis via negative regulation of NRAS expression [34]. Thus, further study is needed to clarify the functions of miR-146a in various cancer and disease settings.
A single miRNA can target multiple protein-coding genes in cells and a single mRNA can be targeted by different miRNAs. Thus, depending on the different miRNA targets available, biological functions of miRNA could vary between different cell types or disease settings [35]. In our current study, we demonstrated that miR-146a binds to the 3’-UTR region of TGF-β1 mRNA and inhibits the expression of TGF-β1 and TGF-β1-regulated genes such as p-Smad2 and PAI-1.
The role of TGF-β as a regulator of adult vascular endothelium is complicated and controversial [36]. The described dual actions of TGF-β in vascular growth are highly dependent on cell context [37] and balance between two distinct TGF-β receptor signaling pathways, namely activin receptor-like kinase 5 (ALK5)-Smad2/3 pathway and the ALK1-Smad1/5 pathway [36,38]. Typically, the TGF-β1/ALK5 pathway leads to inhibition of endothelial cell migration and proliferation, whereas the TGF-β1/ALK1 pathway induces endothelial cell migration and proliferation [38]. Briefly, TGF-β can bind to the TGF-β type II receptor, thus phosphorylating/activating ALK5 [39], which, in turn, phosphorylates Smad2/3 to induce expression of PAI-1 and fibronectin to inhibit endothelial cell migration, proliferation, and tube formation [40,41]. Our current dataon phosphorylation of Smad2 were not in agreement with this description [37-40]. We found only TGF-β1-induced phosphorylation of Smad2, but not Smad3, which is consistent with a previous report [42], and may be partly due to parallel, but distinct, Smad pathways [43]. Altogether, TGF-β signaling is of critical importance in normal vascular development and physiology [36]; TGF-β1 can inhibit endothelial cell proliferation, migration, and proteolytic activity [44]. In our current study, we also used the expression of miR-146a to assess the effect of TGF-β type I receptor inhibitor SB431542 [45] on LPS-induced angiogenesis and on the levels ofPAI-1 and p-Smad2. These findings further confirmed the importance of TGF-β1 in miR-146a-mediated LPS-induced HUVEC angiogenesis.
In our current study, we demonstrated the importance of miR-146a-induced TGF-β1 inhibition in mediating LPS-induced HUVEC angiogenesis in vitro. Future studies will investigate whether targeting miR-146a expression in vivo can modulate angiogenesis in various human diseases such as cancer, heart, and vascular diseases. We will also assess miR-146a-regulated gene pathways (e.g., the NF-κB/CARD10 or the TGF-β1 pathways) during angiogenesis.
Acknowledgements
This study was supported in part by grants from the National Natural Science Foundation of China (Nos. 81572699 and 81501684).
Disclosure of conflict of interest
None.
References
- 1.Wang ZD, Wei SQ, Wang QY. Targeting oncogenic KRAS in non-small cell lung cancer cells by phenformin inhibits growth and angiogenesis. Am J Cancer Res. 2015;5:3339–3349. [PMC free article] [PubMed] [Google Scholar]
- 2.Khurana R, Simons M. Insights from angiogenesis trials using fibroblast growth factor for advanced arteriosclerotic disease. Trends Cardiovasc Med. 2003;13:116–122. doi: 10.1016/s1050-1738(02)00259-1. [DOI] [PubMed] [Google Scholar]
- 3.Goto F, Goto K, Weindel K, Folkman J. Synergistic effects of vascular endothelial growth factor and basic fibroblast growth factor on the proliferation and cord formation of bovine capillary endothelial cells within collagen gels. Lab Invest. 1993;69:508–517. [PubMed] [Google Scholar]
- 4.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 6.Simons M, Bonow RO, Chronos NA, Cohen DJ, Giordano FJ, Hammond HK, Laham RJ, Li W, Pike M, Sellke FW, Stegmann TJ, Udelson JE, Rosengart TK. Clinical trials in coronary angiogenesis: issues, problems, consensus: An expert panel summary. Circulation. 2000;102:E73–86. doi: 10.1161/01.cir.102.11.e73. [DOI] [PubMed] [Google Scholar]
- 7.Borrello MG, Degl’Innocenti D, Pierotti MA. Inflammation and cancer: the oncogene-driven connection. Cancer Lett. 2008;267:262–270. doi: 10.1016/j.canlet.2008.03.060. [DOI] [PubMed] [Google Scholar]
- 8.Li Z, Zheng Z, Ruan J, Li Z, Tzeng CM. Chronic inflammation links cancer and parkinson’s disease. Front Aging Neurosci. 2016;8:126. doi: 10.3389/fnagi.2016.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He W, Li Y, Tian J, Jiang N, Du B, Peng Y. Optimized mixture of As, Cd and Pb induce mitochondria-mediated apoptosis in C6-glioma via astroglial activation, inflammation and P38-MAPK. Am J Cancer Res. 2015;5:2396–2408. [PMC free article] [PubMed] [Google Scholar]
- 10.Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: the role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol. 2016;22:4794–4801. doi: 10.3748/wjg.v22.i20.4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu ZY, Wu B, Guo YS, Zhou YH, Fu ZG, Xu BQ, Li JH, Jing L, Jiang JL, Tang J, Chen ZN. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am J Cancer Res. 2015;5:3174–3185. [PMC free article] [PubMed] [Google Scholar]
- 12.Toral M, Romero M, Jiménez R, Robles-Vera I, Tamargo J, Carmen Martínez M, Pérez-Vizcaíno F, Duarte J. Role of UCP2 in the protective effects of PPARβ/δ activation on lipopolysaccharide-induced endothelial dysfunction. Biochem Pharmacol. 2016;110-111:25–36. doi: 10.1016/j.bcp.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Rau CS, Yang JC, Chen YC, Wu CJ, Lu TH, Tzeng SL, Wu YC, Hsieh CH. Lipopolysaccharide-induced microRNA-146a targets CARD10 and regulates angiogenesis in human umbilical vein endothelial cells. Toxicol Sci. 2014;140:315–326. doi: 10.1093/toxsci/kfu097. [DOI] [PubMed] [Google Scholar]
- 14.Han J, Alvarez-Breckenridge CA, Wang QE, Yu J. TGF-β signaling and its targeting for glioma treatment. Am J Cancer Res. 2015;5:945–955. [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P, Naumann U, Aigner L, Wischhusen J, Beier CP, Beier D. Impaired TGF-β induced growth inhibition contributes to the increased proliferation rate of neural stem cells harboring mutant p53. Am J Cancer Res. 2015;5:3436–3445. [PMC free article] [PubMed] [Google Scholar]
- 16.Tan YY, Xu XY, Wang JF, Zhang CW, Zhang SC. MiR-654-5p attenuates breast cancer progression by targeting EPSTI1. Am J Cancer Res. 2016;6:522–532. [PMC free article] [PubMed] [Google Scholar]
- 17.Xu C, Zhang L, Li H, Liu Z, Duan L, Lu C. MiRNA-1469 promotes lung cancer cells apoptosis through targeting STAT5a. Am J Cancer Res. 2015;5:1180–1189. [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Wang J, Chen Y, Li S, Jin M, Wang H, Chen Z, Yu W. LncRNA MALAT1 exerts oncogenic functions in lung adenocarcinoma by targeting miR-204. Am J Cancer Res. 2016;6:1099–1107. [PMC free article] [PubMed] [Google Scholar]
- 19.He Y, Huang C, Sun X, Long XR, Lv XW, Li J. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell Signal. 2012;24:1923–1930. doi: 10.1016/j.cellsig.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Ren J, Chen H, Geng Q. Inflammation induced-endothelial cells release angiogenesis associated-microRNAs into circulation by microparticles. Chin Med J (Engl) 2014;127:2212–2217. [PubMed] [Google Scholar]
- 21.Lee J, Kim MR, Kim HJ, An YS, Yi JY. TGF-β1 accelerates the DNA damage response in epithelial cells via Smad signaling. Biochem Biophys Res Commun. 2016;476:420–425. doi: 10.1016/j.bbrc.2016.05.136. [DOI] [PubMed] [Google Scholar]
- 22.Ni H, Zhao W, Kong X, Li H, Ouyang J. Celastrol inhibits lipopolysaccharide-induced angiogenesis by suppressing TLR4-triggered nuclear factor-kappa B activation. Acta Haematol. 2014;131:102–111. doi: 10.1159/000354770. [DOI] [PubMed] [Google Scholar]
- 23.Pollet I, Opina CJ, Zimmerman C, Leong KG, Wong F, Karsan A. Bacterial lipopolysaccharide directly induces angiogenesis through TRAF6-mediated activation of NF-kappaB and c-Jun N-terminal kinase. Blood. 2003;102:1740–1742. doi: 10.1182/blood-2003-01-0288. [DOI] [PubMed] [Google Scholar]
- 24.Kihara Y, Mizuno H, Chun J. Lysophospholipid receptors in drug discovery. Exp Cell Res. 2015;333:171–177. doi: 10.1016/j.yexcr.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miura K, Ishioka M, Minami S, Horie Y, Ohshima S, Goto T, Ohnishi H. Toll-like receptor 4 on macrophage promotes the development of steatohepatitis-related hepatocellular carcinoma in mice. J Biol Chem. 2016;291:11504–11517. doi: 10.1074/jbc.M115.709048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu HY, Bai WD, Li C, Zheng Z, Guan H, Liu JQ, Yang XK, Han SC, Gao JX, Wang HT, Hu DH. Knockdown of lncRNA-ATB suppresses autocrine secretion of TGF-β2 by targeting ZNF217 via miR-200c in keloid fibroblasts. Sci Rep. 2016;6:24728. doi: 10.1038/srep24728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boro A, Bauer D, Born W, Fuchs B. Plasma levels of miRNA-155 as a powerful diagnostic marker for dedifferentiated liposarcoma. Am J Cancer Res. 2016;6:544–552. [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Wang J, Chen Y, Li S, Jin M, Wang H, Chen Z, Yu W. LncRNA MALAT1 exerts oncogenic functions in lung adenocarcinoma by targeting miR-204. Am J Cancer Res. 2016;6:1099–1107. [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Q, Zhao X, Liu X, Wang Y, Huang J, Jiang B, Chen Q, Yu J. miR-146a functions as a tumor suppressor in prostate cancer by targeting Rac1. Prostate. 2014;74:1613–1621. doi: 10.1002/pros.22878. [DOI] [PubMed] [Google Scholar]
- 30.De Veirman K, Wang J, Xu S, Leleu X, Himpe E, Maes K, De Bruyne E, Van Valckenborgh E, Vanderkerken K, Menu E, Van Riet I. Induction of miR-146a by multiple myeloma cells in mesenchymal stromal cells stimulates their pro-tumoral activity. Cancer Lett. 2016;377:17–24. doi: 10.1016/j.canlet.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 31.Zhu HY, Bai WD, Liu JQ, Zheng Z, Guan H, Zhou Q, Su LL, Xie ST, Wang YC, Li J, Li N, Zhang YJ, Wang HT, Hu DH. Up-regulation of FGFBP1 signaling contributes to miR-146a-induced angiogenesis in human umbilical vein endothelial cells. Sci Rep. 2016;6:25272. doi: 10.1038/srep25272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu K, Pan Q, Zhang X, Kong LQ, Fan J, Dai Z, Wang L, Yang XR, Hu J, Wan JL, Zhao YM, Tao ZH, Chai ZT, Zeng HY, Tang ZY, Sun HC, Zhou J. MiR-146a enhances angiogenic activity of endothelial cells in hepatocellular carcinoma by promoting PDGFRA expression. Carcinogenesis. 2013;34:2071–2079. doi: 10.1093/carcin/bgt160. [DOI] [PubMed] [Google Scholar]
- 33.Xu B, Wang N, Wang X, Tong N, Shao N, Tao J, Li P, Niu X, Feng N, Zhang L, Hua L, Wang Z, Chen M. MiR-146a suppresses tumor growth and progression by targeting EGFR pathway and in a p-ERK-dependent manner in castration-resistant prostate cancer. Prostate. 2012;72:1171–1178. doi: 10.1002/pros.22466. [DOI] [PubMed] [Google Scholar]
- 34.Halkein J, Tabruyn SP, Ricke-Hoch M, Haghikia A, Nguyen NQ, Scherr M, Castermans K, Malvaux L, Lambert V, Thiry M, Sliwa K, Noel A, Martial JA, Hilfiker-Kleiner D, Struman I. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest. 2013;123:2143–2154. doi: 10.1172/JCI64365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–22. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goumans MJ, Lebrin F, Valdimarsdottir G. Controlling the angiogenic switch: a balance between two distinct TGF-β receptor signaling pathways. Trends Cardiovasc Med. 2003;13:301–307. doi: 10.1016/s1050-1738(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 37.Kumar P, Naumann U, Aigner L, Wischhusen J, Beier CP, Beier D. Impaired TGF-β induced growth inhibition contributes to the increased proliferation rate of neural stem cells harboring mutant p53. Am J Cancer Res. 2015;5:3436–3445. [PMC free article] [PubMed] [Google Scholar]
- 38.Stefansson S, Lawrence DA. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature. 1996;383:441–443. doi: 10.1038/383441a0. [DOI] [PubMed] [Google Scholar]
- 39.Maroni D, Davis JS. TGFB1 disrupts the angiogenic potential of microvascular endothelial cells of the corpus luteum. J Cell Sci. 2011;124:2501–2510. doi: 10.1242/jcs.084558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lebrin F, Deckers M, Bertolino P, Ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65:599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 41.Stefansson S, Petitclerc E, Wong MK, McMahon GA, Brooks PC, Lawrence DA. Inhibition of angiogenesis in vivo by plasminogen activator inhibitor-1. J Biol Chem. 2001;276:8135–8141. doi: 10.1074/jbc.M007609200. [DOI] [PubMed] [Google Scholar]
- 42.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21:1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakagawa T, Li JH, Garcia G, Mu W, Piek E, Bottinger EP, Chen Y, Zhu HJ, Kang DH, Schreiner GF, Lan HY, Johnson RJ. TGF-beta induces proangiogenic and antiangiogenic factors via parallel but distinct Smad pathways. Kidney Int. 2004;66:605–613. doi: 10.1111/j.1523-1755.2004.00780.x. [DOI] [PubMed] [Google Scholar]
- 44.Saksela O, Moscatelli D, Rifkin DB. The opposing effects of basic fibroblast growth factor and transforming growth factor beta on the regulation of plasminogen activator activity in capillary endothelial cells. J Cell Biol. 1987;105:957–963. doi: 10.1083/jcb.105.2.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan W, Liu W, Cai H, Sun X, Yang D, Xu F, Jin C. SB-431542, a specific inhibitor of the TGF-β type I receptor inhibits hypoxia-induced proliferation of pulmonary artery adventitial fibroblasts. Pharmazie. 2016;71:94–100. [PubMed] [Google Scholar]