

Abstract
Pentatricopeptide repeat (PPR) proteins comprise a large family of helical repeat proteins that influence gene expression in mitochondria and chloroplasts. PPR tracts can bind RNA via a modular one repeat–one nucleotide mechanism in which the nucleotide is specified by the identities of several amino acids in each repeat. This mode of recognition, the so-called PPR code, offers opportunities for the prediction of native PPR binding sites and the design of proteins to bind specified RNAs. However, a deep understanding of the parameters that dictate the affinity and specificity of PPR–RNA interactions is necessary to realize these goals. We report a comprehensive analysis of the sequence specificity of PPR10, a protein that binds similar RNA sequences of ∼18 nucleotides (nt) near the chloroplast atpH and psaJ genes in maize. We assessed the contribution of each nucleotide in the atpH binding site to PPR10 affinity in vitro by analyzing the effects of single-nucleotide changes at each position. In a complementary approach, the RNAs bound by PPR10 from partially randomized RNA pools were analyzed by deep sequencing. The results revealed three patches in which nucleotide identity has a major impact on binding affinity. These include 5 nt for which protein contacts were not observed in a PPR10–RNA crystal structure and 4 nt that are not explained by current views of the PPR code. These findings highlight aspects of PPR–RNA interactions that pose challenges for binding site prediction and design.
Keywords: RNA-binding protein, chloroplast, helical repeat protein, bind-n-seq
INTRODUCTION
RNA–protein interactions govern many aspects of gene expression. The ability to predict the repertoire of RNA sequences that will be bound in vivo by a given protein would facilitate the assignment of functions to native RNA-binding proteins as well as the rational design of RNA-binding proteins for specified purposes. Most RNA-binding proteins bind RNA through the combined action of several globular RNA-binding domains, each recognizing several nucleotides and connected by linkers of varying structure (for review, see Chen and Varani 2013; Ban et al. 2015). This architecture is poorly suited to binding site prediction due to the unpredictable arrangement of the different domains with respect to one another. In contrast, RNA-binding proteins from the α-solenoid super family present an elongated RNA-binding surface consisting of regularly spaced helical repeating units (for review, see Rubinson and Eichman 2012; Abil and Zhao 2015; Hall 2016). The best-characterized examples are the Pumilio/fem-3 (PUF) and pentatricopeptide repeat (PPR) proteins (for review, see Wickens et al. 2002; Barkan and Small 2014). Each repeat motif in PUF and PPR proteins binds a single nucleotide whose identity is specified by amino acids at particular positions (Wang et al. 2002; Barkan et al. 2012; Shen et al. 2016). Although the details of RNA recognition by PUFs and PPRs differ, the regularity of their architectures and their predictable amino acid “codes” for nucleotide recognition make these scaffolds especially promising for binding site prediction and protein design (Abil and Zhao 2015; Hall 2016).
The PPR motif is a degenerate ∼35 amino acid sequence that resembles the TPR motif (Small and Peeters 2000). Each repeat forms a pair of anti-parallel α helices, and consecutive repeats stack to form a right-handed super helix (Howard et al. 2012; Yin et al. 2013). PPR proteins are found specifically in eukaryotes where they function almost exclusively in gene expression in mitochondria and chloroplasts (for review, see Barkan and Small 2014). In comparison with the PUF family, the PPR family is notable for enormous variation in family size and for its diversity of protein architectures. The PPR family is particularly large in plants, comprising approximately 450 members in angiosperms (Lurin et al. 2004). The number of PPR motifs per protein varies between two and ∼30, and the motifs fall into several subclasses that differ in length and consensus sequence (Lurin et al. 2004; Cheng et al. 2016). Approximately half of the PPR proteins in land plants are “pure” PPR proteins consisting almost entirely of canonical “P-type” PPR motifs. Most of the remainder are “PLS” proteins, which are formed from alternating P-type, “long,” and “short” PPR motifs, often followed by an accessory domain that is implicated in RNA editing (Barkan and Small 2014; Cheng et al. 2016). PPR tracts bind single-stranded RNA in a “parallel” orientation (N terminus at 5′ end) (Barkan et al. 2012; Yin et al. 2013), contrasting with the anti-parallel arrangement of PUF–RNA complexes (Wang et al. 2002).
Computational analyses revealed correlations between the identities of two amino acids in P-type PPR motifs and the bound nucleotide: the sixth amino acid in one repeat and the first amino acid in the next (referred to here as positions 6 and 1′) (see Fig. 1A; Barkan et al. 2012). This code was validated by recoding the native protein PPR10 to bind novel RNA ligands in vitro (Barkan et al. 2012) and by the analysis of synthetic PPR proteins constructed from consensus repeats (Coquille et al. 2014; Shen et al. 2016). A similar code was predicted for the “S” motifs in PLS proteins (Takenaka et al. 2013; Yagi et al. 2013), and modification of S motifs in PLS editing factors produced the predicted changes in sequence specificity in vivo (Kindgren et al. 2015). The structural basis for nucleotide recognition by several amino acid combinations has been revealed in protein–RNA co-crystal structures of PPR10 (Yin et al. 2013) and synthetic proteins built from a consensus PPR scaffold (Shen et al. 2016).
FIGURE 1.
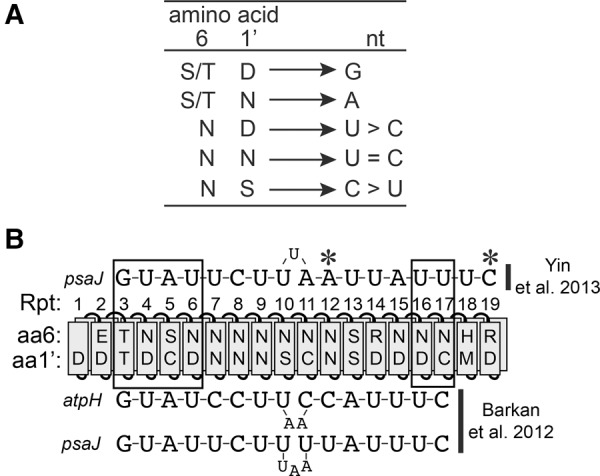
Overview of PPR10–RNA interactions and the PPR code. (A) The PPR code. The nucleotide preference of canonical PPR motifs is determined by the combination of amino acids at positions 6 and 1′ (the first amino acid in the next repeat). These correspond to amino acids 5 and 35 according to the nomenclature of Yin et al. (2013). Only experimentally validated amino acid codes are shown. The specificities of TD, TN, ND, NN, and NS were demonstrated in Barkan et al. (2012). The specificities of SN and SD were demonstrated in Shen et al. (2016). (B) Diagram of PPR10 aligned to its native atpH and psaJ binding sites. PPR motifs are indicated by rectangles representing helix A (front) and helix B (behind). The identities of the specificity-determining amino acids (aa6 and 1′) are indicated (Barkan et al. 2012). The alignment above is extrapolated from the PPR10–psaJ RNA crystal structure (Yin et al. 2013), whereas the alignment below maximizes the number of matches predicted by the canonical PPR code (Barkan et al. 2012). Boxes indicate modular PPR–nucleotide contacts, and asterisks mark nucleotides involved in noncanonical contacts in the PPR10–psaJ crystal structure (Yin et al. 2013).
Despite this progress, many questions remain about the parameters that impact the affinity and specificity of PPR–RNA interactions. That some degree of mismatch along a PPR–RNA interface can be tolerated is apparent in alignments between various native PPR proteins and their RNA ligands (Barkan et al. 2012; Takenaka et al. 2013; Yagi et al. 2013; Kindgren et al. 2015). Indeed, different positions along several PPR–RNA interfaces have been shown to vary in their contributions to RNA binding and in vivo function (Fujii et al. 2013; Yin et al. 2013; Kindgren et al. 2015). Furthermore, the PPR code is degenerate, and different amino acid combinations that specify the same nucleotide may bind with differing affinity. The finding that the native protein PPR10 uses both canonical code-based nucleotide recognition and alternative recognition mechanisms (Yin et al. 2013) adds a further complication.
To accurately predict the binding sites of both native and engineered PPR proteins, it will be necessary to elucidate the tolerance for gaps/mismatches along a PPR–RNA interface, the contributions of specific types of PPR motif-nucleotide pairings to binding affinity, and PPR–RNA interactions outside the modular code-based paradigm. In this study, we address these issues through deep analysis of the sequence specificity of the maize protein PPR10, whose functions, mechanisms, and structure are particularly well understood (Pfalz et al. 2009; Prikryl et al. 2011; Yin et al. 2013; Gully et al. 2015). PPR10 consists of 19 P-type PPR motifs and little else. PPR10 localizes to the chloroplast where it binds to two ∼18 nucleotide (nt) RNA segments of similar sequence: one in the atpI–atpH intergenic region and another in the psaJ–rpl33 intergenic region. These are referred to below as the atpH and psaJ sites, respectively. PPR10 bound to these sites blocks exoribonucleases intruding from either the 5′ or 3′ direction, which stabilizes the adjacent RNA segments and defines the positions of processed 5′ and 3′ RNA termini (Pfalz et al. 2009; Prikryl et al. 2011). In addition, PPR10 increases the translational efficiency of the atpH open reading frame (Zoschke et al. 2013), an effect that correlates with its ability to prevent the formation of an inhibitory RNA structure involving the atpH ribosome-binding region (Prikryl et al. 2011).
Recombinant PPR10 forms a homodimer at high concentrations, but binds atpH RNA in a 1:1 complex (Barkan et al. 2012; Gully et al. 2015). However, an X-ray crystal structure of a modified form of PPR10 bound to psaJ RNA revealed an anti-parallel protein dimer bound to two RNA molecules, each of which formed a loop whose ends bound to different protein monomers (Yin et al. 2013). This structure captured a subset of the predicted modular repeat/nucleotide contacts as well as two “nonmodular” protein/RNA contacts that do not conform to the PPR code. Other evidence supports the view that a 1:1 PPR10:RNA complex reflects the native state (Li et al. 2014; Gully et al. 2015), a view that gains further support from the findings presented here.
Our results revealed three patches of nucleotides within PPR10's atpH binding site whose identities are critical for a high affinity interaction, interspersed with regions in which nucleotide identities have little impact. Two of the critical patches are found at the 5′ and 3′ ends of the binding site; these include all 6 nt that formed canonical modular contacts with PPR10 in the PPR10–psaJ crystal structure as well as two additional nucleotides that form canonical contacts demonstrated here. The third patch includes several nucleotides whose mode of recognition cannot be explained by the canonical code-based mechanism. A high-throughput “bind-n-seq” approach revealed that a diversity of RNA sequence variants can bind PPR10 with high affinity and that the native atpH sequence is among the highest affinity ligands. Analysis of PPR10's sequence specificity by gel mobility shift assays and bind-n-seq resulted in qualitatively similar conclusions. However, in several cases the bind-n-seq assay amplified the effects of small differences in binding affinity and provided a view that is more consistent with inferences based on evolutionary conservation of binding site sequences.
RESULTS
Confirming the register between PPR10's C-terminal PPR motifs and the 3′ end of the atpH binding site
The alignment between PPR10 and the 5′ region of its atpH binding site was established through a compensatory mutation approach (Barkan et al. 2012) and confirmed in the crystal structure of a PPR10–psaJ RNA complex (see Fig. 1B; Yin et al. 2013). However, conflicting models have been proposed for the register between PPR10 and the 3′ end of its binding sites. The original proposed alignment incorporated bulged nucleotides at the center to maximize the number of PPR–nucleotide matches as predicted by the PPR code (Fig. 1B, bottom; Barkan et al. 2012). In contrast, the register observed in the crystal structure of a PPR10–psaJ complex was offset from this prediction by 2 nt (Fig. 1B, top; Yin et al. 2013). That said, this structure captured a quaternary complex of uncertain physiological relevance (Barkan et al. 2012; Li et al. 2014; Gully et al. 2015).
To resolve this issue, we used a compensatory mutation strategy analogous to that used to establish the register in the 5′ region (Barkan et al. 2012). We generated a PPR10 variant in which the specificity-determining amino acids in repeat 16 were changed from 6N,1′D (predicted to bind U) to 6T,1′N (predicted to bind A) (Fig. 2A). We compared the affinity of this protein for the wild-type atpH sequence, the U14A substituted RNA that is predicted to be the preferred ligand according to the register in the crystal structure (Register 2), and the U16A RNA that is predicted to be the preferred ligand according to Register 1 (Fig. 2B). Whereas WT PPR10 bound preferentially to the wild-type RNA, the Rpt16(TN) variant bound preferentially to RNA with the U14A substitution (Fig. 2B). These results provide strong evidence that PPR10 repeat 16 interacts with nucleotide 14 in the atpH binding site, validating the register reported in the PPR10–psaJ RNA crystal structure (Yin et al. 2013). Additional evidence for this register comes from the high-throughput experiment described below that revealed compensatory interactions between Repeat 15 and nucleotide 13 in the atpH site.
FIGURE 2.
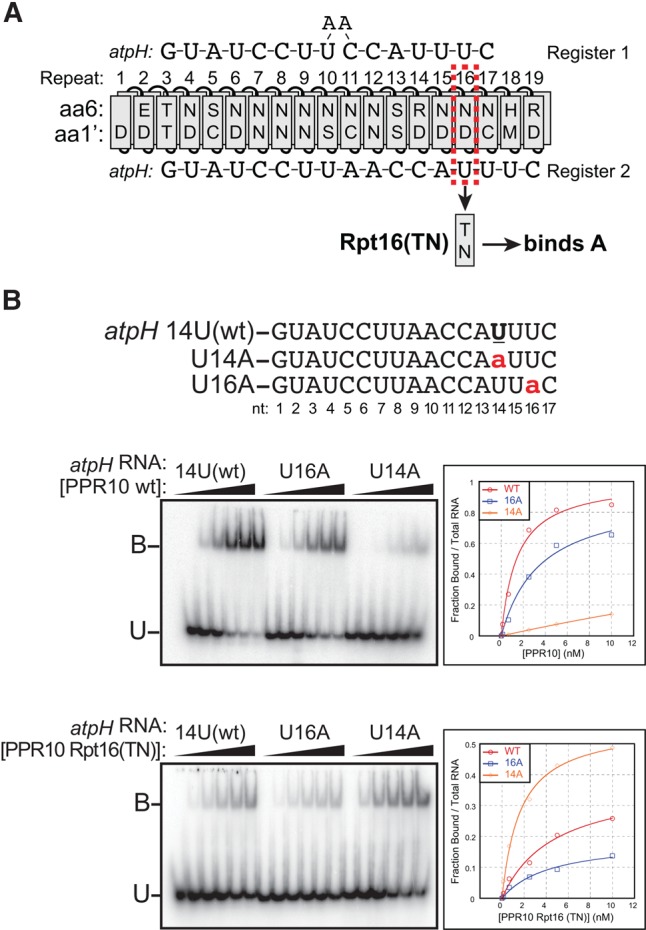
Compensatory mutation experiment to establish the register between the C terminus of PPR10 and the 3′-region of the atpH binding site. (A) Diagram of PPR10 aligned in two proposed registers to the atpH binding site. Register 1 was proposed in Barkan et al. (2012) and Register 2 was proposed in Yin et al. (2013). To distinguish between the registers, the nucleotide specifying amino acids in Repeat 16 were changed from 6N,1′D to 6T,1′N, which is predicted to change the bound nucleotide from U to A. (B) Gel mobility shift assays showing that PPR10 variant Rpt16(TN) binds preferentially to the U14A substituted atpH binding site, supporting Register 2. This assay was repeated three times with similar results. A representative experiment is shown.
Querying the contribution of each nucleotide in the atpH site to PPR10 binding affinity
The minimal atpH binding site spans 17 nt (Prikryl et al. 2011). To explore the contribution of each nucleotide in this site to PPR10 binding affinity, we performed gel mobility shift assays with a set of 17-mers having single-nucleotide changes at each position (Fig. 3). Nucleotides were generally substituted with the nucleotide whose amino acid code is most distinct (see Fig. 1A): adenine was replaced with uracil, guanine with cytosine, uracil with adenine, and cytosine with guanine (Fig. 3B). Each mutant RNA was assayed in parallel with the wild-type RNA to facilitate comparisons among experiments performed on different days. The predicted secondary structures for all of the assayed RNAs have a positive ΔG (Fig. 3B), so differences in secondary structure are unlikely to account for differences in binding affinity.
FIGURE 3.

Effects of single-nucleotide changes in the atpH site on PPR10 binding affinity. (A) Alignment between PPR10 and the atpH binding site. Boxes mark the modular PPR–nucleotide contacts detected in the PPR10–psaJ crystal structure (Yin et al. 2013) and asterisks mark nucleotides that make nonmodular protein contacts in that structure. (B) RNAs used for gel mobility shift assays. Nucleotide substitutions are marked in red. Relative binding affinities were approximated based on the data shown in panel C; mutant RNAs were placed into relative affinity bins by comparing their binding behavior to that of the wild-type RNA when assayed with the identical protein dilutions on the same gel. The thermodynamic stability of RNA secondary structure predicted for each sequence (Mfold prediction [Zuker 2003], 37°C, default parameters) is shown to the right. (C) Gel mobility shift assays. PPR10 was used at the concentrations indicated in the graphs. All assays used the same preparation of PPR10 except the assays with 1U and 1C RNAs. Each assay was repeated either two or three times, with similar results. A representative assay is shown in each case.
The results show that the identity of almost every nucleotide influences PPR10 binding affinity (Fig. 3C). However, the magnitude of this contribution varies dramatically. Three patches of nucleotides whose identities are critical (nucleotides 2–4, 8–11, and 14–15) are interspersed with patches that are more tolerant of mutations. The crystal structure of PPR10 bound to psaJ RNA detected six “modular” contacts between the protein and RNA (corresponding to nucleotides 1–4 and 14–15 in the atpH site assayed here) and two “nonmodular” contacts (corresponding to nucleotides A10 and C17 in the atpH site). Our data support the importance of all of these contacts, except that with C17, and provide evidence for additional nucleotides that contact PPR10 in a sequence-specific fashion.
PPR10's interactions with nucleotides 9, 10, 11, and 13 of the atpH site are puzzling: The identities of these nucleotides strongly impact binding affinity (Fig. 3C), yet these effects cannot be explained by the canonical recognition mode. For example, the adenines at positions 9 and 10 are broadly conserved among atpH sites in different organisms (Hayes and Mulligan 2011) and their mutation to G causes a strong loss of binding (see 9G and 10G in Fig. 3C). In the PPR10–psaJ structure, the second of these adenines forms a noncanonical protein contact: a hydrogen bond between its six-amino group and the aspartate at position 1′ in PPR motif 15. A bond of this nature could not be formed with guanine, which might account for the dramatic loss of PPR10 affinity for the A10G RNA. However, cytosine substitutions at A9 and A10 disrupted binding to a similar extent (see 9C and 9/10CC in Fig. 3C), despite the fact that cytosine seems compatible with the hydrogen bond observed in the structure. In addition, the nucleotides flanking A9 and A10 are intolerant of mutations (Fig. 3C), but these did not contact the protein in the crystal structure (Yin et al. 2013).
Use of bind-n-seq to profile PPR10's RNA sequence specificity
To obtain a comprehensive view of the variety of sequences capable of binding PPR10 with high affinity, we used a modified version of the RNA bind-n-seq assay (Lambert et al. 2014), which uses deep sequencing to analyze the population of RNAs bound by recombinant proteins from a randomized RNA pool. The complexity of a fully randomized pool of 17-mers (the length of PPR10's minimal binding site) would be too high to comprehensively sample by sequencing. To reduce the complexity of the input population, we combined three partially randomized RNA pools, each with 10 contiguous randomized nucleotides corresponding to the 5′, middle, or 3′ region of PPR10's footprint in the atpH 5′UTR (Fig. 4A). Recombinant PPR10 was incubated with a 50-fold molar excess of this RNA pool and the bound and unbound RNAs were separated by native gel electrophoresis. The bound and input RNA populations were then sampled by deep sequencing. This experiment was performed with wild-type PPR10 and with four PPR10 variants harboring amino acid changes at specificity-determining positions in various repeats (Fig. 4B). Three of these variants have modifications in repeats 6 and/or 7, and had been used previously in specificity-swap experiments to validate the PPR code (Barkan et al. 2012). The fourth variant (PPR10 Rpt15[TD]) had not been analyzed previously and was included to gain insight into the enigmatic interactions near the 3′ end of PPR10's binding site.
FIGURE 4.
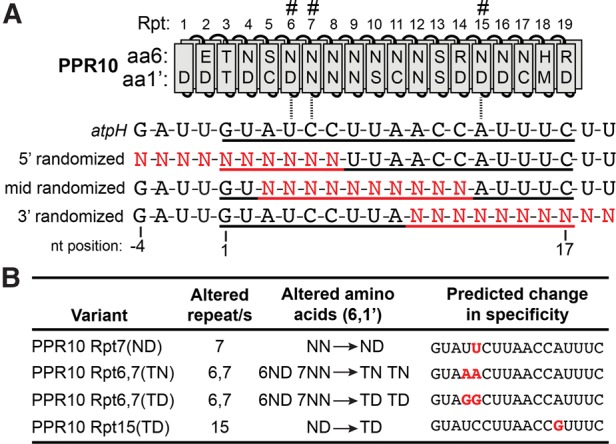
Design of bind-n-seq experiments. (A) RNA pools used for bind-n-seq assays. PPR10 is aligned to the sequence of its in vivo footprint in the atpH 5′UTR, with its minimal binding site underlined (Prikryl et al. 2011). The PPR motifs that were modified in the variants used for the bind-n-seq experiments are marked with hashmarks. The three pools of synthetic oligoribonucleotides diagrammed below were combined in equimolar amounts for use in the binding reactions. The nucleotide positions are numbered based on position in the minimal binding site. (B) PPR10 variants used in bind-n-seq assays. The Rpt7(ND), Rpt6,7(TN), and Rpt 6,7(TD) variants were shown previously to exhibit the predicted changes in sequence specificity in gel mobility shift assays (Barkan et al. 2012). PPR10 Rpt15(TD) had not been studied previously; its predicted specificity is inferred from the specificity of the 6T,1′D code in other contexts (see Fig. 1A).
We obtained approximately 2.2 × 108 sequence reads for the input RNA, almost 100-fold more than the 3.1 million different sequences predicted for the input pool. However, this detected only ∼95% of the expected sequences (Supplemental Fig. S1A) due to biases during synthesis of the “randomized” regions (40% G, 19% A, 15% C, 26% U). Had these nucleotides been randomized, each sequence in the input would be represented an average of ∼70 times in the aliquot that was sequenced; however, the vast majority of sequences were represented only once in this aliquot (Supplemental Fig. S1B). We obtained roughly 3 million sequence reads for each bound fraction (Supplemental Fig. S1A), and clear themes emerged from their analysis despite the incomplete sequence coverage of the input pool.
k-mers enriched in the bound fraction resemble the known binding sites of PPR10 and the predicted binding sites of PPR10 variants
To detect sequence motifs that were enriched in the bound RNA pools, the enrichment of all k-mers at each position within each randomized region was calculated as the frequency of the k-mer at that position in the bound fraction divided by its frequency at the same position in the input library. We analyzed k-values between 3 and 8, but present results only for 7-mers because these most effectively revealed enriched motifs. The frequency distributions of enrichment values for all 7-mers are shown in Figure 5 (wild-type PPR10) and Supplemental Figure S2 (PPR10 variants). The tails of these distributions (e.g., ≥5 SD above the mean or as indicated below) were defined as the enriched fractions for subsequent analysis. In the wild-type PPR10 data set, 7-mers from the 3′ randomized region dominate the enriched subset (Fig. 5), implying a greater tolerance by PPR10 for sequence variants in the 3′ region than in the 5′ region. The number of enriched 7-mers from the middle-randomized pool and the sequences containing these 7-mers were too low for meaningful analysis, so PPR10's nucleotide preferences at three positions in the center of its binding site could not be addressed with this data set.
FIGURE 5.
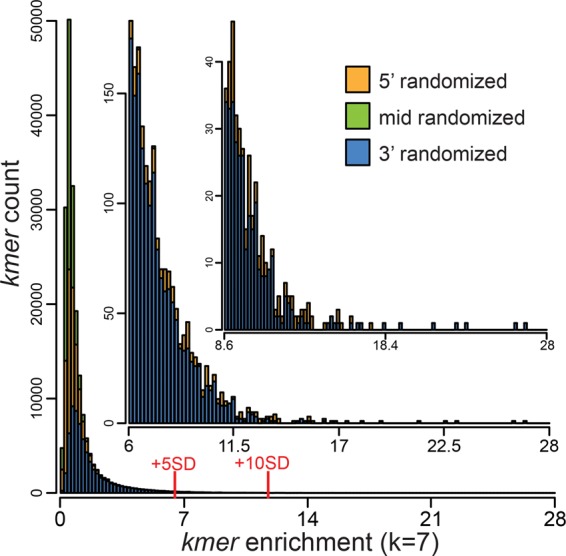
Frequency distribution of 7-mer enrichment values in the PPR10 bind-n-seq experiment. Enrichment values were calculated for each 7-mer at each position as the frequency of that 7-mer in the bound fraction divided by its frequency at the same position in the input library. The graph shows the number of different 7-mers (y-axis) at each enrichment value (x-axis). 7-mers from the 5′-, 3′-, and middle-randomized pools are colored in orange, blue, and green, respectively. Insets show expansions of the data in the tail of the distribution. The subsets of 7-mers that were enriched more than 5 or 10 standard deviations above the mean are marked. Analogous plots for the PPR10 variants that were analyzed by bind-n-seq are shown in Supplemental Figure S2. The frequency distribution of 7-mers in the input pool is plotted in Supplemental Figure S1C.
To display features of the sequences bound by each protein, sequence logos were generated from sequences harboring highly enriched 7-mers (Fig. 6). The contribution of each sequence toward each logo was weighted based on its enrichment value. The logos generated from the WT PPR10 selection showed strong similarity to PPR10's in vivo binding sites (Fig. 6B). Logos generated from sequences harboring enriched 7-mers in the 5′ region (≥5 SD above the mean) are dominated by 5 nt that match the first 5 nt of PPR10's psaJ and atpH binding sites (GUAUY). Strong resemblance to the native binding sites is also apparent in the 3′ region, but only from logos generated from 7-mers that were enriched ≥12 SD above the mean. PPR10's binding sites at atpH and psaJ are very similar at their 5′ and 3′ ends, but the spacing of the conserved regions differs by 1 nt (see Fig. 6A). We observed strong enrichment of the 5′ GUAUY motif only in the same register as that in the atpH binding site, implying that this register results in the highest affinity interaction with PPR10. The sequence logos show further that a G is selected against at the −1 position (Fig. 6B, left); however, results below show that this is likely due to an effect on RNA structure. The −2, −3, and −4 positions show no clear signature of selection, consistent with the known boundaries of PPR10's minimal binding site (Prikryl et al. 2011).
FIGURE 6.
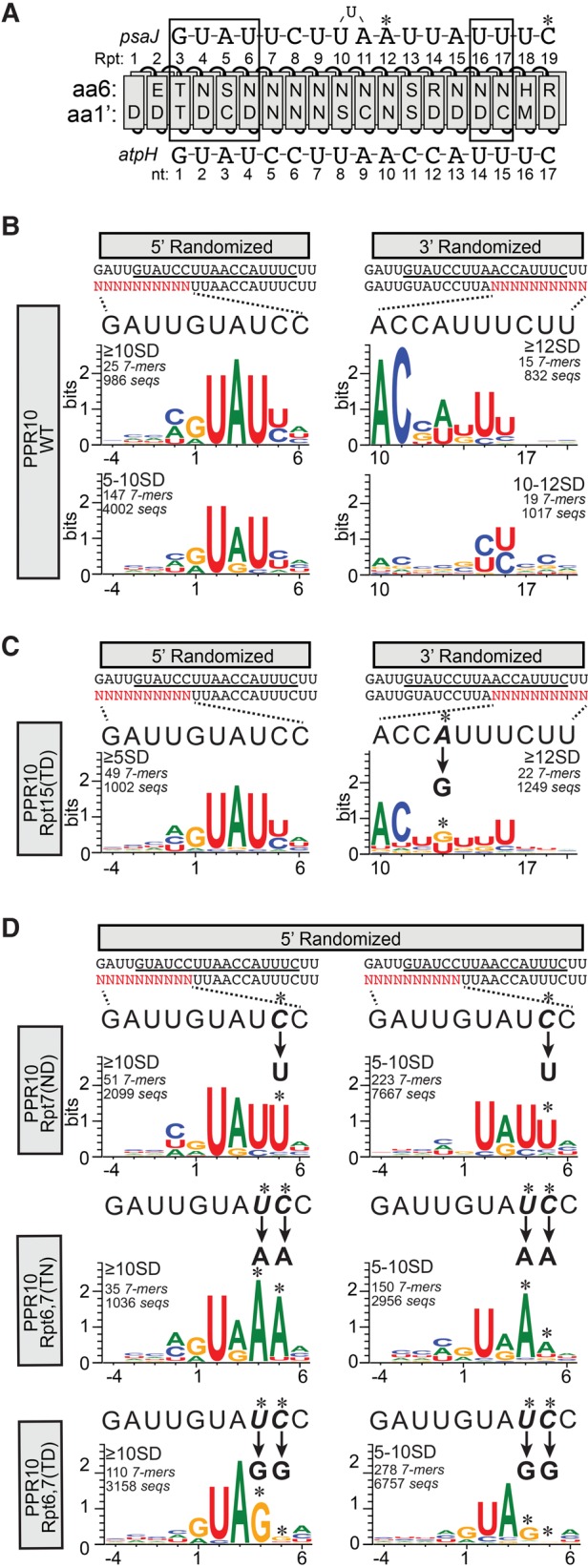
Sequence logos representing sequences harboring 7-mers that were enriched in PPR10 bind-n-seq assays. (A) Diagram of PPR10 aligned with its native atpH and psaJ binding sites. The protein and RNAs are annotated as in Figure 1. (B) Sequence logos representing data from the bind-n-seq assay with wild-type PPR10. Analyses of data from the 5′ and 3′ randomized pools are shown on the left and right, respectively. The oligonucleotide with the randomized region (in red) is displayed beneath the sequence of PPR10's footprint (minimal PPR10 binding site underlined). The wild-type sequence corresponding to each randomized region is expanded above the logos to facilitate comparisons. Position 1 is defined as the start of the minimal atpH binding site based on the register imposed by the constant region of each oligonucleotide. The enrichment cutoffs of the 7-mers used to generate each logo (in standard deviations above the mean), the number of different 7-mers in that subset, and the number of different sequences in that subset are indicated. (C) Sequence logos representing data from PPR10 variant Rpt15(TD). Logos are annotated as described in panel B. The change in sequence specificity predicted by the PPR code for the Rpt15(TD) variant is indicated, and the corresponding position in the logo is marked with an asterisk. (D) Sequence logos representing data from PPR10 variants Rpt7(ND), Rpt6,7(TN), and Rpt6,7(TD). Logos are shown only for data from the 5′ randomized oligonucleotide, which is the region expected to interact with the modified repeats. No substantive differences from the wild-type were observed in the sequences selected from the 3′ region. Logos are annotated as in panel B.
As expected, sequence degeneracy increased when the stringency of the enrichment cutoff was decreased (compare top to bottom panels in Fig. 6B). This comparison showed that either A or G at positions 1 and 3 are compatible with strong enrichment, but the highest affinity results from G1 and A3 as found in native PPR10-binding sites (see Fig. 6A). Gel mobility shift assays confirmed that an A3G substitution caused only a small decrease in binding affinity (Supplemental Fig. S3). It is interesting that positions two and four are represented almost exclusively by U even at the less stringent cutoff because the 6N,1′D amino acid code in the aligned PPR motifs showed only a slight preference for U over C in gel mobility shift assays with PPR10 variant Rpt7(ND) (Barkan et al. 2012). Gel mobility shift assays confirmed that a U2C substitution decreases affinity for PPR10 (Supplemental Fig. S3), but the decrease seems small in comparison to the strong preference for U implied by the bind-n-seq data. This contrast suggests that the competitive nature of the bind-n-seq assay may be particularly effective at detecting modest but physiologically relevant differences in binding affinity.
Logos generated from 3′ randomized sequences at a selection cutoff of ≥12 SD above the mean revealed selection for U residues at positions 14 and 15, consistent with the modular, code-based contacts between PPR10 and the corresponding psaJ nucleotides in the crystal structure (Fig. 6B, right). The logo produced by reducing the selection stringency slightly (10–12 SD above the mean) showed that C's can be accommodated at these positions as well. A selection for U at position 16 is also apparent although this nucleotide did not contact PPR10 in the crystal structure. The amino acids at the specificity-determining positions in the aligning PPR motif (6H,1′M) are unusual, so it is unclear whether position 16 is recognized in a canonical or noncanonical fashion. Positions 10 and 11 are the most constrained nucleotides in the 3′ region and are represented almost exclusively by A and C, respectively, in the highly enriched sequences; these match the nucleotides in the native atpH site in maize and in the atpH ortholog in other angiosperms (Hayes and Mulligan 2011). Gel mobility shift assays showed that G-substitutions at these positions severely disrupt PPR10 binding (Fig. 3). The bind-n-seq data show further that substitution with any nucleotide is disruptive (Fig. 6B, right). The PPR10–psaJ crystal structure does not provide a basis for the strong selection of A and C at these positions (Yin et al. 2013).
In the crystal structure, PPR10 repeats 16 and 17 make modular contacts with a pair of uridines near the 3′ end of the psaJ site (see Fig. 6A; Yin et al. 2013). This suggests that the preceding repeat (Repeat 15) contacts the adenine at the preceding nucleotide position, but experimental evidence for this was lacking. The bind-n-seq data from a PPR10 variant with a modification in Repeat 15 (6N,1′D→6T,1′D) validate this prediction (Fig. 6C). As expected, this variant selected sequences in the 5′ region that were similar to those selected by WT PPR10 (Fig. 6C, left). However, the data revealed a change in the favored nucleotide at position 13, from A/U for WT PPR10 to G/U for PPR10 Rpt15(TD) (Fig. 6C, right). The shift toward G-recognition is as predicted by the PPR code for a modular contact with Repeat 15. That said, recognition of nucleotide 13 is not fully explained by canonical interactions with Repeat 15: Adenine is found at this position in native PPR10 binding sites but the 6N,1′D code in Repeat 15 binds preferentially to uridine in other contexts. In fact, WT PPR10 selected A and U at this position to a similar extent (Fig. 6B, right), but the basis for A recognition remains unknown. Together, these results provide evidence that nucleotide 13 can be specified via either a canonical contact or via an atypical contact. This issue is addressed further below.
The RNAs selected by PPR10 variants Rpt6,7(TN), Rpt6,7(TD), and Rpt7(ND) (Fig. 6D) revealed changes in sequence specificity that are consistent with the PPR code, as observed previously for these proteins in gel mobility shift assays (Barkan et al. 2012). WT PPR10 selects pyrimidines at positions 4 and 5 but these become AA and GG in the Rpt6,7(TN) and Rpt6,7(TD) data sets, respectively. PPR10 Rpt7(ND) shifts the preference at position 5 from either C or U toward U. The nucleotide at position 5 did not contact PPR10 in the PPR10–psaJ crystal structure (Yin et al. 2013). However, the bind-n-seq data show that PPR10's seventh PPR motif binds nucleotide 5 via the standard PPR code: TN, TD, and ND modifications to PPR motif 7 shift the favored nucleotide at position 5 toward A, G, and U, respectively. The bind-n-seq data show further that position 4 is under stronger selection by PPR10 than is position 5.
Parsing the bind-n-seq data based on nucleotide identities at specific positions provides further insight into PPR10–RNA recognition
Sequence logos mask covariations among degenerate positions and do not convey the relative frequencies of different motifs that may contribute to the consensus. To explore features of this type, we sorted sequences that contained enriched 7-mers based on nucleotide identity at positions that showed selection for two different nucleotides. Both A and G were selected by WT PPR10 at positions 1 and 3 (see Fig. 6B). We sorted sequences that contained enriched 7-mers into four bins based on the identity of the nucleotides at those positions: GUA, GUG, AUG, and AUA (Fig. 7A). The fractional representation of these sequence subsets in the enriched fraction was 52% GUA, 25% AUA, 15% GUG, and 7% AUG, which together captured >99% of the sequences. These values presumably reflect the relative affinities of these sequences for PPR10, assuming otherwise identical sequences. Indeed, the native PPR10 binding sites start with GUA, which is the most highly represented combination. These results indicate that AUA, GUG, and possibly AUG at these positions are also compatible with a high affinity interaction. The G1A and A3G substitutions decrease enrichment approximately twofold and ∼3.5-fold, respectively, whereas the two mutations together cause a roughly sevenfold effect as predicted if the PPR interactions with these nucleotides are noncooperative. Sequence logos generated from the RNAs in each bin were otherwise similar, indicating that the presence of A versus G at positions 1 and 3 does not substantially alter nucleotide selectivity at other positions.
FIGURE 7.
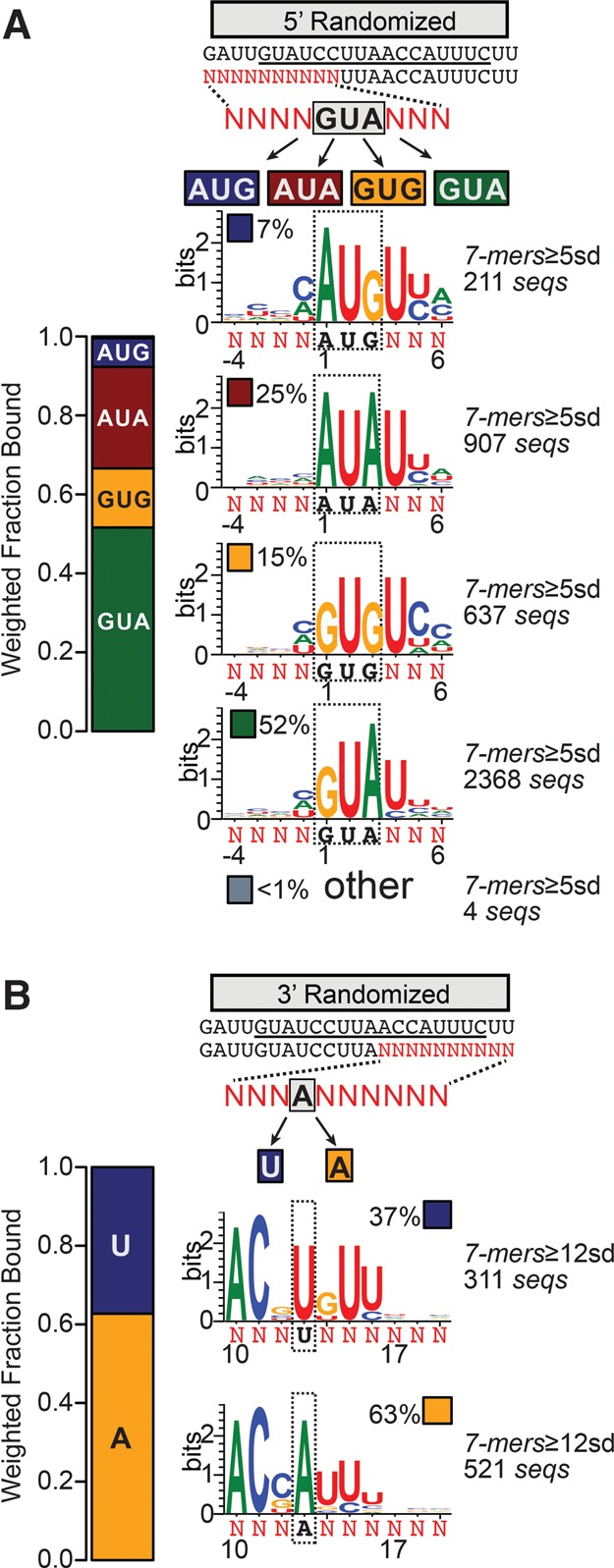
Parsing motifs that contribute to PPR10 bind-n-seq sequence logos. (A) Logos resulting from parsing sequences containing enriched 7-mers (≥5 SD above the mean) from the 5′-randomized pool based on nucleotide identity at positions 1 and 3. Fixed nucleotides (boxed black text) are shown below each logo. The bar plot to the left indicates the fractional contribution of sequences harboring each motif to the sequence set. (B) Logos resulting from parsing sequences containing enriched 7-mers (≥12 SD above the mean) from the 3′-randomized pool based on nucleotide identity at position 13. Fixed nucleotides and their fractional representation are indicated as in panel A.
At the 3′ end, sequences that contained highly enriched 7-mers include either A or U at position 13 with a small preference toward A (Figs. 6B and 7B). As discussed above, recognition of nucleotide 13 is complex: The change in sequence selectivity by PPR10 Rpt15(TD) provided evidence that nucleotide 13 can be recognized via a canonical interaction with Repeat 15 (Fig. 6C); however, this interaction mode cannot explain the adenine at this position in the bind-n-seq data for wild-type PPR10 and in native PPR10 binding sites because the 6N, 1′D code in repeat 15 is predicted to bind uridine. When the highly enriched sequences from the 3′-randomized pool were sorted into two bins based on the presence of either A or U at position 13 (Fig. 7B), it became clear that the identity of nucleotide 13 covaries with those at the flanking positions: 13U is usually flanked by G's, whereas 13A is usually flanked by 12C and 14U as in the native atpH site. These alternative binding elements each include one interaction that is compatible with a canonical PPR–nucleotide interaction and another that is not, albeit at different positions: 13U is compatible with a canonical contact with Repeat 15, but recognition of the flanking G with Repeat 16 is not; 13A is not compatible with a canonical contact with Repeat 15, but the flanking U made a canonical contact with Repeat 16 in the crystal structure (Yin et al. 2013). These results suggest that PPR10 can bind RNA in this region via two different modes, both of which include canonical and alternative recognition mechanisms.
RNA secondary structure influences PPR10's RNA specificity
The bind-n-seq data revealed that PPR10 selects against G at position −1 (Figs. 6B and 8A, left). Selection at this position was unanticipated because it maps outside of PPR10's minimal binding site (Prikryl et al. 2011). Secondary structure modeling suggested that G at position −1 favors the formation of a 3-base pair (bp) stem that sequesters three of the most highly constrained nucleotides in PPR10's minimal binding site (ACC at positions 10–12) (Fig. 8A). We therefore considered the possibility that G is rare at the −1 position due to its impact on RNA structure. To explore this possibility, we generated a sequence logo from the relatively rare sequences harboring highly enriched 7-mers with G at the −1 position (Fig. 8A). This logo revealed strong enrichment of A at position 1, contrasting with the strong bias toward G at this position in the unfiltered set (Fig. 6B). These results strongly suggest that the G1A transition at position 1 compensates for the presence of G at position −1. Indeed, the G1A substitution prevents the formation of the RNA structure that is promoted by G at position −1 (Fig. 8A, right), which presumably underlies its compensatory effect.
FIGURE 8.

Sequence covariations in the bind-n-seq data illustrate the inhibition of PPR10 binding by RNA secondary structure. RNA structures were predicted by M-fold (Zuker 2003). (A) Basis for selection against G at position −1. The bar graph shows the representation of each nucleotide at each indicated position in sequences harboring 7-mers that were enriched in the WT PPR10 assay (≥5 SD above the mean). Frequencies are weighted by the enrichment value of the corresponding sequence. The subset of these sequences harboring G at position −1 was used to generate the sequence logo to the right. The impact of various nucleotide identities at positions 1 and −1 on RNA structure are diagrammed, with nucleotides that differ from the WT site highlighted in red. (B) Basis for selection against G at position 3 by PPR10 variant Rpt6,7(TD). The bar graph shows the representation of each nucleotide at position 3 among sequences harboring enriched 7-mers (≥5 SD above the mean) in bind-n-seq assays with the indicated proteins. The structures predicted for the preferred Rpt6,7(TD) binding site and for the A3G substituted site are diagrammed, with nucleotides that differ from the WT sequence highlighted in red and position 3 indicated by an asterisk.
A similar example comes from PPR10 variant Rpt6,7(TD), which selects G residues at nucleotide positions 4 and 5 rather than the pyrimidines selected by the wild-type protein (Fig. 6D). This variant showed decreased tolerance for G at position 3 (Figs. 6D and 8B), in comparison with WT PPR10 and with variants Rpt7(ND) and Rpt6,7(TN) (which bind U and A, respectively) (Figs. 6D and 8B). RNA structure predictions suggest that the G substitution at position 3 in the context of G4 and G5 substantially increases the likelihood of an RNA hairpin within PPR10's minimal binding site (Fig. 8B). Taken together, these observations add to the evidence that RNA structure inhibits PPR binding (Williams-Carrier et al. 2008; Kindgren et al. 2015; Zoschke et al. 2016) and show that this phenomenon can result in covariations among nucleotides within a PPR binding site.
DISCUSSION
The accurate prediction of sequences bound by native and engineered PPR proteins will require a solid understanding of the parameters that influence the affinity and specificity of canonical PPR–RNA interactions and the elucidation of alternative interaction modes. Toward this end, we comprehensively analyzed the sequence specificity of PPR10, a protein whose biological functions, RNA interactions, and structure were already particularly well characterized. Our results revealed striking differences in the degree to which different canonical PPR–nucleotide contacts contribute to binding affinity, demonstrated differences in nucleotide selectivity among repeats harboring the same amino acid code, provided evidence for multiple noncanonical but sequence-specific interactions, and added to the evidence that selection for unstructured RNA can masquerade as sequence specificity.
One implication of our results is that PPR10's native atpH binding site is likely to be among PPR10's highest affinity RNA ligands (see data summary in Fig. 9). We cannot rule out the possibility that higher affinity RNA ligands might have been detected had we used fully randomized RNAs for bind-n-seq. However, this would have to involve long-range compensatory changes in the RNA, a possibility that seems unlikely given the strong evidence that the RNA binds in single-stranded form along the protein surface, and that altering repeats near both the N and C termini altered specificity only for the corresponding nucleotide. In contrast, the native binding sites of many RNA-binding proteins are less than ideal, which presumably reflects an evolutionary tuning of binding affinity and kinetics to in vivo function (for review, see Campbell and Wickens 2015; Jankowsky and Harris 2015). Evolutionary convergence of PPR10's native binding sites to its highest affinity sequence makes good sense based on PPR10's function as a blockade to exoribonucleases: This blockade function only improves as binding affinity increases, and persistent RNA occupancy in the intergenic regions bound by PPR10 seems unlikely to have negative consequences.
FIGURE 9.
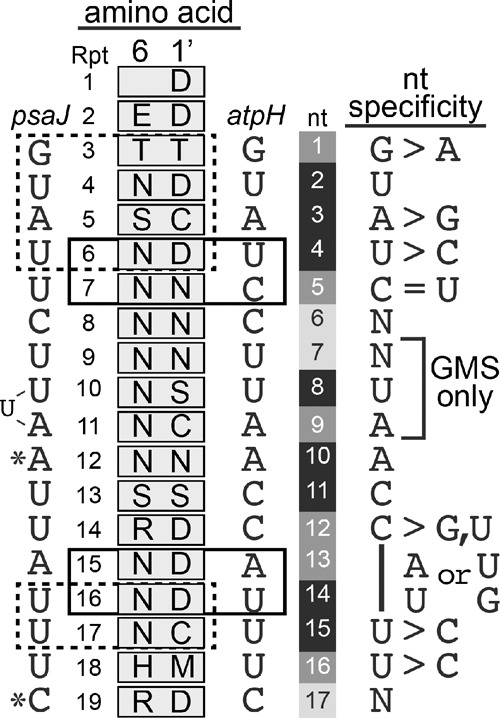
Summary of PPR10's nucleotide preferences within the atpH binding site. Every nucleotide was queried by both gel mobility shift and bind-n-seq with the exception of nucleotides 7, 8, and 9, for which bind-n-seq data are not available. The protein–RNA contacts observed in a PPR10–psaJ crystal structure (Yin et al. 2013) are illustrated to the left, with modular contacts in dashed boxes and nucleotides that make nonmodular contacts marked with asterisks. Modular interactions that can be inferred from data presented here are marked with boxes to the right. Nucleotide positions in the atpH sequence are shaded to reflect the degree to which PPR10 binding is affected by the nucleotide identity at that position. Darker shading indicates increased nucleotide selectivity.
Canonical PPR–nucleotide interactions vary in their degree of nucleotide selectivity and contribution to binding affinity
Our data show that PPR10 distinguishes among nucleotide identities at most positions along its atpH binding site (summarized in Fig. 9). Furthermore, single-nucleotide changes at several positions cause a virtual loss of binding (Fig. 3). These biochemical behaviors are reflected by high sequence conservation along the entirety of the orthologous region of atpH in angiosperms (Hayes and Mulligan 2011). This contrasts with the binding sites of “typical” RNA-binding proteins, within which each nucleotide generally makes an incremental contribution to binding affinity (for review, see Jankowsky and Harris 2015). The identities of 13 nt have a strong impact on PPR10 binding; these include the six modular interactions inferred from the PPR10–psaJ crystal structure (boxed in Fig. 9, left) and two additional modular interactions demonstrated here with compensatory mutations (boxed in Fig. 9, right). Additionally, canonical interactions between nucleotides 6–8 and repeats 8–10 seem likely based on the match between nucleotide identities and the PPR code, but these have not been experimentally confirmed.
Among the five proven modular interactions with the 5′ end of the binding site, matches between nucleotides 2, 3, and 4 and the corresponding PPR motifs (4, 5, and 6) are critical for PPR10 binding (Figs. 3C and 6B), whereas mismatches with nucleotides 1 and 5 can be tolerated (Fig. 3C). These results are consistent with those from experiments involving mutations in individual PPR motifs within PPR10 (Yin et al. 2013). Likewise, mutagenesis of individual PPR motifs in the P-type PPR protein PGR3 and the PLS-type PPR RNA editing factor CLB19 revealed widely varying contributions of different PPR motifs to RNA-binding and/or in vivo function (Fujii et al. 2013; Kindgren et al. 2015). The critical nucleotides in the PPR10 binding site cluster together in patches, suggesting that some number of contiguous PPR–nucleotide matches may be important for establishing an interaction. During microRNA–target RNA recognition, transient pairing of three contiguous nucleotides is required to initiate the interaction and this transitions to a stable interaction if the duplex can be extended to at least 7 bp (Chandradoss et al. 2015). It will be interesting to discover whether an analogous process underlies the selection of high-affinity RNA ligands by PPR proteins.
A related issue concerns the degree to which the nucleotide selectivity of a particular amino acid code varies according to its context in a PPR protein. For example, our data report the nucleotide selectivity of the 6N, 1′D amino acid code in the context of three repeats near PPR10's N terminus that bind RNA in a modular fashion: repeats 4 and 6 of WT PPR10 and repeat 7 in the Rpt7(ND) variant. The 6N,1′D code was highly specific for U in the context of repeat 4 but was somewhat permissive for C in the context of repeats 6 and 7. To improve the accuracy of binding site predictions, it will be necessary to evaluate whether differences in selectivity such as these arise from differences in position along a PPR–RNA interface, differences in amino acid sequence outside the code-bearing positions, or both.
Sequence-specific RNA recognition outside the canonical paradigm
Our data provide evidence for numerous sequence-specific but noncanonical interactions between PPR10 and atpH RNA. For example, single-nucleotide changes at positions 9, 10, and 11 cause a dramatic loss of binding affinity (Fig. 3). The amino acid sequences of the PPR motifs that align with these nucleotides predict binding to a YYR sequence, when in fact the strongly preferred sequence is AAC (Figs. 3 and 6B). The PPR10–psaJ crystal structure revealed a noncanonical contact between A10 and the 1′ aspartate in PPR motif 15 (Yin et al. 2013). Our results imply that the flanking nucleotides (A9 and C11) also contact PPR10 in a sequence-specific but idiosyncratic fashion, although these interactions were not captured in the quaternary complex that crystallized.
Furthermore, our results suggest two alternative modes through which PPR10 binds nucleotides 13 and 14 in the atpH site. These nucleotides align with PPR motifs 15 and 16, both of which harbor the 6N, 1′D amino acid code that predicts binding to U > C (Fig. 9). However, an adenosine is found at position 13 in native PPR10 binding sites and was enriched in the bind-n-seq data (Fig. 6B). That said, the potential for nucleotide 13 to form a canonical interaction with Repeat 15 is shown by the enrichment of the code-predicted uridine in the bind-n-seq data (Fig. 6B) and the selection by PPR10 variant Rpt15(TD) of RNAs with the “expected” G residue at position 13 (Fig. 6C). Analogous evidence suggests both canonical and noncanonical recognition modes for nucleotide 14: A modular contact with Repeat 16 was observed in the PPR10–psaJ crystal structure (Yin et al. 2013) and is supported by the change in specificity of the PPR10 variant Rpt16(TN) (Fig. 2B), but the “unexpected” base guanine is also highly represented at position 14 in the bind-n-seq data (Fig. 6B). Although alternative binding modes have also been reported for PUF proteins, these involve minor variations such as altered spacing between the 5′ and 3′ ends of a binding site or binding to half-sites (Lu and Hall 2011; Campbell et al. 2012; Valley et al. 2012; Prasad et al. 2016). It remains to be seen whether the alternative binding modes implied by our data are idiosyncrasies of PPR10 or are typical of other PPR proteins.
Utility of bind-n-seq to gain insight into PPR–RNA interactions
Several high-throughput assays have been developed to explore the sequence specificity of RNA-binding proteins in vitro (for review, see Campbell and Wickens 2015; Jankowsky and Harris 2015). The assay we used is similar to the bind-n-seq assay that has been used to study several metazoan proteins with globular RNA-binding domains (Lambert et al. 2014; Conway et al. 2016; Kapeli et al. 2016). Because those proteins recognize short motifs (5–7 nt), it was possible to comprehensively analyze enriched k-mers from fully randomized input RNA pools. A similar approach has been used to analyze the sequence specificity of PUF proteins (8–9 nt binding site), but after several rounds of selection to enrich sequences that bound with high affinity from a fully randomized pool (Campbell et al. 2012; Campbell et al. 2014). The much longer length of PPR10's minimal binding site (17 nt) precluded a thorough sampling of a fully randomized input pool, a problem we solved by combining three partially randomized RNA pools. The constant regions in the input RNAs limit the ability to detect synergistic or compensatory effects involving distant nucleotides, and our approach requires some prior knowledge of a protein's sequence specificity. Despite these limitations, the analysis was highly informative.
The bind-n-seq and gel mobility shift assays resulted in qualitatively similar conclusions, but quantitative differences in inferred binding affinities were observed in some instances. For example, the bind-n-seq data revealed a clear preference for G over A at position 1, A over G at position 3, and U over C at positions 2 and 4. These selections match the nucleotides that have been conserved through evolution (Hayes and Mulligan 2011), but were reflected by only small differences in affinity in gel mobility shift assays. Similarly, mismatches between a PPR editing factor and its RNA ligand were less tolerated in vivo than in vitro (Kindgren et al. 2015). It makes intuitive sense that the competitive nature of bind-n-seq would better mimic the in vivo environment than does a gel mobility shift assay involving one RNA ligand. That said, the opposite quantitative relationship between the two assays was observed for the data involving nucleotide positions 12, 13, and 14: The bind-n-seq data implied that PPR10 can accommodate either CAU or GUG at these positions, but gel mobility shift data showed a strong preference for the CAU sequence that is conserved in native PPR10 binding sites (Supplemental Fig. S3). Thus, these two assays provide complementary information and are best used in conjunction with one another.
Implications for the prediction PPR binding sites
Our results highlight limitations of the code-based paradigm for PPR–RNA interactions: Current understanding is insufficient to predict where PPR10 binds in the chloroplast transcriptome, much less where it would bind in the complex nuclear/cytoplasmic sequence space. Use of similar approaches to analyze the sequence specificity of other native PPR proteins will reveal whether PPR10's “idiosyncratic” features represent unrecognized themes. Analogous assays with synthetic proteins built from a uniform PPR scaffold may be particularly informative for clarifying contextual features that influence the nucleotide selectivity and affinity of a particular amino acid code, and for discovering whether canonical PPR tracts exhibit noncanonical nucleotide recognition modes.
MATERIALS AND METHODS
Expression of recombinant PPR10 and PPR10 variants
PPR10 and its variants were expressed as fusion proteins to maltose-binding protein from the pMAL-TEV vector in E. coli Rosetta 2 cells (Novagen), purified by amylose affinity chromatography, cleaved from the MBP and further purified on a size exclusion column as described previously (Pfalz et al. 2009; Barkan et al. 2012). The PPR10 sequence began at amino acid 38, corresponding to its predicted transit peptide cleavage site. The purified protein was dialyzed into 50 mM Tris–HCl, pH 7.5, 400 mM NaCl, 50% glycerol, and 5 mM β-mercaptoethanol and stored at −20°C.
Gel mobility shift assays
Gel mobility shift assays were performed as previously described (Williams-Carrier et al. 2008). Synthetic RNA oligonucleotides (Integrated DNA Technologies) were 5′ end labeled with T4 polynucleotide kinase and [γ-32P]ATP. Binding reactions contained 15 pM RNA, 40 mM Tris–HCl, pH 7.5, 180 mM NaCl, 10% glycerol, 4 mM DTT, 10 U RNAsin, 0.1 mg/mL BSA, 0.5 mg/mL heparin and protein at the indicated concentrations. Binding reactions were incubated for 30 min at 25°C and resolved on 5% polyacrylamide gels in 1× THE (34 mM Tris, 66 mM HEPES, 0.1 mM EDTA, pH 7.5) at 4°C. The data were visualized with a phosphorimager and quantified with ImageQuant. Binding curves were generated with KaleidaGraph software.
Bind-n-seq assays
Our bind-n-seq method is similar to the method described in Lambert et al. (2014), but with two key modifications. (i) We ordered 5′-phosphorylated synthetic oligonucleotides (IDT) that were free of the flanking sequences for library production. (ii) We used a native gel rather than affinity chromatography to separate bound from unbound RNA. Synthesis at the randomized positions used hand-mixed nucleotide pools to decrease sequence bias. The three partially randomized oligonucleotide pools (Fig. 4) were combined in equimolar amounts for use in binding reactions. The RNA pool (12.5 µM total concentration, 52 µL) was heated for 3 min at 95°C snap-cooled on ice and combined with an equal volume of 2.5× BNS buffer (100 mM Tris–HCl, pH 7.5, 250 mM NaCl, 10 mM DTT, 1 U/µL RNAsin [Promega], 0.25 mg/mL BSA, 0.025 mg/mL heparin) and 26 µL of PPR10 (or PPR10 variant) at a concentration of 500 nM. The final concentrations were: 5 µM RNA, 100 nM PPR10/PPR10 variant, 50 mM Tris–HCl, pH 7.5, 180 mM NaCl, 10% glycerol, 4 mM DTT, 0.4 U/µL RNAsin, 0.1 mg/mL BSA, 0.01 mg/mL heparin. Pilot experiments explored a range of PPR10 concentrations (33–300 nM); results are reported for the concentration that was most effective at revealing enriched motifs.
The binding reactions were incubated at 25°C for 30 min, then resolved in a 5% polyacrylamide gel in 1× THE. Electrophoresis was carried out at 4°C for 30 min at 15 W. A separate binding reaction with radiolabeled RNA pool (400,000 cpm) and 250 nM PPR10 was run in a different lane to determine the mobility of the PPR10–RNA complex. A gel slice spanning the expected position of the nonradiolabeled complex was excised and RNA was eluted overnight in 4 mL TESS (10 mM Tris, pH 8, 1 mM EDTA, 100 mM NaCl, 0.1% SDS) at 4°C, purified by phenol–chloroform extraction and concentrated by ethanol precipitation. RNA samples were converted to sequencing libraries using the NEXTflex Small RNA-Seq Kit v2 (Bioo Scientific).
Computational analysis
A sliding window approach was used to count the position specific frequency of kmers (for k = 7) within the randomized regions. Kmer enrichment was calculated as the frequency of a kmer at a specific position in the protein-bound fraction divided by its frequency at that position in the input RNA library. The frequency distribution of all 7-mers in the input pool is plotted in Supplemental Figure S1C. Sequences harboring 7-mers enriched to various degrees (as indicated in the figures) were used for the generation of sequence logos with weblogo 3.4 (Crooks et al. 2004), after weighting the sequences according to their enrichment value (i.e., frequency in the protein-bound RNA pool divided by frequency in the input RNA library). Sequences that were not detected in the input library (<1% of bound sequences) were not used to generate logos due to uncertainty about their enrichment value. Weblogo calculations were performed with a background composition based on the average nucleotide frequencies of the input RNA pool (40% G, 19% A, 15% C, 26% U).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Jim McDermott and Kenny Watkins for helpful comments on the manuscript and useful discussions, Rosalind Williams-Carrier for help with figure preparation, and Nick Stiffler for assistance with the bioinformatics. This work was supported by National Science Foundation grant MCB-1243641 (A.B.) and National Institutes of Health training grant T32-GM007759 (R.G.M.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.059568.116.
REFERENCES
- Abil Z, Zhao H. 2015. Engineering reprogrammable RNA-binding proteins for study and manipulation of the transcriptome. Mol Biosyst 11: 2658–2665. [DOI] [PubMed] [Google Scholar]
- Ban T, Zhu JK, Melcher K, Xu HE. 2015. Structural mechanisms of RNA recognition: sequence-specific and non-specific RNA-binding proteins and the Cas9-RNA-DNA complex. Cell Mol Life Sci 72: 1045–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan A, Small I. 2014. Pentatricopeptide repeat proteins in plants. Annu Rev Plant Biol 65: 415–442. [DOI] [PubMed] [Google Scholar]
- Barkan A, Rojas M, Fujii S, Yap A, Chong YS, Bond CS, Small I. 2012. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet 8: e1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell ZT, Wickens M. 2015. Probing RNA-protein networks: biochemistry meets genomics. Trends Biochem Sci 40: 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell ZT, Bhimsaria D, Valley CT, Rodriguez-Martinez JA, Menichelli E, Williamson JR, Ansari AZ, Wickens M. 2012. Cooperativity in RNA-protein interactions: global analysis of RNA binding specificity. Cell Rep 1: 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell ZT, Valley CT, Wickens M. 2014. A protein-RNA specificity code enables targeted activation of an endogenous human transcript. Nat Struct Mol Biol 21: 732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandradoss SD, Schirle NT, Szczepaniak M, MacRae IJ, Joo C. 2015. A dynamic search process underlies microRNA targeting. Cell 162: 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Varani G. 2013. Engineering RNA-binding proteins for biology. FEBS J 280: 3734–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Gutmann B, Zhong X, Ye Y, Fisher MF, Bai F, Castleden I, Song Y, Song B, Huang J, et al. 2016. Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J 85: 532–547. [DOI] [PubMed] [Google Scholar]
- Conway AE, Van Nostrand EL, Pratt GA, Aigner S, Wilbert ML, Sundararaman B, Freese P, Lambert NJ, Sathe S, Liang TY, et al. 2016. Enhanced CLIP uncovers IMP protein-RNA targets in human pluripotent stem cells important for cell adhesion and survival. Cell Rep 15: 666–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquille S, Filipovska A, Chia T, Rajappa L, Lingford JP, Razif MF, Thore S, Rackham O. 2014. An artificial PPR scaffold for programmable RNA recognition. Nat Commun 5: 5729. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Sato N, Shikanai T. 2013. Mutagenesis of individual pentatricopeptide repeat motifs affects RNA binding activity and reveals functional partitioning of Arabidopsis PROTON gradient regulation3. Plant Cell 25: 3079–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gully BS, Cowieson N, Stanley WA, Shearston K, Small ID, Barkan A, Bond CS. 2015. The solution structure of the pentatricopeptide repeat protein PPR10 upon binding atpH RNA. Nucleic Acids Res 43: 1918–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TM. 2016. De-coding and re-coding RNA recognition by PUF and PPR repeat proteins. Curr Opin Struct Biol 36: 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes ML, Mulligan RM. 2011. Pentatricopeptide repeat proteins constrain genome evolution in chloroplasts. Mol Biol Evol 28: 2029–2039. [DOI] [PubMed] [Google Scholar]
- Howard MJ, Lim WH, Fierke CA, Koutmos M. 2012. Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc Natl Acad Sci 109: 16149–16154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky E, Harris ME. 2015. Specificity and nonspecificity in RNA-protein interactions. Nat Rev Mol Cell Biol 16: 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeli K, Pratt GA, Vu AQ, Hutt KR, Martinez FJ, Sundararaman B, Batra R, Freese P, Lambert NJ, Huelga SC, et al. 2016. Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat Commun 7: 12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindgren P, Yap A, Bond CS, Small I. 2015. Predictable alteration of sequence recognition by RNA editing factors from Arabidopsis. Plant Cell 27: 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, Burge CB. 2014. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell 54: 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Yan C, Xu H, Wang Z, Long J, Li W, Wu J, Yin P, Yan N. 2014. Examination of the dimerization states of the single-stranded RNA recognition protein pentatricopeptide repeat 10 (PPR10). J Biol Chem 289: 31503–31512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Hall TM. 2011. Alternate modes of cognate RNA recognition by human PUMILIO proteins. Structure 19: 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurin C, Andres C, Aubourg S, Bellaoui M, Bitton F, Bruyere C, Caboche M, Debast C, Gualberto J, Hoffmann B, et al. 2004. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16: 2089–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfalz J, Bayraktar O, Prikryl J, Barkan A. 2009. Site-specific binding of a PPR protein defines and stabilizes 5′ and 3′ mRNA termini in chloroplasts. EMBO J 28: 2042–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Porter DF, Kroll-Conner PL, Mohanty I, Ryan AR, Crittenden SL, Wickens M, Kimble J. 2016. The PUF binding landscape in metazoan germ cells. RNA 22: 1026–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prikryl J, Rojas M, Schuster G, Barkan A. 2011. Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc Natl Acad Sci 108: 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson EH, Eichman BF. 2012. Nucleic acid recognition by tandem helical repeats. Curr Opin Struct Biol 22: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Zhang D, Guan Z, Liu Y, Yang Z, Yang Y, Wang X, Wang Q, Zhang Q, Fan S, et al. 2016. Structural basis for specific single-stranded RNA recognition by designer pentatricopeptide repeat proteins. Nat Commun 7: 11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small I, Peeters N. 2000. The PPR motif—a TPR-related motif prevalent in plant organellar proteins. Trends Biochem Sci 25: 46–47. [DOI] [PubMed] [Google Scholar]
- Takenaka M, Zehrmann A, Brennicke A, Graichen K. 2013. Improved computational target site prediction for pentatricopeptide repeat RNA editing factors. PLoS One 8: e65343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valley CT, Porter DF, Qiu C, Campbell ZT, Hall TM, Wickens M. 2012. Patterns and plasticity in RNA-protein interactions enable recruitment of multiple proteins through a single site. Proc Natl Acad Sci 109: 6054–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McLachlan J, Zamore PD, Hall TM. 2002. Modular recognition of RNA by a human pumilio-homology domain. Cell 110: 501–512. [DOI] [PubMed] [Google Scholar]
- Wickens M, Bernstein DS, Kimble J, Parker R. 2002. A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet 18: 150–157. [DOI] [PubMed] [Google Scholar]
- Williams-Carrier R, Kroeger T, Barkan A. 2008. Sequence-specific binding of a chloroplast pentatricopeptide repeat protein to its native group II intron ligand. RNA 14: 1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi Y, Hayashi S, Kobayashi K, Hirayama T, Nakamura T. 2013. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One 8: e57286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Li Q, Yan C, Liu Y, Liu J, Yu F, Wang Z, Long J, He J, Wang HW, et al. 2013. Structural basis for the modular recognition of single-stranded RNA by PPR proteins. Nature 504: 168–171. [DOI] [PubMed] [Google Scholar]
- Zoschke R, Watkins K, Barkan A. 2013. A rapid microarray-based ribosome profiling method elucidates chloroplast ribosome behavior in vivo. Plant Cell 25: 2265–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoschke R, Watkins KP, Miranda RG, Barkan A. 2016. The PPR-SMR protein PPR53 enhances the stability and translation of specific chloroplast RNAs in maize. Plant J 85: 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.