

Abstract
Constitutional mismatch repair deficiency (CMMRD) is a devastating cancer predisposition syndrome for which clinical manifestations, genetic screening, and cancer prevention strategies are limited. We report a case of CMMRD presenting with metachronous colorectal cancer and brain cancer. Oncologists and gastroenterologists should be aware of the CMMRD syndrome as a rare cause of very early-onset colorectal cancer.
Introduction
Constitutional mismatch repair deficiency (CMMRD) is a rare and distinct cancer predisposition syndrome usually with onset in childhood or adolescence that results from biallelic germline mutations in 1 of the 4 mismatch repair (MMR) genes, MLH1, MSH2, MSH6, and PMS2.1 The tumor spectrum can be very broad, including hematological, brain, and gastrointestinal (GI) tract tumors. Patients may also have dermatological findings that are indicative of CMMRD.1
Case Report
A 30-year-old Hispanic male presented with a 2-month history of intermittent left lower quadrant abdominal pain, vomiting, and 10-kg weight loss. He reported a past history significant for colon cancer diagnosed at 13 years old in Mexico and right frontal glioblastoma at 26, both treated successfully. At that time, patient was not offered genetic counseling or colon cancer surveillance due to lack of resources and living in a rural area in Mexico. Besides surgical treatment, the patient did not recall any further information regarding location, stage, or type of colon cancer or whether polyps were present at the time of diagnosis. He reported no family history of cancer, although no information was available regarding his father or his side of the family.
Physical exam was remarkable for several café-au-lait (CAL) macules, axillary freckling, and abdominal tenderness. His hemoglobin was 5.5 g/dL and a fecal occult blood test was positive. Abdominal computed tomography revealed extensive concentric irregular thickening of the descending and sigmoid colon with multiple pericolonic lymph nodes and a 3.8 x 2.4 cm mass abutting the inferior mesenteric artery concerning for neoplastic disease (Figure 1).
Figure 1.
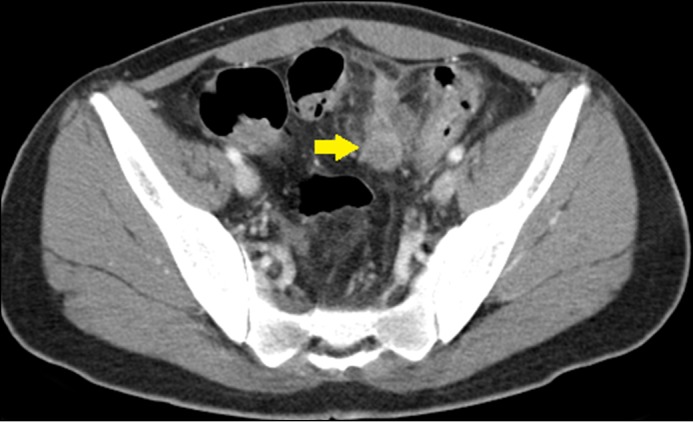
Sagittal computed tomography of the large bowel showing extensive thickening of the distal descending colon with necrotic lymph nodes (arrow).
Colonoscopy revealed a fungating, partially obstructing mass in the descending colon that could not be traversed and 17 polyps (5–30 mm in size) in the rectosigmoid and descending colon (Figure 2). The patient was found to have a poorly differentiated adenocarcinoma of the descending colon (T4aN1bM0) that was treated with palliative surgical resection, chemotherapy, and yearly colonoscopy for surveillance. All polyps were found to be tubular adenomas. Genetic testing (using a multi-gene panel) from a blood sample revealed a homozygous pathogenic mutation in PMS2 (c.137G>T), confirming the diagnosis of CMMRD syndrome. The patient is currently undergoing screening for other CMMRD-related malignancies, including gastric, small bowel, brain, and hematological malignancies.
Figure 2.
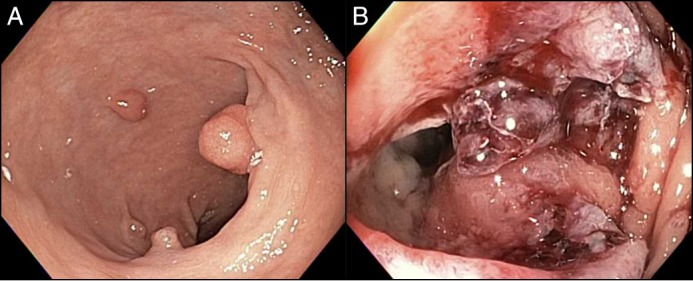
Colonoscopy showing (A) rectosigmoid polyposis and (B) a partially obstructing mass in the descending colon.
Discussion
CMMRD is a rare genetic syndrome caused by biallelic mutations in the MMR genes MLH1, MSH2, MSH6, and PMS2. These genes are in charge of recognizing and repairing erroneous insertion, deletion, and misincorporation of bases that can arise during DNA replication and recombination, as well as repairing some forms of DNA damage. Biallelic mutations in MMR genes are manifested by hematologic malignancies, brain tumors, and various Lynch syndrome (LS)-associated tumors, often occurring during childhood.1 The mean age of onset for hematologic malignancies and brain tumors is 5.5 years and 8 years, respectively.1 The mean age of onset for LS-associated cancers in CMMRD syndrome is 16 years, which is 30 years earlier than the average age of onset seen in LS.2
CMMRD patients who survive their first malignancy or in whom a premalignancy has been removed have a high risk of developing a second, and often different, (pre-)malignancy.3 It is important to determine the molecular cause of CMMRD to allow for predictive testing in at-risk relatives. Individuals with monoallelic MMR mutation in these families should follow LS screening recommendations.
The most frequent underlying gene defects were PMS2 mutations, which were reported in approximately 60% of cases.4 The remaining 40% of cases were equally distributed among MSH6 and MLH1/MSH2 biallelic mutation carriers.5 In general, heterozygous MLH1 and MSH2 mutations are associated with a higher penetrance of LS than heterozygous MSH6 mutations and, especially, PMS2 mutations. Thus, it is not surprising that none of the families with CMMRD patients due to biallelic PMS2 mutations had a clear family history of LS, although some of them showed an increased tumor incidence.1
Tumor tissue testing via immunohistochemistry, which is used to evaluate for LS, is often difficult to interpret in CMMRD cases. In our patient, immunohistochemistry analysis of the colon tissue revealed absent PMS2 staining in tumor and normal colon tissue, MSH6 staining in normal tissue but not in the tumor tissue, and MLH1 and MSH2 staining in both normal and tumor tissue. Genetic testing using a blood sample confirmed a homozygous pathogenic mutation in PMS2.
Another important feature is that the colonoscopy evaluation revealed multiple polyps, as previously reported in adults with CMMRD syndrome.6,7 Herkert et al described >10 adenomas in 18 of the 26 (70%) patients with colorectal cancer (CRC) and multiple CRC in 11 of 29 patients (38%) with CMMRD due to biallelic mutation of PMS2.8 An individual who presents with approximately 20 colonic adenomas could easily be misdiagnosed as having attenuated familial adenomatous polyposis (AFAP) or MUTYH-associated polyposis (MAP). However, CAL spots and skin-fold freckling led us to consider CMMRD syndrome as the most plausible etiology. Of the 78 cases reviewed by Wimmer and Etzler, 57 (73%) were found to have CAL spots.1 This highlights that individuals suspected of a hereditary polyposis or CRC condition should be examined for features suggestive of CMMRD, especially for CAL lesions. The mean age at diagnosis for CRC was 16.4 years (range 8–28 years), 30 years younger than the typical age at diagnosis of CRC in LS, and there was no predilection for a specific site in the colorectum.5-9
In patients with CMMRD with multiple polyps (too many to remove endoscopically and/or presenting with high-grade dysplasia) and patients with CRC, the treatment of choice is colectomy with ileorectal anastomosis or proctocolectomy and construction of an ileal pouch-anal anastomosis. Patients with stage III CRC (and advanced stage II CRC) receive chemotherapy, and those with rectal cancer are treated with radiotherapy. However, the effectiveness and toxicity of chemotherapy and radiotherapy in CMMRD is largely unknown.5 The current surveillance protocol for patients with CMMRD proposed by the European consortium is yearly ileocolonoscopy from age 8 years and virtual capsule endoscopy and upper GI endoscopy yearly from age 10 years, however, the value of surveillance and early detection of other tumors is still unknown.5
The clinical geneticist may play an important role by organizing presymptomatic testing of other family members of patients with CMMRD or LS, and through discussion of the option of prenatal or preimplementation genetic diagnosis. We suspect a biallelic mutation in one of the MMR genes MLH1, MSH2, MSH6, or PMS2 to be the underlying disease cause in children, adolescents, and young adults presenting with malignancies if they present with synchronous colon cancer, CAL lesions, and even negative family history for malignancy. Oncologists and gastroenterologists should be aware of CMMRD as a rare cause of very early-onset CRC. This case illustrates the importance of high suspicion for inherited and genetic-based colon cancer risk syndromes, early referral to a genetic counselor, and the importance of adequate surveillance for cancer prevention.
Disclosures
Author contributions: All authors designed the study, interpreted the data, wrote, and reviewed the article. NJ Samadder is the article guarantor.
Financial disclosure: None to report.
Informed consent was obtained for this case report.
References
- 1.Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: Have we so far seen only the tip of an iceberg? Hum Genet. 2008;(2):105–22. [DOI] [PubMed] [Google Scholar]
- 2.Stoffel E, Mukherjee B, Raymond VM, et al.. Calculation of risk of colorectal cancer among patients with Lynch syndrome. Gastroenterology. 2009; 137(5):1621–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wimmer K, Kratz CP, Vasen HFA, et al.. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium ‘Care for CMMRD’ (C4CMMRD). J Med Genet. 2014; 51(6):355–65. [DOI] [PubMed] [Google Scholar]
- 4.Van der Klift HM, Mensenkamp AR, Dros M, et al.. Comprehensive mutation analysis of PMS2 in a large cohort of probands suspected of Lynch syndrome or constitutional mismatch repair deficiency syndrome. Hum Mutat. 2016; 37(11):1162–79. [DOI] [PubMed] [Google Scholar]
- 5.Vasen HFA, Ghorbanoghli Z, Bourdeaut F, et al.. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium "Care for CMMR-D" (C4MMR-D). J Med Genet. 2014; 51(5):283–93. [DOI] [PubMed] [Google Scholar]
- 6.Jasperson KW, Samowitz WS, Burt RW. Constitutional mismatch repair-deficiency syndrome presenting as colonic adenomatous polyposis: Clues from the skin. Clin Genet. 2011; 80:394–7. [DOI] [PubMed] [Google Scholar]
- 7.Levi Z, Kariv R, Barnes-Kedar I, et al.. The gastrointestinal manifestation of constitutional mismatch repair deficiency syndrome: From a single adenoma to polyposis-like phenotype and early onset cancer. Clin Genet. 2015; 88(5):474–8. [DOI] [PubMed] [Google Scholar]
- 8.Herkert JC, Niessen RC, Olderode-Berends MJ, et al.. Paediatric intestinal cancer and polyposis due to bi-allelic PMS2 mutations: Case series, review and follow up guidelines. Eur J Cancer. 2011; 47(7):965–82. [DOI] [PubMed] [Google Scholar]
- 9.Durno CA, Holter S, Sherman PM, Gallinger S. The gastrointestinal phenotype of germline biallelic mismatch repair gene mutations. Am J Gastroenterol. 2010; 105(11):2449–56. [DOI] [PubMed] [Google Scholar]