

Abstract
Nonsense mutations resulting in a premature stop codon in an open reading frame occur in critical tumor suppressor genes in a large number of the most common forms of cancers and are known to cause or contribute to the progression of disease. Low molecular weight compounds that induce readthrough of nonsense mutations offer a new means of treating patients with genetic disorders or cancers resulting from nonsense mutations. We have identified the nucleoside analog clitocine as a potent and efficacious suppressor of nonsense mutations. We determined that incorporation of clitocine into RNA during transcription is a prerequisite for its readthrough activity; the presence of clitocine in the third position of a premature stop codon directly induces readthrough. We demonstrate that clitocine can induce the production of p53 protein in cells harboring p53 nonsense-mutated alleles. In these cells, clitocine restored production of full-length and functional p53 as evidenced by induced transcriptional activation of downstream p53 target genes, progression of cells into apoptosis, and impeded growth of nonsense-containing human ovarian cancer tumors in xenograft tumor models. Thus, clitocine induces readthrough of nonsense mutations by a previously undescribed mechanism and represents a novel therapeutic modality to treat cancers and genetic diseases caused by nonsense mutations.
Keywords: nonsense suppression, clitocine, nucleoside analog, readthrough, stop codon, p53 therapy
INTRODUCTION
Human diseases often arise as a consequence of mutations that result in the loss of function of a key protein. Nonsense mutations comprise a common type of mutation that can alter gene function by causing a premature translation termination event. Nonsense mutations result in the introduction of a UAA, UAG, or UGA premature stop codon within the open reading frame of mRNA, leading to the production of a truncated nonfunctional polypeptide product. Nonsense mutations have been identified as the basis of many inherited genetic diseases as well as various cancers.
Inactivation of tumor suppressor genes plays a key role in the initiation and progression of cancer. The p53 tumor suppressor protein is a key factor that responds to DNA damage, cellular insult, and oncogenic stress and induces the expression of proteins that either halt cell division to allow for DNA repair or initiate programs of cell death (apoptosis) or senescence (Christophorou et al. 2006; Levine et al. 2006; Vousden and Lane 2007; Levine and Oren 2009). Inactivation of the p53 protein is one of the most prevalent underlying causes of human cancers. Approximately 8% of all known p53 mutations are nonsense mutations (Petitjean et al. 2007). It has been shown that reexpression of the p53 protein within p53 null tumors leads to a rapid regression of established tumors in mice (Ventura et al. 2007; Xue et al. 2007). Additionally, mutations in p53 reduce the efficacy of many commonly used cancer chemotherapeutics (Lowe et al. 1994; El-Deiry 2003; Tchelebi et al. 2014). These results indicate that established tumors remain vulnerable to the tumor suppressor function of the p53 tumor suppressor. Therefore, reactivation or reexpression of p53 will likely have a therapeutic benefit in treating many cancers (Vassilev et al. 2004; Ventura et al. 2007; Brown et al. 2009; Tchelebi et al. 2014; Ribeiro et al. 2016).
One therapeutic approach to treating disease caused by nonsense mutation is the use of agents that promote readthrough of premature stop codons, enabling the synthesis of full-length active proteins. Aminoglycoside antibiotics like gentamicin are well known to read through premature stop codons in human cells and have been used in important proof-of-concept studies to demonstrate the therapeutic potential of nonsense suppression therapy (Bedwell et al. 1997; Barton-Davis et al. 1999; Floquet et al. 2011a,b). We performed high-throughput screens in cell-based and cell-free assays to identify compounds that promote nonsense suppression (Welch et al. 2007). Multiple lead chemical scaffolds were identified and chemical optimization of one scaffold led to the identification of ataluren (PTC124), a new chemical entity that selectively induces ribosomal readthrough of premature but not normal stop codons (Welch et al. 2007; Du et al. 2008; Sermet-Gaudelus et al. 2010; Peltz et al. 2013). Ataluren is clinically active in treating Duchene muscular dystrophy caused by a nonsense mutation in the gene for dystrophin (Finkel et al. 2013; Bushby et al. 2014) and has received conditional marketing approval in the European Union for nonsense mutation DMD (Ryan 2014). In clinical trials of nonsense mutation cystic fibrosis (nmCF), ataluren produces full-length CFTR to improve total chloride transport and has been shown to be well tolerated (Kerem et al. 2008; Sermet-Gaudelus et al. 2010; Wilschanski et al. 2011).
We have characterized other chemical scaffolds identified in our screening programs and report here that the naturally occurring nucleoside analog, clitocine, is a highly active and potent readthrough molecule. We demonstrate that clitocine incorporation into mRNA is required for premature stop codon readthrough activity, and the presence of clitocine at the third position of a premature stop codon is sufficient to promote robust readthrough. Clitocine effectively promotes the production of a full-length p53 protein in both cell culture and in tumors harboring p53 nonsense-containing alleles indicating the potential therapeutic value of clitocine to treat cancers with nonsense-mutated tumor suppressor genes.
RESULTS
The nucleoside analog, clitocine, induces dose-dependent suppression of all three premature nonsense codons
Clitocine, an adenosine nucleoside analog originally isolated from the mushroom Clitocybe inverse (Kubo et al. 1986), was identified in a high-throughput screen for molecules that read through the nonsense luciferase reporter (Fig. 1A,B; Welch et al. 2000). Upon resynthesis of clitocine, readthrough activity was detected in luciferase reporters (LUC-190) each containing one of three nonsense codons (Fig. 1C–F). Gentamicin and G418 were two to three orders of magnitude less potent than clitocine in inducing premature stop codon readthrough in these assays (Fig. 1). The rank order of clitocine readthrough of premature stop codons is UAA >> UGA > UAG. Importantly, clitocine demonstrated a marked preference for readthrough of premature stop codons over that of normal termination codons (Supplemental Fig. S1).
FIGURE 1.

Identification of premature stop codon readthrough activity of clitocine. (A) Schematic of luciferase reporter constructs used in high-throughput screens and assays to identify and characterize compounds with readthrough activity. (B) Structure of clitocine (2R,3R,4S,5R)-2-([6-amino-5-nitropyrimidin-4-yl]amino)-5-(hydroxymethyl) tetrahydrofuran-3,4-diol. Premature stop codon readthrough activity of clitocine in the UAA (C), UGA (D), UAG (E), and wild-type (F) luciferase reporter assays compared to that of the aminoglycosides, gentamicin, and G418. Each point is the average of three determinations and error bars represent the standard deviation of the mean.
Clitocine is incorporated into RNA substituting for adenosine
Since clitocine is a nucleoside analog we hypothesized that clitocine incorporation into RNA is important for readthrough of nonsense mutations. However, we first determined that clitocine was not active in cell-free translation readthrough assays (Supplemental Fig. S2A) and does not bind to the ribosome in vitro (Supplemental Fig. S2B). Furthermore, a clitocine analog lacking a free 5′ hydroxyl (clitocine-5′-F) was not active in cell-based readthrough assays (Supplemental Fig. S2C,D).
To test whether clitocine can be incorporated into RNA, we synthesized the 5′-triphosphate of clitocine (clitocine-TP) and showed that it substituted exclusively for ATP in an in vitro T7 RNA polymerase transcription reaction to produce full-length transcripts (Fig. 2A). To confirm incorporation of clitocine, we digested the RNA products and separated the nucleotides using two-dimensional thin layer chromatography (2D TLC) (Bochner and Ames 1982). When clitocine-TP was used without ATP or when ATP and clitocine-TP were used in an equal molar ratio, a spot corresponding to clitocine-3′-monophosphate (clitocine-3′-p) was detected (Fig. 2B). Similarly, mammalian RNA polymerase II incorporated clitocine in place of adenosine (Supplemental Fig. S3). Taken together, these results confirm that clitocine is incorporated in place of adenosine during transcription.
FIGURE 2.

Clitocine is incorporated into RNA in the position of adenosine. (A) Phosphorimager analysis showing that T7 RNA polymerase transcription of a pre-tRNATyr cDNA labeled with UTP-[α-32P] or CTP-[α-32P] produces full-length transcripts when all four cognate nucleotide triphosphates are present or when ATP is substituted with clitocine-triphosphate. Abortive products are seen in lanes 9 and 10 because the small amount of UTP-[α-32P] or CTP-[α-32P] allows some transcription without UTP or CTP, respectively. (B) Phosphorimager analysis of 2D TLC showing that clitocine is present in in-vitro-transcribed RNAs that were labeled with UTP-[α-32P] (as in panel A), digested with T2 RNase, and separated by 2D TLC. Panels are labeled with nucleotides included in the T7 transcription reactions. A/cl indicates that an equal molar ratio of ATP and clitocine-TP was used. TLC spots corresponding to the monophosphate nucleotides, Ap (adenosine-3′-phosphate), Cp (cytosine-3′-phosphate), Gp (guanidine-3′-phosphate), Up (uridine-3′-phosphate), and clp (clitocine-3′-phosphate), are indicated (Bochner and Ames 1982). The results provided a 2D TLC position reference for clitocine-3′-p.
To determine whether clitocine is incorporated into RNA in cells, we labeled clitocine treated and untreated CALU-6 cells with ortho [32P]phosphate and analyzed purified RNA by 2D TLC. As demonstrated in Figure 3, clitocine was incorporated into RNA as determined by radioactivity associated with the TLC spot corresponding to clitocine-3′-monophosphate. Clitocine was also detected in the RNA isolated from treated fibroblasts and L5178Y/TK+/− mouse lymphoma cells (Supplemental Fig. S4). These results demonstrate that clitocine is phosphorylated by the cellular nucleoside and nucleotide kinases and incorporated into RNA in cells.
FIGURE 3.
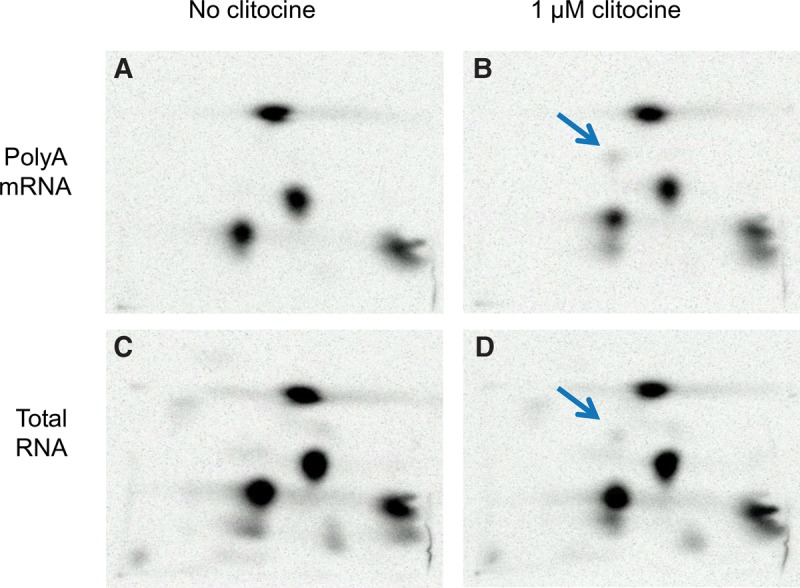
Clitocine is incorporated into RNA in cells. CALU-6 cells were labeled with ortho [32P]phosphate and incubated with 1 µM clitocine (B and D) or without (A and C). After 24 h, total RNA (C and D) and poly(A) RNA (A and B) was isolated and digested with nuclease P1. Nucleotides were purified, separated by 2D TLC, and visualized by Phosphorimager analysis. The arrow in panels B and D indicates clitocine-3′-phosphate. The separation pattern of the 3′-phosphate nucleotides is the same as in Figure 3.
Clitocine incorporation into RNA induces readthrough of premature stop codons
To determine whether the presence of clitocine in mRNA induces premature stop codon readthrough, we transcribed the Luc-190-UGA mRNA with an increasing ratio of clitocine-TP to ATP, purified the resulting mRNAs, and found that readthrough in RRL translation reactions increased when up to 5% clitocine-TP was included in the reaction (Fig. 4A). Next, we used clitocine-TP in coupled transcription–translation reactions and found that readthrough increased with increasing amounts of clitocine-TP (Fig. 4B). These results demonstrate that clitocine incorporation into RNA induces readthrough of premature stop codons.
FIGURE 4.

Clitocine incorporation causes readthrough. (A) Graph showing that incorporation of clitocine results in readthrough of the Luc-190-UGA mRNA. Luc-190-UGA was transcribed by T7 RNA polymerase with varying ratios of clitocine-TP to ATP (x-axis). The resultant RNA was purified, translated using rabbit reticulocyte lysate (RRL), and luciferase enzyme activity was determined (y-axis). Error bars represent standard deviation of two determinations. (B) The Luc-190-UGA cDNA was used in coupled transcription–translation reactions with varying ratios of clitocine-TP and ATP (x-axis). Luciferase activity in the reactions is indicated on the y-axis. Error bars represent the standard deviation of two determinations. (C) Schematic representation of peptides produced from chemically synthesized RNAs (see Materials and Methods). During translation in the RRL system, Transcend tRNA was used to label lysine residues with biotin, and [3H]-leucine was used to label leucine residues with tritium. X indicates different triplet codons synthesized into the three RNAs used in panel D. (D) The indicated RNAs were translated in RRL and labeled as described above. After binding of the resultant peptides to streptavidin magnetic beads, the beads were washed, scintillation fluid was added, and the amount of tritium was quantified (y-axis). Error bars represent the standard deviation of the mean for three determinations.
Since polymerase-mediated incorporation of clitocine would not be restricted to the position of adenosine in a premature stop codon, we developed a method to monitor readthrough of a RNA with clitocine incorporated site-specifically at the third position of a UAA nonsense codon (Fig. 4C). Three RNAs were chemically synthesized containing one variable codon: AUU encoding Ile (WT), UAA, or UAcl (containing clitocine at the third position). The three RNAs were designed such that translation through the variable codon could be measured by capture and tritium counting (see Materials and Methods). Translation of WT RNA led to a significant increase (24-fold) in capture of labeled peptide compared to translation of UAA RNA (Fig. 4D). As a control, UAA RNA yielded a 1.9-fold increase when translated in the presence of gentamicin. Translation of UAcl RNA resulted in a 3.6-fold increase demonstrating that the presence of clitocine in the third position of a nonsense codon induces readthrough.
Characterization of clitocine-induced readthrough products
To investigate whether the presence of clitocine in mRNA influences the recruitment of specific aminoacyl-tRNAs to a nonsense codon, we determined which amino acids were inserted at a UGA nonsense codon upon clitocine-induced readthrough in cells using a previously described system of a HA-LUC(PTCUGA)-SF reporter in conjunction with LC-MS/MS (Roy et al. 2016). Endogenous readthrough in cells treated with DMSO resulted in the incorporation of two amino acids at the premature stop codon: Arg (59 ± 2.3%) and Trp (41 ± 2.3%). Readthrough after treatment with 1 µM clitocine resulted in the incorporation of Arg (74 ± 6.5%), Trp (3 ± 0.3%), and Cys (23 ± 6.5%) (Table 1; Fig. 5).
TABLE 1.
Alteration in amino acid insertion upon treatment with clitocine
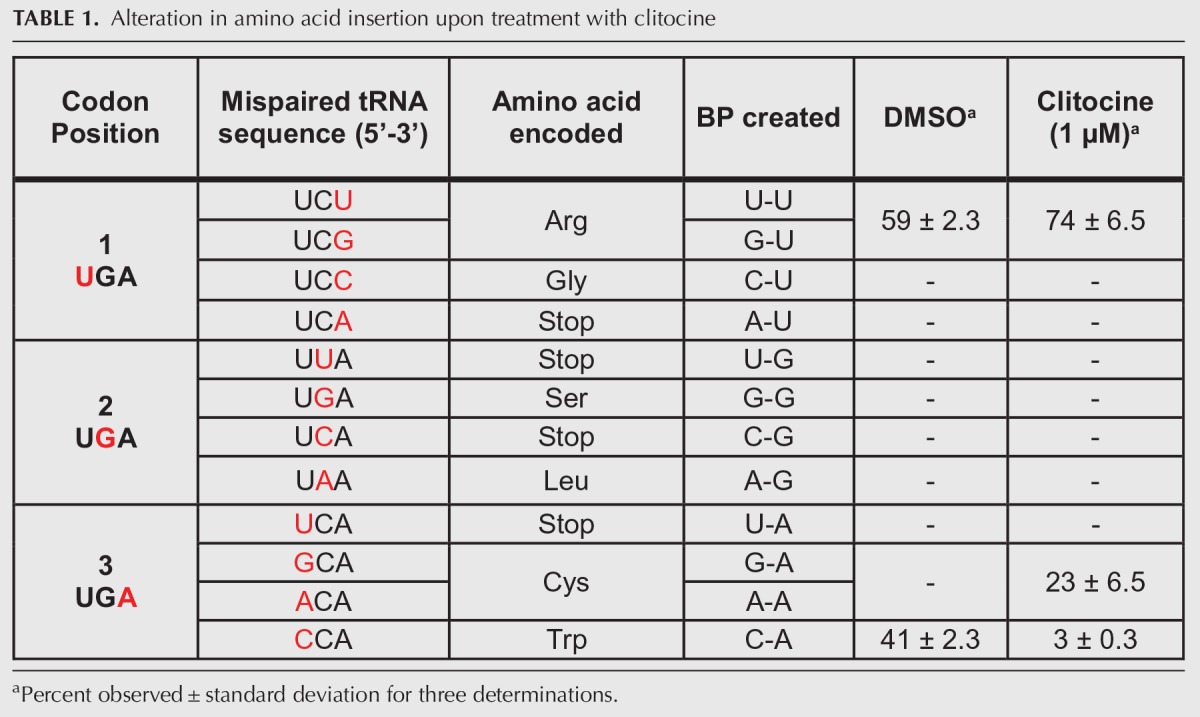
FIGURE 5.
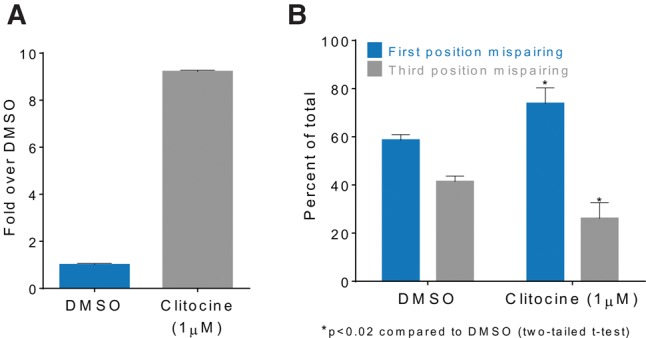
Clitocine alters near-cognate tRNA decoding of nonsense codons. (A) Graph showing that clitocine induces a 9.2-fold increase in luciferase enzyme activity (y-axis) over that in DMSO treated (control) 293H cells stably expressing LUC-(UGA20)-SF. Error bars represent standard deviation of the mean for at least two determinations. (B) Graph showing a change in the ratio of first to third codon position mispairing during near-cognate tRNA decoding of the UGA nonsense codon. Error bars represent the standard deviation of the mean for three determinations. Data are from Table 1.
We have shown previously that readthrough of premature stop codons is facilitated by near-cognate aminoacyl-tRNAs that can mispair independently at the first or third positions of a nonsense codon (Roy et al. 2015, 2016). Upon clitocine treatment, there was an overall decrease in third position mispairing from 41% to 26% (P < 0.02, two-tailed Student's t-test) and an increase in first position mispairing from 59% to 74% (P < 0.02, two tailed t-test) (Table 1; Fig. 5B). Interestingly, the presence of clitocine at third position induced the insertion of cysteine (not seen in the DMSO control) indicating a third codon position base pair formation of either a G:cl or A:cl of Cysteine tRNA isoacceptors. Using LC-MS/MS we also investigated the effect of clitocine on sense codon decoding at specific residues in the luciferase protein. We found that clitocine did not detectably alter sense codon decoding. In contrast, the positive control, G418, did alter sense codon decoding (Supplemental Fig. S5).
Clitocine promotes the production of functional p53 protein
We next investigated the ability of clitocine to read through premature stop codons in the p53 tumor suppressor gene. We utilized multiple cell lines with premature stop codon-containing alleles, including the ovarian cancer cell line CAOV-3, which has a p53 nonsense mutation resulting in the change of codon 136 from CAA to UAA (Yaginuma and Westphal 1992); the lung cancer cell line CALU-6 with a p53 nonsense mutation at codon 196 (CGA to UGA) (Lehman et al. 1991); and the lung cancer cell line H520 with a p53 nonsense mutation at codon 146 (TGG to UGA) (Mitsudomi et al. 1992). We demonstrated that clitocine induces a dose- and time-dependent increase of full-length p53 in all three cell lines (Fig. 6A). In contrast, clitocine did not increase p53 in 293H cells. It is well understood that ribosomal translation through a premature stop codon will remove downstream exon–exon junction complexes (EJCs), which target premature stop codon-containing mRNAs for degradation by the nonsense-mediated decay (NMD) pathway (Maquat 2004). Prompted by this work, we investigated the effect of clitocine on mRNA stability. Surprisingly, and in contrast to G418, clitocine only minimally stabilized the premature stop codon-containing p53 mRNA in CAOV-3p53-UAA136 and CALU-6p53-196UGA cells despite a higher level of readthrough activity (see Discussion and Supplemental Fig. S6).
FIGURE 6.
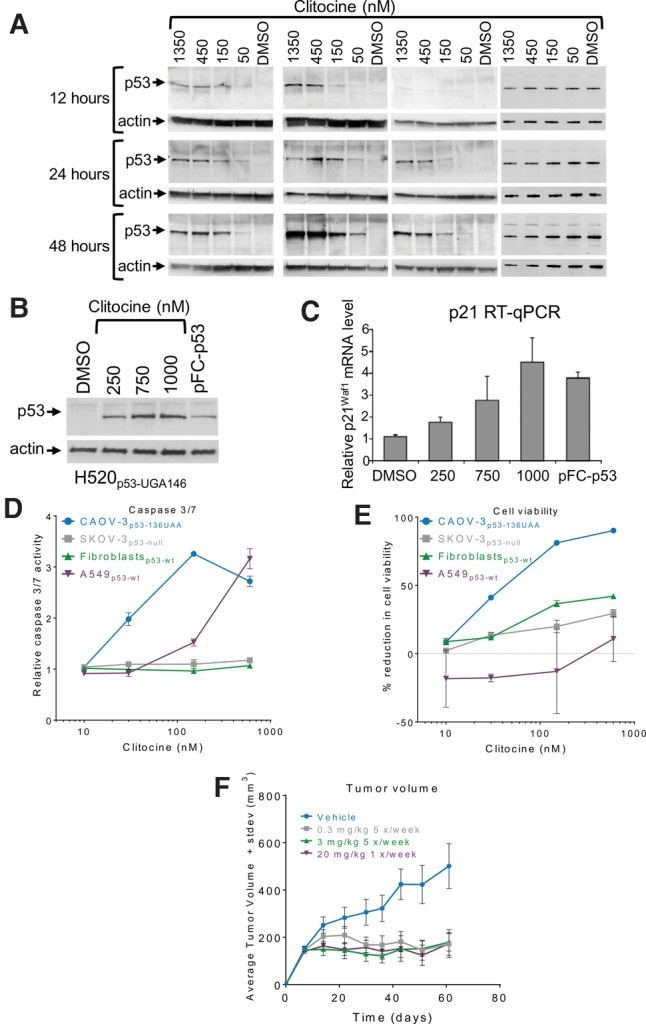
Clitocine induces readthrough of p53 premature stop codons to produce full-length, functional p53 protein. (A) Western blot analysis showing dose response and time course of full-length p53 production after clitocine treatment of cell lines (from left to right) with native p53 nonsense alleles (CAOV-3p53-136UAA, CALU-6p53-196UGA, and H520p53-146UGA) and with wild-type p53 (293H). (B) Western blot (p53) and (C) graph of relative p21waf1 mRNA levels measured by RT-qPCR showing that in H520p53-146UGA cells, clitocine increases p53 protein which, in turn, transactivates p21waf1mRNA. pFC-p53 indicates transient transfection of a plasmid that expresses p53 protein. Error bars represent standard deviation of the mean for three determinations. (D) Caspases 3 and 7 activity and (E) cell viability assays showing that cells containing p53 premature stop codon alleles are more sensitive to the activity of clitocine. Error bars represent standard deviation of the mean for three determinations. (F) Results of a xenograft study showing that clitocine at doses as low as 0.3 mg/kg (five times per week) reduced the growth of CAOV-3p53-UAA136 tumors. Error bars represent standard error of the mean for 10 mice per group.
The activation of p53 has been shown to induce the downstream transcriptional activation of a set of genes involved in regulating cell growth and apoptosis (El-Deiry 2003; Chipuk and Green 2006; Vousden and Lane 2007). Accordingly, we next examined whether the p53 protein produced in clitocine-treated cells promotes a concatenate rise in expression of the p53 target gene p21waf1. In H520p53-UGA146 cells, the levels of p21waf1 mRNA rose with increasing concentrations of clitocine-induced p53 (Fig. 6B,C), indicating that the p53 protein produced by clitocine is functional and can transcriptionally activate the p21waf1 gene. The p53 protein produced exogenously after transfection of a plasmid to express p53 (pFC-p53) or induced by treatment with clitocine (Fig. 6B) resulted in similar increases of p21waf1 mRNA (Fig. 6C). Clitocine-induced p53 also increased p21WAF1 protein in a dose- and time-dependent manner in CAOV-3p53-UAA136 cells (Supplemental Fig. S7A). Taken together these results indicate that p53 expressed as a result of clitocine-induced nonsense suppression is transcriptionally active.
Functional p53 can induce gene expression pathways that result in apoptosis (El-Deiry 2003; Chipuk and Green 2006). The activities of caspases 3 and 7 (Fig. 6D) and the population of cells in subGO/G1 phase increased (Supplemental Fig. S7B) in CAOV-3p53-UAA136 cells upon clitocine treatment, indicating that clitocine-induced p53 can drive cells into apoptosis. In contrast, clitocine treatment did not increase the levels of caspases 3 and 7 activity in p53-null cells (SKOV-3) (Yaginuma and Westphal 1992) or normal fibroblasts. In the case of A549 cells (p53 normal), clitocine induced the activities of caspases 3 and 7 at a 20-fold higher concentration than that required for a response in CAOV-3p53-UAA136 cells. Furthermore, increased levels of caspases 3 and 7 correlated with decreased cell viability in clitocine-treated cells (Fig. 6E). These results demonstrate that clitocine can suppress p53 nonsense mutations to produce full-length p53 that drives cells into apoptosis.
We next determined whether the growth of CAOV-3p53-UAA136 xenograft tumors is inhibited in tumor bearing mice treated with clitocine. Clitocine was administered subcutaneously five times per week at a dose of 3 mg/kg or 0.3 mg/kg or once per week at a dose of 20 mg/kg and CAOV-33p53-UAA136 tumor growth was monitored. The results demonstrated that CAOV-33p53-UAA136 tumor growth was inhibited in mice treated with clitocine when compared to tumor growth in vehicle-treated controls (Fig. 6F). In contrast, clitocine did not affect growth of tumors that did not harbor nonsense-containing p53 alleles (Supplemental Fig. S8). These results indicate that clitocine-induced p53 inhibits CAOV-33p53-UAA136 tumor growth in a xenograft model.
DISCUSSION
Low molecular weight compounds that induce readthrough of premature stop codons represent a novel therapeutic approach to treat genetic disorders and cancers caused by nonsense mutations. Here, we describe the identification and characterization of the nonsense mutation readthrough activity of clitocine. Clitocine is a naturally occurring adenosine nucleoside analog originally isolated from the mushroom Clitocybe inverse (Kubo et al. 1986). We show that clitocine is highly efficient in promoting readthrough of nonsense mutations.
Messenger RNAs that are derived from native intron-containing genes and contain a premature stop codon are subject to nonsense-mediated decay (NMD). Ribosomes that read through premature stop codons stabilize these mRNAs by removal of downstream exon–exon junction complexes (Maquat 2004). Supplemental Figure S6 shows that clitocine marginally increased the levels of nonsense-containing p53 transcripts when compared to much greater (four- to sevenfold) increases with G418. This is consistent with high efficiency readthrough from a relatively small fraction of premature stop codon-containing mRNAs that contain clitocine in the premature stop codon.
We show that clitocine in the third position of a UAA stop codon induces readthrough (Fig. 4) and that the rank order of clitocine readthrough of premature stop codons is UAA >> UGA > UAG (Fig. 1). The fact that clitocine induces readthrough of an UAG nonsense codon also indicates that clitocine in the second position causes premature stop codon readthrough. Perhaps, the rare occurrence of clitocine in both the second and third positions of a UAA stop codon accounts for the considerably higher readthrough seen with this codon.
It has been shown that pseudouridylation of the first position of a nonsense codon causes robust readthrough, and that pseudouridine (Ψ) enhances the insertion of serine and threonine (Karijolich and Yu 2011). Interestingly, decoding of ΨAA and ΨAG by serine and threonine tRNAs requires an A:G mispair at the second position. Previous reports have shown that low levels of native readthrough and higher levels of compound-induced readthrough occur through near-cognate tRNAs, primarily through mispairing at the first and third codon positions (Roy et al. 2015, 2016). Unlike ataluren and aminoglycosides that act directly on the translation machinery, Ψ and clitocine are similar in that they must be present in the nonsense codon of an mRNA to cause readthrough. They differ, however, in that clitocine is incorporated by RNA polymerase and causes near-cognate tRNA decoding of the nonsense codon while Ψ is a post-transcriptional modification and causes noncognate decoding (Karijolich and Yu 2011). It is still unclear whether Ψ-codons cause readthrough via reduction in release factor binding or an increase in noncognate tRNA binding as the former appears to be unperturbed by the presence of Ψ-codons (Svidritskiy et al. 2016) and the latter appears to be stable in structural analysis (Fernandez et al. 2013). The molecular determinants for clitocine-induced readthrough on the ribosome await further investigation.
Interestingly, the presence of clitocine at the third position of a UGA nonsense codon caused a nearly complete shift from tryptophan to cysteine incorporation (Table 1) indicating that a base pair at the third codon position of either a G:cl or A:cl from the near-cognate tRNA Cys UGU or UGC is preferred over a C:cl base pair with tRNA Trp UGG. Interestingly, we also saw an increase in the recruitment of first position mispaired arginine tRNA, possibly due to the ability of clitocine to pair with A and/or G in the third position, allowing recruitment of additional arginine isoacceptor tRNAs (tRNA Arg GCG, GCA, and GCU). These results suggest that readthrough promoted by clitocine may benefit from enhanced recruitment of near-cognate tRNA to the ribosome to decode the nonsense codon. However, initial experiments demonstrated that clitocine does not disrupt sense codon decoding (Supplemental Fig. S5), thereby suggesting that clitocine preferentially base pairs with uracil during the process of ribosomal tRNA decoding. Therefore, during translation elongation, clitocine appears to base pair preferentially with uracil to recruit cognate tRNA consistent with the recognition of this nucleoside analog as adenosine during transcription.
In addition to enhancement of the recruitment of a near-cognate tRNA to stop codons, it is possible that the UGcl codon is poorly recognized by the eRF1 translation termination factor, preventing efficient termination, and thus increasing the level of near-cognate tRNA recruitment and increased readthrough. It is known that the presence of cytosine at the +4 position (immediately downstream from the termination codon) results in greater permissiveness of all three nonsense codons for readthrough. Based on genetic interaction studies in yeast, it has been proposed that +4 cytosine compromises the ability of eRF1 to recognize appropriately a stop codon, thus providing precedence for the importance of nucleotide positioning for eRF1-mediated termination efficiency (Beznoskova et al. 2016). It would be of interest to conduct further studies to determine the kinetics of the interaction of clitocine/RNA with release factors.
We demonstrate that p53 produced as a result of clitocine-induced readthrough is functional and inhibits growth of tumors harboring the p53 nonsense mutation. These results are particularly relevant in light of studies demonstrating that reactivation of the p53 protein by genetic manipulation in established tumors leads to tumor regression (Kaeser et al. 2004; Ventura et al. 2007; Xue et al. 2007). More recently, peptides and small molecules have been identified that reactivate mutant p53 (Yu et al. 2012; Parrales and Iwakuma 2015; Tal et al. 2016).
Our data show that clitocine-produced p53 is functional as evidenced by its ability to transactivate the p53 target gene p21waf1 and drive cells into apoptosis (Fig. 6; Supplemental Fig. S7). Others have presented results showing that clitocine can induce apoptosis in cancer cell lines and may inhibit Mcl-1 (Ren et al. 2008; Sun et al. 2014). Here, in contrast to those studies a defined molecular mechanism for p53-premature stop codon readthrough is presented in conjunction with p53 reactivation. Our data demonstrate that the anti-cancer effects of clitocine are driven by p53 reexpression. Taken together, our results are important since they point to the potential clinical utility of clitocine and other small molecules that can induce readthrough of p53 nonsense mutations as a molecular targeted strategy for cancer therapy.
Other tumor suppressor genes harboring nonsense mutations are also potential targets for a nonsense readthrough therapeutic agent. For example, the adenomatous polyposis coli (APC) gene containing nonsense mutations would be a clinically relevant target since the lack of functional APC protein can result in colorectal and other cancers (Jaiswal et al. 2005). The majority of APC gene mutations associated with colon cancer result in truncated APC protein, and 32% of colon cancer-associated APC gene mutations are nonsense mutations (Laurent-Puig et al. 1998; Floquet et al. 2011b). Therefore, investigation of the potential use of nonsense suppressing drugs to restore full-length APC protein in cancers is warranted and has been shown to be conceptually feasible using aminoglycosides (Floquet et al. 2011b).
Clitocine-induced readthrough is unique in that it requires the incorporation of clitocine in mRNA through the process of transcription. Although, clitocine is a natural product found in the mushroom Clitocybe inverse and was first described 30 years ago (Kubo et al. 1986), the function of clitocine in the mushroom is still unknown. It is possible that C. inverse uses clitocine to regulate gene expression via readthrough. The natural biological role for clitocine awaits further investigation.
The small-molecule drug ataluren promotes readthrough of premature stop codons and is conditionally approved by the European Medicines Agency's Committee for Medicinal Products for Human Use to treat nonsense-mutated DMD (Ryan 2014). Because of the key role that p53 plays in cancer initiation and progression there is significant interest in therapeutic reactivation of mutated p53 (Ribeiro et al. 2016). Clitocine with its ability to read through premature stop codons resulting in the production of functional p53 may one day provide therapeutic benefit to cancer patients.
MATERIALS AND METHODS
Cell culture
All cell lines used in this study were obtained from the American Type Culture Collection (ATCC) and maintained according to their recommendations.
Organic synthesis
Clitocine, (2R,3R,4S,5R)-2-([6-amino-5-nitropyrimidin-4-yl]amino)-5-(hydroxymethyl) tetrahydrofuran-3,4-diol, (Fig. 1B) was synthesized according to a published procedure (Moss et al. 1988) in which 4,6-diamino-5-nitropyrimidine was coupled with 1-oacetyl-2,3,5-tri-O-benzoyl-D-ribofuranose following the procedure of Vorbrueggen (Niedballa and Vorbruggen 1974). Removal of the benzoyl protecting groups resulted in the nucleoside clitocine in a 92:8 ratio of β/α anomers. We found that these could be separated by chromatography to give the respective β and α anomers each in >99.9% purity. Clitocine 5′-F (6-amino-5-nitro-4-[5-deoxy-5-fluoro-β-D-ribofuranosylamino]-pyrimidine) was synthesized according to a published method (Wilde et al. 2007). The method used to resynthesize clitocine (Moss et al. 1988) resulted in a 92:8 mixture of β- to α-clitocine anomers. We separated the two anomers and measured the activity of each using the LUC-190-UAA reporter. The β-clitocine anomer demonstrated significantly greater activity and potency than did the α-clitocine anomer (Supplemental Fig. S10). Therefore, the purified β-clitocine anomer was used in the present studies.
LUC constructs and cell-based luciferase assay
All LUC constructs were made and cell-based luciferase assays were conducted as previously described (Welch et al. 2007).
Western blot analysis
Antibodies used in this study included: anti-p53 (DO-1) (Santa Cruz Biotechnology), anti-p21waf1 (Oncogene Research Products), and anti-actin (Oncogene Research Products). For Western blot analysis, cell pellets were boiled directly in SDS sample buffer. Extracts containing ∼30 µg of total protein were separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE) on 12% Tris HCl gels (Bio-Rad) and transferred to nitrocellulose membranes. The membranes were blocked in Li-Cor blocking buffer for 1 h, incubated with manufacturer's recommended dilution of primary antibody for 1.5 h (Li-Cor blocking buffer), washed twice for 5 min each (PBS, 1% tween-20), incubated with an infrared-800 or -680 conjugated secondary antibody, washed once for 5 min and twice for 10 min each (PBS, 1% tween-20), and visualized on an Odyssey Imager.
Meso scale discovery (MSD) assays
All incubations were done on a Bellco Biotechnology orbital shaker at 1600 revolutions per minute. Washes were done with 200 µL of PBS-T (phosphate-buffered saline [pH = 7.4] with 0.05% tween-20). Plate coating was done in PBS. Blocking was done with 150 µL Blocker Casein in TBS (ThermoFisher) for 1 h at room temperature or overnight at 4°C. All antibody incubations were done in blocking buffer. Cells (30,000 cells per well in a final volume of 100 µL) were plated in 96-well tissue culture plates that had previously received 10 nL per well of compound diluted in DMSO. Cells were lysed in 60 µL of lysis buffer (20 mM Tris–HCl [pH 7.5], 0.5% Triton-X100, 150 mM NaCl, 1 mM EDTA, and 1 mM EGTA) per well for 1 h at room temperature. For the V5-xpress assay, anti-xpress (ThermoFisher) was used as capture at 2 µg/mL and SULFO-TAG labeled anti-V5 (Abcam #SV-Pk1) was used as detection at 0.25 µg/mL. For the full-length p53 MSD ELISA, anti-p53 (Abcam #ab59243) was used as capture at a 1:2000 dilution and SULFO-TAG labeled anti-p53 antibody from the Total p53 Whole Cell Lysate Kit (Meso Scale Discovery #K150DBD-1) was used as detection. MSD plates (96-well, MSD #L15XA-3/L11A-3) were coated with antibody overnight at 4°C, washed three times and blocked for 1 h at room temperature. A volume of 5 µL of detection antibody and 20 µL lysate per well were added and incubated 2 h at room temperature. Plates were washed three times, 150 µL of read buffer added, and plates were read on a SECTOR Imager 6000 (Meso Scale Discovery).
Real-time reverse transcription quantitative PCR (RT-qPCR)
Cells were seeded in 96-well plates (10,000 cells per well) and compounds were added 18 h later. After the indicated incubation period, cells were lysed in 50 µL iScript lysis buffer (Bio-Rad), diluted 1:5 in water, and 2 µL was used for RT-qPCR with AgPath One-Step RT-PCR reagent (Applied Biosystems #AM1005). Pre-Developed Assay Reagent (PDAR) (Applied Biosystems) primers and probes were used for p53 and p21waf1. For fLuc RT-qPCR the following were used, forward primer, caattctttatgccggtgtt; reverse primer, ggctgcgaaatgttcatact; and probe, FAMttatcggagttgcagttgcgcMGB. Internal control reactions included either a VIC-labeled MGB quenched Human PPIA (cyclophilin A) PDAR or a VIC-labeled MGB quenched Human ACTB (β actin) PDAR. Reactions were cycled on an ABI 7500 Real-Time PCR System: one cycle of 48°C for 15 min, 95°C for 10 min and 40 cycles of 95°C for 15 sec and 60°C 1 min. Relative mRNA levels were calculated as 2−ΔΔCT or 2, where CT (cycle threshold) is the cycle number in the linear amplification range, and ΔCT = CT(experimental) − CT(control) and ΔΔCT = ΔCT(treatment) − ΔCT(vehicle) (Bustin 2002).
Apoptosis analysis
Levels of caspase 3/7 activity were quantified using the Apo-ONE Homogeneous Caspase-3/7 Assay (Promega) according to the manufacturer's recommendations. Cell viability was determined using the CellTiter-Glo Luminescent Cell Viability Assay kit (Promega) according to the manufacturer's recommendations. For FACS analysis, cells were harvested with trypsin-EDTA, washed with 1 mL of PBS, and fixed in 100% cold ethanol for 15 min on ice. After centrifugation, cells were resuspended in 1 mL of PBS at room temperature, incubated at 37°C for 30 min with 200 µg/mL RNase A. Cells were then stained with 1 mg/mL propidium iodide (PI) at room temperature for 15 min and analyzed on a Becton Dickenson FACSCalibur flow cytometer using CellQuest software.
Xenograft studies
All animal studies were performed under IACUC-approved protocols at AAALAC-certified animal facilities. Cells were removed from plates with PBS, EDTA (1 mM), 1.5 mL trypsin/EDTA, centrifuged and resuspended at a concentration of 2.0 × 108 cells/mL in ice-cold Hank's buffered salt solution supplemented with 10 mM HEPES. An equal volume of matrigel was added and 200 µL were injected subcutaneously into nu/nu mice with a 27-gauge needle. After the majority of tumors reached 150–250 mm3 (as measured with digital calipers), mice were randomized into treatment groups (at least 10 mice per group). Only mice with tumors within the range of 150–250 mm3 were used. Clitocine was dosed subcutaneously in phosphate-buffered saline as indicated in the figure legend.
Microscale thermophoresis (MST) analysis
Binding of clitocine to HeLa 80S ribosome was determined using microscale thermophoresis (MST) (Beckert et al. 2015). Purified 80S ribosomes (Khatter et al. 2014) were diluted to a final concentration of 20 pM in 1× MST buffer (NanoTemper Technologies). Clitocine or G418 was titrated with concentration ranges of 50–0.003 µM and 10 mM to 0.3 µ, respectively. The 80S ribosomes were then incubated at 25°C for 20 min and 10 µL from each reaction was loaded into premium coated capillaries (NanoTemper Technologies). The thermophoretic response was then measured by monitoring the change in tryptophan fluorescence from the ribosomes with a Monolith NT label free instrument (NanoTemper Technologies) at 20% and 10% infrared (IR) laser power. Change in fluorescence due to binding upon IR laser-mediated heating was measured and plotted against clitocine or G418 concentration. Binding constants for G418 were calculated as averages from three independent experiments.
Clitocine incorporation in cells
Cells (80% confluent) in T-175 flasks were grown in the presence of 250 µCi ortho [32P]phosphate. After 48 h incubation at 37°C, cells were harvested by trypsinization, pelleted, washed with PBS, and total RNA was isolated using the RNeasy Mini Kit followed by the Oligotex mRNA Mini Kit (QIAGEN) according to the manufacturer's recommendation. RNA (50 ng) was digested with RNAse T2 (Boca Scientific) for 6 h to overnight at 37°C in 10 mM ammonium acetate buffer (pH 4.5). Reaction volume was reduced using a vacuum concentrator, resuspended in 5 µL and approximately one-fifth of the purified material (1 µL) was separated by 2D TLC on PEI Cellulose. First dimension solvent was isobutyric acid:25% ammonium hydroxide:water (66:1:33), and second dimension solvent was 2-propanol:HCL:water (66:17.6:14.4) (Bochner and Ames 1982). TLC plates were air dried and analyzed on a Phosphorimager.
In vitro translation and capture
The following RNA oligos were synthesized by Dharmacon: CUUCCGAGGAGACGCCGACG AUG AAA GGC AAA AUU AAA AAA AAA XXX CUG CUA CUU CUU CUG CUU CUU CUG CUA UAG CCCGACAAAAAAAAA. For the UAA-oligo, XXX = UAA. For the UAcl-oligo, XXX = UAclitocine. For the WT-oligo, XXX = AUU. The RNAs contain five lysine codons upstream of the variable position to allow for biotin labeling and nine leucine codons downstream to allow for incorporation of 3H-Leu to monitor translation readthrough. A 20-nucleotide leader sequence (CUUCCGAGGAGACGCCGACG), derived from tobacco mosaic virus, was included in each oligonucleotide to enhance translation (Sleat et al. 1987). Oligonucleotides were translated at 25°C with the Flexi Rabbit Reticulocyte Lysate System (Promega) according to the manufacturer's recommendation and reactions included 4 µL ε-labeled biotinylated lysine-tRNA complex (Promega), 1 µL of amino acid mix (1 mM) minus lysine and leucine and 4 µL L-[4,5-3H]leucine (1 mCi/mL). After 2 h, 500 µL of RIPA buffer was added and labeled peptides were captured with 10 µL Pierce streptavidin magnetic beads for 30 min at 4°C. Magnetic beads were washed five times for 1 min each, mixed with 25 mL scintillation fluid and analyzed on a scintillation counter.
Analyses of luciferase readthrough products
The full-length luciferase readthrough products resolved on SDS-polyacrylamide gel were excised after silver staining of the gel and the destained gel slices were subjected to endoproteinase Lys-C (Roche Diagnostics) digestion followed by LC-MS/MS analyses as described previously (Roy et al. 2015, 2016). Raw data files were subjected to database searching with Mascot Server (version 2.4) against human SwissProt index that contain the sequences of all 20 potential mutations of the fLuc protein at each position investigated. The relative abundance of each amino acid at position 20 of luciferase was calculated by adding the corresponding precursor intensity of individual endo Lys-C peptide containing the codon 20 to yield a total peptide abundance that was then used to calculate the percentage of insertion of each amino acid (Roy et al. 2015, 2016).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
W.J.F., C.R.T., Y.T., J.Z., B.J., B.R., M.W., J.H., J.S., J.T., Y.-C.M., S.B., R.B., J.M.C., S.W.P., and E.M.W. are financially compensated as employees of PTC Therapeutics Inc. (PTCT). J.D.L. is financially compensated as a consultant to PTCT.
Supplementary Material
Acknowledgments
Author contributions: W.J.F., C.R.T., E.M.W., T.W.D., M.W., J.T., J.M.C., and S.W.P. designed the experiments; W.J.F., C.R.T., Y.T., J.Z., B.J., B.R., S.B., R.B., J.H., J.S., J.T., and J.D.L. carried out the experiments; W.J.F., C.R.T., E.M.W., T.W.D., M.W., and S.W.P. interpreted the data; and W.J.F. and C.R.T. wrote the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.060236.116.
REFERENCES
- Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. 1999. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 104: 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckert B, Kedrov A, Sohmen D, Kempf G, Wild K, Sinning I, Stahlberg H, Wilson DN, Beckmann R. 2015. Translational arrest by a prokaryotic signal recognition particle is mediated by RNA interactions. Nat Struct Mol Biol 22: 767–773. [DOI] [PubMed] [Google Scholar]
- Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ. 1997. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 3: 1280–1284. [DOI] [PubMed] [Google Scholar]
- Beznoskova P, Gunisova S, Valasek LS. 2016. Rules of UGA-N decoding by near-cognate tRNAs and analysis of readthrough on short uORFs in yeast. RNA 22: 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochner BR, Ames BN. 1982. Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J Biol Chem 257: 9759–9769. [PubMed] [Google Scholar]
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. 2009. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 9: 862–873. [DOI] [PubMed] [Google Scholar]
- Bushby K, Finkel R, Wong B, Barohn R, Campbell C, Comi GP, Connolly AM, Day JW, Flanigan KM, Goemans N, et al. 2014. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 50: 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA. 2002. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29: 23–39. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. 2006. Dissecting p53-dependent apoptosis. Cell Death Differ 13: 994–1002. [DOI] [PubMed] [Google Scholar]
- Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. 2006. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 443: 214–217. [DOI] [PubMed] [Google Scholar]
- Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. 2008. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci 105: 2064–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry WS. 2003. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 22: 7486–7495. [DOI] [PubMed] [Google Scholar]
- Fernandez IS, Ng CL, Kelley AC, Wu G, Yu YT, Ramakrishnan V. 2013. Unusual base pairing during the decoding of a stop codon by the ribosome. Nature 500: 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel RS, Flanigan KM, Wong B, Bonnemann C, Sampson J, Sweeney HL, Reha A, Northcutt VJ, Elfring G, Barth J, et al. 2013. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One 8: e81302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floquet C, Deforges J, Rousset JP, Bidou L. 2011a. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res 39: 3350–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floquet C, Rousset JP, Bidou L. 2011b. Readthrough of premature termination codons in the adenomatous polyposis coli gene restores its biological activity in human cancer cells. PLoS One 6: e24125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal AS, Balusu R, Narayan S. 2005. Involvement of adenomatous polyposis coli in colorectal tumorigenesis. Front Biosci 10: 1118–1134. [DOI] [PubMed] [Google Scholar]
- Kaeser MD, Pebernard S, Iggo RD. 2004. Regulation of p53 stability and function in HCT116 colon cancer cells. J Biol Chem 279: 7598–7605. [DOI] [PubMed] [Google Scholar]
- Karijolich J, Yu YT. 2011. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 474: 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M, et al. 2008. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet 372: 719–727. [DOI] [PubMed] [Google Scholar]
- Khatter H, Myasnikov AG, Mastio L, Billas IM, Birck C, Stella S, Klaholz BP. 2014. Purification, characterization and crystallization of the human 80S ribosome. Nucleic Acids Res 42: e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo I, Kim M, Wood WF, Naoki H. 1986. Clitocine, a new insecticidal nucleoside analog from the mushroom Clitocybe inversa. Tetrahedron Lett 27: 4277–4280. [Google Scholar]
- Laurent-Puig P, Beroud C, Soussi T. 1998. APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res 26: 269–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman TA, Bennett WP, Metcalf RA, Welsh JA, Ecker J, Modali RV, Ullrich S, Romano JW, Appella E, Testa JR, et al. 1991. p53 mutations, ras mutations, and p53-heat shock 70 protein complexes in human lung carcinoma cell lines. Cancer Res 51: 4090–4096. [PubMed] [Google Scholar]
- Levine AJ, Oren M. 2009. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9: 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Hu W, Feng Z. 2006. The P53 pathway: what questions remain to be explored? Cell Death Differ 13: 1027–1036. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. 1994. p53 status and the efficacy of cancer therapy in vivo. Science 266: 807–810. [DOI] [PubMed] [Google Scholar]
- Maquat LE. 2004. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5: 89–99. [DOI] [PubMed] [Google Scholar]
- Mitsudomi T, Steinberg SM, Nau MM, Carbone D, D'Amico D, Bodner S, Oie HK, Linnoila RI, Mulshine JL, Minna JD, et al. 1992. p53 gene mutations in non-small-cell lung cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene 7: 171–180. [PubMed] [Google Scholar]
- Moss RJ, Petrie CR, Meyer RB Jr, Nord LD, Willis RC, Smith RA, Larson SB, Kini GD, Robins RK. 1988. Synthesis, intramolecular hydrogen bonding, and biochemical studies of clitocine, a naturally occurring exocyclic amino nucleoside. J Med Chem 31: 786–790. [DOI] [PubMed] [Google Scholar]
- Niedballa U, Vorbruggen H. 1974. A general synthesis of N-glycosides. IV. Synthesis of nucleosides of hydroxy and mercapto N-heterocycles. J Org Chem 39: 3668–3671. [DOI] [PubMed] [Google Scholar]
- Parrales A, Iwakuma T. 2015. Targeting oncogenic mutant p53 for cancer therapy. Front Oncol 5: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltz SW, Morsy M, Welch EM, Jacobson A. 2013. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med 64: 407–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. 2007. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28: 622–629. [DOI] [PubMed] [Google Scholar]
- Ren G, Zhao YP, Yang L, Fu CX. 2008. Anti-proliferative effect of clitocine from the mushroom Leucopaxillus giganteus on human cervical cancer HeLa cells by inducing apoptosis. Cancer Lett 262: 190–200. [DOI] [PubMed] [Google Scholar]
- Ribeiro CJ, Rodrigues CM, Moreira R, Santos MM. 2016. Chemical variations on the p53 reactivation theme. Pharmaceuticals (Basel) 9: E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Leszyk JD, Mangus DA, Jacobson A. 2015. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc Natl Acad Sci 112: 3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Friesen WJ, Tomizawa Y, Leszyk JD, Zhuo J, Johnson B, Dakka J, Trotta CR, Xue X, Mutyam V, et al. 2016. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc Natl Acad Sci 113: 12508–12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan NJ. 2014. Ataluren: first global approval. Drugs 74: 1709–1714. [DOI] [PubMed] [Google Scholar]
- Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L, Hirawat S, et al. 2010. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med 182: 1262–1272. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Gallie DR, Jefferson RA, Bevan MW, Turner PC, Wilson TM. 1987. Characterisation of the 5′-leader sequence of tobacco mosaic virus RNA as a general enhancer of translation in vitro. Gene 60: 217–225. [DOI] [PubMed] [Google Scholar]
- Sun JG, Li H, Li X, Zeng X, Wu P, Fung KP, Liu FY. 2014. Clitocine targets Mcl-1 to induce drug-resistant human cancer cell apoptosis in vitro and tumor growth inhibition in vivo. Apoptosis 19: 871–882. [DOI] [PubMed] [Google Scholar]
- Svidritskiy E, Madireddy R, Korostelev AA. 2016. Structural basis for translation termination on a pseudouridylated stop codon. J Mol Biol 428: 2228–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal P, Eizenberger S, Cohen E, Goldfinger N, Pietrokovski S, Oren M, Rotter V. 2016. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 7: 11817–11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchelebi L, Ashamalla H, Graves PR. 2014. Mutant p53 and the response to chemotherapy and radiation. Subcell Biochem 85: 133–159. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848. [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. 2007. Restoration of p53 function leads to tumour regression in vivo. Nature 445: 661–665. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. 2007. p53 in health and disease. Nat Rev Mol Cell Biol 8: 275–283. [DOI] [PubMed] [Google Scholar]
- Welch EM, Wang W, Peltz SW. 2000. Translation termination: it's not the end of the story. In Translational control of gene expression (ed. Sonenberg N, et al. ), pp. 467–486. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, et al. 2007. PTC124 targets genetic disorders caused by nonsense mutations. Nature 447: 87–91. [DOI] [PubMed] [Google Scholar]
- Wilde RG, Kennedy PD, Almstead NG, Welch EM, Takasugi JJ, Friesen WJ. 2007. Nucleoside compounds and their use for treating cancer and diseases associated with somatic mutations. PTC Therapeutics, U.S. patent no. 7,291,603. [Google Scholar]
- Wilschanski M, Miller LL, Shoseyov D, Blau H, Rivlin J, Aviram M, Cohen M, Armoni S, Yaakov Y, Pugatsch T, et al. 2011. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J 38: 59–69. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaginuma Y, Westphal H. 1992. Abnormal structure and expression of the p53 gene in human ovarian carcinoma cell lines. Cancer Res 52: 4196–4199. [PubMed] [Google Scholar]
- Yu X, Vazquez A, Levine AJ, Carpizo DR. 2012. Allele-specific p53 mutant reactivation. Cancer Cell 21: 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.