

Abstract
Mitochondrial DNA (mtDNA) genes are long known to be cotranscribed in polycistrones, yet it remains impossible to study nascent mtDNA transcripts quantitatively in vivo using existing tools. To this end, we used deep sequencing (GRO-seq and PRO-seq) and analyzed nascent mtDNA-encoded RNA transcripts in diverse human cell lines and metazoan organisms. Surprisingly, accurate detection of human mtDNA transcription initiation sites (TISs) in the heavy and light strands revealed a novel conserved transcription pausing site near the light-strand TIS. This pausing site correlated with the presence of a bacterial pausing sequence motif, with reduced SNP density, and with a DNase footprinting signal in all tested cells. Its location within conserved sequence block 3 (CSBIII), just upstream of the known transcription–replication transition point, suggests involvement in such transition. Analysis of nonhuman organisms enabled de novo mtDNA sequence assembly, as well as detection of previously unknown mtDNA TIS, pausing, and transcription termination sites with unprecedented accuracy. Whereas mammals (Pan troglodytes, Macaca mulatta, Rattus norvegicus, and Mus musculus) showed a human-like mtDNA transcription pattern, the invertebrate pattern (Drosophila melanogaster and Caenorhabditis elegans) profoundly diverged. Our approach paves the path toward in vivo, quantitative, reference sequence-free analysis of mtDNA transcription in all eukaryotes.
Mitochondrial ATP production via the oxidative phosphorylation system (OXPHOS) is the major energy resource in eukaryotes. Because of its central role for life, OXPHOS dysfunction leads to devastating disorders and plays a major role in common multifactorial diseases (Dowling 2014) such as type 2 diabetes (Gershoni et al. 2014) and Parkinson's disease (Coskun et al. 2012). In the vast majority of eukaryotes, OXPHOS is operated by genes encoded by two genomes: most in the nuclear genome (nDNA) and 37 in the short circular mitochondrial genome (mtDNA). This bigenomic division is accompanied by a profoundly different transcription regulatory system: Whereas nDNA-encoded genes are transcribed individually by RNA polymerase II and the general nuclear transcription machinery, mtDNA transcription is long known to be regulated mainly by a dedicated RNA polymerase (POLRMT) and mtDNA-specific transcription factors (TFAM and TFB2M) (Shutt and Shadel 2010). Moreover, mtDNA genes are cotranscribed in a strand-specific manner (Aloni and Attardi 1971): the heavy-strand (i.e., 12 mRNAs, 14 tRNAs, and two ribosomal RNAs) and light-strand (one mRNA and eight tRNAs) polycistrones, relics of the mitochondrial ancient bacterial ancestor (Zollo et al. 2012). However, as mtDNA transcription was mostly studied in vitro, little remains known about the mode and tempo of in vivo OXPHOS genes’ transcription residing on the mtDNA.
During the early 1980s, human mtDNA transcription initiation sites (TISs) were identified at a single-nucleotide resolution within the light- and heavy-strand promoters (LSP and HSP, respectively) (Montoya et al. 1982; Chang and Clayton 1984). These findings led to precise identification of mtDNA TISs in mouse (Chang and Clayton 1986a,b), Xenopus (Bogenhagen et al. 1986), chicken (L'Abbe et al. 1991), and the crustacean Artemia franciscana (Carrodeguas and Vallejo 1997). Although such studies provided insights into the location of mtDNA promoters in the mentioned organisms, the techniques used were typically low throughput, only semiquantitative, challenging to apply, and required prior sequence knowledge. These include S1 nuclease protection and primer extension (Chang and Clayton 1984), as well as in vitro capping (Yoza and Bogenhagen 1984). These obstacles interfered with comparative in vivo investigation of mtDNA transcription in diverse conditions and hampered expanding the study of mtDNA nascent transcripts to organisms lacking an mtDNA reference sequence. Finally, mtDNA transcription termination sites have been either mapped in vitro or were associated with MTERF binding sites (Christianson and Clayton 1986), thus, again, limiting the capability to in vivo map transcription termination sites in diverse organisms. It is thus imperative to develop alternative approaches.
Recently, global and precision-global run-on transcription and sequencing assays (GRO-seq and PRO-seq, respectively) enabled high-throughput detection of nascent transcripts (Core et al. 2008; Kwak et al. 2013). Such assays can be used to resolve the genome-wide landscape of transcription start, pausing, and termination sites (Kwak et al. 2013; Danko et al. 2015). In these techniques, run-on transcription reaction is performed in the presence of a tag that is affinity-purified to specifically isolate nascent RNA. As the cell nucleus is long known to be attached to a subset of the mitochondria (Barer et al. 1960), we reasoned that they will be copurified with the isolated nuclei, thus potentially generating mtDNA reads. Furthermore, RNA polymerase II (Core et al. 2008) and T7 polymerase (an ortholog of POLRMT) were successfully used to measure transcription in similar conditions (Mentesana et al. 2000), with the latter and POLRMT having conserved nucleotide incorporation mechanism (Kuhl et al. 2016). Here, we analyzed mtDNA reads generated by GRO-seq and PRO-seq experiments from 11 human cell types and seven metazoan species. We developed a bioinformatics pipeline that identifies candidate TISs, transcription pausing and termination sites with extremely high accuracy. Such analysis revealed, for the first time, precise quantitative differences in light- versus heavy-strand TIS ratios between human cell types and other organisms and identified candidate transcription pausing and termination sites for both the light and heavy strands in diverse organisms. Our analysis paves the path toward investigating mtDNA transcription in diverse physiological conditions, and in any given eukaryote.
Results
Adapting PRO-seq and GRO-seq data to analyze mtDNA transcription
GRO-seq and PRO-seq are based on massive parallel sequencing of nascent RNA extracted from either permeabilized cells or isolated cell nuclei. Since a subset of the mitochondrial population physically interacts with the nuclear membrane (Barer et al. 1960), it is reasonable to assume that some of the GRO-seq/PRO-seq reads will correspond to mtDNA transcription. To test for this possibility, we analyzed GRO-seq (nascent RNA labeled incorporating only bromo-uridine) and PRO-seq data (nascent RNA labeled by incorporating a biotinylated set of all 4 nucleotides [nt]) from 11 different human cell types (Table 1; Supplemental Table S1). First, we mapped the reads using the human revised Cambridge reference mtDNA sequence (rCRS) as a scaffold, which is included as ChrM within the human GRCh38 reference genome. Since the human mtDNA sequence is highly variable, and hence the dense SNP map could reduce the amount of mapped reads, we used the mapped mtDNA reads to reconstruct the mtDNA sequence for each of the analyzed samples separately. Moreover, to further increase the amount of accurately mapped reads, we took into account that the mtDNA is a circular molecule during the mapping procedure (see Methods).
Table 1.
Mitochondrial DNA initiation and termination sites across human cell lines
Analysis of NUMTs confirmed mtDNA mapping specificity
The isolation of cell nuclei during sample preparation for both GRO-seq and PRO-seq raised the possibility that a subset of the identified mtDNA reads reflect contamination by mtDNA-like pseudogenes that have been transferred to the cell nucleus during the course of evolution (NUMTs) (Hazkani-Covo et al. 2003; Mishmar et al. 2004). To control for this possibility, we focused our analyses on regions encompassing the light- and heavy-strand mtDNA promoters (LSP and HSP, respectively). Because GRO-seq and PRO-seq data sequence cDNA generated from nascent RNA extracts, we first BLAST searched our identified mtDNA reads against the entire Homo sapiens RefSeq RNA database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). To increase sensitivity of our NUMT screen while taking into account the relatively short read length generated by GRO-seq and PRO-seq (i.e., a minimum of 30 bases), we focused our screen on nuclear genomic BLAST hits that were >28 bp. This screen did not reveal any candidate NUMTs that mapped within the promoter regions of either mtDNA strand (Supplemental Fig. S1). To increase our stringency, we expanded our BLAST analysis to DNA reads of the entire human genome (GRCh38). This screen revealed three BLAST hits: one from Chromosome 5, within the region spanning the LSP (hereby referred as the light-strand NUMT), and an additional two BLAST hits from Chromosome 5 and Chromosome 11, respectively, that span the HSP (hereby referred as heavy-strand NUMT 1 and 2). The light-strand NUMT diverged from the rCRS in three mtDNA positions (i.e., 369, 377, 401); the heavy-strand NUMTs diverged from the rCRS in eight mtDNA positions (i.e., 572, 573, 576, 592, 596, 686, 710, 711). Analysis of the DNA sequences in mtDNA mapped reads indicated that only 0.21% of the reads encompassing the LSP could be explained by NUMT contamination (SD = 0.007). Similarly, only 0.6% of the heavy-strand reads corresponded to candidate NUMTs in the region encompassing HSP1 (SD = 0.017), and only 0.013% of the reads (SD = 0.047) corresponded to NUMTs within the region encompassing HSP2 (Table 2; Supplemental Table S2). Since the proportion of NUMT reads was very low and hence is expected to have only negligible impact on our transcription analysis, we avoided unique mtDNA mapping in further analyses.
Table 2.
Identification of NUMts in all human cells lines tested
Identification of mtDNA TISs at the light and heavy strands in diverse human cell types
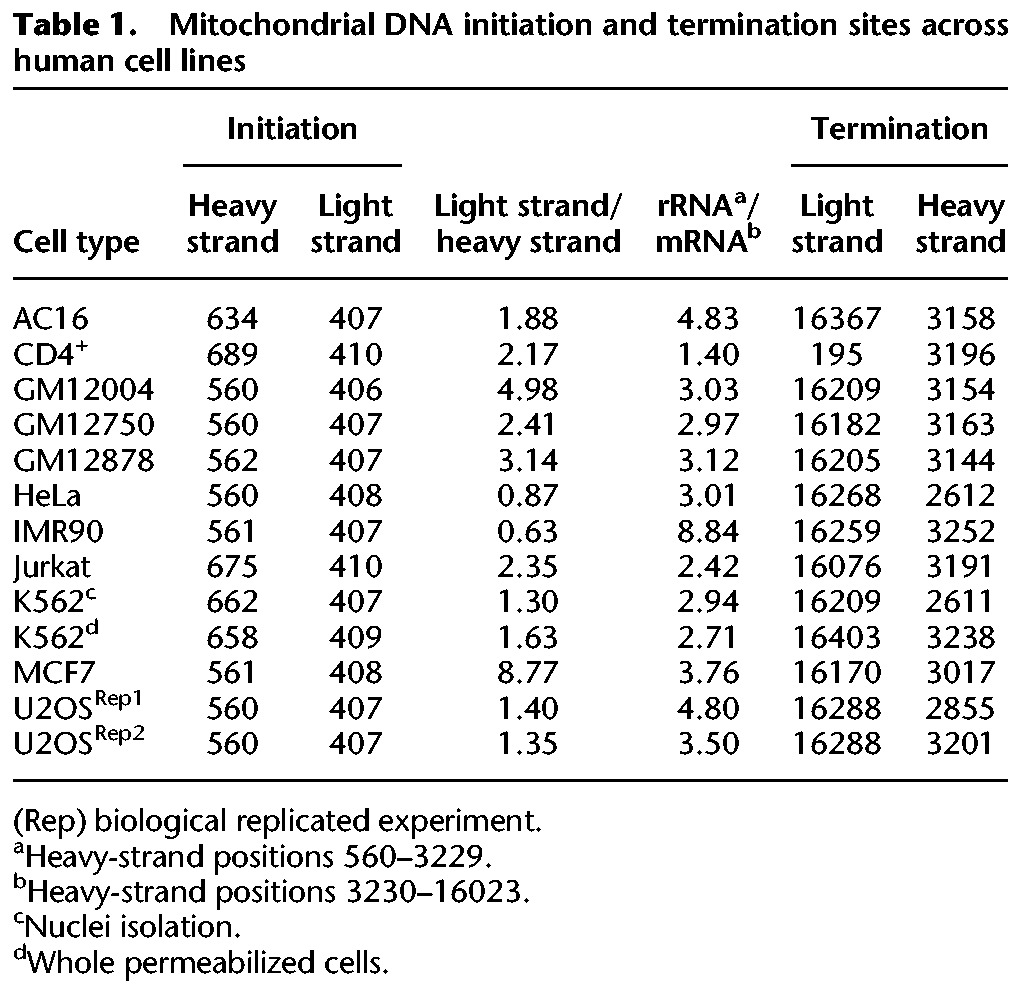
Having shown that PRO-seq and GRO-seq can be used to analyze mtDNA transcription, we next sought to identify candidate TISs. We screened mtDNA for regions harboring no mapped reads followed by a sudden increase in downstream reads (Fig. 1). To increase our sensitivity, we used a two-step approach (Fig. 1). The first step (step 1) was aimed toward crude identification of the best candidate TIS: We normalized the sequence coverage of each nucleotide position to the average in sliding 200-bp windows. Next, we searched for mtDNA nucleotide positions with the following characteristics: an upstream (200- to 1000-base window) read coverage of <5% of the average read coverage across the entire mtDNA in combination with a downstream (500 bases) read coverage >5% of the mtDNA average read coverage. In samples lacking nucleotide positions that passed these criteria, the read coverage threshold was increased by 1% increments until such positions were identified. The score for each of these sites was the ratio of downstream (50 bases) to upstream (500 bases) read coverage. Notably, if the distance between the two positions was >1 kb, we divided them into separate units. Finally, scores were calculated for the candidate TIS of each transcription unit (if there were more than one). The second analysis step (step 2) was employed to sort for the best TIS among the candidates identified in step 1. To this end, we reanalyzed the read coverage per nucleotide and recalculated the downstream (50 bases) versus upstream (250 bases) ratio for positions ±100 nt relative to the candidate positions listed in step 1. The nucleotide position with the highest score served as the best candidate TIS.
Figure 1.

Workflow of analysis. (A) GRO-seq and PRO-seq experiments generate genome-wide nascent transcript data. The extracted mtDNA sequences enable reconstruction of sample-specific mtDNA sequence, which is used in turn as a circular-like mapping reference. This allows counting the sequencing read coverage in a strand-specific manner. (B) Analysis of mtDNA transcription initiation and termination sites. Two steps were designed: (1) a crude step for candidate transcription initiation site (TIS) identification, identifying abrupt increase in read coverage in a nucleotide resolution within 200 bp sliding windows; and (2) fine analysis, focusing on highest scoring regions to identify the best TIS candidate. Notably, the identification of transcription termination sites utilizes the same approach, yet instead of an abrupt increase in reads, an abrupt decrease in read coverage is identified.
First, we applied our approach to analyze PRO-seq data from whole permeabilized K562 cells. Our analysis indicated that the TISs of both human mtDNA strands were consistent with known mtDNA promoters (Table 1). Specifically, consistent with previous findings (Chang and Clayton 1984), the TIS at position 409 was exactly within the known LSP, and the major heavy-strand TIS was within position 658, right downstream to position 645, exactly within the identified heavy-strand promoter 2 (HSP2) (Lodeiro et al. 2012; Zollo et al. 2012). Second, previous estimates of higher transcription signal intensity near the promoter of the light strand (Chang and Clayton 1984) were corroborated using PRO-seq; i.e., the read density of the entire light strand was 1.63-fold higher than the read density of the entire heavy strand. We used the entire human genome (GRCh38) as a reference while performing unique mapping. While applying unique mapping, reads that resulted in more than a single high-quality similarity hit (i.e., were mapped to both the mtDNA and to the nuclear genome) were excluded from further analysis. However, while employing unique mtDNA mapping against the entire human genome, the region encompassing the light-strand TIS was precisely identified, yet several candidate heavy-strand TISs emerged, thus preventing precise identification of the best TIS candidate. Hence, unique mapping against the entire human genome did not improve our precision. Finally, when we applied both GRO-seq (using isolated nuclei) and PRO-seq (using permeabilized cells) assays to the K562 cell line, the TIS was identified in identical positions, suggesting high similarity between the sequencing techniques and cell fractions analyzed.
mtDNA mode of transcription diverges across human cell types
Encouraged by our precise TIS identification in K562 cells, we tested for possible variability of mtDNA transcription among human cell types. Since we analyzed primary transcription, read coverage is expected to differ across the mtDNA sequence. Sequencing read coverage not only diverged across the mtDNA but also varied among 11 tested cell types (Supplemental Table S3). For the entire heavy strand, the range of total coverage per nucleotide position was 2.71–14900 (mean = 1395.69, SD = 3431.15), and the number of positions with coverage >1% of the total sequence coverage ranged between 6822 and 16402 in the different cell lines (mean = 14777.94, SD = 2371.39). Considering the entire light strand, the range of total coverage was 24.21–24317.09 (mean = 2453.07, SD = 5581.18), and the number of positions with coverage >1% of the total coverage ranged between 12623 and 15191 between the cell lines (mean = 14191.88, SD = 629.78). As such differences were consistent between experimental replicates, we hypothesized that our results reflect quantitative variation in mtDNA transcription levels among human tissues. To test for this hypothesis and to avoid the impact of known tissue variability in mitochondrial mass, we asked whether we could identify differences in the ratio of reads mapping to the HSP versus LSP between cell lines. Our analysis of the tested human cell lines uncovered a surprising amount of variability in the location of the TIS between different cell lines. First, we found that similar to K562 cells, the light-strand TIS of all samples was located within mtDNA nucleotide positions 406–410, which matches the known human LSP. Seven out of the 11 tested cell lines revealed a heavy-strand mtDNA TIS within nucleotide positions 560–562, exactly within the known HSP1. Samples with very high read coverage tended to provide highly reproducible results in duplicate experiments as can be seen in U2OS cells. Considering the other four samples, heavy-strand TIS was located within positions 634 and 689, downstream from the known HSP1 and closer to HSP2. Notably, patterns did not vary between experiments that used isolated cell nuclei or whole permeabilized cells (Supplemental Table S3), thus partially controlling for possible overrepresentation of certain subcellular mitochondrial populations. Nevertheless, we cannot exclude possible variation between whole cells and isolated nuclei experiments in other cells.
Differences between the read density in the heavy and light strands are also consistent with previously shown higher activation of light-strand compared with heavy-strand promoters(Chang and Clayton 1984) in most of the tested cells (nine out of 11). Nevertheless, while calculating the read density of the entire light strand and heavy strand, respectively, such ratios differed among cell types (Table 1; Fig. 2). Specifically, the highest light-strand/heavy-strand transcription ratio (approximately ninefold) was calculated for MCF7 cells, and the lowest light-strand/heavy-strand read density (about 0.6) was calculated for IMR90 cells. For the remaining nine cell lines, the calculated light-strand/heavy-strand read density ratios ranged between 1.3- to fivefold. Notably this ratio did not vary between biological experimental replicates of the K562 and U2OS cell lines (Table 1). Finally, while considering the heavy strand, we noticed higher transcription in the region encompassing the 12S–16S rRNAs compared with the rest of the heavy strand (Table 1), suggesting higher level of nascent rRNA transcripts. Notably, this ratio between the rRNA and the rest of the heavy-strand transcripts varied among cell lines (1.3- to 8.84-fold). This suggests, for the first time, profound quantitative variation in mtDNA transcription initiation patterns among human tissues.
Figure 2.

Accurate identification of human mtDNA TIS. (A) Sequencing read coverage around the mtDNA TIS in two cell lines. (Top) Sequencing read coverage pattern of the mtDNA heavy strand (red); (bottom) sequencing read coverage of the light strand (blue). Putative TIS is designated by black triangle. (y-axis) Sequencing read coverage; (x-axis) mtDNA position. (Left) PRO-seq experiment of the K562 cell line. (Right) GRO-seq experiment from the U2OS cell line. (B) Ratio of sequencing coverage between the light and heavy strands. (y-axis) Ratio of read density between the light and heavy strands. Black dots correspond to the calculated ratios for each tested cell line (indicated near the dots). (C) Summary of mtDNA transcription pattern: PRO-seq and GRO-seq experiments in 11 human cell types: (TIS) Light-strand TIS was identified in all tested human cell types (N = 11) in positions 407–410. In most of the tested cells (seven of 11), the major heavy-strand TIS was mapped in positions 560–562 (TIS 1). In the remaining cell types (four), the major heavy-strand TIS was located in positions 634–689 (TIS 2). (Termination sites) Light-strand transcription termination was identified within the 16S rRNA gene, in the region encompassing positions 2612–3252 (dark blue arrow). Heavy-strand transcription termination was identified within the D-Loop, between positions 16,076 and 195 (dark red arrow).
Transcription pausing occurs immediately downstream from the light-strand TIS
The sequencing read pattern encompassing the light-strand TIS appeared very different from that of the heavy-strand TIS. Unexpectedly, in the light strand we observed a sharp peak of read coverage immediately downstream (∼50-nt distance) from the TIS (Fig. 2), whereas the read pattern right downstream from the heavy-strand TIS appears to be ragged (Fig. 3). PRO-seq peaks in the nuclear genome, which had similar pattern to the sharp light-strand peak, were previously interpreted as pausing sites of the transcription machinery (see below) (Kwak et al. 2013). To determine whether a detectable enrichment of transcriptionally competent RNA polymerase was found in either HSP or LSP, we assessed the read coverage in a 10-nt sliding window downstream from the TIS in all tested human cell lines. This revealed a significant enrichment of reads near in the light-strand TIS, which was consistently located in positions 356–380, 30–50 nt from the TIS. In the heavy strand, we found that only in two out of 11 cell lines was there significantly enriched pausing peaks: In MCF7 and IMR90 cells, the candidate pausing peaks were located at positions 597 and 653, respectively. These results were consistent between the GRO-seq and PRO-seq experiments.
Figure 3.
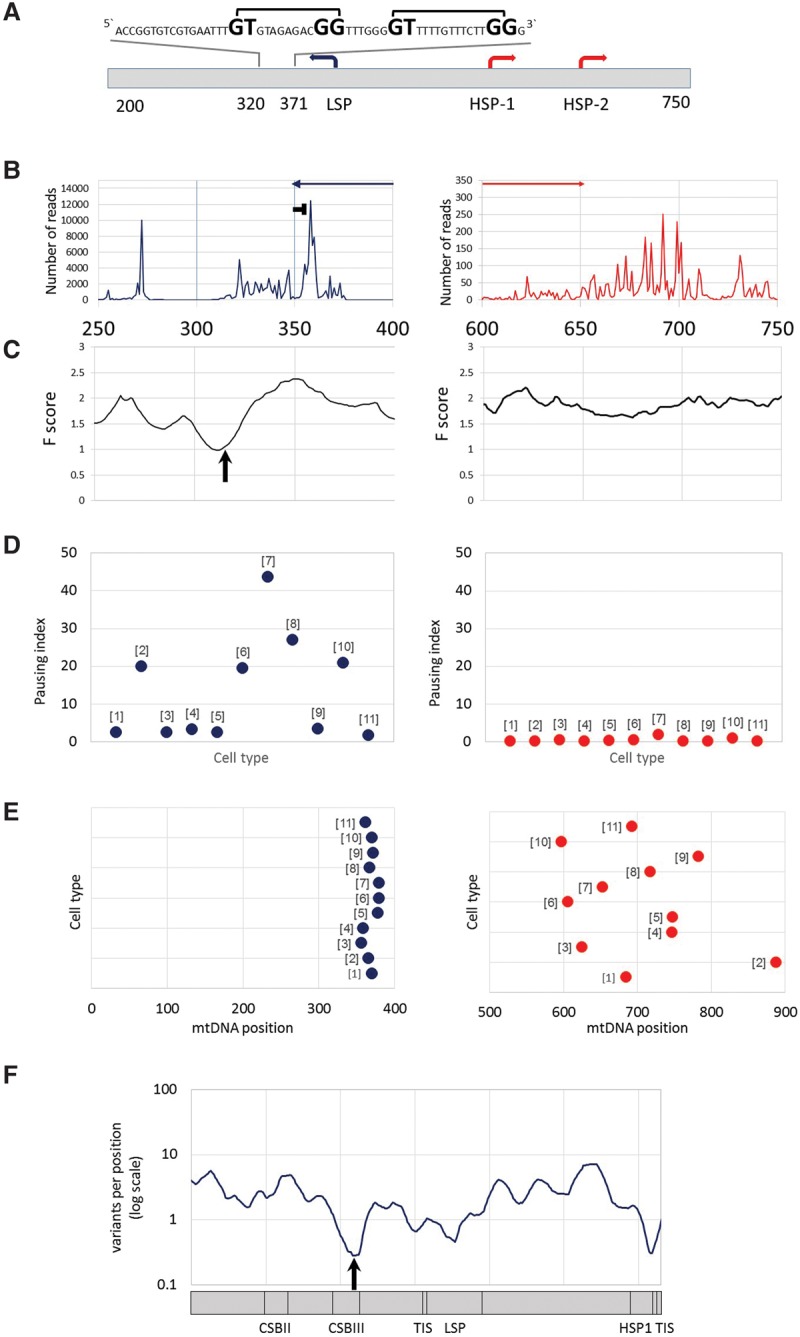
mtDNA transcription consistently pauses at distinct sites near the heavy- and light-strand TIS. (A) mtDNA transcriptional regulation elements. Presented is the complementary human sequence of the light mtDNA strand. The mtDNA sequence around the pausing peak is above the illustrated graph. (Square bracket) The bacterial pausing motif. The mandatory nucleotides within the motif are highlighted by a larger font size. (B) Coverage of the 3′ end of the PRO-seq experiment from K562 cell line. (x-axis) mtDNA nucleotide position; (y-axis) number of reads in the 3′. (Blue and red arrows) The direction of the light- and heavy-strand transcription, respectively. The “horizontal T” sign represents the pausing site. (C) DNase-seq experiment from K562 cell line. (x-axis) mtDNA nucleotide position; (y-axis) F-score of DNase-seq analysis. The lower the score, the more protected is the DNA by proteins. The black arrow points to the DGF site. (D) Pausing index across human cell types. (Left) Light strand; (right) heavy strand. (y-axis) Pausing index values. Dots correspond to the calculated pausing index for each tested cell line (indicated as numbers in brackets near the dots: [1] AC16; [2] CD4+; [3] GM12004; [4] GM 12750; [5] GM12878; [6] HeLa; [7] IMR90; [8] Jurkat; [9] K562; [10] MCF7; [11] U2OS). (E) Pausing site nucleotide position across human cell types. (Left) Light strand; (right) heavy strand; (x-axis) mtDNA position. Dots correspond to the pausing site nucleotide position of each tested cell line (numbering as in D). (F) Human population SNPs density. (x-axis) mtDNA position; (y-axis) SNPs density measured as variants per position (log scale).
To determine the precise coordinates of the mitochondrial RNA polymerase near the transcription active site, we focused further analysis on PRO-seq experiments, which are designed to resolve RNA polymerase progression at a single-nucleotide resolution in three cell lines (K562, Jurkat, and CD4+ T cells). We mapped the position of each read using only the precise coordinates of the 3′ end. We found that the light-strand pause site occurred within mtDNA positions 355–361. Since the peak morphology of the heavy strand was ragged and the pausing index was very low (less than 0.1), mapping the candidate pausing site was less accurate (mtDNA positions 677–715). Second, such mapping was limited only to the K562 cell line, since Jurkat and CD4+ cells had lower sequence coverage at these positions (less than 10×) and hence were less informative. We interpret these results to mean little or no transcription pausing near the heavy-strand TIS but a robust pause on the light-strand TIS. Notably, the pausing index in the light strand varied approximately 44-fold between the tested cell lines (Fig. 3; Supplemental Fig. S2; Supplemental Table S4), which considerably exceeds the variation between experimental replicates (available for U2OS and K562 cells). This suggested quantitative tissue-specific mtDNA transcription pausing differences.
We next sought to address the mechanism that underlies pausing near the light-strand TIS. Although metazoan systems establish nDNA transcription pausing by specific protein complexes, including DSIF and NELF (Kwak and Lis 2013), these protein complexes are strictly nuclear localized and have not been characterized in the mitochondria. As mitochondria originated from an ancient bacterial symbiont, we hypothesized that the transcription pausing sites may harbor bacterial-like attributes. To test this hypothesis, we searched for the presence of a ∼15-bp sequence motif responsible for transcription pausing in Escherichia coli by destabilizing bacterial RNA polymerase (Larson et al. 2014; Vvedenskaya et al. 2014). We found two such tandem motifs within the light-strand pausing peak (Fig. 3). As an alternative model, we also tested whether pausing occurs because POLRMT encounters a DNA-bound protein. We analyzed available DNase-seq data for six cell lines (Supplemental Table S4). This analysis revealed a DNase protected site, termed digital genomic footprinting (DGF), right downstream from the light-strand TIS (Supplemental Table S4), suggesting that an mtDNA-bound protein is involved in the light-strand transcription pausing. In contrast, no DGF was identified downstream from the heavy-strand TIS, which also lacked a pausing site as mentioned above.
We next sought to assess the functional importance of mutation in the TIS at both strands, as well as that of the light-strand pausing site. Since genome editing technology has yet to be established for the mtDNA in cells, alternative approaches must be used to assess the functional importance of mtDNA sequences. Hence, to assess whether the DNA sequence putatively responsible for paused polymerase is important for mtDNA genome function, we asked whether DNA sequence encoding the pausing site is conserved during the course of evolution. We studied SNP density in humans at the pausing site and TIS as a proxy for signatures of selection. We found that the frequency of mutational events within the light-strand pausing site was significantly lower than the rest of the D-Loop (positions 358–360, 0.28 variants per position, normalized to 10-base windows; P = 0.037) (Fig. 3). Similarly, the frequency of mutational events around HSP1 was also significantly lower (positions 558–559, 0.31 variants per position, normalized to 10 bases; P = 0.045), although the reduced frequency of mutational events at the LSP was only marginally significant (position 427, 0.46 variants per position, normalized to 10 bases; P = 0.072). These results imply that in vivo mtDNA transcription pausing at the light strand is not only common to all cell lines tested but also negatively selected and hence likely to be functionally significant. Interestingly, while screening for additional putative pausing sites throughout the mtDNA (internal pausing sites), apart from the above described site, we identified a single internal pausing site in the heavy strand (positions 5787–5835), which partially overlapped the light-strand origin of replication (Ori-L; positions 5721–5798). Notably, the bacterial pausing motif was absent from this additional site. The association of both pausing sites with a replication regulatory mtDNA element underlines the connection between transcription and replication dynamics in the mitochondrial genome.
Identification of mtDNA transcription termination sites
To date, mtDNA transcription termination sites have been determined in vitro (Christianson and Clayton 1986) and were correlated with the mtDNA binding sites of the transcription termination factors of the mTERF family (Park et al. 2007). Given our successful precise identification of TIS in both mtDNA strands, we attempted to identify candidate mtDNA transcription termination sites. To this end, we employed the similar set of criteria as those applied while identifying TIS; yet instead of looking for regions in which the sequencing read coverage was dramatically increased, we looked for the opposite—regions in which the read coverage had dramatically dropped. We found that mapping heavy and light strand termination sites was less precise than TIS, although all tested samples revealed candidate termination sites within the same regions (Table 1; Fig. 2). This argues for gradual rather than an abrupt transcription termination process. Specifically, we found that transcription termination of the light strand occurred between mtDNA positions 2619 and 3259, corresponding to the 3′ end of the 16S rRNA gene. The heavy-strand termination was identified between positions 16,076 and 195 within the D-Loop in all cell lines tested.
Human mtDNA RNA–DNA differences
GRO-seq and PRO-seq experiments were recently used to estimate the timing at which RNA–DNA differences (RDDs) occur during transcription of nDNA genes (Wang et al. 2014). Recently, we found A-to-U and A-to-G RDDs in human mtDNA position 2617 (Bar-Yaacov et al. 2013) and were curious whether they appeared already at the early stages of transcription. Analysis of all human GRO-seq and PRO-seq data available to us revealed that the RDDs were represented by <1% of reads encompassing mtDNA position 2617 in all samples (Supplemental Table S5) as opposed to >40% of the steady-state mitochondrial transcripts (Bar-Yaacov et al. 2013). Hence our data are consistent with likely post-transcriptional accumulation of the 2617 RDD in humans.
Identification of mtDNA transcription initiation and termination sites in divergent metazoans
Our successful identification of transcription initiation and termination sites in humans urged us to test our approach on nonhuman organisms, lacking previous experimental data and accurate mtDNA TIS mapping. As the first step, we analyzed available PRO-seq data generated by us and others from CD4+ lymphocytes from mammals (i.e., Pan troglodytes [chimpanzee], Macaca mulatta [rhesus macaque], Rattus norvegicus [rat], and Mus musculus [mouse]). We found that the general mammalian pattern of mtDNA transcription initiation and termination was quite similar to humans. Specifically, in chimpanzee, rat, and mouse transcription initiation, termination, and pausing exhibited similar pattern to humans (Fig. 4A): a distinct pausing peak 28–36 bases downstream from the light-strand TIS, a light-strand transcription termination around the 3′ end of 16S rRNA gene, and a heavy-strand TIS (Fig. 4B). In rhesus macaque, the pattern was somewhat different: The pausing site was more than 100 bases downstream from the light-strand TIS (Fig. 4B). The mtDNA TIS and termination pattern of the heavy strand in the chimpanzee, rat, and mouse was generally similar to that of humans, with transcription initiation occurring right upstream of the tRNA−phe gene (corresponding to the putative HSP1) and termination within the D-Loop. The rhesus macaque heavy-strand TIS mapped downstream from tRNA−phe and likely correspond to HSP2. The light-strand/heavy-strand TIS ratio (calculated as described for human samples) varied among species (Supplemental Table S6). Moreover, the ratio between the overall read coverage across the coding regions of the light and heavy strands notably varied among species: While the read coverage of the mtDNA heavy strand was twice the coverage of the light strand in rat and mouse, this ratio became nearly one to one in rhesus macaque. In humans and chimpanzee, an opposite pattern emerged, with twofold higher coverage of the light-strand compared with the heavy-strand coding regions (Supplemental Table S6).
Figure 4.
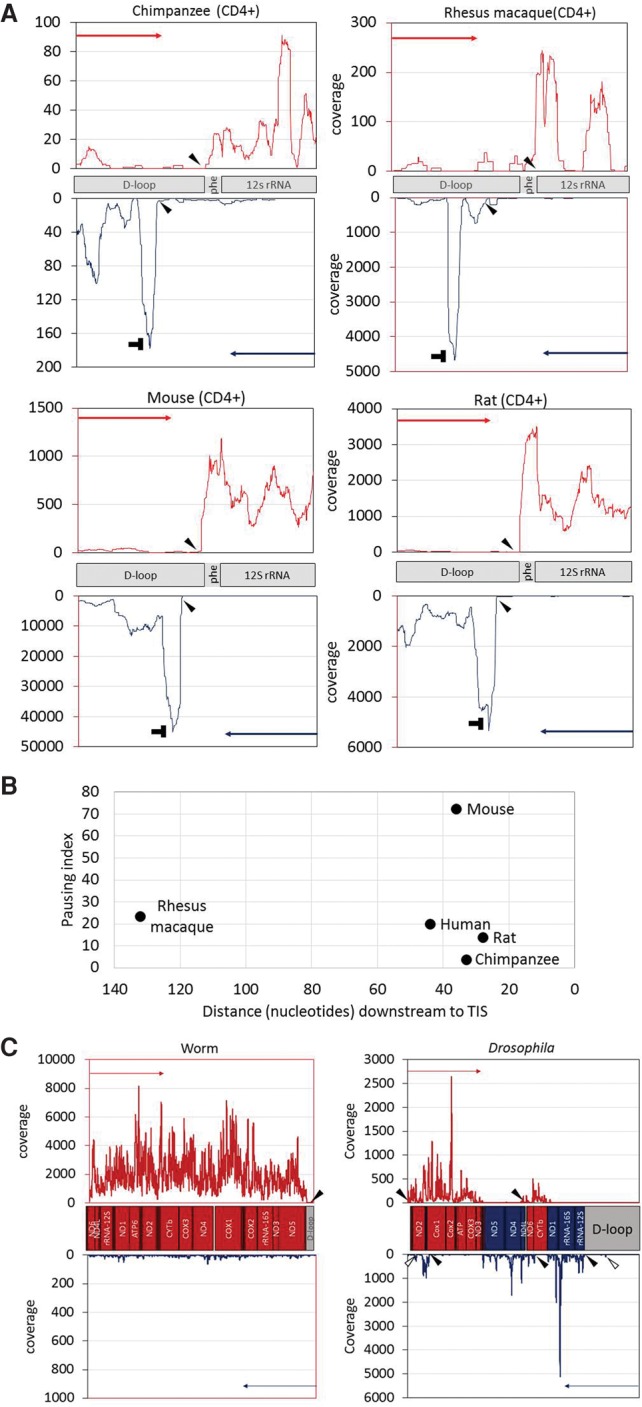
Identification of mtDNA nascent transcript across evolution: (A) PRO-seq experiment performed in four mammalian CD4+ cells: chimpanzee, rhesus macaque, rat, and mouse. x- and y-axes are identical to those in Figure 2, and “horizontal T” sign designates the pausing site. Filled arrowheads in all panels point to the calculated identified TIS. Notably, in three species (chimpanzee, rat, and mouse), the major heavy-strand TIS was identified downstream from the tRNA phenylalanine gene, similar to the human heavy-strand TIS 1. (B) Pausing site of light-strand transcription in mammals. (x-axis) Distance (in nucleotides) of the pausing site from the light-strand TIS; (y-axis) pausing index value of each species. The name of each species is indicated to the right of each dot. (C, left) Analysis of GRO-seq data from worm. In this species, there is a single TIS for a single transcription unit, present only at the heavy strand. (C, right) Analysis of PRO-seq data from Drosophila. Five candidate TISs were identified: two in the heavy strand and three in the light strand. Two minor additional TISs are marked by empty arrowheads.
We next employed our approach to identify mtDNA TIS and termination sites in invertebrates: Drosophila melanogaster (Drosophila) and Caenorhabditis elegans (worm). Our analysis revealed a single mtDNA TIS in worm, only in the heavy strand (Fig. 4C), which matches the gene content: In the worm, all genes are encoded by the heavy mtDNA strand. In Drosophila, the mtDNA genes are alternately encoded by the light and heavy strands, presumably in five transcription units (Torres et al. 2009). Our nascent RNA analysis revealed two TISs for the heavy strand and three for the light strand (Fig. 4C), which exactly correspond to the previously described five transcription units. Notably, two additional minor light-strand TIS were identified in the Drosophila mtDNA. One of these minor TISs mapped at mtDNA position 17110 within the control region and the other at position 3012 within the first heavy-strand transcription unit (according to GenBank accession NC_024511.2). These minor Drosophila TISs did not correspond to any previously described transcription unit.
GRO-seq and PRO-seq data enabled de novo assembly of the mtDNA in nonhuman organisms
Since the mtDNA is present in high copy number across all studied eukaryotes, sequence coverage is expected to be sufficiently high to enable de novo mtDNA sequence assembly. As this might be very useful for organisms lacking a reference genome, we assessed our capability to de novo assemble the mtDNA sequence using GRO-seq and PRO-seq data from Drosophila and worm as test cases. For the assembly of the mtDNA in both Drosophila and worm, we used the mtDNA of phylogenetically related species as a scaffold (Bactrocera arecae and Litoditis aff. Marina Pml, respectively). Drosophila mtDNA sequence contigs encompassed 80.4% of the used mtDNA scaffold, in comparison to 86.8% coverage, when the species-related scaffold was replaced by the known Drosophila mtDNA reference sequence. In worm, the reconstructed sequence contigs encompassed most (97%) of the species-related scaffold compared with 98.3% coverage when we used the known worm mtDNA as a reference sequence (Supplemental Fig. S3). The gaps in both studied species mostly corresponded to noncoding mtDNA regions, which are known to vary in length among species (Supplemental Fig. S3).
Discussion
Here, we analyzed modes of early mtDNA transcription in diverse cell lines and organisms by focusing on nascent transcripts. By adapting PRO-seq and GRO-seq experimental data to analyze the mitochondrial genome, we accurately identified the mtDNA TIS and transcription termination sites of both mtDNA strands in a variety of cell types and organisms, and unearthed quantitative variation in the transcription initiation of the two mtDNA strands. Additionally, we in vivo mapped, for the first time, a transcription pausing site at the light mtDNA strand of humans and other organisms. Finally, our analysis of GRO-seq and PRO-seq data that others and we generated from nonhuman animals enabled de novo assembly of the entire mtDNA sequence regardless of the availability of species-specific reference genomes. Thus, our approach paves the path toward functional mtDNA genomic studies of nonmodel animals, far beyond RNA-seq–based studies of steady-state gene expression.
Similar to genome-wide promoters, 40–50 bp downstream from the mtDNA light-strand TIS there was a read coverage peak in all human cells and most mammals. Since, the major advantage of PRO-seq and GRO-seq is exploring the dynamic of transcription rather than describing the steady-state RNA level, such peaks in the nuclear genome were interpreted as RNA polymerase pausing sites during the elongation process. This pausing site in human, chimpanzee, mouse, and rat overlapped a known bacterial transcription pausing motif (Larson et al. 2014; Vvedenskaya et al. 2014), though unlike bacteria this motif cannot be connected to translation, which is in line with the uncoupling of transcription and translation in the mitochondria (Small et al. 2013). Since we identified pausing downstream from the light-strand TIS, and not in the heavy-strand TIS, we are inclined to interpret the pausing as functionally related to the spatially adjacent transcription–replication transition point at CSB II in human cells (Chang and Clayton 1985; Xu and Clayton 1996; Pham et al. 2006; Shutt et al. 2010; Agaronyan et al. 2015). More precisely, the human transcription pausing overlapped conserved sequence block 3 (CSB III), which is upstream of the previously interpreted transcription–replication transition point (CSBII). In consistence with this correlation, we found the mouse light-strand transcription pausing site downstream from the LSP, just upstream of previously mapped RNA–DNA transition site in the mouse (Chang et al. 1985). Together, we interpret these results to mean that light-strand transcription pausing may serve to allow sufficient time and hence enable successful transcription-to-replication transition (Fig. 5). Consistent with this interpretation, we identified another pausing site, this time on the heavy strand, which was adjacent to the origin of replication of the light strand. The location of these two transcription pausing sites (i.e., in the light and heavy strands) led us to speculate that pausing of the transcription machinery enables sufficient time for the replication machinery to assemble, an interpretation that requires further experimental support. It is worth noting, however, that we cannot exclude different functional roles of the two pausing sites, due to differences in their location and associated attributes (lack of known sequence motif in the heavy-strand pausing site). Furthermore, the identification of TEFM, an mtDNA transcription elongation factor orthologous to the nuclear elongation factor Spt6 (Minczuk et al. 2011), suggests that mtDNA transcription pausing involves a mechanism similar to the nucleus. All this suggests that the mtDNA RNA polymerase (POLRMT) and the entire mitochondrial transcription machinery resemble the dynamics of RNA pol II.
Figure 5.

A model offering a mechanistic explanation for the role of light-strand transcription pausing. Presented are the stages right after transcription initiation of the light strand, as well as the suggested role for our discovered transcription pausing site. (Alternative products) Replication-based and transcription-based products (i.e., DNA and RNA products, respectively) of the light-strand promoter, as both require the same light-strand RNA primer (∼120 nt in length).
Whereas the light-strand TIS was within the same mtDNA region in all tested human cells, heavy-strand TIS divided the cells into two groups: seven cell lines in which the TIS was identified within the HSP1 region (GM12004, GM12750, GM12878, HeLa, IMR90, MCF7, and U2OS) and four cell lines (K562, AC16, CD4+, and Jurkat) in which the heavy-strand TIS was within the region corresponding to HSP2. HSP2 was first identified during the early 1980s by Attardi and colleagues (Montoya et al. 1982, 1983). Although, its presence was supported by others (Yoza and Bogenhagen 1984; Martin et al. 2005), the existence of two functional heavy-strand promoters was questioned (Chang and Clayton 1984; Litonin et al. 2010). This controversy remained even when an in vitro transcription assay was applied: Whereas some observations did not support transcription initiation from HSP2 (Litonin et al. 2010), others strongly supported the functionality of HSP2, especially when utilizing templates, which excluded the HSP1 sequence (Lodeiro et al. 2012; Zollo et al. 2012). Our results show that at least in some cell lines, the major heavy-strand transcription initiation overlapped HSP2, thus supporting the functional activity of HSP2 in vivo. Since in all 11 cell lines tested an increase in read coverage was observed around the HSP1 region, it is possible that in many cases our ability to detect the activity of HSP2 is masked by HSP1. Alternatively, differences in heavy-strand TIS pattern among the tested cell lines may reflect the relative strength of the two heavy-strand mtDNA promoters.
Analysis of a variety of human cell types revealed varying ratios between the light-strand and heavy-strand TIS. This may reflect differences in rates of transcription initiation, or differential proportion of pausing, similar to findings in the nuclear genome (Kwak et al. 2013). Since these cell types also differed in their mtDNA genetic backgrounds (haplogroups), we could not determine whether the physiological differences, mtDNA sequence, or even nuclear genetic variants contributed most to the observed quantitative variation in transcription initiation. As the number of analyzed samples was low, and since previous analysis of mtDNA gene expression patterns in mtDNA haplogroups revealed only subtle differences (Gomez-Duran et al. 2010; Kenney et al. 2014; Cohen et al. 2016), association of genetic backgrounds with mtDNA TIS/pausing patterns, as well as controlled assessment of mtDNA transcription in a variety of physiological conditions, still awaits a larger sample size collected in a controlled manner.
We used our approach also to get a glimpse into the evolution of mtDNA transcription in metazoans. We found that the pattern of mtDNA transcription (considering both initiation and termination) was very similar among the tested mammals, although quantitative differences were evident. While applying our approach to invertebrates (Drosophila and C. elegans), a completely different transcription pattern emerged, which correlated with the strand coding capacity. This observation raises the possibility that mtDNA gene arrangement correlated with transcription regulatory changes. This could be tested once PRO-seq/GRO-seq data become available from larger collection of metazoans.
In summary, we for the first time provided accurate and quantitative analysis of mitochondrial nascent transcripts without dependence on prior sequence knowledge. We found a previously unknown evolutionarily conserved transcription pausing site downstream from the mitochondrial LSP, with likely regulatory importance for the transition between mtDNA transcription and replication. Nevertheless, we found staggering diversity in mtDNA transcription patterns among metazoans. Our approach presents previously unmatched capabilities to analyze mitochondrial transcription and assess quantitative mtDNA regulatory differences among humans, cells, physiological conditions, and a variety of organisms. De novo assembly of our new data provides a means to assay the mtDNA in nonmodel organisms. Our findings underline the ability to measure mitochondrial transcription using the same molecular tool as is becoming more and more widely used for measuring nuclear transcription.
Methods
Data generation and initial analysis
CD4+ T-cell PRO-seq libraries were prepared from nonhuman primate and rodents species as described in established protocols (Kwak et al. 2013; Danko et al. 2015). Blood samples (80–100 mL) were obtained from three rhesus macaque and chimpanzee individuals in compliance with Cornell University IACUC guidelines. We used density gradient centrifugation to isolate peripheral blood mononuclear cells and positive selection for CD4+ cells using CD4 microbeads from Miltenyi Biotech (130-045-101 chimpanzee; 130-091-102 rhesus macaque). Mouse and rat CD4+ T cells were isolated from splenocytes using species-specific reagents from Miltenyi Biotech (130-049-201 mouse; 130-090-319 rat). In all cases, enriched CD4+ T-cell nuclei were prepared by resuspending cells in 1 mL lysis buffer (10 mM Tris-Cl at pH 8, 300 mM sucrose, 10 mM NaCl, 2 mM MgAc2, 3 mM CaCl2, and 0.1% NP-40), washed in a wash buffer (10 mM Tris-Cl at pH 8, 300 mM sucrose, 10 mM NaCl, and 2 mM MgAc2), and subsequently resuspended in 50 µL of storage buffer (50 mL Tris-Cl at pH 8.3, 40% glycerol, 5 mM MgCl2, and 0.1 mM EDTA) as described (Danko et al. 2015). For CD4+ T cells in all species, PRO-seq was performed as previously described (Kwak et al. 2013) and sequenced using an Illumina HiSeq 2000 or a NextSeq 500 at the Cornell University Biotechnology Resource Center. In addition, available GRO-seq and PRO-seq SRA files were downloaded from the GEO data set. Accession numbers of each sample are listed in Supplemental Table S1. SRA files were converted into FASTQ format using sratoolkit (http://www.ncbi.nlm.nih.gov/Traces/sra/?view=toolkit_doc). The sequencing adaptors were trimmed by Trim-galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to reach a minimum reads length of 30 nt.
Sample-specific mtDNA sequence reconstruction and mapping
As a first step to map PRO-seq and GRO-seq reads to the mtDNA, FASTQ files were uniquely mapped to the rCRS (GenBank accession no. NC_012920.1), which is included as ChrM in the GRCh38 human reference genome sequence, using BWA-aln (−q = 5, −l = 20, −k = 2, −t = 1) (Li and Durbin 2009). BWA (Li and Durbin 2009) was used to convert SAI into SAM format, which in turn was converted into a BAM file and sorted using SAMtools (Li et al. 2009). Next, SAMtools was used to generate VCF files of each sample (mpileup (-uf) command). Then, sample-specific mtDNA sequence was reconstructed for each of the analyzed samples using bcftools call (-c) (SAMtools) in combination with vcf2fq from the vcfutils.pl package. The resulting FASTQ files were uniquely remapped to the reconstructed sample-specific mtDNA using BWA-aln (−q = 5, −l = 32, −k = 2, −t = 1), and BAM files were generated again. Removal of low MAPQ reads was performed using the SAMtools “view” command (−F = 1804, −q = 30). When analyzing nonhuman species, we used publicly available relevant mtDNA sequences (Supplemental Table S7).
Coverage calculation
Coverage per base was calculated for a given sequence interval (separately for each strand) using BEDTools (Quinlan and Hall 2010). Specifically, we employed the command “genomecov” using the “-d” and “-strand” options. For the stringent identification of pausing sites, coverage of the 3′ end of the reads was calculated using the BEDTools “genomecov” command, with “-d,” “-strand,” and “-3” options.
Circular-like mapping of sample-specific mtDNA sequence
Since the mtDNA is a circular molecule and some reads may have been erroneously excluded, we reanalyzed the FASTQ files. To this end, we remapped the reads to the sample-specific mtDNA sequence that was rearranged such that the last 500 nt of the standard mtDNA sequence was cut and pasted at the beginning of the sequence. Mapping was performed, and read coverage at the former circle junction of the rearranged sequence was calculated and added to the previous mapping results.
Pausing site identification
Pausing sites were analyzed throughout the mtDNA similarly to a method described previously (Core et al. 2014) with some modifications. We used the following equation: IPI = (T + 1)/(GB + 1), where IPI represents internal pausing index, T represents density of reads in 20 bases of the tested position, and GB represents the density of reads in the gene body. In order to minimize putative reciprocal influence of close internal pausing sites, we modified the calculation so that “gene body” of each position was calculated in sliding windows of 10–1000 bases that flank each of the tested positions (both upstream and downstream). The highest IPI value for each position was considered as the optimal value for the tested position. For each PRO-seq/GRO-seq experiment, positions exhibiting with higher IPI values than the average plus 1 SD, were considered pausing sites. Finally, we focused our analysis only on pausing sites that were identified in at least 10 out the 11 studied cell lines.
Pausing index
Pausing index was calculated as the ratio between the density average across 10-nt sliding windows, with each position divided by the density average across the “gene body.” Since mtDNA genes are transcribed in polycistrones in a strand-specific manner, the “gene body” was defined as the transcription unit governed by each of the strand-specific promoters. Specifically, the human heavy-strand “gene body” was defined as the region between 100 bases downstream from the relevant identified heavy-strand TIS and the end of the coding region (mtDNA position 16024). The light-strand “gene body” was defined as the region between mtDNA positions 3250-16024. In nonhuman species, “gene body” was defined based on the same logic, depending on differences in the identified transcription units.
DNase-seq analysis
DNase-seq FASTQ files of the GM12878, HeLa, Jurkat, K562, and MCF7 cell lines were downloaded from the ENCODE Project Consortium website (hgdownload-test.cse.ucsc.edu/goldenPath/hg19/encodeDCC/) (The ENCODE Project Consortium 2012). DNase-seq FASTQ files of the CD4+ and IMR90 cell lines were downloaded from the Roadmap Epigenomics Consortium website (http://egg2.wustl.edu/roadmap/web_portal/index.html) (Roadmap Epigenomics Consortium 2015). DNase protected sites were identified as previously described (Blumberg et al. 2014). Briefly, for each nucleotide mtDNA position, the F-score was calculated in sliding read windows of ∼120 bp using the following equation: F = (C + 1)/L + (C + 1)/R, where C represents the average number of reads in the central fragment, L represents the average reads’ count in the proximal fragment, and R represents the average reads’ count in the distal fragment. The lowest F scores were interpreted as a DNase protected site (DGF site).
NUMTs identification
NUMTs diagnosis was performed in three steps: (1) BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) screen (Wheeler et al. 2008) the mtDNA (rCRS) as a query in order to search for NUMT hits; (2) collect variants that distinguish the active mtDNA from the candidate NUMTs identified in step 1; and (3) identify and count mtDNA mapped PRO-seq/GRO-seq reads (within BAM files) that contain NUMT variants using bam-readcount (https://github.com/genome/bam-readcount). Correlation was estimated between NUMT variants and the nucleotide content. Since the FASTQ reads were trimmed to a length of 30 bases, BWA-aln mapability parameters were restricted to a single mismatch. The three steps of NUMTs identification were employed using two types of reference data sets: First, since PRO-seq and GRO-seq are based on RNA, we focused our first analysis only on “RNA NUMTs” and performed our initial BLAST screen against the human RefSeq RNA database; second, we extended our screen to the entire human genome, utilizing the whole genome (GRCh38) and corresponding RNA as a reference.
Assignment of sample mtDNA sequence to known genetic backgrounds: haplogroups
PRO-seq and GRO-seq reads covered a mean of 89.19% of the human heavy mtDNA strand, and 85.65% of the light mtDNA strand. Thus, assignment of samples to known mtDNA genetic backgrounds (haplogroups) was plausible. To this end, each sample-specific mtDNA sequence was compared to the rCRS, and a set of sample-specific SNPs list was generated. These data were analyzed by HaploGrep (Kloss-Brandstatter et al. 2011), and mtDNA haplogroups were assigned (Supplemental Table S8).
De novo mtDNA sequence assembly
We aimed at assessing whether GRO-seq and PRO-seq data were sufficient to extract the majority of the mtDNA sequence in a given species. To this end, we employed CLC Genomics Workbench (https://www.qiagenbioinformatics.com/) to PRO-seq data from two species (D. melanogaster and C. elegans). Specifically, the clc_assembler command was used to de novo assemble FASTQ data, employing default parameters. BWA-mem (parameters use:−B = 2) was employed to map the generated contigs to the mtDNA of a phylogenetically related species that served as a scaffold (Supplemental Table S2).
Identifying RNA–DNA differences
Previously, we identified a prominent RNA–DNA difference site common to all human samples analyzed to date (Bar-Yaacov et al. 2013). We aimed toward assessing the presence of such differences during the early stages of mtDNA transcription. To this end, BAM files generated from all tested human samples that were indexed by SAMtools (the index command) and analyzed by bam-readcount were used to generate metrics of nucleotide content in mtDNA nucleotide position 2617.
Assessment of SNPs frequency
We utilized a previously published assembly of human mtDNA population variants, which stem from the analysis of nearly 10,000 individuals from diverse worldwide populations (Levin et al. 2013; Blumberg et al. 2014). The frequency of variants events was calculated only considering the D-Loop (mtDNA position 16024-576). The number of variants was normalized to the average of mutational events in sliding windows of 10 bases.
Data access
The raw and processed sequencing files from this study (PRO-seq data from CD4+ T-cells) and BED-graph files have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/), under accession numbers GSE85337 and GSE85747, respectively.
Supplementary Material
Acknowledgments
This study was supported by an Israeli Science Foundation grant (610-12), US Army Life Sciences Division grant 67993LS, and a United States–Israel Binational Science Foundation grant (2013060) awarded to D.M., with the latter in collaboration with A.K. The study gained additional support from the National Heart, Lung, and Blood Institute grant (UHL129958A) awarded to C.G.D. We also thank the Harbor Foundation for a scholarship for excellent PhD students awarded to A.B.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.209924.116.
References
- Agaronyan K, Morozov YI, Anikin M, Temiakov D. 2015. Mitochondrial biology. Replication–transcription switch in human mitochondria. Science 347: 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloni Y, Attardi G. 1971. Symmetrical in vivo transcription of mitochondrial DNA in HeLa cells. Proc Natl Acad Sci 68: 1757–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barer R, Joseph S, Meek G. 1960. Membrane interrelationships during meiosis. In Vierter Internationaler Kongress für Elektronenmikroskopie/Fourth International Conference on Electron Microscopy/Quatrième Congrès International de Microscopie Électronique, pp. 233–236. Springer-Verlag, Berlin, Heidelberg. [Google Scholar]
- Bar-Yaacov D, Avital G, Levin L, Richards AL, Hachen N, Rebolledo Jaramillo B, Nekrutenko A, Zarivach R, Mishmar D. 2013. RNA–DNA differences in human mitochondria restore ancestral form of 16S ribosomal RNA. Genome Res 23: 1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg A, Sailaja BS, Kundaje A, Levin L, Dadon S, Shmorak S, Shaulian E, Meshorer E, Mishmar D. 2014. Transcription factors bind negatively-selected sites within human mtDNA genes. Genome Biol Evol 6: 2634–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenhagen DF, Yoza BK, Cairns SS. 1986. Identification of initiation sites for transcription of Xenopus laevis mitochondrial DNA. J Biol Chem 261: 8488–8494. [PubMed] [Google Scholar]
- Carrodeguas JA, Vallejo CG. 1997. Mitochondrial transcription initiation in the crustacean Artemia franciscana. Eur J Biochem 250: 514–523. [DOI] [PubMed] [Google Scholar]
- Chang DD, Clayton DA. 1984. Precise identification of individual promoter for transcription of each strand of human mitochondrial DNA. Cell 36: 635–643. [DOI] [PubMed] [Google Scholar]
- Chang DD, Clayton DA. 1985. Priming of human mitochondrial DNA replication occurs at the light-strand promoter. Proc Natl Acad Sci 82: 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA. 1986a. Precise assignment of the heavy-strand promoter of mouse mitochondrial DNA: Cognate start sites are not required for transcriptional initiation. Mol Cell Biol 6: 3262–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA. 1986b. Precise assignment of the light-strand promoter of mouse mitochondrial DNA: A functional promoter consists of multiple upstream domains. Mol Cell Biol 6: 3253–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Hauswirth WW, Clayton DA. 1985. Replication priming and transcription initiate from precisely the same site in mouse mitochondrial DNA. EMBO J 4: 1559–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson TW, Clayton DA. 1986. In vitro transcription of human mitochondrial DNA: Accurate termination requires a region of DNA sequence that can function bidirectionally. Proc Natl Acad Sci 83: 6277–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen T, Levin L, Mishmar D. 2016. Ancient out-of-Africa mitochondrial DNA variants associate with distinct mitochondrial gene expression patterns. PLoS Genet 12: e1006407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. 2014. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46: 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, Laferla F, Wallace DC. 2012. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta 1820: 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hyland SL, Core LJ, Martins AL, Waters CT, Lee HW, Cheung VG, Kraus WL, Lis JT, Siepel A. 2015. Identification of active transcriptional regulatory elements from GRO-seq data. Nat Methods 12: 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling DK. 2014. Evolutionary perspectives on the links between mitochondrial genotype and disease phenotype. Biochim Biophys Acta 1840: 1393–1403. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni M, Levin L, Ovadia O, Toiw Y, Shani N, Dadon S, Barzilai N, Bergman A, Atzmon G, Wainstein J, et al. 2014. Disrupting mitochondrial–nuclear coevolution affects OXPHOS complex I integrity and impacts human health. Genome Biol Evol 6: 2665–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Duran A, Pacheu-Grau D, Lopez-Gallardo E, Diez-Sanchez C, Montoya J, Lopez-Perez MJ, Ruiz-Pesini E. 2010. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum Mol Genet 19: 3343–3353. [DOI] [PubMed] [Google Scholar]
- Hazkani-Covo E, Sorek R, Graur D. 2003. Evolutionary dynamics of large Numts in the human genome: rarity of independent insertions and abundance of post-insertion duplications. J Mol Evol 56: 169–174. [DOI] [PubMed] [Google Scholar]
- Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, Tarek M, Del Carpio JC, Nesburn AB, Boyer DS, et al. 2014. Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: implications for population susceptibility to diseases. Biochim Biophys Acta 1842: 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss-Brandstatter A, Pacher D, Schonherr S, Weissensteiner H, Binna R, Specht G, Kronenberg F. 2011. HaploGrep: a fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum Mutat 32: 25–32. [DOI] [PubMed] [Google Scholar]
- Kuhl I, Miranda M, Posse V, Milenkovic D, Mourier A, Siira SJ, Bonekamp NA, Neumann U, Filipovska A, Polosa PL, et al. 2016. POLRMT regulates the switch between replication primer formation and gene expression of mammalian mtDNA. Sci Adv 2: e1600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Lis JT. 2013. Control of transcriptional elongation. Annu Rev Genet 47: 483–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, Lis JT. 2013. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339: 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Abbe D, Duhaime JF, Lang BF, Morais R. 1991. The transcription of DNA in chicken mitochondria initiates from one major bidirectional promoter. J Biol Chem 266: 10844–10850. [PubMed] [Google Scholar]
- Larson MH, Mooney RA, Peters JM, Windgassen T, Nayak D, Gross CA, Block SM, Greenleaf WJ, Landick R, Weissman JS. 2014. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science 344: 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin L, Zhidkov I, Gurman Y, Hawlena H, Mishmar D. 2013. Functional recurrent mutations in the human mitochondrial phylogeny: dual roles in evolution and disease. Genome Biol Evol 5: 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litonin D, Sologub M, Shi Y, Savkina M, Anikin M, Falkenberg M, Gustafsson CM, Temiakov D. 2010. Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J Biol Chem 285: 18129–18133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodeiro MF, Uchida A, Bestwick M, Moustafa IM, Arnold JJ, Shadel GS, Cameron CE. 2012. Transcription from the second heavy-strand promoter of human mtDNA is repressed by transcription factor A in vitro. Proc Natl Acad Sci 109: 6513–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Cho J, Cesare AJ, Griffith JD, Attardi G. 2005. Termination factor–mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell 123: 1227–1240. [DOI] [PubMed] [Google Scholar]
- Mentesana PE, Chin-Bow ST, Sousa R, McAllister WT. 2000. Characterization of halted T7 RNA polymerase elongation complexes reveals multiple factors that contribute to stability. J Mol Biol 302: 1049–1062. [DOI] [PubMed] [Google Scholar]
- Minczuk M, He J, Duch AM, Ettema TJ, Chlebowski A, Dzionek K, Nijtmans LG, Huynen MA, Holt IJ. 2011. TEFM (c17orf42) is necessary for transcription of human mtDNA. Nucleic Acids Res 39: 4284–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini E, Brandon M, Wallace DC. 2004. Mitochondrial DNA-like sequences in the nucleus (NUMTs): insights into our African origins and the mechanism of foreign DNA integration. Hum Mutat 23: 125–133. [DOI] [PubMed] [Google Scholar]
- Montoya J, Christianson T, Levens D, Rabinowitz M, Attardi G. 1982. Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrial DNA. Proc Natl Acad Sci 79: 7195–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya J, Gaines GL, Attardi G. 1983. The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 34: 151–159. [DOI] [PubMed] [Google Scholar]
- Park CB, Asin-Cayuela J, Camara Y, Shi Y, Pellegrini M, Gaspari M, Wibom R, Hultenby K, Erdjument-Bromage H, Tempst P, et al. 2007. MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell 130: 273–285. [DOI] [PubMed] [Google Scholar]
- Pham XH, Farge G, Shi Y, Gaspari M, Gustafsson CM, Falkenberg M. 2006. Conserved sequence box II directs transcription termination and primer formation in mitochondria. J Biol Chem 281: 24647–24652. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt TE, Shadel GS. 2010. A compendium of human mitochondrial gene expression machinery with links to disease. Environ Mol Mutagen 51: 360–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt TE, Lodeiro MF, Cotney J, Cameron CE, Shadel GS. 2010. Core human mitochondrial transcription apparatus is a regulated two-component system in vitro. Proc Natl Acad Sci 107: 12133–12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small ID, Rackham O, Filipovska A. 2013. Organelle transcriptomes: products of a deconstructed genome. Curr Opin Microbiol 16: 652–658. [DOI] [PubMed] [Google Scholar]
- Torres TT, Dolezal M, Schlotterer C, Ottenwalder B. 2009. Expression profiling of Drosophila mitochondrial genes via deep mRNA sequencing. Nucleic Acids Res 37: 7509–7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vvedenskaya IO, Vahedian-Movahed H, Bird JG, Knoblauch JG, Goldman SR, Zhang Y, Ebright RH, Nickels BE. 2014. Interactions between RNA polymerase and the “core recognition element” counteract pausing. Science 344: 1285–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IX, Core LJ, Kwak H, Brady L, Bruzel A, McDaniel L, Richards AL, Wu M, Grunseich C, Lis JT, et al. 2014. RNA–DNA differences are generated in human cells within seconds after RNA exits polymerase II. Cell Rep 6: 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, et al. 2008. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 36: D13–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Clayton DA. 1996. RNA–DNA hybrid formation at the human mitochondrial heavy-strand origin ceases at replication start sites: an implication for RNA–DNA hybrids serving as primers. EMBO J 15: 3135–3143. [PMC free article] [PubMed] [Google Scholar]
- Yoza BK, Bogenhagen DF. 1984. Identification and in vitro capping of a primary transcript of human mitochondrial DNA. J Biol Chem 259: 3909–3915. [PubMed] [Google Scholar]
- Zollo O, Tiranti V, Sondheimer N. 2012. Transcriptional requirements of the distal heavy-strand promoter of mtDNA. Proc Natl Acad Sci 109: 6508–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}