

Abstract
The current WHO classification system for tumors of the head and neck has made few changes from the previous edition with regards to tumors of the nasopharynx. The classification system is discussed here with particular attention to nasopharyngeal carcinoma, nasopharyngeal papillary adenocarcinoma, salivary gland anlage tumor, hairy polyp, juvenile angiofibroma, and other tumors.
Keywords: Nasopharynx, Carcinoma, Squamous cell carcinoma, Papillary, Adenocarcinoma, Angiofibroma, Salivary gland, Anlage, Angiofibroma, WHO
Introduction
Owing to the restricted anatomic area of the nasopharynx and correlating limited number of disease/tumor entities, the 4th edition of the World Health Organization (WHO) Classification of Head and Neck Tumors has few changes (e.g., addition of new entities, substantial change in terminology, etc.) from the previous (3rd) edition. Indeed, only minor semantic changes, mentioned below, have been made.
Nasopharyngeal Carcinoma
In spite of the relatively non-specific nature of the appellation, “nasopharyngeal carcinoma” remains the diagnostic term of choice for all squamous cell carcinomas (SCCs) of the nasopharynx. The WHO then subtypes these cancers as non-keratinizing and keratinizing types, as well as basaloid SCC. Non-keratinizing tumors are further sub-categorized as undifferentiated or differentiated (Fig. 1). This classification system is in keeping with the previous (3rd) edition.
Fig. 1.

Nonkeratinizing nasopharyngeal carcinoma: These tumors are further sub-classified as differentiated (a) and undifferentiated (b) based upon whether epithelial differentiation is more or less apparent
There are fewer than 100,000 cases of nasopharyngeal carcinoma in the world every year [1]. The majority of these tumors arise in areas of southeastern Asia. Indeed, geographical incidence varies considerably as some southeastern Asian populations have an incidence rate of more than 15 per 100,000 person years whereas in the United States the incidence rate is less than 1 per 100,000 person years. This is believed to be caused by differences in population susceptibilities and exposures.
The most obvious differences regarding the disease reflect the tumors’ relationship to Epstein–Barr virus (EBV). In parts of the world with so-called “endemic” nasopharyngeal carcinoma, the vast majority (>95%) of cases are associated with EBV as can be shown by a variety of methodologies, most often in situ hybridization for EBV-encoded small RNAs (EBERs) [2]. It is believed that people from these areas are more genetically susceptible to the disease with a number of human leukocyte antigens (HLA) haplotypes being linked to the disease (e.g., HLA-A2, AW19, BW46 and B17) [3, 4]. Other genetic susceptibilities, such as mutations of the cytochrome P450s involved with metabolism of nitrosamines have been postulated [5]. Such theories are potentially credible as salted fish and other preserved foods are considered environmental risk factors for the disease [6]. It should be noted that while EBV appears necessary for the development of most non-keratinizing SCCs of the nasopharynx, viral infection in itself is insufficient to cause the malignancy.
EBV plays much less of a role in the development of keratinizing SCC, especially in areas where the malignancy is not considered to be endemic [2]. Keratinizing SCC appears to be related to smoking, akin to the keratinizing SCC at other sites within the upper aerodigestive tract [7]. Keratinizing SCC is more likely than non-keratinizing SCC to be locally advanced at its presentation and less likely to be metastatic to locoregional lymph nodes [8].
As keratinizing and non-keratinizing nasopharyngeal carcinomas develop in vastly different proportions in different parts of the world, present in patients with different ages and risk factors for the disease, and at different clinical stages, it is not hard to imagine that showing EBV or even keratinization as an independent prognostic factor has been difficult, although many have done so [8, 9]. Still, as studies have been mixed, there is not a consensus regarding the prognostic significance of the histologic subtype of nasopharyngeal carcinoma or of the tumors’ EBV status. The most powerful prognostic factor of nasopharyngeal carcinoma is stage at presentation [10].
It is perhaps due only the entrenchment of name “nasopharyngeal carcinoma” that the WHO did not suggest classification of the disease either by keratinization or EBV status (e.g., EBV-associated nasopharyngeal carcinoma). This is in contrast to high-risk human papillomavirus (HR-HPV) status in the oropharynx. At that site, however, HR-HPV status has been universally accepted to be a significant prognostic factor engendering consideration of using the designation HPV-associated carcinoma for such oropharyngeal cancers.
Finally, it should be noted that some SCCs centered in the nasopharynx are secondary to HR-HPV infection [11]. This should not be surprising as the majority of oropharyngeal SCCs are secondary to HR-HPV infection as well as approximately a quarter of sinonasal carcinomas. As at those sites, in the nasopharynx such SCCs are typically non-keratinizing and can appear similar to tumors associated with EBV infection. Differentiation is predicated on testing and identifying of HPV (e.g., p16 immunohistochemical staining, in situ hybridization and/or polymerase chain reaction) or EBV (e.g., EBER). Although the prognostic meaning of EBV viral infection at this site is debated (see above), some have suggested that the prognosis for HR-HPV-related tumors may be between that of EBV-related SCCs and those tumors not related to oncogenic viruses [11].
Other Tumors
Among malignant salivary gland tumors, adenoid cystic carcinoma is the most common type to occur in the nasopharynx, is histologically identical to its more common salivary gland counterpart, shows the presence of MYB-NFIB gene fusion, and shares overall similar biologic behavior. Two other epithelial tumors appear unique or relatively unique to the nasopharynx, nasopharyngeal papillary adenocarcinoma and salivary gland anlage tumor. A few other neoplasms or pseudoneoplasms occur solely or relatively commonly in the nasopharynx and are mentioned in this section of the classification system. These include hairy polyps, ectopic pituitary adenomas, and craniopharyngioma (discussed within the category of “other lesions”), as well as chordoma and juvenile angiofibroma (the sole lesions discussed within the sections devoted to “bone and cartilage tumors” and “soft tissue tumors,” respectively).
As the name implies, nasopharyngeal papillary adenocarcinomas occur in the nasopharynx and are papillary. They are, invariably, low grade. The tumors are very rare and represent less than 1% of nasopharyngeal malignancies. They occur over a wide age range (reported ages: 9–64 years), equally in both sexes, and can involve any part of the nasopharynx. Most patients present with obstruction, although rhinorrhea, bleeding and otitis media or hearing-related issues occur. Most patients have been treated with surgery alone, although some have also received radiation therapy. No patients have developed recurrences or metastases.
Nasopharyngeal papillary adenocarcinomas are composed of complex arborizing papillae with hyalinized fibrovascular cores and glands (Fig. 2). The lesions are invasive and typically involve the surface epithelium, focally merging with non-neoplastic epithelium. Papillae are lined by a single layer of cuboidal to columnar cells that have a moderate amount of eosinophilic cytoplasm. Nuclei vary from round to oval and have moderate membrane irregularity with vesicular to clear chromatin, similar to the nuclei seen with papillary thyroid carcinomas. Psammomatoid calcifications are seen in one-third of cases. Mitotic figures are uncommon and necrosis is rarely seen. Perineural and angiolymphatic invasion are not seen.
Fig. 2.
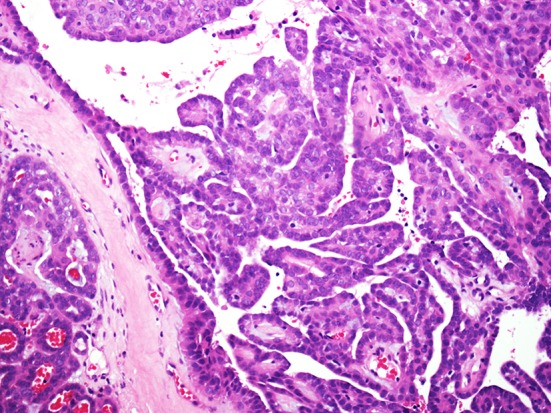
Nasopharyngeal papillary adenocarcinoma: these tumors are invariably low grade and composed of delicate arborizing papillae and tubules lined by a single layer of bland columnar to cuboidal epithelial cells
Tumours express EMA, CK5/6 and often CK7 [12, 13]. A subset of cases express CK19 and TTF1 and has been referred to as “thyroid-like low-grade nasopharyngeal papillary adenocarcinoma.” These cases do not express thyroglobulin. S100 protein expression is seen focally in many cases.
Salivary gland anlage tumor is a very rare neoplasm that typically arises on the posterior nasopharyngeal wall or posterior nasal septum [14, 15]. Tumors are usually diagnosed in patients less than 3 months of age, with boys affected approximately 3 times more commonly than girls. Tumors contain an admixture of epithelial and stromal elements (Fig. 3). Epithelial elements consist of both papillary and tubular gland proliferations with solid and cystic squamous nests. Stromal tissue consists of bland spindled cells arranged at variable cellularity. These lesions are benign and do not recur after resection. This tumor is now discussed within the section dedicated to salivary gland tumors rather than in the “benign epithelial tumors” section.
Fig. 3.

Salivary gland anlage tumor: this tumor appeared cellular and somewhat nodular from low power (a). Bland squamous nests and small cysts were present admixed with spindled cells (b) and tubules (c). Other areas were composed mostly of bland tubules with immature epithelial and stromal elements (d)
Hairy polyps are likely developmental abnormalities of the 1st or 2nd branchial cleft. They are thus considered by some to be accessory auricles [16]. The lesions predominately present in infants, but may occasionally be detected in older children and adolescents. The tumors present most frequently in the nasopharynx but may also present in the oropharynx or middle ear and Eustachian tube. Symptoms are secondary to local mass effect. The lesions are grossly polypoid. Histologically, they are covered by a dermis with keratinizing, stratified squamous epithelium and appendages. The underlying soft tissue consists of adipose tissue with admixed fibrous tissue. Mature cartilage and bone may also be present.
Pituitary adenomas can also present as nasopharyngeal masses, usually through the direct extension of tumor from the sella turcica or, rarely, through the development of tumor from presumed ectopic pituitary tissue (ectopic pituitary adenomas) [17]. The tumors typically present as mass lesions, however endocrine disturbances such as Cushing’s syndrome or acromegaly also may be present. The tumors typically have features of lower grade neuroendocrine epithelial neoplasms with nests, rosettes, ribbons and pseudoglandular structures composed of monomorphic epithelioid or plasmacytoid cells. Mitotic figures are usually infrequent and the chromatin pattern is typically granular. Nuclear atypia and nucleoli may sometimes be seen and tumors may even resemble poorly differentiated carcinomas. Immunohistochemistry can be helpful and most tumors (but not all) will react with antibodies to cytokeratins and endocrine antigens such as synaptophysin and chromogranin. Staining for specific pituitary hormones such as prolactin, ACTH, human growth hormone, luteinizing hormone, thyroid stimulating hormone, and follicle stimulating hormone can also be helpful. Any given tumor may express more than one pituitary hormone marker and may be referred to as plurihormonal (ectopic) pituitary adenoma, although a subset may not express any pituitary hormone markers and are referred to as (ectopic) null cell pituitary adenomas.
Rarely, craniopharyngiomas will present in the nasopharynx [18]. Patients are usually younger than 30 years old and will present with nonspecific findings, occasionally with visual problems. The tumors are composed of nests and cords of maturing squamous cells that keratinize and have a reticular appearance akin to that of ameloblastoma.
The WHO has changed the terminology from nasopharyngeal angiofibroma to juvenile angiofibroma. Juvenile angiofibromas are tumors of male adolescents [19–22]. They almost universally arise during the second decade of life consistent with the now widely accepted theory that they are androgen dependent. The tumors extend from the posterior lateral nasal wall of the nasopharynx and often lead to nasal obstruction. Epistaxis and nasal drainage are also typically seen. Nasopharyngeal angiofibromas may sometimes be locally aggressive and can extend into surrounding tissues. Most, however, will behave in a benign fashion, and only 20% will recur after resection and eventually those patients will be cured with more surgery. Patients with familial adenomatous polyposis (FAP) have been found to be 25 times more likely to have nasopharyngeal angiofibromas than the general public, implying that mutations of the adenomatous polyposis coli gene may be involved in the pathogenesis of these tumors [23, 24].
Grossly, juvenile angiofibromas are polypoid and firm [19–22]. Histologically, they are composed of a rich network of variably sized, irregularly shaped vessels within a low to moderately cellular stroma (Fig. 4). The vessel wall thickness can vary from a single layer of endothelium to vessels with multiple layers of smooth muscle that can appear hyalinized or “pad-like”. The vessels can be thrombosed either naturally or, in the case of resected specimens, with preoperatively injected material. The stromal tissue is composed mostly of thick and thin fibrils of collagen and can appear variably cellular. Numerous oval to spindled to stellate stromal cells are arranged haphazardly throughout the lesions. These cells are bland and have a small to moderate amount of eosinophilic cytoplasm and bland oval nuclei with fine chromatin and small, indistinct nucleoli. Occasional atypical and multinucleated cells can be seen, but mitotic figures are rare. The overlying respiratory-type epithelium can be intact or eroded and, when intact, may undergo squamous metaplasia. Background mast cells are invariably present, often in large numbers. Rarely, these tumors undergo sarcomatous “transformation,” almost invariably after the patients have received radiation therapy [25, 26].
Fig. 4.
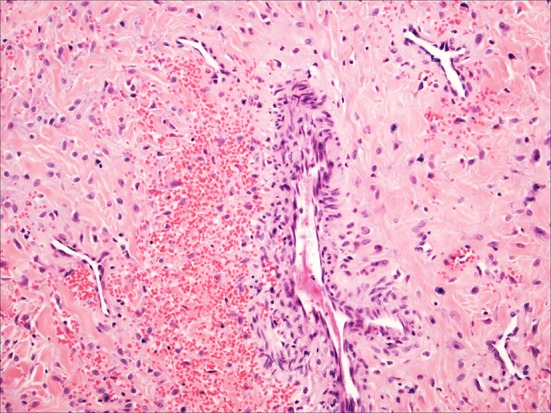
Juvenile Angiofibroma: a typical case with small delicate vessels, occasional larger more muscular vessels and bland spindled stromal cells
Immunohistochemically, the lesions show non-specific but expected findings [27]. The endothelial cells react with antibodies to typical endothelial antigens such as CD31, CD34 or factor VIII-related antigen. The focal smooth muscle surrounding these vessels typically shows immunoreactivity with antibodies to SMA and can react with antibodies to desmin. The stromal cells are usually noted to show immunoreactivity only with antibodies to vimentin. Androgen receptors have been identified by immunohistochemistry in both the stromal and endothelial cells, whereas most cases will show no immunoreactivity with antibodies to estrogen or progesterone receptors [28]. Supporting a possible relationship to FAP, nuclear localization of beta-catenin has also been reported [29].
Chordomas develop along the spinal axis and are believed by some to arise from notochordal remnants [30–34]. The tumors most often involve the sacrococcygeal areas, although up to one quarter develop at the base of the skull. As such, projection into the nasopharynx, nasal cavity or sinuses occurs in up to a quarter of these cases and the tumors may be sampled through these regions [31, 35].
Chordomas are more common in men and can develop at any age. In one large series of tumors involving the spheno-occipital area, the mean age at presentation was 38 years [31]. Patients with chordomas involving the base of the skull frequently present with diplopia or other visual defects. The tumors are notoriously difficult to resect in their entirety and frequently can recur, eventually leading to the death of their patients. Children with chordomas fare somewhat better, while patients with dedifferentiation within their tumors fare considerably worse [32, 36]. Newer treatments using proton beam therapy, allow for increased radiation doses to the tumor while reducing doses to surrounding tissues [37].
Grossly, chordomas are lobulated and gelatinous [30, 31]. Microscopically, tumors are composed of lobules of mucoid material and neoplastic cells separated by fibrous bands. The cellularity varies from lobule to lobule. Tumor cells are arranged singly, in cords or in sheets and are suspended in the myxoid substance. These cells vary in size and character. They range from smaller, more plasmacytoid cells with little or no cytoplasmic vacuolization to large, multivacuolated physalipherous cells. Cellular atypia and mitotic activity vary from case to case and some examples show frank anaplasia with numerous mitotic figures. In some tumors, the myxoid material focally becomes chondroid and the neoplastic cells reside in apparent lacunae (chondroid chordoma) [31, 38]. Areas of more obvious conventional chordoma are usually seen with these tumors. Rare chordomas are associated with areas of pleomorphic sarcoma (dedifferentiated chordoma), usually at the time of a later recurrence [36].
The neoplastic cells of chordoma are usually immunoreactive with antibodies to cytokeratins, EMA, S100 protein, brachyury and vimentin [33, 38, 39]. This unique immunoreactivity helps distinguish chordoma from both epithelial malignancies, such as mucinous carcinomas and mesenchymal neoplasms, especially cartilaginous tumors (i.e., chondrosarcoma). It is typically retained in both the conventional and chondroid areas of chordomas but is diminished in dedifferentiated areas [36].
Haematolymphoid tumors of the nasopharynx include neoplasms of lymphoid, plasma cell, or myeloid origin arising in the nasopharynx [40, 41]. Diffuse large B-cell lymphoma is most common, followed by NK/T-cell lymphoma and peripheral T-cell lymphoma, NOS. Other lymphomas are rare and may include MALT lymphoma, follicular lymphoma, Burkitt lymphoma, and mantle cell lymphoma and anaplastic large cell lymphoma. The tumors are discussed in depth elsewhere.
Summary
The nasopharynx is home to a limited number of tumor types with nasopharyngeal carcinoma being the most common. In the 4th edition of the WHO Classification of Head and Neck Tumors there are only minor changes in terminology compared with the 3rd edition.
Compliance with Ethical Standards
Conflict of interest
Drs. Wenig and Stelow have no conflict of interest.
Human and Animal Participants
This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Petersson F. Nasopharyngeal carcinoma: A review. Semin Diagn Pathol. 2015;32:54–73. doi: 10.1053/j.semdp.2015.02.021. [DOI] [PubMed] [Google Scholar]
- 2.Niedobitek G, Hansmann ML, Herbst H, Young LS, Dienemann D, Hartmann CA, Finn T, Pitteroff S, Welt A, Anagnostopoulos I, Friedrich R, Lobeck H, Sam CK, Araujo I, Rickinson AB, Stein H. Epstein–Barr-Virus and Carcinomas—undifferentiated carcinomas but not squamous-cell carcinomas of the nasopharynx are regularly associated with the virus. J Pathol. 1991;165:17–24. doi: 10.1002/path.1711650105. [DOI] [PubMed] [Google Scholar]
- 3.Goldsmith DB, West TM, Morton R. HLA associations with nasopharyngeal carcinoma in southern Chinese: a meta-analysis. Clin Otolaryngol. 2002;27:61–67. doi: 10.1046/j.0307-7772.2001.00529.x. [DOI] [PubMed] [Google Scholar]
- 4.Chan SH, Chew CT, Prasad U, Wee GB, Srinivasan N, Kunaratnam N. Hla and nasopharyngeal carcinoma in Malays. Brit J Cancer. 1985;51:389–392. doi: 10.1038/bjc.1985.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hildesheim A, Chen CJ, Caporaso NE, Cheng YJ, Hoover RN, Hsu MM, Levine PH, Chen IH, Chen JY, Yang CS, Daly AK, Idle JR. Cytochrome P4502e1 genetic polymorphisms and risk of nasopharyngeal carcinoma—results from a case-control study conducted in Taiwan. Cancer Epidemiol Biomar. 1995;4:607–610. [PubMed] [Google Scholar]
- 6.Zou XN, Lu SH, Liu B. Volatile N-nitrosamines and their precursors in Chinese salted fish—a possible etiologic factor for NPC in China. Int J Cancer. 1994;59:155–158. doi: 10.1002/ijc.2910590202. [DOI] [PubMed] [Google Scholar]
- 7.Ji XM, Zhang WD, Xie CH, Wang BC, Zhang G, Zhou FX. Nasopharyngeal carcinoma risk by histologic type in central China: impact of smoking, alcohol and family history. Int J Cancer. 2011;129:724–732. doi: 10.1002/ijc.25696. [DOI] [PubMed] [Google Scholar]
- 8.Reddy SP, Raslan WF, Gooneratne S, Kathuria S, Marks JE. Prognostic-significance of keratinization in nasopharyngeal carcinoma. Am J Otolaryngol. 1995;16:103–108. doi: 10.1016/0196-0709(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 9.Shanmugaratnam K, Chan SH, Dethe G, Goh JEH, Khor TH, Simons MJ, Tye CY. Histo-pathology of nasopharyngeal carcinoma—correlations with epidemiology, survival rates and other biological characteristics. Cancer. 1979;44:1029–1044. doi: 10.1002/1097-0142(197909)44:3<1029::AID-CNCR2820440335>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Sun XM, Su SF, Chen CY, Han F, Zhao C, Xiao WW, Deng XW, Huang SM, Lin CG, Lu TX. Long-term outcomes of intensity-modulated radiotherapy for 868 patients with nasopharyngeal carcinoma: an analysis of survival and treatment toxicities. Radiother Oncol. 2014;110:398–403. doi: 10.1016/j.radonc.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 11.Robinson M, Suh YE, Paleri V, Devlin D, Ayaz B, Pertl L, Thavaraj S. Oncogenic human papillomavirus-associated nasopharyngeal carcinoma: an observational study of correlation with ethnicity, histological subtype and outcome in a UK population. Infect Agents Cancer. 2013;8:30. doi: 10.1186/1750-9378-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wenig BM, Hyams VJ, Heffner DK. Nasopharyngeal papillary adenocarcinoma. a clinicopathologic study of a low-grade carcinoma. Am J Surg Pathol. 1988;12:946–953. doi: 10.1097/00000478-198812000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Pineda-Daboin K, Neto A, Ochoa-Perez V, Luna MA. Nasopharyngeal adenocarcinomas: a clinicopathologic study of 44 cases including immunohistochemical features of 18 papillary phenotypes. Ann Diagn Pathol. 2006;10:215–221. doi: 10.1016/j.anndiagpath.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Dehner LP, Valbuena L, Perezatayde A, Reddick RL, Askin FB, Rosai J. Salivary-gland anlage tumor (congenital pleomorphic adenoma)—a clinicopathological, immunohistochemical and ultrastructural-study of 9 cases. Am J Surg Pathol. 1994;18:25–36. doi: 10.1097/00000478-199401000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Herrmann BW, Dehner LP, Lieu JEC. Congenital salivary gland anlage tumor: a case series and review of the literature. Int J Pediatr Otorhinolaryngol. 2005;69:149–156. doi: 10.1016/j.ijporl.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Heffner DK, Thompson LD, Schall DG, Anderson V. Pharyngeal dermoids (“hairy polyps”) as accessory auricles. Ann Otol Rhinol Laryngol. 1996;105:819–824. doi: 10.1177/000348949610501010. [DOI] [PubMed] [Google Scholar]
- 17.Luk IS, Chan JK, Chow SM, Leung S. Pituitary adenoma presenting as sinonasal tumor: pitfalls in diagnosis. Hum Pathol. 1996;27:605–609. doi: 10.1016/S0046-8177(96)90170-7. [DOI] [PubMed] [Google Scholar]
- 18.Byrne MN, Sessions DG. Nasopharyngeal craniopharyngioma. Case report and literature review. Ann Otol Rhinol Laryngol. 1990;99:633–639. doi: 10.1177/000348949009900809. [DOI] [PubMed] [Google Scholar]
- 19.Apostol JV, Frazell EL. Juvenile nasopharyngeal angiofibroma. A clinical study. Cancer. 1965;18:869–878. doi: 10.1002/1097-0142(196507)18:7<869::AID-CNCR2820180715>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 20.Hubbard EM. Nasopharyngeal angiofibromas. AMA Arch Pathol. 1958;65:192–204. [PubMed] [Google Scholar]
- 21.Neel HB, III, Whicker JH, Devine KD, Weiland LH. Juvenile angiofibroma. review of 120 cases. Am J Surg. 1973;126:547–556. doi: 10.1016/S0002-9610(73)80048-0. [DOI] [PubMed] [Google Scholar]
- 22.Sternberg SS. Pathology of juvenile nasopharyngeal angiofibroma; a lesion of adolescent males. Cancer. 1954;7:15–28. doi: 10.1002/1097-0142(195401)7:1<15::AID-CNCR2820070104>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 23.Giardiello FM, Hamilton SR, Krush AJ, Offerhaus JA, Booker SV, Petersen GM. Nasopharyngeal angiofibroma in patients with familial adenomatous polyposis. Gastroenterology. 1993;105:1550–1552. doi: 10.1016/0016-5085(93)90164-8. [DOI] [PubMed] [Google Scholar]
- 24.Guertl B, Beham A, Zechner R, Stammberger H, Hoefler G. Nasopharyngeal angiofibroma: an APC-gene-associated tumor? Hum Pathol. 2000;31:1411–1413. doi: 10.1016/S0046-8177(00)80012-X. [DOI] [PubMed] [Google Scholar]
- 25.Chen KT, Bauer FW. Sarcomatous transformation of nasopharyngeal angiofibroma. Cancer. 1982;49:369–371. doi: 10.1002/1097-0142(19820115)49:2<369::AID-CNCR2820490226>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 26.Spagnolo DV, Papadimitriou JM, Archer M. Postirradiation malignant fibrous histiocytoma arising in juvenile nasopharyngeal angiofibroma and producing alpha-1-antitrypsin. Histopathology. 1984;8:339–352. doi: 10.1111/j.1365-2559.1984.tb02346.x. [DOI] [PubMed] [Google Scholar]
- 27.Beham A, Fletcher CD, Kainz J, Schmid C, Humer U. Nasopharyngeal angiofibroma: an immunohistochemical study of 32 cases. Virchows Arch A Pathol Anat Histopathol. 1993;423:281–285. doi: 10.1007/BF01606891. [DOI] [PubMed] [Google Scholar]
- 28.Hwang HC, Mills SE, Patterson K, Gown AM. Expression of androgen receptors in nasopharyngeal angiofibroma: an immunohistochemical study of 24 cases. Mod Pathol. 1998;11:1122–1126. [PubMed] [Google Scholar]
- 29.Abraham SC, Montgomery EA, Giardiello FM, Wu TT. Frequent beta-catenin mutations in juvenile nasopharyngeal angiofibromas. Am J Pathol. 2001;158:1073–1078. doi: 10.1016/S0002-9440(10)64054-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dahlin DC, Maccarty CS. Chordoma. Cancer. 1952;5:1170–1178. doi: 10.1002/1097-0142(195211)5:6<1170::AID-CNCR2820050613>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 31.Heffelfinger MJ, Dahlin DC, MacCarty CS, Beabout JW. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973;32:410–420. doi: 10.1002/1097-0142(197308)32:2<410::AID-CNCR2820320219>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 32.Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE. Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol. 2006;30:811–818. doi: 10.1097/01.pas.0000209828.39477.ab. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell A, Scheithauer BW, Unni KK, Forsyth PJ, Wold LE, McGivney DJ. Chordoma and chondroid neoplasms of the spheno-occiput. An immunohistochemical study of 41 cases with prognostic and nosologic implications. Cancer. 1993;72:2943–2949. doi: 10.1002/1097-0142(19931115)72:10<2943::AID-CNCR2820721014>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 34.O’Connell JX, Renard LG, Liebsch NJ, Efird JT, Munzenrider JE, Rosenberg AE. Base of skull chordoma. A correlative study of histologic and clinical features of 62 cases. Cancer. 1994;74:2261–2267. doi: 10.1002/1097-0142(19941015)74:8<2261::AID-CNCR2820740809>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 35.Perzin KH, Pushparaj N. Nonepithelial tumors of the nasal cavity, paranasal sinuses, and nasopharynx. A clinicopathologic study. XIV: Chordomas. Cancer. 1986;57:784–796. doi: 10.1002/1097-0142(19860215)57:4<784::AID-CNCR2820570418>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 36.Meis JM, Raymond AK, Evans HL, Charles RE, Giraldo AA. “Dedifferentiated” chordoma. A clinicopathologic and immunohistochemical study of three cases. Am J Surg Pathol. 1987;11:516–525. doi: 10.1097/00000478-198707000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Mendenhall WM, Mendenhall CM, Lewis SB, Villaret DB, Mendenhall NP. Skull base chordoma. Head Neck. 2005;27:159–165. doi: 10.1002/hed.20144. [DOI] [PubMed] [Google Scholar]
- 38.Wojno KJ, Hruban RH, Garin-Chesa P, Huvos AG. Chondroid chordomas and low-grade chondrosarcomas of the craniospinal axis. An immunohistochemical analysis of 17 cases. Am J Surg Pathol. 1992;16:1144–1152. doi: 10.1097/00000478-199212000-00002. [DOI] [PubMed] [Google Scholar]
- 39.Sangoi AR, Karamchandani J, Lane B, Higgins JP, Rouse RV, Brooks JD, McKenney JK. Specificity of brachyury in the distinction of chordoma from clear cell renal cell carcinoma and germ cell tumors: a study of 305 cases. Mod Pathol. 2011;24:425–429. doi: 10.1038/modpathol.2010.196. [DOI] [PubMed] [Google Scholar]
- 40.Hanna E, Wanamaker J, Adelstein D, Tubbs R, Lavertu P. Extranodal lymphomas of the head and neck—a 20-year experience. Arch Otolaryngol. 1997;123:1318–1323. doi: 10.1001/archotol.1997.01900120068011. [DOI] [PubMed] [Google Scholar]
- 41.Jaffe ES. Lymphoid lesions of the head and neck: A model of lymphocyte homing and lymphomagenesis. Modern Pathol. 2002;15:255–263. doi: 10.1038/modpathol.3880521. [DOI] [PubMed] [Google Scholar]