

Stimulation of S2 cortex, but not that of an adjacent cortical area, induced descending heat antinociception in rats with the spinal nerve ligation-induced model of neuropathy. Antinociception was bilateral, and it involved suppression of pronociceptive medullary cells and activation of serotonergic pathways that act on the spinal 5-HT1A receptor. S2 stimulation failed to induce descending antinociceptive effect in sham-operated controls or in nerve-ligated animals that had not developed mechanical hypersensitivity.
Keywords: descending inhibition, neuropathic hypersensitivity, SII cortex, 5-HT1A receptor, rostroventromedial medulla, spinal wide dynamic range neuron
Abstract
Stimulation of the secondary somatosensory cortex (S2) has attenuated pain in humans and inflammatory nociception in animals. Here we studied S2 stimulation-induced antinociception and its underlying mechanisms in an experimental animal model of neuropathy induced by spinal nerve ligation (SNL). Effect of S2 stimulation on heat-evoked limb withdrawal latency was assessed in lightly anesthetized rats that were divided into three groups based on prior surgery and monofilament testing before induction of anesthesia: 1) sham-operated group and 2) hypersensitive and 3) nonhypersensitive (mechanically) SNL groups. In a group of hypersensitive SNL animals, a 5-HT1A receptor agonist was microinjected into the rostroventromedial medulla (RVM) to assess whether autoinhibition of serotonergic cell bodies blocks antinociception. Additionally, effect of S2 stimulation on pronociceptive ON-cells and antinociceptive OFF-cells in the RVM or nociceptive spinal wide dynamic range (WDR) neurons were assessed in anesthetized hypersensitive SNL animals. S2 stimulation induced antinociception in hypersensitive but not in nonhypersensitive SNL or sham-operated animals. Antinociception was prevented by a 5-HT1A receptor agonist in the RVM. Antinociception was associated with decreased duration of heat-evoked response in RVM ON-cells. In spinal WDR neurons, heat-evoked discharge was delayed by S2 stimulation, and this antinociceptive effect was prevented by blocking spinal 5-HT1A receptors. The results indicate that S2 stimulation suppresses nociception in SNL animals if SNL is associated with tactile allodynia-like hypersensitivity. In hypersensitive SNL animals, S2 stimulation induces antinociception mediated by medullospinal serotonergic pathways acting on the spinal 5-HT1A receptor, and partly through reduction of the RVM ON-cell discharge.
NEW & NOTEWORTHY Stimulation of S2 cortex, but not that of an adjacent cortical area, induced descending heat antinociception in rats with the spinal nerve ligation-induced model of neuropathy. Antinociception was bilateral, and it involved suppression of pronociceptive medullary cells and activation of serotonergic pathways that act on the spinal 5-HT1A receptor. S2 stimulation failed to induce descending antinociceptive effect in sham-operated controls or in nerve-ligated animals that had not developed mechanical hypersensitivity.
in the clinic, electrical stimulation of the primary motor cortex (M1) has been used for treatment of chronic pain since 1991 (Nguyen et al. 1999; Tsubokawa et al. 1991). Additionally, noninvasive repetitive transcranial magnetic stimulation (rTMS) of M1 is frequently used to alleviate intractable chronic pain in the clinic [for a recent review see Moisset et al. (2016)]. Since only 30 to 70% of pain patients benefit from M1 stimulation, other cortical areas have also been tested as potential new stimulation targets. Among them the secondary somatosensory cortex (S2), a brain site activated by painful stimulation (Talbot et al. 1991) and involved, e.g., in fibromyalgia (Lim et al. 2016), has proved promising. This is indicated by the finding that TMS of S2 reduced experimental pain in healthy controls (Lockwood et al. 2013; Valmunen et al. 2009). Importantly, the S2-induced reduction of pain was more pronounced than that produced by rTMS of M1 or dorsolateral prefrontal cortex (Valmunen et al. 2009). In chronic pain patients, rTMS of S2 has attenuated visceral pain (Fregni et al. 2011) and relieved neuropathic orofacial pain more effectively than rTMS of somatomotor cortex (S1/M1; Lindholm et al. 2015). However, analgesic effect induced by rTMS of S2 was weaker in interictal migraineurs than healthy controls (Uglem et al. 2016). A recent study indicated that the analgesic effect of S2 stimulation may not be explained indirectly by improvement of comorbid psychiatric or sleep disorders (Lindholm et al. 2016).
While many earlier experimental animal studies have assessed descending modulation of sensory signals by stimulation of somatomotor cortex in physiological conditions (e.g., Coulter et al. 1974; Dubner and Sessle 1971; Hagbarth and Kerr 1954; Senapati et al. 2005) or in various experimental animal models of pain hypersensitivity (e.g., Lucas et al. 2011; Pagano et al. 2011, 2012; Vaculín et al. 2008; Viisanen and Pertovaara 2010a, 2010b; Viisanen et al. 2012), there are only a few animal studies assessing the analgesic effect of S2 stimulation and its underlying mechanisms. It has been shown that electrical stimulation of the rat S2 fails to influence behavioral responses evoked by heat or mechanical stimulation in healthy control rats (Kuroda et al. 2000). In rats with formalin-induced pain, S2 stimulation, however, produces weak antinociception alone and strong antinociception when combined with a subeffective dose of neuronal NO synthase inhibitor (7-nitro-indazole). Such combination is associated with a reduction in the formalin-induced expression of c-Fos in the superficial laminae of the spinal dorsal horn (SDH) (Kuroda et al. 2001). By itself S2 stimulation attenuated c-Fos expression in the superficial medullary dorsal horn, unlike M1 stimulation (Gojyo et al. 2002). Systemic administration of a µ-opioid receptor or α1/2-adrenoceptor antagonist failed to attenuate synergistic antinociceptive action of S2 stimulation and 7-nitro-indazole, whereas intrathecal nonselective 5-HT antagonist in part prevented it (Kuroda et al. 2001). These findings in experimental animals indicate that among mechanisms contributing to the S2 stimulation-induced reduction of sustained pain behavior is recruitment of descending pathways involving spinal 5-HT receptors, but not noradrenergic or opioidergic systems.
It is not yet known whether and through which mechanisms S2 stimulation attenuates nociception or hypersensitivity in experimental neuropathy models. Here we assessed the effect of electrical S2 stimulation on pain-related behavior, medullary pain control neurons, and spinal pain relay neurons in animals with an experimental model of peripheral neuropathy. We hypothesized that the descending serotoninergic pathway acting on the spinal 5-HT1A receptor contributes to the S2-induced descending effect. Furthermore, we tested the hypothesis that nerve injury per se may not produce changes in S2 stimulation-induced antinociception, unless it is accompanied by neuronal plasticity that causes hypersensitivity that can be most prominently observed in awake animals as tactile allodynia-like behavior.
For the invasive electrophysiological recordings of the present study, the experiments had to be performed under light anesthesia. To allow meaningful comparisons to neuronal data, behavioral (withdrawal reflex) responses were also assessed under light anesthesia. Since even a low level of anesthesia markedly attenuates mechanical hypersensitivity while it has only a minor, if any, effect on heat nociception (Pertovaara et al. 2001), noxious heat was used as the test stimulus in the parallel assessments of behavioral and neuronal correlates of S2 stimulation-induced descending antinociception.
METHODS
Animals
Experiments were performed with male RccHan:WIST rats (Envigo Laboratories, Horst, the Netherlands) that resided in cages with free access to food and water. In the animal room, light and darkness alternated every 12 h. The weight of the rats was 150 to 200 g preceding spinal nerve ligation (SNL) or sham operation. The experimental protocol was approved by the Experimental Animal Ethics Committee of the Provincial Government of Southern Finland (Hämeenlinna, Finland; permission ESAVI/7863/04.10.07/2013) and followed the European Community Council Directive 2010/63/EU concerning the use of animals for scientific purposes. All efforts were made to minimize animal suffering and to use only the number of animals necessary to produce reliable scientific data.
Anesthesia Procedure
For all surgical and electrophysiological procedures, anesthesia was induced through intraperitoneal administration of pentobarbital sodium (60 mg/kg; Mebunat, OrionPharma, Espoo, Finland). Anesthesia level was assessed by observation of pupil size, general muscle tone, and by assessing withdrawal responses to noxious pinching; anesthesia was maintained by administering additional doses of pentobarbital sodium (15–20 mg·kg−1·h−1 or more as required). A deep level of anesthesia was present during SNL and preliminary surgical procedures for S2 simulation. Rats were naturally breathing, a warming blanket maintained the body temperature within a physiological range, and a pink color of the ears indicated normal blood circulation. During the actual experiments (when the effect of S2 stimulation was assessed), anesthesia was kept at a level at which no spontaneous movement of extremities was observed, but noxious limb stimulation evoked a withdrawal reflex in the stimulated limb only. While the anesthesia level was kept as stable as possible, it cannot be excluded that anesthesia or a change in its level may have had an influence on withdrawal or neuronal responses. Importantly, however, since the anesthesia procedure was identical in all experimental conditions, the possible effect by anesthesia or a change in its level cannot explain differences between the effects induced by real and fake S2 stimulation, differences in the effect of studied drug vs. vehicle, differences between nerve-injured and sham-operated animals, or differences in the effect of S2 stimulation among studied neuronal populations (such as that between medullary ON- vs. OFF-cells). Animals were allowed to recover from the preliminary surgical operation, in which SNL or a corresponding sham operation was performed. However, animals were not allowed to recover from the anesthesia in actual experiments that were carried out 2 wk later. After completion of the experiment assessing the effect of S2 stimulation, the animals were given a lethal dose of pentobarbital sodium (150 mg/kg).
Induction of Experimental Neuropathy and Determination of Mechanical Hypersensitivity
When comparing various experimental models of neuropathy (Honoré et al. 2011), among advantages of the model induced by unilateral ligation of spinal nerves L5 and L6 is that it provides a possibility to use the easily accessible plantar skin as test site and, therefore, the SNL model of neuropathy described in detail elsewhere (Kim and Chung 1992) was used in the present study. Briefly, after induction of a deep level of anesthesia as described above, separation of left paraspinal muscles from spinous processes provided access to the spinal segment L6. The following removal of a transverse process allowed identification of spinal nerves L5 and L6 on the left side. Ligation of the nerves with silk thread was necessary to produce neuropathy. In the group of sham-operated animals, the surgical procedure was identical, except that the spinal nerve was not ligated. After surgery, rats received 0.03 mg/kg of buprenorphine (RB Pharmaceuticals Limited, Slough, UK) subcutaneously every 12 h for 3 consecutive days for treatment of postoperative pain.
Animals were allowed to recover for 2 wk from the initial surgery (nerve ligation or sham operation) during which time they were habituated at least for 2 days to the experimental conditions. Then, their mechanical sensitivity was determined before induction of anesthesia for the actual experiments in which the effect of S2 stimulation was assessed. For assessment of mechanical sensitivity, paw withdrawal response evoked by repetitive monofilament stimulation of the hind paw ipsilateral to SNL or sham surgery was determined. During the sensitivity assessment, the animal was standing on a metal grid, while its hind paw was stimulated by calibrated monofilaments of varying forces (North Coast Medical, Morgan Hill, CA). If the animal had three to five withdrawal responses to five repetitive stimulations of the paw at an intensity of ≤4 g, the animal was classified as (mechanically) hypersensitive. Nerve-injured rats that failed to develop hypersensitivity formed a study group classified as nonhypersensitive SNL animals. Sham-operated animals had three to five withdrawal responses to five repetitive monofilament stimulations only at the stimulus force of more than 10 g. In this study, 8 of the 42 SNL animals (i.e., 19%) were classified as mechanically nonhypersensitive; this rate is close to that in our earlier study (17%; Röyttä et al. 1999).
Heat Stimulation and Assessment of the Heat-Evoked Limb Withdrawal
Heat stimuli were applied to the plantar skin of the left (nerve injured or sham operated) hind limb in anesthetized rats. A feedback-controlled Peltier thermode (82.8 mm2; LTS-3 Stimulator, Thermal Devices, Golden Valley, MN; Wilcox and Giesler 1984) was used for thermal stimulation. Contact thermostimulator maintained the baseline temperature at 35°C, to minimize changes in the baseline skin temperature that might provide a significant confounding factor when assessing heat-evoked withdrawal latencies (Hole and Tjølsen 1993). In this study, heat stimuli had a rise rate of 10°C/s, a peak temperature of 53°C, and duration of 10 s. A miniature piezoelectric movement detector (Siemens Elema Ab, Solna, Sweden) over gastrocnemius muscle was used to detect the heat-evoked limb withdrawal.
Electric Stimulation of S2
For implantation of a concentric bipolar stimulus electrode (Rhodes NE-100, David Kopf Instruments, Tujunga, CA) into the S2, the anesthetized rat was placed into a standard stereotaxic frame according to the atlas of Paxinos and Watson (1998). A small hole was drilled for placement of the electrode through the skull at a location that was 8.0 mm anterior to the interaural line and 6.2 mm lateral to the midline. In most animals, the location was in the right hemisphere (i.e., contralateral to the nerve-injured or sham-operated limb), but in one group it was in the left (ipsilateral to nerve injury) hemisphere. The tip of the electrode extended 4.5 mm ventrally from the brain surface.
For S2 stimulation, square pulses of 0.1-ms duration were applied at a frequency of 300 Hz for 15 s by using a constant current unit that was driven by a stimulus generator (PSIU6 and Grass S88, Grass Instruments, Quincy, MA). Depending on the experimental condition, stimulation was applied at the intensity of 0 µA (fake stimulation), 30, 50, or 70 µA. These stimulation parameters were chosen on the basis of our earlier studies assessing descending antinociception induced by motor cortex stimulation; in motor cortex stimulation, maximum antinociceptive effect was obtained at 30–50 µA, and a further increase of intensity to 70 µA failed to increase antinociception (Viisanen and Pertovaara 2010a). The onset of S2 stimulation was 5 s before the onset of cutaneous heat stimulation of 10-s duration, since our previous findings in the motor cortex indicated that starting cortical stimulation 5 s before the onset of the heat stimulus was enough to produce a significant antinociception (Viisanen et al. 2012). S2 and heat stimulation were set off at the same time.
RVM Injections
In one group of hypersensitive SNL rats, 63 ng of 8-OH-DPAT (a 5-HT1A receptor agonist; Sigma-Aldrich, St. Louis, MO) or saline control was microinjected into the rostroventromedial medulla (RVM) before S2 stimulation to assess whether inhibition of serotonergic cell bodies in the RVM, through activation of their 5-HT1A autoinhibitory receptors (Rogawski and Aghajanian 1981), attenuates descending antinociception induced by S2 stimulation. This approach has been used earlier to study, e.g., the involvement of raphe-spinal serotonergic neurons in descending modulation of pain behavior induced by the amygdala (Sagalajev et al. 2015). For RVM injections, a guide cannula (C315G, 26GA, Plastics One, Roanoke, VA) was implanted through a hole drilled in the skull (anterior to the interaural line: −2.3 mm, lateral to the midline: 0.0 mm; ventral from the brain surface: 9.5 mm; Paxinos and Watson 1998) under anesthesia at the beginning of the experiment, before installation of the cortical stimulus electrode. The guide cannula was held in place with a stereotaxic micromanipulator. RVM injections were performed with an internal cannula (C315I, 33GA, Plastics One) that protruded 1 mm from the tip of the guide cannula and that was connected with a polyethylene tubing (Intramedic PE-10, Becton Dickinson and Company, Sparks, MD) to a 10-µl syringe (Hamilton, Bonaduz, Switzerland). A small air bubble in the tubing indicated the flow of the injection. The injection volume was 0.5 µl, and the injection lasted 30 s. After completing the injection, the internal cannula was kept an additional 30 s in the RVM before removal to minimize retrograde diffusion.
Electrophysiological Recordings of Single Neurons
Electrophysiological recordings of single RVM or SDH neurons were performed with lacquer-coated tungsten microelectrodes that had tip impedance 5–10 MOhm (FHC, Bowdoinham, ME). Neural signals were amplified with a custom-made amplifier, filtered (Krohn-Hite filter, Model 3700R, Brockton, MA), digitized (Micro 1401, Cambridge Electronic Design, Cambridge, UK), stored by using a standard computer, and thereafter analyzed offline (Spike2 software, Cambridge Electronic Design). Spike2 software classifies waveform shapes based on full wave templating, and in the offline analysis the template matching can be complemented by clustering using principal component analysis, because of which it was possible to evaluate separately the discharge of more than one neuron in single recording sessions.
Single-Unit Recordings in the RVM
After induction of anesthesia and placement of the animal in the stereotaxic frame, a hole was drilled in the skull for placement of the recording electrode in the RVM (AP: −2.3 mm; ML: 0.0 mm; DV: 10.0–10.5 mm; Paxinos and Watson 1998). Next, the cortical stimulus electrode was placed in the right S2, as described above. Before starting the actual recording of neurons, the deep level of anesthesia needed for surgical procedures was allowed to wear off to the level where the animal did not have any spontaneous limb movements but noxious stimulation caused a brief flexion reflex but no other behavioral responses. RVM neurons were classified based on the concurrently assessed neuronal and limb flexion response to noxious heat (see Heat Stimulation and Assessment of the Heat-evoked Limb Withdrawal). Neurons that gave an excitatory discharge just before the heat-evoked limb withdrawal were classified as presumably pronociceptive RVM ON-cells, whereas neurons in which the ongoing discharge was inhibited just before the heat-evoked limb withdrawal were classified as presumably antinociceptive OFF-cells (Heinricher et al. 1989). Neurons that did not respond to noxious heat were considered NEUTRAL-cells, and they were not considered in the present study.
Because of electric noise induced by S2 stimulation that masked concurrent recording of heat-evoked responses in RVM neurons, primary analysis of the heat-evoked discharge in RVM neurons was made based on the response to heat stimulus presented 2 min after the offset of S2 stimulation. Among discharge parameters assessed in RVM cells were the mean ongoing discharge rate before heat or S2 stimulation, the mean burst discharge rate evoked by heat stimulation, the latency to the onset of the heat-evoked burst discharge in ON-cells or the latency to the heat-evoked discharge inhibition in OFF-cells, and the duration of the heat-evoked burst discharge in ON-cells or discharge inhibition in OFF-cells (Fig. 1). Additionally, since the electric noise disappeared immediately after the offset of the S2 stimulus, it was possible to record delayed heat-evoked responses also in the condition in which real S2 stimulus was applied concurrently with the heat stimulus. In this additional analysis, only the heat-evoked delayed discharge rate and duration (i.e., afterdischarge occurring immediately after offset of the S2 and heat stimulation) could be determined; this stimulus condition was defined as “during” S2 stimulation (see interval E in Fig. 1).
Fig. 1.
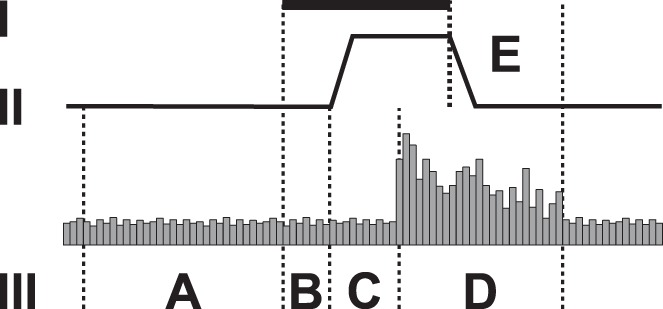
Measurements in recordings of neuronal responses, Traces I-III. I: duration of S2 stimulation (15 s). II: temperature of the heat stimulus (baseline 35°C, peak temperature 53°C). III: peristimulus time histogram of the heat-evoked neuronal discharge. Intervals A–E. A: ongoing mean discharge rate. B: mean discharge rate during S2-stimulation. C: latency to heat-evoked burst discharge. D: duration and mean rate of the heat-evoked burst discharge. E: duration and rate of the late heat-evoked burst discharge. In RVM recordings, electric noise induced by S2 stimulation prevented assessing intervals B, C, and D, but C and D were assessed in the post-S2 condition. In RVM recordings, measurements “during S2” were performed in interval E.
Single-Unit Recordings in the Spinal Dorsal Horn
To study SDH neurons, laminectomy was performed in deeply anesthetized rats. Laminectomy was performed at the level of the T12-L2 vertebrae to expose the L4-L6 segments of the spinal cord. The dura was cut and a pool of skin formed and filled with warm saline to prevent dehydration. The animal was placed in a standard stereotaxic frame and two spinal clamps, one rostral and one caudal to the laminectomy, were used to stabilize the preparation. Before starting spinal recordings, a cortical stimulation electrode was placed in the right S2 as described above.
When searching for SDH neurons, mechanical and heat stimuli were used to classify the neurons. Neurons responding to both innocuous mechanical and noxious heat stimulation were classified as wide dynamic range (WDR) neurons, neurons responding only to noxious heat stimulation were classified as nociceptive-specific neurons, and neurons responding only to innocuous mechanical stimulation were classified as low-threshold mechanoreceptive neurons (Willis and Coggeshall 2004). Only WDR neurons that are considered to be involved in coding of nociception (Coghill et al. 1993) and that were located in the SDH according to the recording depth from the cord surface (<1,000 µm; Willis and Coggeshall 2004) were considered further.
Electromagnetic noise induced by S2 stimulation did not mask spinal recordings and therefore, it was possible to record spinal neuronal responses also during S2 stimulation. The ongoing discharge and the heat-evoked response of spinal WDR neurons were assessed without and with real or fake S2 stimulation. The ongoing discharge rate was assessed during the first 5-s period after the onset of fake and real S2 stimulation and preceding the onset of the heat stimulus. Additionally, the studied parameters were the mean burst discharge rate evoked by heat stimulation, the onset latency of the heat-evoked burst discharge, and the duration of the heat-evoked burst discharge (Fig. 1). When assessing the involvement of the spinal 5-HT1A receptor in descending antinociception, saline control or WAY-100635 (5-HT1A receptor antagonist; Sigma-Aldrich) was administered onto the exposed spinal cord at the dose of 2.5 µg/50 µl. Assessment of the effect of WAY-100635 started 10 min after its administration. At this dose range, WAY-100635 alone has not produced changes in nociception in nerve-injured animals (Wei and Pertovaara 2006), nor did it influence baseline responses of WDR neurons in the present study as shown by comparison of Pre-S2 responses after WAY-100635 treatment with the Pre-S2 responses after saline treatment (see Effect of S2 stimulation on discharge of spinal WDR neurons in results). The current injection volume (50 µl) is commonly used when applying drugs directly onto the spinal cord in laminectomized animals (Stanfa and Dickenson 2004).
Course of the Study
SNL or sham operation was performed two weeks before the actual S2 experiment. Before induction of anesthesia for the actual S2 experiments, mechanical hypersensitivity was assessed in the operated hind limb for classification of animals into hypersensitive and nonhypersensitive groups.
Course of the Behavioral Study
The descending antinociceptive effect of S2 stimulation was behaviorally characterized in lightly anesthetized animals. In the first behavioral series of experiments, heat-evoked withdrawal latency and its modulation by S2 stimulation was studied in six experimental groups: contralateral real S2 stimulation in hypersensitive SNL animals (n = 12), contralateral real S2 stimulation in sham-operated animals (n = 6), contralateral real S2 stimulation in nonhypersensitive SNL animals (n = 6), ipsilateral real S2 stimulation in hypersensitive SNL animals (n = 6), contralateral fake S2 stimulation in hypersensitive SNL animals (n = 6), and contralateral fake stimulation in sham-operated animals (n = 6). Since postmortem histological analyses indicated that in some animals the cortical stimulation site was outside of S2, hypersensitive SNL animals with a misplaced cortical electrode formed an additional experimental group (n = 6). The animals with misplaced cortical electrodes in other groups were replaced by testing additional animals so that the number of animals in the group with the S2 electrode in the correct place was not reduced.
When performing the experiment, the baseline withdrawal latency evoked by noxious heat stimulation was first determined without S2 stimulation. Then, the antinociceptive effect of S2 stimulation applied for 15 s was determined. Onset of S2 stimulation preceded the onset of heat stimulation by 5 s, and S2 stimulation continued until the end of heat stimulation (53°C, 10 s). Antinociceptive effect of S2 stimulation was tested at an ascending order of intensity (30, 50, and 70 µA) at 2-min intervals. Since S2 stimuli were presented only in an ascending order, one should be cautious when assessing the variation of the magnitude of antinociception with the intensity of cortical stimulation. However, since testing was performed in identical fashion in all experimental groups, the differences among experimental groups cannot be explained by the order of testing or anesthesia. Moreover, comparisons of real S2 stimulation conditions with the fake S2 stimulation condition allow excluding the potential effect induced by the repetition of the testing session per se.
Course of the Behavioral Study Assessing Involvement of Medullary Serotonergic Neurons
The role of medullary serotonergic neurons in the S2 stimulation-induced antinociception was assessed behaviorally in hypersensitive SNL animals (n = 6) as described in the above section, except for the following differences. The animals had a chronic guide cannula for RVM injections that was installed at the beginning of the experiment. Before S2 stimulation sessions, the heat-evoked withdrawal latency was determined twice: before and 10 min after RVM injection of 8-OH-DPAT (63 ng) or saline. After that, antinociceptive effect of S2 stimulation at an ascending series of intensities was determined at 2-min intervals. Two minutes after testing S2 stimulus intensities 30, 50, and 70 µA, effect of Post-S2 stimulation (stimulus intensity: 0 µA) was tested. Then, after a pause that lasted 1 h, the experiment was repeated. In the first test round, half of the animals received intramedullary injection of 8-OH-DPAT and another half received saline. In the second test round, the treatment was changed to have a counterbalanced testing of the drug/saline. At the currently used dose, the effect of 8-OH-DPAT had disappeared in an hour as indicated by comparison of the results between animals treated first with 8-OH-DPAT with those treated first with saline.
Course of the Study Assessing Involvement of RVM Neurons
In the RVM recordings, only hypersensitive SNL animals were studied. Recordings were performed 2 wk after SNL surgery. For the preliminary surgery of the RVM recording session (see above Single-Unit Recordings in the RVM) the animal had a deep level of anesthesia. After recovery from deep level of anesthesia, the recordings were started when anesthesia level was low enough as indicated by reproducible heat-evoked limb withdrawal responses. After finding an RVM neuron, it was classified based on its response properties (Heinricher et al. 1989). If the cell was classified as ON- or OFF-cell, then its response to heat (53°C, 10 s) was determined at 2-min intervals. First, without S2 stimulation (Pre-S2), then at 2-min intervals with concurrent S2 stimulation in an ascending series of stimulus intensities (30, 50, and 70 µA for 15 s each), and again without S2 stimulation (Post-S2) 2 min after the end of the ascending series of S2 stimulations. As in all other experiments, the onset of S2 stimulation preceded by 5 s the onset of heat stimulus and continued until the end of the heat stimulus. In all RVM recording sessions, S2 electrode was contralateral to the nerve-injured limb. In RVM ON-cells (but not OFF-cells), the effect of fake S2 stimulation was also tested in a similar fashion (fake S2 stimulation at 0 µA was delivered with the accompanying heat stimulus consecutively three times at 2-min intervals to mimic delivery of ascending series of three different S2 stimulus intensities). The interval between testing fake and real S2 stimulation in the same ON-cell was at least 15 min, and the order of testing real and fake S2 stimulation was varied between the cells. After completing the testing of an RVM cell, the recording depth was changed to find a new cell. The interval between studying cells in two different recording depths in the same animal was 1 h. At the end of the RVM recording session, an electrolytic lesion was made in the recording site. In one recording site it was possible to record responses of several different cells at the same time, and in one animal recordings were performed at 1–3 recording depths. Therefore, it was possible to study several cells in one animal.
Course of the Study Assessing Involvement of Spinal WDR Neurons
SDH recordings were performed only in hypersensitive SNL animals, and recordings were performed 2 wk after SNL operation. After completion of the surgical operations needed for the recordings, single neurons were searched and classified based on their response properties (Willis and Coggeshall 2004). When finding a WDR neuron, its response to heat (53°C, 10 s) was determined before S2 stimulation (Pre-S2) and 2 min later during S2 stimulation (real stimulation at 30 µA or fake stimulation at 0 µA for 15 s; S2 stimulation was applied contralateral to the recorded WDR neuron and S2 stimulation preceded onset of heat stimulation by 5 s, as in all other experiments). Unlike in behavioral or RVM studies, S2 stimulation was not applied at the intensities of 50 or 70 µA when studying SDH neurons. WDR recordings involved two experiments. 1) Effect of real vs. fake S2 stimulation, in which the two successive heat stimulations were given as above, i.e., the second heat stimulus was accompanied by real or fake S2 stimulation. When testing real and fake S2 stimulation in the same rat, the order was varied and the interval between the conditions was 60 min. 2) Effect of pretreatment of the exposed spinal cord with saline or WAY-100635 (2.5 µg/50 µl 10 min before the testing). Here, heat-evoked response was first assessed without S2 stimulation (Pre-S2) and 2 min later with concurrent S2 stimulation (30 µA; during S2). In half of the WDR neurons, only one drug condition (saline or WAY-100635) was tested, whereas in the second half of the WDR neurons saline condition was tested first, followed by testing the effect of WAY-100635 60 min later; the results were pooled, since the results were not different but independent whether only one or two drug conditions were tested in the same rat. Sessions assessing the effect of WAY-100635, however, were always performed last to avoid the confounding effect of WAY-100635 accumulation in the spinal cord on results of the following sessions. As in the RVM recordings, it was possible to record several neurons at the same time. After completing the recording session at one recording depth, the recordings could be continued 1 h later at another depth.
Completion of the Experiments
S2 stimulation sessions lasted no more than 8 h, during which animals were all the time under anesthesia. After completion of the experiment, the anesthetized animals received a lethal dose of pentobarbital sodium (150 mg/kg) intraperitoneally. The brain was removed, fixed in formalin, and sliced. The brain slices were stained with hematoxylin and eosin. Finally, medullary injection sites and electrolytically marked medullary recording and cortical stimulation sites were determined microscopically in the brain slices (Fig. 2). Recording sites in the SDH were determined based on the depth of the recording site from the cord surface.
Fig. 2.

Brain injection and recording sites. A: stimulation sites within the secondary somatosensory cortex (S2). B: recording and injection sites in the rostral ventromedial medulla (RVM). In A, circles = sham-operated rats, squares = hypersensitive spinal nerve-ligated (SNL) rats, stars = nonhypersensitive SNL rats, asterisks = stimulation site outside S2 in hypersensitive SNL rats. In B, squares = recording sites in hypersensitive SNL rats, triangles = recording sites in hypersensitive SNL rats. S1, primary somatosensory cortex (orofacial area); S2, secondary somatosensory cortex; GI, granular insular cortex; RMg, raphe magnus nucleus; GiA, gigantocellular reticular nucleus alpha; Contra, contralateral to the operated and tested hind limb; Ipsi, ipsilateral to the operated and tested hind limb. Each symbol represents one to two rats. Numbers represent distances in millimeters from interaural line (IA). In both graphs, photos demonstrate examples of brain slices stained with hematoxylin and eosin; arrows point to a stimulation site in A and recording site in B. [Adapted from Paxinos and Watson (1998) with permission. Copyright Academic Press].
Statistics
Data are shown as means and SD. Data were analyzed using one-way repeated measures analysis of variance (ANOVA) or two-way mixed design or repeated measures ANOVA followed by t-test with a Bonferroni correction for multiple comparisons. Paired t-test was used when comparing less than three groups. Statistical software used for the analyses was Prism 6 for Windows (GraphPad Software, La Jolla, CA). P < 0.05 was considered to represent a significant difference.
RESULTS
Behavior: Influence of Various Experimental Factors on S2 Stimulation-Induced Descending Antinociception Assessed by Heat-Evoked Limb Withdrawal Reflex
Heat-evoked baseline response before S2 stimulation.
The assessment of the noxious heat-evoked limb withdrawal response was performed in lightly anesthetized rats that were divided into three experimental groups based on the surgical procedure they had undergone and their mechanical sensitivity before induction of anesthesia: SNL animals with mechanical hypersensitivity, SNL animals without mechanical hypersensitivity, and sham-operated animals. Baseline latency of the heat-evoked response did not vary significantly among these three groups (F2,48 = 1.46; Fig. 3A).
Fig. 3.
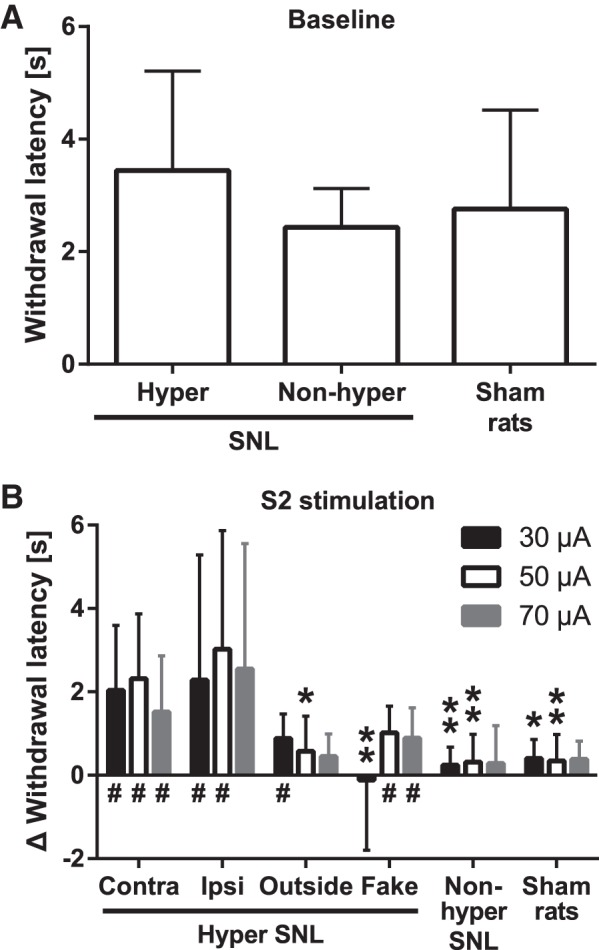
Noxious heat-evoked limb withdrawal and its modulation by S2 stimulation in various experimental conditions. A: baseline response latency in spinal nerve-ligated (SNL) animals that have mechanical hypersensitivity (hyper) or no hypersensitivity (nonhyper) and in sham-operated animals. B: increase of the heat-evoked response latency induced by S2 stimulation in various experimental groups. Intensity of S2 stimulation is indicated in the upper right corner of the graph. Contra, S2 stimulation contralateral to nerve injury; Ipsi, S2 stimulation ipsilateral to nerve injury; Outside, cortical stimulation outside of S2; Fake, contralateral S2 stimulation at the intensity of 0 µA. The bars show mean values, and the error bars represent SD (in A, nhyper = 29, nnonhyper = 8, nsham = 14; in B, n = 12 for the Contra group, and n = 6 for all other groups). For illustrative purposes the latency data shown in B is standardized in reference to the corresponding pre-S2 latencies, but all the statistical analyses are based on raw data. In B, y-axis shows the cortical stimulation-induced increase in the withdrawal latency; 0 s represents the baseline latency before cortical stimulation and values >0 s represent increase of the latency. *P < 0.05, **P < 0.01 (Bonferroni-corrected t-test; reference: the corresponding value in the Contra group); #95% confidence interval of the cortical stimulation-induced latency does not overlap with the corresponding baseline latency.
Influence of peripheral nerve injury vs. sham operation.
Electrical stimulation of the right S2 (contralateral to the operated left limb) increased the response latency to heat significantly more in the (mechanically) hypersensitive SNL group than in the sham-operated group (main effect of neuropathy: F1,16 = 11.9, P = 0.003; Fig. 3B). The increase in the heat-evoked response latency did not vary significantly when the intensity of S2 stimulation was increased from 30 to 70 µA (interaction between neuropathy and stimulus intensity: F2,32 = 0.5). Post hoc testing indicated that the S2 stimulation-induced attenuation of the noxious heat-evoked withdrawal response was significantly stronger in hypersensitive SNL rats than sham-operated ones at S2 stimulation intensities of 30 and 50 µA, but not at the intensity of 70 µA (Fig. 3B).
In the (mechanically) hypersensitive SNL group, the 95% confidence interval (CI) of the heat-evoked response latency during right S2 stimulation did not overlap with the corresponding baseline latency at any of the S2 stimulation intensities, whereas in the sham-operated group the 95% CI of the response latency assessed during S2 stimulation overlapped at all intensities of S2 stimulation with the corresponding baseline latency (Fig. 3B).
Is the descending antinociceptive effect uni- or bilateral?
To assess whether the descending effect of S2 stimulation is stronger contra- or ipsilaterally, we compared the thermal antinociceptive effect of contralateral S2 stimulation with that of ipsilateral S2 stimulation in two separate groups of animals, both of which had mechanical hypersensitivity induced by SNL in the left hind limb. The magnitude of heat antinociception induced by stimulation of the (right) contralateral or the (left) ipsilateral S2 were not significantly different (main effect of cortical stimulation side: F1,16 = 0.5; Fig. 3B), independent of the increase of S2 stimulation intensity from 30 to 70 µA (interaction between stimulation side and stimulus intensity: F2,32 = 0.5). The 95% CI of the heat-evoked response latency during stimulation of the ipsilateral S2 did not overlap with the corresponding baseline latency, except at the intensity of 70 µA (Fig. 3B).
Descending antinociception induced by stimulation of S2 vs. an adjacent cortical site.
While in all experiments the aim was to stimulate S2, histological analyzes indicated that in six (mechanically) hypersensitive SNL animals the tip of the stimulation electrode was outside of the S2, in five of the animals the mislocalized electrode was in the granular insular cortex, and in one in the external capsule (Fig. 2). In four animals, the mislocation was in the right (contralateral) hemisphere and in two animals in the left (ipsilateral) hemisphere. Since the antinociceptive effects induced by contralateral and ipsilateral S2 stimulations were not different (Fig. 3B), the data with mislocated S2 stimulation sites were pooled. The magnitude of heat antinociception induced by electrical stimulation of the right S2 was significantly stronger than that of cortical stimulation adjacent to the S2 (main effect of cortical stimulation site: F1,16 = 8.1, P = 0.01; Fig. 3B), independent of the increase of S2 stimulation intensity from 30 to 70 µA (interaction between cortical stimulation site and stimulus intensity: F2,32 = 0.4). Post hoc tests revealed a significant difference in the magnitude of heat antinociception between the groups when cortical stimulation was applied at the intensity of 50 µA in S2 vs. an adjacent site (Fig. 3B). The 95% CI of the heat-evoked response latency during stimulation of the cortical site adjacent to S2 overlapped with the corresponding baseline latency, except at the intensity of 30 µA (Fig. 3B).
Influence by fake stimulation of S2 in nerve-injured animals.
Fake stimulation of the right S2 (stimulus intensity 0 µA) at three successive time points (that correspond to those when real S2 stimulation was applied at three different intensities) had a significantly weaker antinociceptive effect than real stimulation of the right S2 in (mechanically) hypersensitive SNL animals (main effect of cortical stimulus condition: F1,16 = 8.8, P = 0.009; Fig. 3B), independent of time point of testing/intensity of S2 stimulation (interaction between cortical stimulus condition and stimulus intensity: F2,32 = 1.7). Post hoc testing indicated that the magnitude of antinociception between the groups treated with real vs. fake S2 stimulation was significant when real S2 stimulation at the intensity of 30 µA was compared with the corresponding fake S2 stimulation (Fig. 3B). The 95% CI of the heat-evoked response latency overlapped with the corresponding baseline latency during the first but not the last two fake S2 treatments.
Influence of nerve injury-induced hypersensitivity.
Electrical stimulation of the right S2 (contralateral to the operated and tested left limb) had a significantly stronger effect on the heat-evoked response latency in the (mechanically) hypersensitive SNL group than in the (mechanically) nonhypersensitive SNL group (main effect of hypersensitivity: F1,18 = 16.8, P = 0.0007; Fig. 3B). The change in S2 stimulus intensity from 30 to 70 µA did not influence the magnitude of heat antinociception (interaction between hypersensitivity and stimulus intensity: F2,36 = 0.8). Post hoc tests indicated that the attenuation of heat-evoked withdrawal response was significantly stronger in the hypersensitive than the nonhypersensitive SNL group at S2 stimulation intensity of 30 and 50 µA, but not at the intensity of 70 µA. Unlike in the hypersensitive SNL group, the S2 stimulation-induced increase of the response latency in the nonhypersensitive SNL group overlapped the corresponding baseline latency at all cortical stimulus intensities (Fig. 3B).
Influence by fake stimulation of S2 in sham-operated animals.
As an additional control experiment, we compared the antinociceptive effect of contralateral fake S2 stimulation (n = 6) with contralateral real S2 stimulation (n = 6) in sham-operated animals. In these rats, withdrawal latencies during real S2 stimulation at intensities of 30, 50, and 70 µA were not significantly different from those assessed during fake S2 stimulation in sham-operated animals (main effect of cortical stimulus condition: F1,16 = 0.4), independent of cortical stimulus intensity (interaction between cortical stimulus condition and intensity: F2,32 = 0.6; not shown).
Behavior: involvement of medullary 5-HT cells in descending antinociception induced by S2 stimulation.
To assess whether medullary 5-HT cells are involved in mediating the descending antinociceptive effect induced by contralateral S2 stimulation, saline (control) or 8-OH-DPAT (5-HT1A receptor agonist; 63 ng) was microinjected into the RVM of (mechanically) hypersensitive SNL animals. It was expected that 8-OH-DPAT would inhibit 5-HT cell-mediated descending antinociception, due to action on 5-HT1A autoinhibitory receptors expressed by cell bodies of medullary 5-HT cells (Rogawski and Aghajanian 1981). While the main effect of treatment group was not significant (8-OH-DPAT vs. saline: F1,5 = 3.0; Fig. 4), heat-evoked response latency was significantly changed when the experiment proceeded to the application of S2 stimulation (main effect of elapsed time: F5,25 = 3.2, P = 0.023); this change varied significantly with the medullary treatment (interaction between elapsed time and treatment: F5,25 = 3.2, P = 0.023). Post hoc testing indicated that during S2 stimulation at the intensity of 30 µA, the heat-evoked response latency was significantly longer in animals receiving saline than 8-OH-DPAT in the RVM (Fig. 4).
Fig. 4.
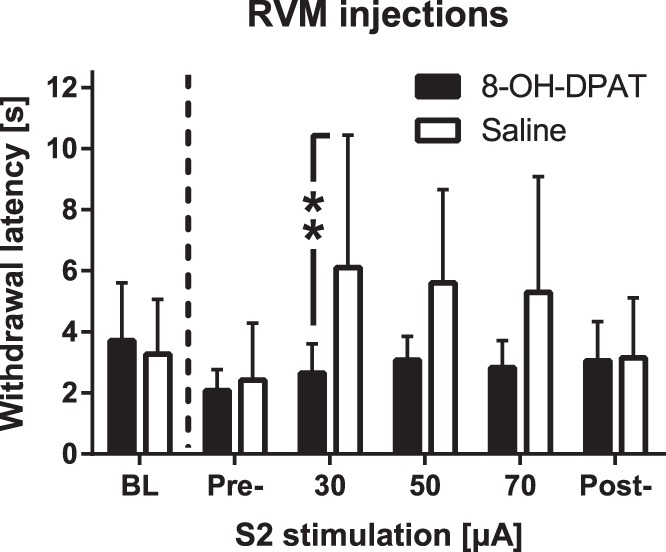
Effect of medullary administration of a 5-HT1A receptor agonist on antinociception induced by S2 stimulation. Saline or 8-OH-DPAT (a 5-HT1A receptor agonist; 63 ng) was microinjected into the rostroventromedial medulla (RVM) of hypersensitive rats with a spinal nerve ligation-induced model of neuropathy. Bars show mean values and error bars represent SD (n = 6). **P < 0.01 (Bonferroni-corrected t-test).
Single-unit recordings of RVM neurons.
Single-unit recordings of presumed pain control neurons of RVM were performed to assess their involvement in the S2 stimulation-induced descending antinociceptive effect. Recordings were performed with and without stimulation of the right (contralateral) S2 while noxious heat stimulation was applied to the paw of the left (nerve injured) hind limb in lightly anesthetized (mechanically) hypersensitive SNL animals (see Fig. 5 for an original example of the RVM recordings). Since the antinociceptive effect induced by S2 stimulation at the intensity of 50 or 70 µA was not stronger than at the intensity of 30 µA (Figs. 3B and 5) and since electric noise masked RVM recordings during real S2 stimulation (Fig. 5), the results of RVM recordings at the S2 intensity of 50 and 70 µA are not reported here. We report here only the discharge before S2 stimulation (Pre-S2), the late heat-evoked discharge immediately after the end of S2 stimulation at the intensity 30 µA (the effect of which was expected to be more locally restricted that induced by higher stimulus intensities, “during S2” condition), and the heat-evoked discharge post-S2 stimulation (i.e., 2 min after presenting the ascending series of S2 stimuli).
Fig. 5.
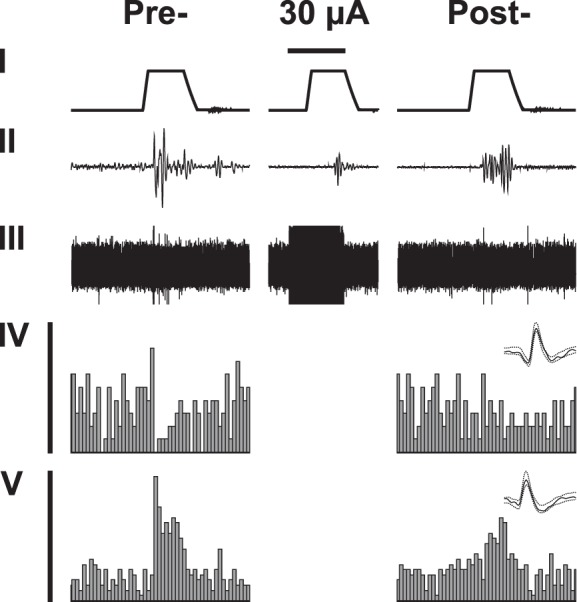
An example of original recordings of neurons in the rostral ventromedial medulla of an animal with neuropathic hypersensitivity. I: the temperature of the contact stimulator (baseline 35°C, peak 53°C) applied to the left (nerve injured) hind limb. II: withdrawal of the stimulated hind limb as assessed by a piezoelectric device. III: raw data of neuronal recordings (due to long time scale and high discharge frequency; the consecutive spikes are adjacent to each other). IV: peristimulus time histograms of an OFF-cell discharge. V: peristimulus time histogram of an ON-cell discharge. Templates of the simultaneously recorded OFF- and ON-neuron are shown above the peristimulus time histograms. Graphs in the left column show recording before S2 stimulation (Pre-), and graphs in the middle column show recording during S2 stimulation (the horizontal bar above the stimulus temperature trace represents 15 s, and it indicates the duration of S2 stimulation at the intensity of 30 µA; note the S2 stimulation-induced electric noise that masks concurrent neuronal recordings). The graphs in the right column show recording 2 min after the end of the ascending series of S2 stimulations (Post-). S2 stimuli were applied at 2-min intervals at the intensity of 30 µA (shown), 50 µA (not shown), and 70 µA (not shown).
As in behavioral experiments described in the above sections, contralateral S2 stimulation had a significant effect on the concurrently assessed heat-evoked limb withdrawal latency in the experiment in which single-unit recordings of RVM neurons were performed in lightly anesthetized SNL animals (F2,14 = 7.6, P = 0.006; Fig. 6A). Post hoc testing indicated that S2 stimulation at the intensity of 30 µA induced a significant prolongation of the limb withdrawal latency (Fig. 6A). At the currently used stimulus parameters, S2 stimulation induced no long-term behavioral antinociception as shown by the finding that the heat-evoked withdrawal latency returned to the baseline level when the test was replicated without concurrent S2 stimulation 2 min after the previous delivery of the S2 stimulus (at time point indicated as post-S2 in Figs. 5 and 6A).
Fig. 6.

Effect of S2 stimulation on discharge of rostroventromedial medullary (RVM) neurons in animals with neuropathic hypersensitivity. A: heat-evoked limb withdrawal latency before, during, and 2 min following the end of contralateral S2 stimulation determined concurrently when performing recordings of RVM neurons. B: heat-evoked change in discharge rates of ON-cells before (Pre) or 2 min after (Post) the end of real or fake stimulation of the right S2. C: latency to the heat-evoked discharge inhibition (OFF-cells) or burst discharge (ON-cells) before and after real or fake S2 stimulation. D: duration of the heat-evoked discharge inhibition (OFF-cells) or burst discharge (ON-cells) before and after real or fake S2 stimulation. E: heat-evoked change in the late discharge rate (left) or burst discharge duration (right) of ON-cells “during” S2 stimulation (immediately after offset of S2 stimulus). F: heat-evoked change in the late discharge rate (left) or the duration of discharge inhibition (right) of OFF-cells “during” S2 stimulation (immediately after offset of S2 stimulus). In B, 0 Hz (broken horizontal line) represents the corresponding baseline discharge rate and values <0 Hz represent heat-evoked decreases in the rate, while values >0 Hz represent heat-evoked increases in the rate. In E and F, 0 represents the corresponding Pre-S2 value, and values <0 represent decreases induced by S2 stimulation, real S2 = 30 µA, fake S2 = 0 µA. In all graphs, bars show the mean values and the error bars represent SD (in A, n = 8; in B–D, nOFF = 17, nONrealS2 = 24, nONfakeS2 = 16; in E, nreal = 21, nfake = 19; in F, nreal = 7). *P < 0.05, ***P < 0.005 (in A–D, Bonferroni-corrected t-test; in E, paired t-test).
Heat-evoked response magnitude in RVM cells after S2 stimulation.
S2 stimulation induced electromagnetic noise that masked the concurrent neuronal discharge in the RVM (Fig. 5). Therefore, the primary data analysis about the effect of S2 stimulation on heat-evoked responses of RVM cells was not performed during S2 stimulation, but 2 min after completing the delivery of an ascending series of S2 stimuli (stimulus condition “Post S2” in Fig. 5). Heat-evoked discharge rates were studied in 17 presumably antinociceptive RVM OFF-cells that were recorded in five hypersensitive SNL animals with and without (i.e., Pre- and Post-) real S2 stimulation (fake S2 stimulation condition was not studied in RVM OFF-cells). In RVM OFF-cells, the (absolute) ongoing discharge rate (i.e., before heat or S2 stimulation) was 8.0 ± 6.6 Hz (±SD; n = 17). Heat stimulation evoked a marked inhibition of the ongoing discharge rate in RVM OFF-cells. However, the decrease of the heat-evoked discharge inhibition in RVM OFF-cells was not significantly different before vs. 2 min after S2 stimulation (t16 = 1.4, paired t-test; Fig. 6B).
Effect of real S2 stimulation was studied in 24 presumably pronociceptive RVM ON-cells that were sampled in seven SNL animals, while the effect by fake stimulation of S2 (0 µA) was studied in 16 RVM ON-cells recorded in five SNL animals. In RVM ON-cells, the (absolute) ongoing discharge rate (before heat or S2 stimulation) was 2.2 ± 2.4 Hz (n = 24). Heat stimulation induced a marked increase in the discharge rate of ON-cells. Statistical assessments of the S2 stimulation-induced changes in RVM ON-cell discharge rate were performed by using mixed design two-way ANOVA with main factors cortical stimulation condition (fake vs. real S2 stimulation) and time (before vs. after S2 stimulation). Heat-evoked increase in the discharge rate of RVM ON-cells was not influenced by S2 stimulation as indicated by nonsignificant main effects of S2 stimulation (real vs. fake S2 stimulation: F1,38 = 0.7) and time point of testing (before vs. after S2 stimulation: F1,38 = 2.8), or their interaction (F1,38 = 0.2; Fig. 6B).
Heat-evoked response latency in RVM cells after S2 stimulation.
In RVM OFF-cells, the heat-evoked response latency (i.e., the latency to discharge inhibition) was increased in post-S2 condition (t16 = 2.8, P = 0.01; Fig. 6C). In RVM ON-cells, main effect of cortical stimulus condition (post-fake vs. post-real S2 stimulation) on the latency of the heat-evoked burst discharge was not significant (F1,38 = 1.9). The main effect of time (pre- vs. post-cortical stimulation) on ON-cell response latency was close to significance (F1,38 = 3.4, P = 0.07), while the interaction between cortical stimulus condition and time was not significant (F1,38 = 1.7; Fig. 6C). Post hoc testing indicated that the heat-evoked response latency of RVM ON-cells was significantly prolonged following real S2 stimulation but not following fake S2 stimulation (Fig. 6C).
Heat-evoked response duration in RVM cells after S2 stimulation.
In RVM OFF-cells, the duration of the heat-evoked pause was not significantly different when comparing values assessed before vs. after S2 stimulation (t16 = 1.4). In RVM ON-cells, main effect of cortical stimulus condition (post-real vs. post-fake S2 stimulation) on the duration of the heat-evoked burst discharge was short of significance (F1,36 = 2.8). However, the main effect of time (before vs. after cortical stimulation) on discharge duration of RVM ON-cells was highly significant (F1,36 = 11.6, P = 0.002), and the effect varied with the cortical stimulus condition (interaction: F1,36 = 5.6, P = 0.02; Fig. 6D). Post hoc testing indicated that the decrease in the duration of the heat-evoked burst discharge in RVM ON-cells was significant only after real S2 stimulation but not after fake S2 stimulation (Fig. 6D).
Heat-evoked late responses of RVM cells “during” S2 stimulation.
Behavioral (above) and spinal neuronal (below) analyses on the antinociceptive effect of S2 stimulation were performed during S2 stimulation, whereas because of S2 stimulation-induced electric noise, real and fake S2 stimulation-induced modulations of heat-evoked responses of RVM neurons shown in Fig. 6, B–D, were assessed from the response to a heat stimulus that was delivered 2 min after the end of series of real or fake S2 stimulations (Fig. 5). During the 2-min delay the effect of S2 stimulation on the successive heat-evoked behavioral response was attenuated (Fig. 5 and 6A). To assess whether also the neuronal response in the RVM was attenuated during this delay, we assessed the late response to the heat stimulus (i.e., the delayed heat-evoked discharge determined immediately after the offset of the heat stimulus and the S2 stimulus of 30 µA, which was a stimulus condition defined as “during” S2 stimulation; see Fig. 5 and interval E in Fig. 1). The late response “during” real S2 stimulation was compared with the corresponding late response in the Pre-S2 and fake S2 conditions (Fig. 5). The comparisons of the late responses in RVM ON-cells indicated that the duration of the late response was significantly shorter “during” real than fake S2 stimulation (t38 = 4.9, P < 0.0001), whereas the magnitude of the late response in RVM ON-cells was not different between real and fake S2 conditions (t38 = 0.4; Fig. 6E). In RVM OFF-cells, the magnitude or the duration of the late discharge inhibition “during” real S2 stimulation was not significantly different from the corresponding value in the Pre-S2 condition (rate: t7 = 1.0; duration of inhibition: t7 = 1.0; Fig. 6F).
Effect of S2 stimulation on discharge of spinal WDR neurons.
Effect of (right) contralateral S2 stimulation at the intensity of 30 µA (real S2 stimulation) or 0 µA (fake S2 stimulation) on ongoing activity and heat-evoked responses of nociceptive SDH neurons were assessed only in (mechanically) hypersensitive SNL rats (see Fig. 7 for original examples of recordings). All SDH neurons were located in the deep SDH (recording depth from the cord surface 0.4–1.0 mm) and they had their receptive fields in the paw of the nerve-injured (left) hind limb. All SDH neurons reported here could be classified as WDR neurons based on their excitatory response to innocuous mechanical stimulation as well as noxious heat. Unlike with RVM recordings, real S2 stimulation did not induce electric noise masking neuronal data sampled in the SDH. Therefore, all the S2-induced effects on spinal WDR neurons are assessed during real (30 µA) or fake (0 µA) S2 stimulation.
Fig. 7.
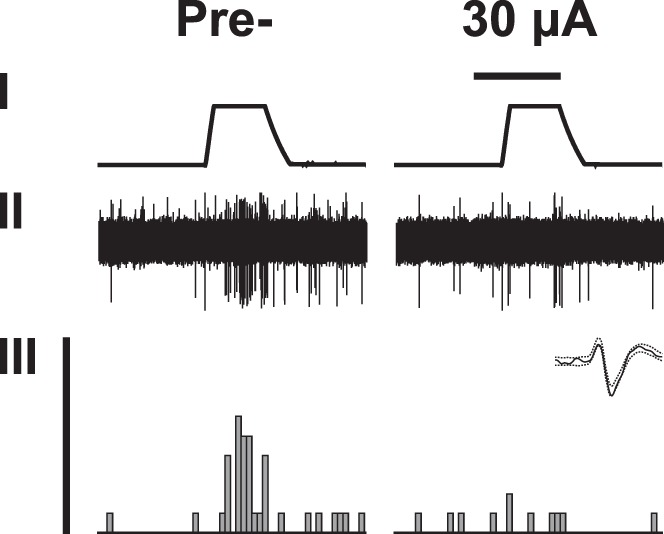
An example of original recording of a wide dynamic range neuron in the spinal dorsal horn of an animal with neuropathic hypersensitivity. Trace I shows the temperature of the contact thermode applied to the hind paw of the nerve-injured limb (baseline 35°C, peak 53°C). Trace II shows raw data of neuronal recording. Trace III shows the peristimulus time histogram, above which is shown the template of the recorded WDR neuron. In the left column, recording before S2 stimulation (Pre-). In the right column, recording during stimulation of S2 contralateral to the tested limb. Horizontal bar above the temperature trace represents 15 s and indicates the duration of S2 stimulation at the intensity of 30 µA.
Ongoing discharge of spinal WDR neurons during S2 stimulation.
According to two-way ANOVA, main effect of stimulation group (real vs. fake stimulation of the contralateral S2) on ongoing (“spontaneous”) discharge rate (assessed during the first 5 s of S2 stimulation that preceded the onset of heat stimulation) was not significant (F1,14 = 0.2; nrealS2 = 9 neurons/5 animals, nfakeS2 = 7 neurons/3 animals). Nor did the time point of testing have a significant effect on ongoing discharge rate (before or during cortical stimulation: F1,14 = 0.3), independent of the stimulation group (interaction: F1,14 = 1.5; Fig. 8A).
Fig. 8.

Influence of contralateral S2 stimulation on ongoing and heat-evoked discharge of spinal wide dynamic range neurons in the spinal dorsal horn of hypersensitive neuropathic animals. A: ongoing discharge rate. B: heat-evoked discharge rate. C: latency of the heat-evoked discharge. D: duration of the heat-evoked discharge. Pre = before S2 stimulation, Real S2 = during S2 stimulation at the intensity of 30 µA, Fake S2 = during S2 stimulation at the intensity of 0 µA. Error bars represent SD (nRealS2 = 9, nFakeS2 = 7), *P < 0.05 (Bonferroni-corrected t-test).
Heat-evoked response magnitude in spinal WDR neurons during S2 stimulation.
Heat-evoked discharge rate of WDR neurons was not significantly influenced by the treatment group (real vs. fake S2 stimulation: F1,14 = 0.2; Fig. 8B), while main effect of time (before vs. during S2 stimulation: F1,14 = 7.5, P = 0.02) but not the interaction between time and treatment group (F1,14 = 1.1) had a significant effect on the heat-evoked discharge rate of WDR neurons (Fig. 8B). Post hoc testing indicated that the heat-evoked discharge rate of WDR neurons was significantly attenuated during real S2 stimulation (Fig. 8B).
Heat-evoked response latency in spinal WDR neurons during S2 stimulation.
Main effect of treatment group (real vs. fake S2) on the latency of the heat-evoked discharge in WDR neurons was not significant (F1, 14 = 1.5), while the response latency was significantly influenced by time (before vs. during S2 stimulation: F1,14 = 29.7, P < 0.0001; Fig. 8C), and this effect varied with the treatment group (interaction: F1,14 = 26.0, P = 0.0002). Post hoc testing indicated that the latency of the heat-evoked burst discharge in spinal WDR neurons was significantly prolonged during real S2 stimulation but not during fake stimulation of S2 (Fig. 8C).
Heat-evoked response duration in spinal WDR neurons during S2 stimulation.
The duration of the heat-evoked discharge in WDR neurons was not significantly influenced by the stimulation group (real S2 vs. fake S2 stimulation: F1,14 = 0.1) or time point of testing (before vs. during S2 stimulation: F1,14 = 0.4), while interaction between these two main factors was significant (F1,14 = 11.5, P = 0.004). Post hoc testing indicated that the duration of the heat-evoked discharge was significantly reduced by real but not fake S2 stimulation (Fig. 8D).
Attempt to Prevent the S2 Stimulation-Induced Descending Antinociception by Blocking the Spinal 5-HT1A Receptor
Ongoing discharge rate of spinal WDR neurons after block of the spinal 5-HT1A receptor.
To assess whether the spinal 5-HT1A receptor is involved in mediating the S2 stimulation-induced antinociception, we assessed whether spinally administered WAY-100635 (a 5-HT1A receptor antagonist; 2.5 µg) vs. saline prevents the effect of contralateral S2 stimulation (30 µA) on spinal WDR neurons in (mechanically) hypersensitive SNL animals (nSal = 9 neurons/5 animals, nWAY-100635 = 9 neurons/4 animals). Main effect of drug treatment (WAY-100635 vs. saline: F1,16 = 2.6) or S2 stimulation (before or during S2 stimulation: F1,16 = 0.1) on ongoing discharge rate of WDR neurons was not significant, nor was there any significant interaction between these two main factors (F1,16 = 2.2; Fig. 9A).
Fig. 9.

Effect of a spinally administered 5-HT1A receptor antagonist on the S2 stimulation-induced modulation of the discharge in spinal dorsal horn wide dynamic range neurons in hypersensitive neuropathic animals. A: ongoing discharge rate. B: heat-evoked discharge rate. C: latency of the heat-evoked discharge. D: duration of the heat-evoked discharge. Pre = before S2 stimulation, WAY-100635 = 5-HT1A receptor antagonist (2.5 µg). Error bars represent SD (nWAY-100635 = 9, nSaline = 9), *P < 0.05, **P < 0.01, ***P < 0.005 (Bonferroni-corrected t-test).
Heat-evoked response magnitude in spinal WDR neurons after block of the 5-HT1A receptor.
WAY-100635 treatment as a main factor had a close to significant effect on the heat-evoked discharge rate of WDR neurons (F1,16 = 3.8, P = 0.07). The accompanying S2 stimulation as a main factor had a significant effect on the discharge rate (F1,16 = 5.6, P = 0.03), whereas interaction between these two main factors was not significant (F1,16 = 1.7; Fig. 9B). Post hoc testing indicated that the heat-evoked discharge rate was significantly attenuated by S2 stimulation in the saline-treated but not the WAY-100635-treated group (Fig. 9B).
Heat-evoked response latency in spinal WDR neurons after block of the 5-HT1A receptor.
Main factors WAY-100635 treatment (F1,16 = 6.2, P = 0.02; Fig. 9C) and accompanying S2 stimulation (F1,16 = 17.6, P = 0.007) had significant effects on the heat-evoked response latency of spinal WDR neurons. Interaction between these two main factors was also significant (F1,16 = 9.0, P = 0.009). Post hoc testing indicated that the heat-evoked response latency was significantly increased by S2 stimulation in the saline group only, and that the response latency assessed during S2 stimulation was significantly longer in the saline than WAY-100635 group (Fig. 9C).
Heat-evoked response duration in spinal WDR neurons after block of the 5-HT1A receptor.
WAY-100635 did not have a significant main effect on the duration of the heat-evoked discharge in WDR neurons (F1,16 = 1.8; Fig. 9D), while the main effect of accompanying S2 stimulation was significant (F1,16 = 5.8, P = 0.03). Interaction between these two main factors was not significant (F1,16 = 0.0). Post hoc testing failed to find significant differences between the experimental conditions (Fig. 9D).
DISCUSSION
The present results indicate that S2 stimulation attenuates heat nociception in animals with the SNL model of experimental neuropathy, but only if SNL is associated with mechanical hypersensitivity that presumably reflects chronic pain. S2 stimulation, however, was ineffective in sham-operated rats and, importantly, in (mechanically) nonhypersensitive SNL rats. This finding suggests that the nerve injury-induced neuroplasticity underlying hypersensitivity rather than nerve injury itself determines the descending antinociceptive efficacy of S2 stimulation. In line with our findings, an earlier experimental animal study indicated that S2 stimulation attenuated pain-related behavior in animals with sustained inflammatory pain but not in healthy controls (Kuroda et al. 2000, 2001). The antinociceptive effect induced by S2 stimulation was bilateral, and the maximal antinociceptive effect was obtained already at the lowest stimulus current of the present study, whereas fake stimulation of S2 failed to influence nociception. The optimal site for inducing antinociception was within S2 rather than adjacent to it, as indicated by the finding that stimulation in cortical sites adjacent to the S2 had a significantly weaker antinociceptive effect. The finding that stimulation of S2 at a high intensity did not produce stronger effects than stimulation at a low intensity supports the proposal that antinociception in the present study was due to local action on S2 rather than spread of the stimulus to adjacent cortical areas. In line with this, the adjacent primary somatosensory cortical area represented orofacial region, and the effect of cortical stimulation in the present study was assessed in the hind paw, while somatotopic presentation area of the painful region is typically stimulated in human TMS studies to suppress pain (Moisset et al. 2016). Concerning the other adjacent cortical area, the granular insular cortex, spread of current to it is not expected to induce antinociception, since its inhibition or lesion has produced antinociception (Burkey et al. 1999; Coffeen et al. 2011; Jasmin et al. 2003), while its activation in humans has evoked painful perceptions (Mazzola et al. 2009). This proposal is further supported by the finding that five of six of the mislocated cortical stimulation sites in the present study were in the granular insular cortex, the stimulation of which had a significantly weaker antinociceptive effect than that of S2.
The duration of the S2 stimulation-induced antinociceptive effect was short; behaviorally, spinal nociception was back to baseline level in 2 min, while in RVM neurons the effect was still significant 2 min after the offset of cortical stimulation. However, it should be noted that in the present protocol the duration of S2 stimulation was short, only 15 s. In earlier human studies describing longer analgesia effects, the duration of S2 stimulation was an order of magnitude longer (Lindholm et al. 2015; Valmunen et al. 2009), suggesting that prolongation of S2 stimulation might have produced a longer lasting effect also in the present study.
S2 Stimulation-Induced Descending Antinociception: Roles of RVM ON- and OFF-Cells
A large number of cortical and subcortical structures contribute to descending pain control through projections to the midbrain periaqueductal gray that further projects to the RVM, a major final relay in descending control of nociception (Heinricher et al. 2009; Millan 2002; Pertovaara and Almeida 2006). In the RVM, pronociceptive ON-cells that have an excitatory response just before a nocifensive reflex and antinociceptive OFF-cells that have a pause in their discharge before a nocifensive reflex are known to contribute to descending control of nociception (Heinricher et al. 1989). In the present study, S2 stimulation induced a significant decrease in the duration of the heat-evoked RVM ON-cell discharge, and this was associated with the antinociceptive action of S2 stimulation. This finding suggests that suppression of RVM ON-cells contributed to the S2 stimulation-induced antinociception. In RVM OFF-cells, S2 stimulation delayed the onset of the heat-evoked pause while it did not induce significant changes in pause duration. The delay in the onset of the RVM OFF-cell pause might have contributed to the descending antinociceptive effect induced by S2 stimulation, since RVM OFF-cell discharge is expected to promote antinociception (Heinricher et al. 1989). However, one should be cautious with interpretations of this finding, since in RVM OFF-cell recordings the effect of real S2 stimulation was not compared with that of fake S2 stimulation. Additionally, the cortical stimulus-induced noise prevented assessing the delay in the onset of OFF-cell pause when delivering concurrently S2 and heat stimuli. Therefore, the delay in the onset of the response pause in RVM OFF-cells had to be assessed later when delivering heat stimulus alone at a time point when the behavioral effect of S2 stimulation was not anymore significant. In contrast, in RVM ON-cell recordings the effect of real S2 stimulation was compared with that of fake S2 stimulation. Moreover, the shortening of the heat-evoked RVM ON-cell discharge by S2 stimulation could be observed during concomitant delivery of S2 and heat stimuli as a significantly decreased duration of the heat-evoked afterdischarge. This finding is in line with the hypothesis that suppression of RVM ON-cell discharge is among mechanisms contributing to the S2 stimulation-induced antinociception.
S2 Stimulation-Induced Descending Antinociception: Role of the Medullospinal Serotonergic Pathway Acting on the Spinal 5HT1A Receptor
There is abundant evidence indicating that spinally projecting serotonergic RVM cells are involved in descending control of nociception that may vary from anti- to pronociception, depending on the type of the 5-HT receptor that is activated in the SDH (Millan 2002; Suzuki et al. 2002). Neither the RVM ON- or OFF-cell is serotonergic, but serotonergic RVM cells belong to a subgroup of NEUTRAL-cells that have distinct discharge properties and that give only a weak if any response to noxious stimulation (Mason 1997). Since serotonergic RVM cells were not recorded in the present study, the results do not give any direct electrophysiological evidence on the involvement of serotonergic RVM cells in the S2 stimulation-induced antinociception. However, behavioral results obtained with microinjection of a selective 5-HT1A receptor agonist into the RVM provide indirect evidence indicating that serotonergic RVM cells were involved in mediating the descending antinociceptive effect induced by S2 stimulation. This is indicated by the finding that a 5-HT1A agonist in the RVM at a dose that alone had no effect prevented the descending antinociceptive effect induced by S2 stimulation. A potential underlying mechanism for this disinhibitory action is activation of autoinhibitory 5-HT1A receptors on cell bodies of serotoninergic RVM neurons. Autoinhibitory 5-HT1A receptors are expected to inhibit medullary serotonergic neurons (Rogawski and Aghajanian 1981), the spinal projections of which are the main source of spinal serotonin (Kwiat and Basbaum 1992). However, because of the complexity of serotonergic innervation of the RVM, one needs to be cautious with interpretations. Serotonergic appositions have been demonstrated on various types of RVM cells, including nonserotonergic ON- and OFF-cells as well as serotonergic NEUTRAL-cells (Potrebic et al. 1995). The functional role of these serotonergic connections within the RVM is still somewhat puzzling, as indicated by the finding that RVM administration of 5-HT or its agonist has produced effects that vary from activation to inhibition in all cell types (Roychowdhury and Heinricher 1997). Present findings in the SDH give more straightforward evidence than those in the RVM for the involvement of descending serotonergic pathways in the S2 stimulation-induced antinociception.
Earlier, it was shown that blocking spinal 5-HT receptors with a nonselective antagonist attenuates antinociceptive effect of S2 stimulation (Kuroda et al. 2001). The present study extends this earlier finding by showing that selective blocking of the spinal 5-HT1A receptor prevents the S2 stimulation-induced antinociceptive action on spinal WDR neurons. This result supports the proposal that medullospinal serotonergic neurons acting on spinal 5-HT1A receptors were involved in descending antinociception induced by S2 stimulation. Interestingly, descending antinociception induced by motor cortex stimulation (MCS) in nerve-injured rats has also been suppressed following pharmacological block of the spinal 5-HT1A receptor (Viisanen and Pertovaara 2010b), indicating that in animals with experimental neuropathy the medullospinal serotonergic pathway contributes to descending antinociception induced by MCS as well as S2 stimulation.
Significant prolongation of the heat-evoked response latency in spinal WDR neurons by concurrent S2 stimulation indicates that the prolongation of the heat-evoked limb withdrawal by S2 stimulation in the behavioral part of the study is predominantly due to inhibition of spinal sensory rather than motor neurons. The finding that S2 stimulation failed to suppress ongoing discharge of WDR neurons is in line with the interpretation that the descending inhibitory action was pre- rather than postsynaptic to the studied WDR neurons, although the low level of ongoing discharge may have contributed to the failure to observe significant suppression of ongoing discharge by S2 stimulation. Nevertheless, spinally administered 5-HT1A receptor antagonist prevented the S2 stimulation-induced prolongation of the heat-evoked response latency in the present study, and a recent patch clamp study in spinal cord slices demonstrated that serotonin suppresses nociceptive excitatory transmission from primary afferent nerve fibers due to action on presynaptic 5-HT1A receptors (Tomoyose et al. 2014). Together, these results support the proposal that presynaptic inhibition of nociceptive afferent barrage in the SDH contributed to the present findings. The reversal of the S2 stimulation-induced antinociception with a spinally administered 5-HT1A receptor antagonist is in line with the earlier evidence indicating that 5-HT1A receptor agonists administered spinally or systemically have proved effective in suppressing pain behavior in various experimental animal models of chronic pain (Colpaert 2006).
Other Mechanisms Contributing to Antinociception Induced by S2 Stimulation
An earlier anatomical study suggested that S2 might influence spinal trigeminal nucleus (that corresponds to the SDH at the lumbar spinal cord level) through direct corticospinal pathways (Gojyo et al. 2002). Therefore, we cannot exclude the possibility that direct corticospinal projections contributed to the S2 stimulation-induced antinociception in the present study. In analogy, it has been shown that spinal antinociception induced by stimulation of the sensorimotor cortex is at least partly mediated by direct spinal projections from the cortical stimulation area (Rojas-Piloni et al. 2010).
In addition to direct S2 projections to the medullary SDH (Gojyo et al. 2002), S2 may influence spinal nociception through descending projections to various relay nuclei, although there is only a limited amount of data of these connections. For example, retrograde tract-tracing studies have demonstrated that S2 projects to an antinociceptive brainstem site, the lateral caudal ventrolateral medulla (Cobos et al. 2003), and to a pronociceptive medullary site, the medullary dorsal reticular nucleus (Almeida et al. 2002), that further projects to the RVM (Leite-Almeida et al. 2006) thereby providing one potential link to medullospinal serotonergic pathways. However, it still remains to be studied whether these or some other pathways mediated the descending antinociceptive effect induced by S2 stimulation. Moreover, further studies are needed to characterize plastic mechanisms explaining that S2 stimulation produced significant antinociception in hypersensitive SNL animals at an intensity that failed to influence nociception in nonhypersensitive SNL animals or sham-operated controls.
The earlier failure to block S2 stimulation-induced antinociception by an antagonist of the α1,2-adrenoceptor or µ-opioid receptor suggests that noradrenergic or opioidergic systems may not have a key role in antinociception induced by S2 stimulation (Kuroda et al. 2001). Similarly, it has been shown that the noradrenergic system is not critical for antinociception induced by MCS in neuropathic animals (Viisanen and Pertovaara 2010a), whereas previous experimental animal studies suggest that opioidergic system contributes to antinociception induced by MCS both in physiological and neuropathic conditions (Fonoff et al. 2009; Silva et al. 2015). It still remains to be studied whether other mechanisms associated with MCS-induced antinociception such as the dopaminergic system (Lapirot et al. 2011; Viisanen et al. 2012), activation of the thalamic zona incerta nucleus (Lucas et al. 2011) or various other subcortical structures (Pagano et al. 2011), and actions on the spinal cannabinoid and antineuroinflammatory systems (Silva et al. 2015) might contribute to the S2 stimulation-induced effects.
Caveats
In the present study, the experiments were performed under anesthesia. While anesthesia is an important confounding factor and the results might have been at least partly different in awake animals, anesthesia may not explain the differences in the results among the experimental groups, since the anesthetic conditions were identical among groups.
The present sample of nerve-injured animals that had mechanical hypersensitivity did not have heat hypersensitivity. While the currently used rat model of neuropathy frequently produces both mechanical and heat hyperalgesia (Kim and Chung 1992), there are also studies reporting that mechanical hypersensitivity is not invariably associated with heat hypersensitivity in this model (Kontinen et al. 1998; Röyttä et al. 1999). It should also be noted that a decrease in response latency to radiant heat reported in animals with peripheral neuropathy may in some cases be explained by a nerve injury-associated change in skin temperature rather than heat nociception (Luukko et al. 1994). In the present study, heat nociception was assessed with a contact thermostimulator that clamped the baseline skin temperature to 35°C, which excluded a change in skin temperature as a confounding factor. Additionally or alternatively, among possible explanations for the dissociation of heat and mechanical hypersensitivity in the present study is that mechanical hypersensitivity was assessed in awake animals, whereas heat sensitivity was assessed under anesthesia. Nevertheless, in clinical conditions, as in the present study, neuropathy is typically associated with mechanical rather than heat hypersensitivity (Scadding and Koltzenburg 2006).
It should be noted that the present study assessed S2 stimulation-induced effect on heat-evoked spinal nociception. Therefore, one should be cautious when applying the present results to supraspinally organized nociception or to responses evoked by other submodalities of noxious test stimulation. When comparing the present results in experimental animals with results in humans, it is important to note that supraspinal mechanisms influencing perception or the emotional appraisal of pain are likely to exert a key role in cortical stimulation-induced pain relief in human studies (Garcia-Larrea and Peyron 2007).
Conclusions
S2 stimulation attenuates spinal heat nociception in animals that have mechanical hypersensitivity (and thereby presumably chronic pain) induced by experimental model of peripheral neuropathy. Among mechanisms underlying the S2 stimulation-induced descending antinociception are suppression of pronociceptive RVM ON-cells and activation of medullospinal serotonergic pathways that suppress nociceptive transmission to spinal WDR neurons because of action on presumably presynaptic 5-HT1A receptors in the SDH.
GRANTS
The funding for this study was provided by the Sigrid Jusélius Foundation, Helsinki, Finland (to A. Pertovaara).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
B.S., H.V., and H.W. performed experiments; B.S. and A.P. conception and design of research, analysis of the data, interpretation of results, preparation of figures, editing and revision of manuscript; B.S., H.V., H.W., and A.P. approved the final version of manuscript.
REFERENCES
- Almeida A, Cobos A, Tavares I, Lima D. Brain afferents to the medullary dorsal reticular nucleus: a retrograde and anterograde tracing study in the rat. Eur J Neurosci 16: 81–95, 2002. doi: 10.1046/j.1460-9568.2002.02058.x. [DOI] [PubMed] [Google Scholar]
- Burkey AR, Carstens E, Jasmin L. Dopamine reuptake inhibition in the rostral agranular insular cortex produces antinociception. J Neurosci 19: 4169–4179, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos A, Lima D, Almeida A, Tavares I. Brain afferents to the lateral caudal ventrolateral medulla: a retrograde and anterograde tracing study in the rat. Neuroscience 120: 485–498, 2003. doi: 10.1016/S0306-4522(03)00209-4. [DOI] [PubMed] [Google Scholar]
- Coffeen U, Manuel Ortega-Legaspi J, López-Muñoz FJ, Simón-Arceo K, Jaimes O, Pellicer F. Insular cortex lesion diminishes neuropathic and inflammatory pain-like behaviours. Eur J Pain 15: 132–138, 2011. doi: 10.1016/j.ejpain.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Coghill RC, Mayer DJ, Price DD. Wide dynamic range but not nociceptive-specific neurons encode multidimensional features of prolonged repetitive heat pain. J Neurophysiol 69: 703–716, 1993. [DOI] [PubMed] [Google Scholar]
- Colpaert FC. 5-HT(1A) receptor activation: new molecular and neuroadaptive mechanisms of pain relief. Curr Opin Investig Drugs 7: 40–47, 2006. [PubMed] [Google Scholar]
- Coulter JD, Maunz RA, Willis WD. Effects of stimulation of sensorimotor cortex on primate spinothalamic neurons. Brain Res 65: 351–356, 1974. doi: 10.1016/0006-8993(74)90049-3. [DOI] [PubMed] [Google Scholar]
- Dubner R, Sessle BJ. Presynaptic excitability changes of primary afferent and corticofugal fibers projecting to trigeminal brain stem nuclei. Exp Neurol 30: 223–238, 1971. doi: 10.1016/S0014-4886(71)80003-1. [DOI] [PubMed] [Google Scholar]
- Fonoff ET, Dale CS, Pagano RL, Paccola CC, Ballester G, Teixeira MJ, Giorgi R. Antinociception induced by epidural motor cortex stimulation in naive conscious rats is mediated by the opioid system. Behav Brain Res 196: 63–70, 2009. doi: 10.1016/j.bbr.2008.07.027. [DOI] [PubMed] [Google Scholar]
- Fregni F, Potvin K, Dasilva D, Wang X, Lenkinski RE, Freedman SD, Pascual-Leone A. Clinical effects and brain metabolic correlates in non-invasive cortical neuromodulation for visceral pain. Eur J Pain 15: 53–60, 2011. doi: 10.1016/j.ejpain.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Larrea L, Peyron R. Motor cortex stimulation for neuropathic pain: from phenomenology to mechanisms. Neuroimage 37, Suppl 1: S71–S79, 2007. doi: 10.1016/j.neuroimage.2007.05.062. [DOI] [PubMed] [Google Scholar]
- Gojyo F, Sugiyo S, Kuroda R, Kawabata A, Varathan V, Shigenaga Y, Takemura M. Effects of somatosensory cortical stimulation on expression of c-Fos in rat medullary dorsal horn in response to formalin-induced noxious stimulation. J Neurosci Res 68: 479–488, 2002. doi: 10.1002/jnr.10227. [DOI] [PubMed] [Google Scholar]
- Hagbarth KE, Kerr DI. Central influences on spinal afferent conduction. J Neurophysiol 17: 295–307, 1954. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Barbaro NM, Fields HL. Putative nociceptive modulating neurons in the rostral ventromedial medulla of the rat: firing of on- and off-cells is related to nociceptive responsiveness. Somatosens Mot Res 6: 427–439, 1989. doi: 10.3109/08990228909144685. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: specificity, recruitment and plasticity. Brain Res Brain Res Rev 60: 214–225, 2009. doi: 10.1016/j.brainresrev.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hole K, Tjølsen A. The tail-flick and formalin tests in rodents: changes in skin temperature as a confounding factor. Pain 53: 247–254, 1993. doi: 10.1016/0304-3959(93)90220-J. [DOI] [PubMed] [Google Scholar]
- Honoré PH, Basnet A, Eljaja L, Kristensen P, Munkholm Andersen L, Neustrup S, Møllgaard P, Bjerrum OJ. Neuropathic pain models in the development of analgesic drugs. Scand J Pain 2: 172–177, 2011. doi: 10.1016/j.sjpain.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Rabkin SD, Granato A, Boudah A, Ohara PT. Analgesia and hyperalgesia from GABA-mediated modulation of the cerebral cortex. Nature 424: 316–320, 2003. doi: 10.1038/nature01808. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50: 355–363, 1992. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kontinen VK, Paananen S, Kalso E. The effects of the α2-adrenergic agonist, dexmedetomidine, in the spinal nerve ligation model of neuropathic pain in rats. Anesth Analg 86: 355–360, 1998. [DOI] [PubMed] [Google Scholar]
- Kuroda R, Kawabata A, Kawao N, Umeda W, Takemura M, Shigenaga Y. Somatosensory cortex stimulation-evoked analgesia in rats: potentiation by NO synthase inhibition. Life Sci 66: PL271–PL276, 2000. doi: 10.1016/S0024-3205(00)00525-7. [DOI] [PubMed] [Google Scholar]
- Kuroda R, Kawao N, Yoshimura H, Umeda W, Takemura M, Shigenaga Y, Kawabata A. Secondary somatosensory cortex stimulation facilitates the antinociceptive effect of the NO synthase inhibitor through suppression of spinal nociceptive neurons in the rat. Brain Res 903: 110–116, 2001. doi: 10.1016/S0006-8993(01)02446-5. [DOI] [PubMed] [Google Scholar]
- Kwiat GC, Basbaum AI. The origin of brainstem noradrenergic and serotonergic projections to the spinal cord dorsal horn in the rat. Somatosens Mot Res 9: 157–173, 1992. doi: 10.3109/08990229209144768. [DOI] [PubMed] [Google Scholar]
- Lapirot O, Melin C, Modolo A, Nicolas C, Messaoudi Y, Monconduit L, Artola A, Luccarini P, Dallel R. Tonic and phasic descending dopaminergic controls of nociceptive transmission in the medullary dorsal horn. Pain 152: 1821–1831, 2011. doi: 10.1016/j.pain.2011.03.030. [DOI] [PubMed] [Google Scholar]
- Leite-Almeida H, Valle-Fernandes A, Almeida A. Brain projections from the medullary dorsal reticular nucleus: an anterograde and retrograde tracing study in the rat. Neuroscience 140: 577–595, 2006. doi: 10.1016/j.neuroscience.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Lim M, Roosink M, Kim JS, Kim HW, Lee EB, Son KM, Kim HA, Chung CK. Augmented pain processing in primary and secondary somatosensory cortex in fibromyalgia: a magnetoencephalography study using intra-epidermal electrical stimulation. PLoS One 11: e0151776, 2016. doi: 10.1371/journal.pone.0151776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm P, Lamusuo S, Taiminen T, Pesonen U, Lahti A, Virtanen A, Forssell H, Hietala J, Hagelberg N, Pertovaara A, Parkkola R, Jääskeläinen S. Right secondary somatosensory cortex-a promising novel target for the treatment of drug-resistant neuropathic orofacial pain with repetitive transcranial magnetic stimulation. Pain 156: 1276–1283, 2015. doi: 10.1097/j.pain.0000000000000175. [DOI] [PubMed] [Google Scholar]
- Lindholm P, Lamusuo S, Taiminen T, Virtanen A, Pertovaara A, Forssell H, Hagelberg N, Jääskeläinen S. The analgesic effect of therapeutic rTMS is not mediated or predicted by comorbid psychiatric or sleep disorders. Medicine (Baltimore) 95: e5231, 2016. doi: 10.1097/MD.0000000000005231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood PL, Iannetti GD, Haggard P. Transcranial magnetic stimulation over human secondary somatosensory cortex disrupts perception of pain intensity. Cortex 49: 2201–2209, 2013. doi: 10.1016/j.cortex.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JM, Ji Y, Masri R. Motor cortex stimulation reduces hyperalgesia in an animal model of central pain. Pain 152: 1398–1407, 2011. doi: 10.1016/j.pain.2011.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukko M, Konttinen Y, Kemppinen P, Pertovaara A. Influence of various experimental parameters on the incidence of thermal and mechanical hyperalgesia induced by a constriction mononeuropathy of the sciatic nerve in lightly anesthetized rats. Exp Neurol 128: 143–154, 1994. doi: 10.1006/exnr.1994.1122. [DOI] [PubMed] [Google Scholar]
- Mason P. Physiological identification of pontomedullary serotonergic neurons in the rat. J Neurophysiol 77: 1087–1098, 1997. [DOI] [PubMed] [Google Scholar]
- Mazzola L, Isnard J, Peyron R, Guénot M, Mauguière F. Somatotopic organization of pain responses to direct electrical stimulation of the human insular cortex. Pain 146: 99–104, 2009. doi: 10.1016/j.pain.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol 66: 355–474, 2002. doi: 10.1016/S0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Moisset X, de Andrade DC, Bouhassira D. From pulses to pain relief: an update on the mechanisms of rTMS-induced analgesic effects. Eur J Pain 20: 689–700, 2016. doi: 10.1002/ejp.811. [DOI] [PubMed] [Google Scholar]
- Nguyen JP, Lefaucheur JP, Decq P, Uchiyama T, Carpentier A, Fontaine D, Brugières P, Pollin B, Fève A, Rostaing S, Cesaro P, Keravel Y. Chronic motor cortex stimulation in the treatment of central and neuropathic pain. Correlations between clinical, electrophysiological and anatomical data. Pain 82: 245–251, 1999. doi: 10.1016/S0304-3959(99)00062-7. [DOI] [PubMed] [Google Scholar]
- Pagano RL, Assis DV, Clara JA, Alves AS, Dale CS, Teixeira MJ, Fonoff ET, Britto LR. Transdural motor cortex stimulation reverses neuropathic pain in rats: a profile of neuronal activation. Eur J Pain 15: 268.e1–268.e14, 2011. doi: 10.1016/j.ejpain.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Pagano RL, Fonoff ET, Dale CS, Ballester G, Teixeira MJ, Britto LR. Motor cortex stimulation inhibits thalamic sensory neurons and enhances activity of PAG neurons: possible pathways for antinociception. Pain 153: 2359–2369, 2012. doi: 10.1016/j.pain.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press, 1998. [Google Scholar]
- Pertovaara A, Almeida A. Chapter 13 Descending inhibitory systems. Handb Clin Neurol 81: 179–192, 2006. doi: 10.1016/S0072-9752(06)80017-5. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Keski-Vakkuri U, Kalmari J, Wei H, Panula P. Response properties of neurons in the rostroventromedial medulla of neuropathic rats: attempted modulation of responses by [1DMe]NPYF, a neuropeptide FF analogue. Neuroscience 105: 457–468, 2001. doi: 10.1016/S0306-4522(01)00187-7. [DOI] [PubMed] [Google Scholar]
- Potrebic SB, Mason P, Fields HL. The density and distribution of serotonergic appositions onto identified neurons in the rat rostral ventromedial medulla. J Neurosci 15: 3273–3283, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA, Aghajanian GK. Serotonin autoreceptors on dorsal raphe neurons: structure-activity relationships of tryptamine analogs. J Neurosci 1: 1148–1154, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Piloni G, Martínez-Lorenzana G, Condés-Lara M, Rodríguez-Jiménez J. Direct sensorimotor corticospinal modulation of dorsal horn neuronal C-fiber responses in the rat. Brain Res 1351: 104–114, 2010. doi: 10.1016/j.brainres.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Roychowdhury SM, Heinricher MM. Effects of iontophoretically applied serotonin on three classes of physiologically characterized putative pain modulating neurons in the rostral ventromedial medulla of lightly anesthetized rats. Neurosci Lett 226: 136–138, 1997. doi: 10.1016/S0304-3940(97)00270-X. [DOI] [PubMed] [Google Scholar]
- Röyttä M, Wei H, Pertovaara A. Spinal nerve ligation-induced neuropathy in the rat: sensory disorders and correlation between histology of the peripheral nerves. Pain 80: 161–170, 1999. doi: 10.1016/S0304-3959(98)00199-7. [DOI] [PubMed] [Google Scholar]
- Sagalajev B, Bourbia N, Beloushko E, Wei H, Pertovaara A. Bidirectional amygdaloid control of neuropathic hypersensitivity mediated by descending serotonergic pathways acting on spinal 5-HT3 and 5-HT1A receptors. Behav Brain Res 282: 14–24, 2015. doi: 10.1016/j.bbr.2014.12.052. [DOI] [PubMed] [Google Scholar]
- Scadding JW, Koltzenburg M. Painful peripheral neuropathies, in Wall and Melzacks’s Textbook of Pain (5th ed.), edited by McMahon SB and Koltzenburg M. Philadelphia, PA: Elsevier, 2006. doi: 10.1016/B0-443-07287-6/50067-9. [DOI] [Google Scholar]
- Senapati AK, Huntington PJ, Peng YB. Spinal dorsal horn neuron response to mechanical stimuli is decreased by electrical stimulation of the primary motor cortex. Brain Res 1036: 173–179, 2005. doi: 10.1016/j.brainres.2004.12.043. [DOI] [PubMed] [Google Scholar]
- Silva GD, Lopes PS, Fonoff ET, Pagano RL. The spinal anti-inflammatory mechanism of motor cortex stimulation: cause of success and refractoriness in neuropathic pain? J Neuroinflammation 12: 10, 2015. doi: 10.1186/s12974-014-0216-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfa LC, Dickenson AH. In vivo electrophysiology of dorsal-horn neurons. Methods Mol Med 99: 139–153, 2004. doi: 10.1385/1-59259-770-X:139. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci 5: 1319–1326, 2002. doi: 10.1038/nn966. [DOI] [PubMed] [Google Scholar]
- Talbot JD, Marrett S, Evans AC, Meyer E, Bushnell MC, Duncan GH. Multiple representations of pain in human cerebral cortex. Science 251: 1355–1358, 1991. doi: 10.1126/science.2003220. [DOI] [PubMed] [Google Scholar]
- Tomoyose O, Kodama D, Ono H, Tanabe M. Presynaptic inhibitory effects of fluvoxamine, a selective serotonin reuptake inhibitor, on nociceptive excitatory synaptic transmission in spinal superficial dorsal horn neurons of adult mice. J Pharmacol Sci 126: 136–145, 2014. doi: 10.1254/jphs.14127FP. [DOI] [PubMed] [Google Scholar]
- Tsubokawa T, Katayama Y, Yamamoto T, Hirayama T, Koyama S. Chronic motor cortex stimulation for the treatment of central pain. Acta Neurochir Suppl (Wien) 52: 137–139, 1991. doi: 10.1007/978-3-7091-9160-6_37. [DOI] [PubMed] [Google Scholar]
- Uglem M, Omland PM, Engstrøm M, Gravdahl GB, Linde M, Hagen K, Sand T. Non-invasive cortical modulation of experimental pain in migraine. Clin Neurophysiol 127: 2362–2369, 2016. doi: 10.1016/j.clinph.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Vaculín S, Franek M, Yamamotová A, Rokyta R. Motor cortex stimulation in rats with chronic constriction injury. Exp Brain Res 185: 331–335, 2008. doi: 10.1007/s00221-007-1158-y. [DOI] [PubMed] [Google Scholar]
- Valmunen T, Pertovaara A, Taiminen T, Virtanen A, Parkkola R, Jääskeläinen SK. Modulation of facial sensitivity by navigated rTMS in healthy subjects. Pain 142: 149–158, 2009. doi: 10.1016/j.pain.2008.12.031. [DOI] [PubMed] [Google Scholar]
- Viisanen H, Ansah OB, Pertovaara A. The role of the dopamine D2 receptor in descending control of pain induced by motor cortex stimulation in the neuropathic rat. Brain Res Bull 89: 133–143, 2012. doi: 10.1016/j.brainresbull.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Viisanen H, Pertovaara A. Antinociception by motor cortex stimulation in the neuropathic rat: does the locus coeruleus play a role? Exp Brain Res 201: 283–296, 2010a. doi: 10.1007/s00221-009-2038-4. [DOI] [PubMed] [Google Scholar]
- Viisanen H, Pertovaara A. Roles of the rostroventromedial medulla and the spinal 5-HT(1A) receptor in descending antinociception induced by motor cortex stimulation in the neuropathic rat. Neurosci Lett 476: 133–137, 2010b. doi: 10.1016/j.neulet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Wei H, Pertovaara A. 5-HT(1A) receptors in endogenous regulation of neuropathic hypersensitivity in the rat. Eur J Pharmacol 535: 157–165, 2006. doi: 10.1016/j.ejphar.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Wilcox GL, Giesler GJ Jr. An instrument using a multiple layer Peltier device to change skin temperature rapidly. Brain Res Bull 12: 143–146, 1984. doi: 10.1016/0361-9230(84)90227-2. [DOI] [PubMed] [Google Scholar]
- Willis WD, Coggeshall RE. Sensory Mechanisms of the Spinal Cord: Volume 1 Primary Afferent Neurons and the Spinal Dorsal Horn (3rd ed.). New York: Springer, 2004. [Google Scholar]