

Abstract
Idiopathic pulmonary fibrosis (IPF) has attracted extensive attention for its unexplained progressive lung scarring, short median survival and its unresponsiveness to traditional therapies. Despite extensive studies, the mechanisms underlying IPF pathoetiologies, however, remain poorly understood. Recent advances delineated a potential function of endoplasmic reticulum (ER) stress in meeting the need of fibrotic response, which pinpointed a critical role for the unfolded protein response (UPR) pathways in IPF pathogenesis. In this review, we highlight the effect of ER stress and the activation of UPR on the survival, differentiation, function and proliferation of major profibrotic cells in lung tissues during the course of IPF, and discuss the feasibility whether targeting UPR components could be an orientation for developing effective therapeutic strategies against this devastating disorder in clinical settings.
Keywords: IPF, ER stress, UPR, profibrotic cells, pathogenesis
Introduction
Idiopathic pulmonary fibrosis (IPF) is a disease associated with severe lung dysfunction by affecting gas exchange that often bears fatal consequences [1]. The survival median range for the affected patients is about three years after the initial diagnosis [2]. The incidence of IPF has been recognized with a profound increase in elderly patients (50-70 years old), particularly in those with a history of cigarette smoking [3,4]. There is evidence that males are more vulnerable to IPF than females [1,5]. Despite past extensive studies, the mechanisms underlying IPF, however, remained enigmatic.
The endoplasmic reticulum (ER) is a special organelle in eukaryotic cells characterized by an interconnected network of flattened sacs or tubes encased in membranes. In general, ER is responsible for the correct folding and transport of synthesized proteins in vesicles to the Golgi apparatus. However, factors like calcium depletion, redox homeostatic alteration, nutrient deprivation and environmental insults (e.g., viral infection) can affect the folding processes of synthesized proteins, leading to an increase of unfolded proteins, a state of ER stress that triggers the activation of the unfolded protein response (UPR) [6]. To date, ER stress and UPR signaling have been recognized implicated in the pathogenesis of many complex disorders such as diabetes and neurodegenerative diseases. However its involvement in diseases associated with fibrotic remodeling of internal organs including heart, kidneys, liver, gastrointestinal tract and lungs is just recently emerging [6-11]. We, therefore, in this review, will discuss with focus on its impact on major profibrotic cells that contribute to the pathogenesis of pulmonary fibrosis. Our purpose is to deepen and expand our understanding of the mechanisms underlying the initiation and progression of IPF, thereby developing better therapeutic strategies against this devastating disorder in clinical settings.
The initiation and progression of IPF
In general, a wound-healing response is consisted of three distinct stages: injury, inflammation and repair. In the current paradigm for IPF pathogenesis, pulmonary fibrosis progresses as a final pathological outcome of aberrant wound healing responses to persistent lung injury (Figure 1), as it is characterized by the excessive extracellular matrix (ECM) deposition in the lungs [12-15]. In the early stage, events such as ER stress, and molecular mediators such as excessive transforming growth factor β (TGF-β) activation and a variety of chemokines release, induce epithelial cell dysfunction or apoptosis, which then activate resident fibroblasts to proliferate for injury repair. Epithelial-to-mesenchymal transition (EMT) along with fibrocyte recruitment and differentiation is also considered as a pivotal feature relevant to pulmonary fibrotic remodeling [16-20]. Epithelial death would also recruit inflammatory cells such as macrophages, which then produce cytokines or chemokines to generate a microenvironment in favor of fibrosis for injury repair. As this process continues, abnormal quantities of matrix components would be produced, which then trigger an excessive deposition of scars in the lung tissues [18,21]. Furthermore, the pathologically remodeled matrix or epigenetic changes within fibroblasts may lead to a feed-forward loop of mesenchymal cell activation and progressive fibrosis [22,23]. Collectively, persistent irritants contribute to a cascade of abnormal regulatory mechanisms to cause vast pulmonary epithelial apoptosis, continuous fibroblast activation and increased myofibroblast differentiation, which then lead to excessive ECM deposition and distort lung tissue architecture, ultimately, resulting in pulmonary fibrosis and respiratory failure.
Figure 1.
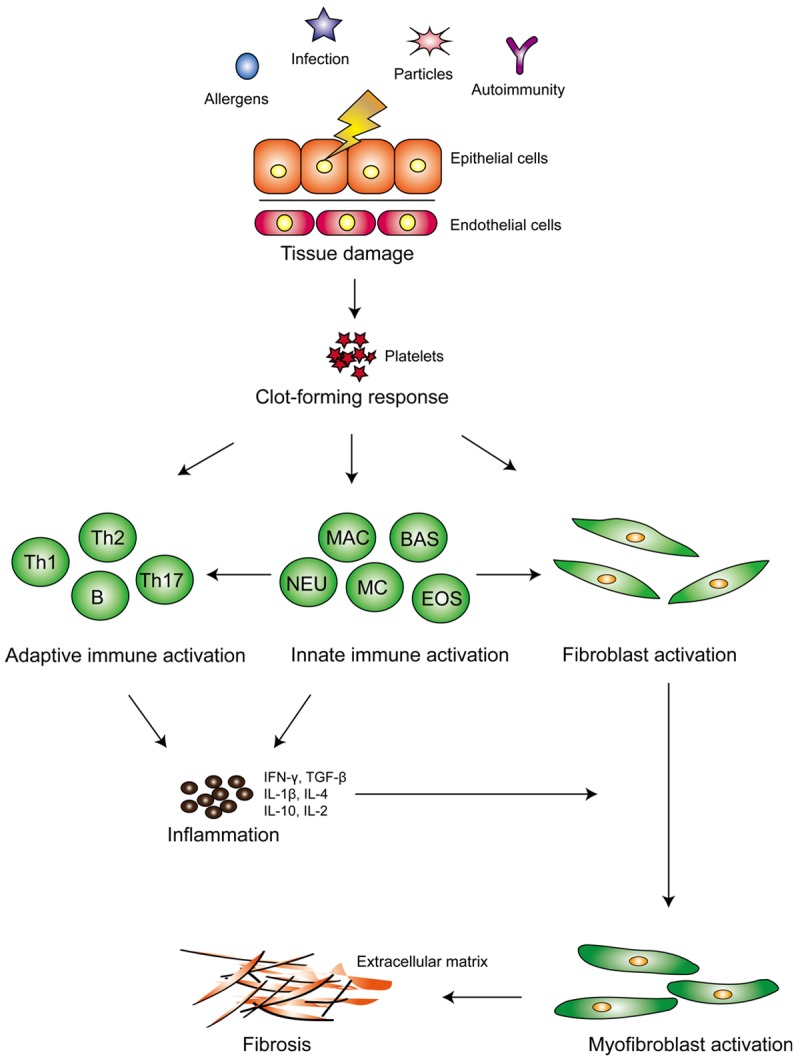
Overview of fibrogenesis in IPF. In the early phase of fibrogenesis, epithelial and/or endothelial damage caused by a variety of irritants can initiate an anti-fibrinolytic coagulation cascade, temporarily plugging the damaged vessel with platelets and fibrin-rich clots to quickly restore homeostasis. Meanwhile, thrombin and the injured epithelium can directly evoke fibroblast activation and promote fibroblast differentiation into collagen-producing myofibroblasts. After a short period of time, clot-forming responses rapidly progress into a phase, in which many inflammatory cells such as macrophages, neutrophils, and lymphocytes are recruited into the injured site, where they secretecopious amount of cytokines to eliminate the inciting factor whilst activating the resident quiescent fibroblasts into myofibroblasts. However, once an imbalance in cytokine production coupled with dysregulated cellular recruitment occurs, a normal wound-healing response can switch into a pathological fibrotic reaction, ultimately resulting in pulmonary fibrosis. MAC = macrophages, BAS = basophils, NEU = neutrophils, MC = mast cells, EOS = eosinophils.
ER stress and the unfolded protein response (UPR)
As mentioned above, ER is a organelle responsible for proper folding of membrane and secreted proteins, lipid biosynthesis, glycogen production and storage, as well as intracellular calcium homeostasis [24,25]. Under physiological condition, the nascent proteins enter ER lumen where they undergo a chaperone-based folding, together with intricate polypetide modifications, including N-linked glycosylation, disulfide bond formation, proline cis-trans isomerization and so on [26-28]. The proper function of proteins requires correct folding and post-translation modification, which is assisted in part by chaperone proteins such as calnexin, calreticulin (CRT), the Hsp70 family member immunoglobulin heavy-chain-binding protein (BiP, also named as glucose regulated protein 78, GRP78). However, disturbances in redox regulation, glucose deprivation, viral infection or calcium metabolism are proved to bring about aggregation of unfolded or misfolded proteins along with ER stress [29-31]. Upon the initiation of ER stress cascade, unfolded protein response (UPR), integrated stress response (ISR) and ER-associated degradation (ERAD) are activated, which are aimed to halt protein translation, improve protein folding, sustain cellular homeostasis, and avoid cell death from accumulation of unfolded or misfolded proteins [32-35]. Nevertheless, growth arrest and cell death through apoptosis would occur once the disruption is overwhelming that these objectives could not be achieved within a certain time span [36,37].
Generally, the UPR pathways are governed by the coordinated action of three ER transmembrane stress sensors: PKR-like ER kinase (PERK; also known as EIF2ZAK3), activating transcription factor 6α (ATF6α) and inositol-requiring enzyme 1α (IRE1α; also known as ERN1) [25,38]. PERK and IRE-1 share similar ER luminal domain structures and a cytosolic Ser/Thr kinase domain, and are activated by autophosphorylation. In contrast, ATF6α contains a cytosolic cyclic AMP response element-binding protein (CREB)-ATF basic leucine zipper domain, and is activated by proteases [39]. Under unstressed homeostatic conditions, these three proteins maintain each in an inactive state through binding to the molecular chaperone BiP (GRP78) [40]. Once aberrant proteins accumulated in ER, more available BiP is in demand to dissociate from the ER stress sensors to interact with the exposed hydrophobic regions of these proteins [41,42], thereby releasing the stress sensors to activate the cascade of events designed to protect the cell from ER stress (Figure 2).
Figure 2.
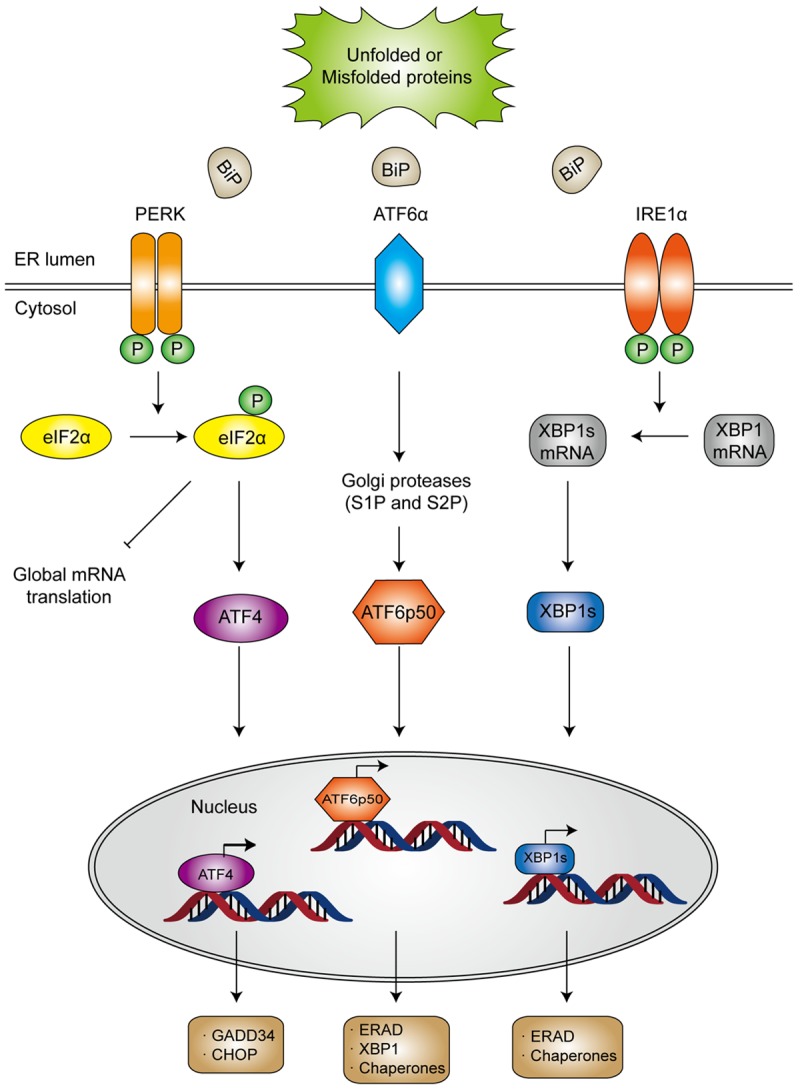
Schematic illustration of ER stress and the activation of three UPR pathways. Under stressed condition, BiP dissociates from the ER stress sensors owing to aggregation of unfold or misfolded proteins in ER lumen, which releases the stress sensors to initiate downstream signaling. Activated PERK undergoes autophosphorylation and dimerization and subsequently inhibits ribosome assembly by phosphorylating the α-subunit of eukaryotic translational initiation factor 2 (eIF2α). Once becomes phosphorylated, eIF2α not only suppresses protein translation, but also upregulates the expression of activating transcription factor 4 (ATF4), which would induce the transcription of protective genes DNA damage-inducible protein 34 (GADD34) as well as the pro-apoptotic gene encoding C/EBP homologous protein (CHOP). In general, CHOP is often produced in the terminal unfolded protein response (UPR) to induce apoptosis. After dissociating from BiP, ATF6α translocates to the Golgi apparatus, where it is cleaved by site 1 protease (S1P) and S2P into an NH2 terminal domain and a cytosolic fragment (ATF6p50). ATF6p50 is then transported into the nucleus and activates the transcription of several ER proteins such as X-box binding protein 1 (XBP1), calreticulin, calnexin, disulfide isomerase and CHOP. Upon activation, IRE-1α dimerizes and cleaves XBP1 into its spliced form, which then acts as a transcription factor of many stress proteins to enlarge the protein-folding capacity of ER, and to induce the expression of ER associated degradation (ERAD)-related proteins such as ER degradation enhancing α-mannosidase-like protein (EDEM).
Although all three UPR pathway sensors are activated to alleviate ER stress and sustain the cellular homeostasis, cell death pathways such as caspase-4 (and its murine homolog caspase-12) and C/EBP homologous protein (CHOP), are activated by prolonged or overloaded ER stress [25,43-45]. Caspase-4 is found in the ER membrane and activates cell death pathways through caspase-3 and caspase-9 [45]. CHOP, a major transcriptional factor for regulation of cell death under ER stress, can be activated by all three UPR pathways [46,47], resulting in the reduction of anti-apoptotic mitochondrial protein Bcl-2, which would favor a pro-apoptotic intention at mitochondria associated with mitochondrial damage, cytochrome c release and caspase 3 activation [48].
The implication of profibrotic cellular ER stress during the course of IPF
Lung parenchyma constitutes all sorts of cell types including alveolar epithelial cells, macrophages, (myo)fibroblasts, and so forth that contributes to the progression of IPF. The past decade witnessed convincing potential links related to ER stress in these profibrotic cell types during the course of IPF [49]. In this section, we seek to bring all evidence together supporting the potential role of ER stress in those critical cells during the course of IPF development and progression.
Alveolar epithelial cells (AECs)
The alveolar surface is covered by large flat type I alveolar epithelial cells (AECIs) and small fraction of type II alveolar epithelial cells (AECIIs). The former are the main cell type to mediate lung gas exchange function based on their location in approximation to the pulmonary capillary endothelium, while the latter are responsible for the biosynthesis of pulmonary surfactant and for the maintenance of alveolar integrity due to its capability in cellular division [50-53]. Early reports suggest that ER stress-induced epithelial cell dysfunction is perceived as a considerable aspect in the pathophysiology of IPF.
AEC apoptosis
Several lines of evidence convincingly show that AECs appear to be particularly sensitive to apoptosis following lung injury in IPF [54-56]. Increasing data support that apoptosis of AECs is considered as a key incident initiating and propagating pulmonary fibrosis in the lung parenchyma [57-59]. Indeed, ER stress in AECs can be induced by numerous stimuli, among which exon4 deletion and L188Q substitution of SFTPC as well as SFTPA2 are classically identified in IPF patients [46,60-63], and AECs are more prone to apoptosis after bleomycin treatment. In 2008, studies conducted by Mulugeta and colleagues for the first time provided evidence that a severe ER stress in AECIIs lining the areas of fibrosis was likely to underlie the execution of the intrinsic apoptosis pathway in patients with IPF [64]. Intriguingly, herpesvirus proteins (CMV, EBV, and KSHV) could be also detected in the same AECs that show evidence of ER stress and UPR activation, implicating a potential role of herpesviruses in IPF progression through induction of this pathway [65]. However, no typical IPF characteristics can be noted under conditions that AECIIs only possess the L188Q substitution of SFTPC or treat with tunicamycinalone [66]. Other than the classical bleomycin-induced pulmonary fibrosis animal model, the murine model for amiodarone (AD)-induced lung fibrosis has also revealed that intratracheal aerosol administration of AD causes an interstitial fibrosis in C57Bl/6 mice accompanied by increased AEC apoptosis, lysosomal stress and ER stress [67]. Furthermore, treatment of A549 cells, a human lung adenocarcinoma epithelial cell line, with amosite asbestos fibers induces AEC ER stress as evidenced by the increased expression of ER stress-related proteins (IRE-1, spliced XBP1, and CHOP), and similarly, ER Ca2+ release along with intrinsic apoptosis were manifested in rat primary-isolated AECIIs following the treatment [68].
As noted above, IPF is a hard-to-diagnose fatal interstitial lung disease with a poor response to traditional therapies, and therefore, early diagnostic biomarkers are urgently needed. There is evidence that circulating caspase-cleaved cytokeratin-18 (Cck-18), a cytoskeletal protein originally found in pseudostratified and simple epithelia [69], could serve as a marker of AEC apoptosis and UPR activation in IPF patients, and elevated circulating levels of Cck-18 in patients also portrays it as a useful IPF diagnostic biomarker [70]. More recently, studies revealed that ER stress-induced intrinsic apoptosis in human AECs is mediated by the generation of angiotensin (ANG) II, which can be attenuated by its counter regulatory antiapoptotic peptide ANG1-7 [71], implicating ANG1-7 possibly held therapeutic potential for the treatment of ER stress-induced pulmonary fibrosis [72]. Similarly, inhibition of CaMKII [73] and inhibitor of synoviolin (LS-102) [74] have been found to provide protection against pulmonary fibrosis through suppressing ER stress and AEC apoptosis. Moreover, DNA damage in AECIIs caused by bleomycin or doxorubicin, and ER stress caused by tunicamycin, upregulate the expression of osteopontin (OPN) in AECIIs in an ERK-dependent manner, indicating the probable function of OPN as a survival factor for AECIIs during the early stage of IPF [75]. As the massive target spots emerge, due to several critical shortages such as insufficiency in sample capacity and the lack of robust clinical trials, further studies are still required in order to confirm these therapeutic potentials.
Epithelial-to-mesenchymal transition (EMT)
EMT is defined as a process by which epithelial cells lose their cellular polarity and cell to cell adhesion, and then gradually convert to mesenchymal stem cells following activation by specific growth factors, of which TGF-β is the prototype [19]. Recently, EMT, in which AECs undergo transition to ECM-producing (myo)fibroblasts, has received intense attention in the pathogenesis of IPF. The response to TGF-β in EMT during pulmonary fibrosis is mediated predominantly via Smad-dependent pathways, although non-Smad signaling pathways have also been discovered under certain conditions [76]. Upon TGF-β stimulation, the complex composed of phosphorylated Smad2, Smad3 and Smad4 translocate into the nucleus, where it binds to transcriptional factors and modulate transcriptional responses. As a result, the inhibitory Smad, Smad7, can reverse fibrosis in renal and lens epithelia in a Smad-dependent manner [76-79]. In contrast, the mitogen-activated protein kinase (p38MAPK), Src family protein kinases, phosphatidylinositol 3’-OH kinase (PI3K/Akt), Rho/Rac, Wnt/β-catenin, as well as ERK have been suggested implicated in TGF-β-induced non-Smad-dependent EMT [76,80,81]. However, the distinction between Smad-dependent and nondependent pathways is difficult due to their significant cross talk between each other.
There is emerging evidence that accumulation of mutant SP-C protein or thapsigargin- or tunicamycin-induced ER stress in A549 and RLE-6TN cells contributes to pulmonary fibrosis through EMT at least in part dependent of Smad2/3 or Src-related pathways [82,83]. It has also been noted that the IRE1/XBP1 pathway promotes EMT by means of mediating snail expression in pulmonary fibrosis [84]. Furthermore, Bax inhibitor-1 (Bl-1) [85], an anti-apoptosis protein capable of inhibiting Bax activation [86], phenylbutyric acid (PBA), a low molecular weight fatty acid [87], and melatonin, a potent antioxidant mainly secreted by pineal gland [88], are also found to inhibit ER stress and EMT during bleomycin-induced pulmonary fibrosis in mice. They regulate the Ca2+ dynamic status and the expression of calnexin, and thereby modulating NF-κB signaling, and attenuating the activation of ER stress-related proteins. Collectively, ER stress is likely implicated in EMT during IPF development, additional studies, however, would be necessary to reach a conclusive remark.
Macrophages
Macrophages are present in almost all tissues of the body and play a critical role in innate and adaptive immunity in response to the change of microenvironment [89]. Generally, pulmonary macrophages can be divided into alveolar macrophages (AMs) that strategically distributed in the airways, and interstitial macrophages (IMs) that positioned in the lung parenchymal tissues [90,91]. Macrophages that mainly produce pro-inflammatory cytokines are called classically activated macrophages (M1), which can be activated either by IFN-γ or LPS [92], whereas those activated by IL-4 or IL-13 associated with attenuation of inflammation and enhancement of tissue repair are referred as alternatively activated macrophages (M2) [93,94]. During activated immune responses of IPF, different classes of macrophages with distinct functions are now recognized.
Over past few years, our understanding of the mechanisms underlying pulmonary fibrosis has been further expanded by the discovery of ER stress in pulmonary macrophages [95]. It was initially noted that mesenchymal stem cells (MSCs) hold promise as a novel treatment in IPF through the secretion of stanniocalcin-1 (STC1). Specifically, STC1 regulates oxidative and ER stress along with reduced TGF-β production in pulmonary macrophages to attenuate fibrosis in the lung [96]. Indeed, it has been well recognized that predominant infiltration of M2 macrophages in the areas of lung fibrosis acts as a vital regulator of fibrogenesis during IPF development and progression [97-99]. Similarly, the role of IL-4 and IL-13-mediated signaling in M2 macrophage polarization has been well established both in vitro and in vivo [100]. IL-4 receptor-α (IL-4Rα) signals through a JAK-STAT6 pathway to regulate the expression of numerous geneses sential for M2 polarization such as arginase 1 (Arg1), macrophage mannose receptor 1 (also known as Cd206), resistin-like-α (also known as Fizz1) and chitinase 3-like 3 (also known as Ym1) [101,102]. IL-4 may also induce the activation of phosphoinositide 3-kinase (PI3K), as evidenced by that phosphatidylinositol-3,4,5-trisphosphate, a product of PI3K, can be dephosphorylated by the phosphatase SHIP, and mice deficient in Ship manifest impaired M2 polarization [103].
Interestingly, recent evidence indicates that ER stress probably modulates the activation of M2 macrophages [104]. During the past few years, our laboratory has been focused on the effect of ER stress on fibrogenesis. In a model of unilateral ureteral obstruction (UUO)-induced renal fibrosis, we first noted that mice deficient in Chop were protected from UUO-induced renal fibrosis [105]. We demonstrated evidence that Chop deficiency provides protection for tubular cells against UUO-induced apoptosis and secondary necrosis along with attenuated Hmgb1 passive release and active secretion. As a result, loss of Chop repressed Hmgb1/TLR4/NF-κB signaling, thereby inhibiting UUO-induced IL-1β production. Subsequently, the IL-1β downstream Erk1/2 activity and its related c-Jun activity were reduced, which led to attenuated production of TGF-β1 along with repressed renal fibrosis following UUO insult. Consistent results were reported by Tanaka and colleagues [106]. We, therefore, next expanded our discoveries into the pathogenesis of IPF [107]. We first demonstrated evidence that pulmonary fibrosis manifests altered Chop expression and ER stress in both IPF patients and animals with bleomycin-induced pulmonary fibrosis. In consistent with these observations, mice deficient in Chop were protected from bleomycin-induced lung injury and fibrosis. Specifically, loss of Chop significantly attenuated TGF-β production along with reduced M2 macrophage infiltration in the lung following bleomycin induction. Mechanistic studies revealed that Chop deficiency suppressed M2 program in macrophages, which then attenuated TGF-β secretion. Loss of Chop enhanced the expression of SOCS1 and SOCS3, thereby inhibiting STAT6/PPARγ signaling that is essential for macrophage M2 program [107]. In sharp contrast to the above reported data, Ayauband colleagues held the viewpoint that Chop plays a role in bleomycin triggered macrophage apoptosis, which then protects Grp78+/- micefrom bleomycin-induced lung injury and fibrosis [108]. Nevertheless, no matter what point of view is more persuasive, both of which suggested the implication of ER stress in modulating macrophage polarization, which contributes to the pathogenesis of pulmonary fibrosis.
(Myo)fibroblasts
New discoveries in rodent model have revealed that mesenchymal fibroblasts are essential for forming vascular network, sensing damage, recruiting inflammatory cells, as well as remodeling the extracellular matrix of body organs, which are beneficial by maintaining physiological tissue homeostasis. However, injuries, infections and cellular damage trigger differentiation of fibroblasts into activated myofibroblasts that drive pathological inflammation and excessive extracellular matrix deposition, ultimately leading to tissue fibrosis with progressive scarring [109].
The pathologic hallmarks of IPF are the activation and proliferation of fibroblast-like cells and differentiation of myofibroblasts in the lung tissues [110], which not only arise from lung resident fibroblasts, but also derive from circulating fibrocytes and bone marrow-derived progenitor cells [111-113]. Fibroblast migration, proliferation, myofibroblast differentiation and ECM accumulation, are triggered by epithelial cell dysfunction and aberrant epithelial-mesenchymal signaling, and regulated by various cytokines, especially TGF-β [112]. Although advances have been made in understanding of the pathogenesis in (myo)fibroblasts during IPF, many crucial mechanisms underlying disease etiologies remain unclear.
In 2012, Baek and colleagues reported that altered GRP78 expression was noted not only in AECs but also in fibroblasts in lung tissues from IPF patients [114], implying ER stress may be also critical in fibroblastic differentiation over the evolvement of pulmonary fibrosis. Indeed, studies both in mouse and human fibroblasts revealed that TGF-β1 substantially facilitated the production of ER stress-associated proteins (GRP78, XBP-1, and ATF6α) along with high levels of α-SMA and collagen type I expression. In line with these crosslinks, the 150-kDa oxygen-regulated protein (ORP150), one of ER chaperones, could promote bleomycin-induced pulmonary fibrosis via augmenting pulmonary levels of TGF-β1 and myofibroblasts [115]. In support of this notion, mouse embryonic fibroblasts (MEFs) isolated from Crt-/- mouse and human IPF lung fibroblasts with knockdown of CRT by siRNA impaired TGF-β-induced collagen trafficking and matrix assembly in a Ca2+-dependent manner [116]. Together, those data establish a novel mechanism in which ER stress modulates fibroblast proliferation and myofibroblast differentiation contributing to the development of pulmonary fibrosis.
Summary and perspectives
We reviewed recent studies relevant to major profibrotic cells in lung tissues and collected evidence that ER stress may act as a critical player during the initiation and progression of pulmonary fibrosis (Figure 3). Those studies demonstrated feasibility that targeting ER stress could be a viable therapeutic strategy against IPF in clinical settings. While these discoveries are exciting, many questions remain unsolved regarding the exact mechanisms of ER stress and UPR pathway in IPF pathogenesis. However, the development of therapeutic agents that interfere with specific components of the UPR pathway would be useful tools to fully establish the role of ER stress in IPF pathoetiology, which would ultimately pave new therapeutic avenues for this devastating disorder.
Figure 3.
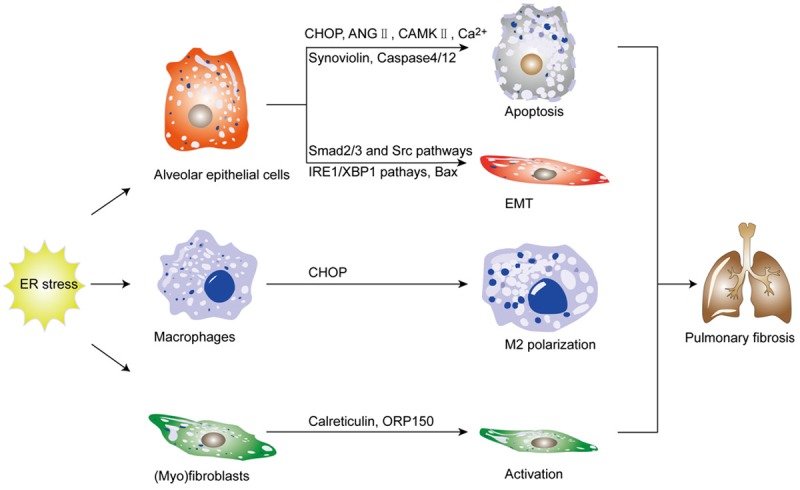
ER stress modulates the function and phenotype of profibrotic cells during the development of pulmonary fibrosis. This schematic paradigm demonstrates how ER stress modulates the function and phenotype of profibrotic cells contributing to IPF pathogenesis. It is believed that ER stress can drive alveolar epithelial cell apoptosis, epithelial-to-mesenchymal transition (EMT), macrophages polarization, and (myo)fibroblasts activation, by which it promotes pulmonary fibrosis.
Acknowledgements
Our work is supported by the National Natural Science Foundation of China (81530024, 81471046, 81130014 and 81470988), the Chinese Ministry of Science & Technology (2016YFC1305002), and the Innovative Funding for Translational Research from Tongji Hospital.
Disclosure of conflict of interest
None.
References
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 4.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61:980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartert M, Senbaklavacin O, Gohrbandt B, Fischer BM, Buhl R, Vahld CF. Lung transplantation: a treatment option in end-stage lung disease. Dtsch Arztebl Int. 2014;111:107–116. doi: 10.3238/arztebl.2014.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Back SH, Lee K, Vink E, Kaufman RJ. Cytoplasmic IRE1 alpha-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J Biol Chem. 2006;281:18691–18706. doi: 10.1074/jbc.M602030200. [DOI] [PubMed] [Google Scholar]
- 7.Asselah T, Bieche I, Mansouri A, Laurendeau I, Cazals-Hatem D, Feldmann G, Bedossa P, Paradis V, Martinot-Peignoux M, Lebrec D, Guichard C, Ogier-Denis E, Vidaud M, Tellier Z, Soumelis V, Marcellin P, Moreau R. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J Pathol. 2010;221:264–274. doi: 10.1002/path.2703. [DOI] [PubMed] [Google Scholar]
- 8.Ayala P, Montenegro J, Vivar R, Letelier A, Aranguiz Urroz P, Copaja M, Pivet D, Humeres C, Troncoso R, Miguel Vicencio J, Lavandero S, Diaz-Araya G. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp Mol Pathol. 2012;92:97–104. doi: 10.1016/j.yexmp.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 9.Bertolotti A, Zhang YH, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 10.Bonner JC. Lung fibrotic responses to particle exposure. Toxicol Pathol. 2007;35:148–153. doi: 10.1080/01926230601060009. [DOI] [PubMed] [Google Scholar]
- 11.Bosio A, Knorr C, Janssen U, Gebel S, Haussmann HJ, Muller T. Kinetics of gene expression profiling in Swiss 3T3 cells exposed to aqueous extracts of cigarette smoke. Carcinogenesis. 2002;23:741–748. doi: 10.1093/carcin/23.5.741. [DOI] [PubMed] [Google Scholar]
- 12.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 13.Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knipe RS, Tager AM, Liao JK. The Rho kinases: critical mediators of multiple profibrotic processes and rational targets for new therapies for pulmonary fibrosis. Pharmacol Rev. 2015;67:103–117. doi: 10.1124/pr.114.009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–179. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aoki Y, Maeno T, Aoyagi K, Ueno M, Aoki F, Aoki N, Nakagawa J, Sando Y, Shimizu Y, Suga T, Arai M, Kurabayashi M. Pioglitazone, a peroxisome proliferator-activated receptor gamma ligand, suppresses bleomycin-induced acute lung injury and fibrosis. Respiration. 2009;77:311–319. doi: 10.1159/000168676. [DOI] [PubMed] [Google Scholar]
- 17.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 18.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr Opin Pulm Med. 2012;18:517–523. doi: 10.1097/MCP.0b013e3283566721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis--a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014;10:390–402. doi: 10.1038/nrrheum.2014.53. [DOI] [PubMed] [Google Scholar]
- 22.Strieter RM, Keeley EC, Hughes MA, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J Leukoc Biol. 2009;86:1111–1118. doi: 10.1189/jlb.0309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maharaj SS, Baroke E, Gauldie J, Kolb MR. Fibrocytes in chronic lung disease--facts and controversies. Pulm Pharmacol Ther. 2012;25:263–267. doi: 10.1016/j.pupt.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 26.Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- 27.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol. 2013;14:630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bi MX, Naczki C, Koritzinsky M, Fels D, Blais J, Hu NP, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG, Koumenis C. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. Embo J. 2005;24:3470–3481. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bialik S, Kimchi A. DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin Cancer Biol. 2004;14:283–294. doi: 10.1016/j.semcancer.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. 2008;118:3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu Y, Mao T, Zhang Y, Shao M, You J, Ding Q, Chen Y, Wu D, Xie D, Lin X, Gao X, Kaufman RJ, Li W, Liu Y. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1alpha activation in pancreatic beta cells. Sci Signal. 2010;3:ra7. doi: 10.1126/scisignal.2000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bettigole SE, Lis R, Adoro S, Lee AH, Spencer LA, Weller PF, Glimcher LH. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol. 2015;16:829–837. doi: 10.1038/ni.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 37.Tashiro E, Hironiwa N, Kitagawa M, Futamura Y, Suzuki S, Nishio M, Imoto M. Trierixin, a novel Inhibitor of ER stress-induced XBP1 activation from Streptomyces sp. 1. Taxonomy, fermentation, isolation and biological activities. J Antibiot (Tokyo) 2007;60:547–553. doi: 10.1038/ja.2007.69. [DOI] [PubMed] [Google Scholar]
- 38.Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol. 2005;79:6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 40.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 41.Tanjore H, Lawson WE, Blackwell TS. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta. 2013;1832:940–947. doi: 10.1016/j.bbadis.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016;16:469–484. doi: 10.1038/nri.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tessitore A, del P Martin M, Sano R, Ma Y, Mann L, Ingrassia A, Laywell ED, Steindler DA, Hendershot LM, d’Azzo A. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell. 2004;15:753–766. doi: 10.1016/j.molcel.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 44.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2 alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol. 2001;21:5018–5030. doi: 10.1128/MCB.21.15.5018-5030.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 47.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 49.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L721–729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res. 2001;2:33–46. doi: 10.1186/rr36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rozycki HJ. Potential contribution of type I alveolar epithelial cells to chronic neonatal lung disease. Front Pediatr. 2014;2:45. doi: 10.3389/fped.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao Z, Lis R, Ginsberg M, Chavez D, Shido K, Rabbany SY, Fong GH, Sakmar TP, Rafii S, Ding BS. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat Med. 2016;22:154–162. doi: 10.1038/nm.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mulugeta S, Nureki S, Beers MF. Lost after translation: insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2015;309:L507–525. doi: 10.1152/ajplung.00139.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 55.Gunther A, Lubke N, Ermert M, Schermuly RT, Weissmann N, Breithecker A, Markart P, Ruppert C, Quanz K, Ermert L, Grimminger F, Seeger W. Prevention of bleomycin-induced lung fibrosis by aerosolization of heparin or urokinase in rabbits. Am J Respir Crit Care Med. 2003;168:1358–1365. doi: 10.1164/rccm.2201082. [DOI] [PubMed] [Google Scholar]
- 56.Kuwano K. Involvement of epithelial cell apoptosis in interstitial lung diseases. Intern Med. 2008;47:345–353. doi: 10.2169/internalmedicine.47.0713. [DOI] [PubMed] [Google Scholar]
- 57.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta. 2013;1832:1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cui Y, Robertson J, Maharaj S, Waldhauser L, Niu J, Wang J, Farkas L, Kolb M, Gauldie J. Oxidative stress contributes to the induction and persistence of TGF-beta 1 induced pulmonary fibrosis. Int J Biochem Cell Biol. 2011;43:1122–1133. doi: 10.1016/j.biocel.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 59.Lekkerkerker AN, Aarbiou J, van Es T, Janssen RA. Cellular players in lung fibrosis. Curr Pharm Des. 2012;18:4093–4102. doi: 10.2174/138161212802430396. [DOI] [PubMed] [Google Scholar]
- 60.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A. 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mulugeta S, Maguire JA, Newitt JL, Russo SJ, Kotorashvili A, Beers MF. Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am J Physiol Lung Cell Mol Physiol. 2007;293:L720–L729. doi: 10.1152/ajplung.00025.2007. [DOI] [PubMed] [Google Scholar]
- 62.Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol. 2005;32:521–530. doi: 10.1165/rcmb.2005-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem. 2010;285:22103–22113. doi: 10.1074/jbc.M110.121467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, Weaver TE, Guenther A. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 66.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A. 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mahavadi P, Henneke I, Ruppert C, Knudsen L, Venkatesan S, Liebisch G, Chambers RC, Ochs M, Schmitz G, Vancheri C, Seeger W, Korfei M, Guenther A. Altered surfactant homeostasis and alveolar epithelial cell stress in amiodarone-induced lung fibrosis. Toxicol Sci. 2014;142:285–297. doi: 10.1093/toxsci/kfu177. [DOI] [PubMed] [Google Scholar]
- 68.Kamp DW, Liu G, Cheresh P, Kim SJ, Mueller A, Lam AP, Trejo H, Williams D, Tulasiram S, Baker M, Ridge K, Chandel NS, Beri R. Asbestos-induced alveolar epithelial cell apoptosis. The role of endoplasmic reticulum stress response. Am J Respir Cell Mol Biol. 2013;49:892–901. doi: 10.1165/rcmb.2013-0053OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leers MP, Kolgen W, Bjorklund V, Bergman T, Tribbick G, Persson B, Bjorklund P, Ramaekers FC, Bjorklund B, Nap M, Jornvall H, Schutte B. Immunocytochemical detection and mapping of a cytokeratin 18 neo-epitope exposed during early apoptosis. J Pathol. 1999;187:567–572. doi: 10.1002/(SICI)1096-9896(199904)187:5<567::AID-PATH288>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 70.Cha SI, Ryerson CJ, Lee JS, Kukreja J, Barry SS, Jones KD, Elicker BM, Kim DS, Papa FR, Collard HR, Wolters PJ. Cleaved cytokeratin-18 is a mechanistically informative biomarker in idiopathic pulmonary fibrosis. Respir Res. 2012;13:105. doi: 10.1186/1465-9921-13-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uhal BD, Li X, Xue A, Gao X, Abdul-Hafez A. Regulation of alveolar epithelial cell survival by the ACE-2/angiotensin 1-7/Mas axis. Am J Physiol Lung Cell Mol Physiol. 2011;301:L269–L274. doi: 10.1152/ajplung.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uhal BD, Nguyen H, Dang M, Gopallawa I, Jiang J, Dang V, Ono S, Morimoto K. Abrogation of ER stress-induced apoptosis of alveolar epithelial cells by angiotensin 1-7. Am J Physiol Lung Cell Mol Physiol. 2013;305:L33–41. doi: 10.1152/ajplung.00001.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Winters CJ, Koval O, Murthy S, Allamargot C, Sebag SC, Paschke JD, Jaffer OA, Carter AB, Grumbach IM. CaMKII inhibition in type II pneumocytes protects from bleomycin-induced pulmonary fibrosis by preventing Ca2+-dependent apoptosis. Am J Physiol Lung Cell Mol Physiol. 2016;310:L86–94. doi: 10.1152/ajplung.00132.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakajima F, Aratani S, Fujita H, Yagishita N, Ichinose S, Makita K, Setoguchi Y, Nakajima T. Synoviolin inhibitor LS-102 reduces endoplasmic reticulum stress-induced collagen secretion in an in vitro model of stress-related interstitial pneumonia. Int J Mol Med. 2015;35:110–116. doi: 10.3892/ijmm.2014.1984. [DOI] [PubMed] [Google Scholar]
- 75.Kato A, Okura T, Hamada C, Miyoshi S, Katayama H, Higaki J, Ito R. Cell stress induces upregulation of osteopontin via the ERK pathway in type II alveolar epithelial cells. PLoS One. 2014;9:e100106. doi: 10.1371/journal.pone.0100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 77.Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saika S, Ikeda K, Yamanaka O, Flanders KC, Ohnishi Y, Nakajima Y, Muragaki Y, Ooshima A. Adenoviral gene transfer of BMP-7, Id2, or Id3 suppresses injury-induced epithelial-to-mesenchymal transition of lens epithelium in mice. Am J Physiol Cell Physiol. 2006;290:C282–289. doi: 10.1152/ajpcell.00306.2005. [DOI] [PubMed] [Google Scholar]
- 79.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 80.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 81.Ulianich L, Garbi C, Treglia AS, Punzi D, Miele C, Raciti GA, Beguinot F, Consiglio E, Di Jeso B. ER stress is associated with dedifferentiation and an epithelial-to-mesenchymal transition-like phenotype in PC Cl3 thyroid cells. J Cell Sci. 2008;121:477–486. doi: 10.1242/jcs.017202. [DOI] [PubMed] [Google Scholar]
- 82.Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, Blackwell TS. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2015;290:3277. doi: 10.1074/jbc.A110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhong Q, Zhou B, Ann DK, Minoo P, Liu Y, Banfalvi A, Krishnaveni MS, Dubourd M, Demaio L, Willis BC, Kim KJ, duBois RM, Crandall ED, Beers MF, Borok Z. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am J Respir Cell Mol Biol. 2011;45:498–509. doi: 10.1165/rcmb.2010-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mo XT, Zhou WC, Cui WH, Li DL, Li LC, Xu L, Zhao P, Gao J. Inositol-requiring protein 1 - X-box-binding protein 1 pathway promotes epithelial-mesenchymal transition via mediating snail expression in pulmonary fibrosis. Int J Biochem Cell Biol. 2015;65:230–238. doi: 10.1016/j.biocel.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 85.Lee MR, Lee GH, Lee HY, Kim DS, Chung MJ, Lee YC, Kim HR, Chae HJ. BAX inhibitor-1-associated V-ATPase glycosylation enhances collagen degradation in pulmonary fibrosis. Cell Death Dis. 2014;5:e1113. doi: 10.1038/cddis.2014.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reimers K, Choi CY, Bucan V, Vogt PM. The Bax Inhibitor-1 (BI-1) family in apoptosis and tumorigenesis. Curr Mol Med. 2008;8:148–156. doi: 10.2174/156652408783769562. [DOI] [PubMed] [Google Scholar]
- 87.Zhao H, Qin HY, Cao LF, Chen YH, Tan ZX, Zhang C, Xu DX. Phenylbutyric acid inhibits epithelial-mesenchymal transition during bleomycin-induced lung fibrosis. Toxicol Lett. 2015;232:213–220. doi: 10.1016/j.toxlet.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 88.Zhao H, Wu QQ, Cao LF, Qing HY, Zhang C, Chen YH, Wang H, Liu RY, Xu DX. Melatonin inhibits endoplasmic reticulum stress and epithelial-mesenchymal transition during bleomycin-induced pulmonary fibrosis in mice. PLoS One. 2014;9:e97266. doi: 10.1371/journal.pone.0097266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Varin A, Gordon S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology. 2009;214:630–641. doi: 10.1016/j.imbio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 90.Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70:1189–1196. doi: 10.1136/thoraxjnl-2015-207020. [DOI] [PubMed] [Google Scholar]
- 91.Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: A new therapeutic pathway in fibrosing lung disease? Trends Mol Med. 2016;22:303–316. doi: 10.1016/j.molmed.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 92.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mills CD. M1 and M2 macrophages: Oracles of health and disease. Crit Rev Immunol. 2012;32:463–488. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 94.Rhee I. Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res. 2016;39:1588–1596. doi: 10.1007/s12272-016-0820-y. [DOI] [PubMed] [Google Scholar]
- 95.Ryan AJ, Larson-Casey JL, He C, Murthy S, Carter AB. Asbestos-induced disruption of calcium homeostasis induces endoplasmic reticulum stress in macrophages. J Biol Chem. 2014;289:33391–33403. doi: 10.1074/jbc.M114.579870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ono M, Ohkouchi S, Kanehira M, Tode N, Kobayashi M, Ebina M, Nukiwa T, Irokawa T, Ogawa H, Akaike T, Okada Y, Kurosawa H, Kikuchi T, Ichinose M. Mesenchymal stem cells correct inappropriate epithelial-mesenchyme relation in pulmonary fibrosis using stanniocalcin-1. Mol Ther. 2015;23:549–560. doi: 10.1038/mt.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tao B, Jin W, Xu J, Liang Z, Yao J, Zhang Y, Wang K, Cheng H, Zhang X, Ke Y. Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J Immunol. 2014;193:2801–2811. doi: 10.4049/jimmunol.1303463. [DOI] [PubMed] [Google Scholar]
- 98.Gharib SA, Johnston LK, Huizar I, Birkland TP, Hanson J, Wang Y, Parks WC, Manicone AM. MMP28 promotes macrophage polarization toward M2 cells and augments pulmonary fibrosis. J Leukoc Biol. 2014;95:9–18. doi: 10.1189/jlb.1112587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 101.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 102.Brombacher F, Arendse B, Peterson R, Holscher A, Holscher C. Analyzing classical and alternative macrophage activation in macrophage/neutrophil-specific IL-4 receptor-alpha-deficient mice. Methods Mol Biol. 2009;531:225–252. doi: 10.1007/978-1-59745-396-7_15. [DOI] [PubMed] [Google Scholar]
- 103.Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, Huxham L, Minchinton AI, Mui A, Krystal G. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–374. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 104.Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, Cella M, Bernal-Mizrachi C. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem. 2012;287:11629–11641. doi: 10.1074/jbc.M111.338673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang M, Guo Y, Fu H, Hu S, Pan J, Wang Y, Cheng J, Song J, Yu Q, Zhang S, Xu JF, Pei G, Xiang X, Yang P, Wang CY. Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFkappaB/IL-1beta signaling. Cell Death Dis. 2015;6:e1847. doi: 10.1038/cddis.2015.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tanaka Y, Ishitsuka Y, Hayasaka M, Yamada Y, Miyata K, Endo M, Kondo Y, Moriuchi H, Irikura M, Tanaka K, Mizushima T, Oike Y, Irie T. The exacerbating roles of CCAAT/enhancer-binding protein homologous protein (CHOP) in the development of bleomycin-induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol Res. 2015;99:52–62. doi: 10.1016/j.phrs.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 107.Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, He X, Cheng Z, Ao Q, Cao Y, Yang P, Su Y, Zhao J, Zhang S, Yu Q, Ning Q, Xiang X, Xiong W, Wang CY, Xu Y. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. 2016;24:915–925. doi: 10.1038/mt.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ayaub EA, Kolb PS, Mohammed-Ali Z, Tat V, Murphy J, Bellaye PS, Shimbori C, Boivin FJ, Lai R, Lynn EG, Lhotak S, Bridgewater D, Kolb MR, Inman MD, Dickhout JG, Austin RC, Ask K. GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J Pathol. 2016;239:411–425. doi: 10.1002/path.4738. [DOI] [PubMed] [Google Scholar]
- 109.Barron L, Gharib SA, Duffield JS. Lung pericytes and resident fibroblasts: Busy multitaskers. Am J Pathol. 2016;186:2519–2531. doi: 10.1016/j.ajpath.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lama VN, Phan SH. The extrapulmonary origin of fibroblasts: stem/progenitor cells and beyond. Proc Am Thorac Soc. 2006;3:373–376. doi: 10.1513/pats.200512-133TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Andersson-Sjoland A, de Alba CG, Nihlberg K, Becerril C, Ramirez R, Pardo A, Westergren-Thorsson G, Selman M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–2140. doi: 10.1016/j.biocel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 112.Gharaee-Kermani M, Gyetko MR, Hu B, Phan SH. New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis: a potential role for stem cells in the lung parenchyma and implications for therapy. Pharm Res. 2007;24:819–841. doi: 10.1007/s11095-006-9216-x. [DOI] [PubMed] [Google Scholar]
- 113.Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–1321. doi: 10.1378/chest.06-2568. [DOI] [PubMed] [Google Scholar]
- 114.Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol. 2012;46:731–739. doi: 10.1165/rcmb.2011-0121OC. [DOI] [PubMed] [Google Scholar]
- 115.Tanaka KI, Shirai A, Ito Y, Namba T, Tahara K, Yamakawa N, Mizushima T. Expression of 150-kDa oxygen-regulated protein (ORP150) stimulates bleomycin-induced pulmonary fibrosis and dysfunction in mice. Biochem Biophys Res Commun. 2012;425:818–824. doi: 10.1016/j.bbrc.2012.07.158. [DOI] [PubMed] [Google Scholar]
- 116.Zimmerman KA, Graham LV, Pallero MA, Murphy-Ullrich JE. Calreticulin regulates transforming growth factor-beta-stimulated extracellular matrix production. J Biol Chem. 2013;288:14584–14598. doi: 10.1074/jbc.M112.447243. [DOI] [PMC free article] [PubMed] [Google Scholar]