

Abstract
Fasting elicits transcriptional programs in hepatocytes leading to glucose and ketone production. This transcriptional program is regulated by many transcription factors (TFs). To understand how this complex network regulates the metabolic response to fasting, we aimed at isolating the enhancers and TFs dictating it. Measuring chromatin accessibility revealed that fasting massively reorganizes liver chromatin, exposing numerous fasting-induced enhancers. By utilizing computational methods in combination with dissecting enhancer features and TF cistromes, we implicated four key TFs regulating the fasting response: glucocorticoid receptor (GR), cAMP responsive element binding protein 1 (CREB1), peroxisome proliferator activated receptor alpha (PPARA), and CCAAT/enhancer binding protein beta (CEBPB). These TFs regulate fuel production by two distinctly operating modules, each controlling a separate metabolic pathway. The gluconeogenic module operates through assisted loading, whereby GR doubles the number of sites occupied by CREB1 as well as enhances CREB1 binding intensity and increases accessibility of CREB1 binding sites. Importantly, this GR-assisted CREB1 binding was enhancer-selective and did not affect all CREB1-bound enhancers. Single-molecule tracking revealed that GR increases the number and DNA residence time of a portion of chromatin-bound CREB1 molecules. These events collectively result in rapid synergistic gene expression and higher hepatic glucose production. Conversely, the ketogenic module operates via a GR-induced TF cascade, whereby PPARA levels are increased following GR activation, facilitating gradual enhancer maturation next to PPARA target genes and delayed ketogenic gene expression. Our findings reveal a complex network of enhancers and TFs that dynamically cooperate to restore homeostasis upon fasting.
Transcriptional activation is facilitated by transcription factor (TF) binding at DNA regulatory elements (i.e., enhancers), eventually increasing the rate of gene transcription (Shlyueva et al. 2014). Active enhancers show three key characteristics: (1) an increase in specific histone marks such as H3K27 acetylation (Creyghton et al. 2010); (2) enrichment of TF binding events; and (3) increased chromatin accessibility, measured by hypersensitivity to DNase I digestion followed by sequencing (DNase-seq) (Shlyueva et al. 2014). Much of the accessible chromatin structure of enhancers is established during differentiation, allowing cell-type–specific gene expression (Thurman et al. 2012). However, accessibility of enhancers and enhancer marks also dynamically change in terminally differentiated tissues to accommodate signal-dependent gene expression (Grøntved et al. 2013, 2015; Ostuni et al. 2013). Therefore, the established enhancer landscape of the tissue is not static, but is rather dynamic and partly mediated by signal-activated TFs affecting chromatin accessibility (Voss and Hager 2014).
It is well established that some TF-bound DNA elements are relatively protected from DNase I digestion, presumably due to physical hindrance of DNase I by the TF (Galas and Schmitz 1978), leading to the presence of “TF footprints.” These footprints are identified genome-wide in DNase I hypersensitive sites (DHS) by calculating the relative decrease in DNase I digestion at TF motifs (Hesselberth et al. 2009; Sung et al. 2016). Thus, in addition to characterizing enhancers genome-wide, DNase-seq can provide a map of TF footprints within enhancers in an unbiased manner (i.e., not necessitating prior knowledge of an involved TF). Although the identification of TF footprints genome-wide is a valuable and promising technique, it is not amenable for all TFs because some of them do not leave a footprint (Sung et al. 2014).
The prevalent dogma assumes continuous TF binding from the point of upstream TF activation to the completion of gene induction, a time period ranging from minutes to hours. Widely used techniques such as chromatin immunoprecipitation followed by sequencing (ChIP-seq) have propagated this concept (Spitz and Furlong 2012). With a static model in mind, it was assumed that many TFs bind enhancers simultaneously and reside there for long periods of time. Yet, evidence from single-cell imaging experiments have accumulated to show many TFs reside on DNA for only a few seconds, cycling on and off repeatedly following activation (McNally et al. 2000; Bosisio et al. 2006; Sharp et al. 2006). Recently, single-molecule tracking (SMT) allowing imaging of single TFs in real-time was applied to examine TF residence time (Mueller et al. 2013). The handful of TFs tracked to date all exhibit DNA residence times in the ranges of seconds (Chen et al. 2014; Izeddin et al. 2014; Morisaki et al. 2014; Swinstead et al. 2016). This important aspect of TF biology has critical ramifications regarding gene expression. By frequently exchanging with DNA, TFs continuously reassess their environment and can respond quickly to changes in activating signals, enabling the TF to achieve a fine-tuned transcriptional response (Stavreva et al. 2009).
Dynamic binding endows TFs with the ability to indirectly facilitate the binding of other TFs. Two TFs that bind the same DNA element were shown not only to lack competition for DNA binding, but rather augment each other's binding through increasing DNA accessibility, a phenomenon termed “dynamic assisted loading” (Voss et al. 2011). Such an assisted loading relationship between TFs has been described in several systems (Biddie et al. 2011; Grøntved et al. 2013; Miranda et al. 2013; Madsen et al. 2014; Soccio et al. 2015; Zhu et al. 2015; Swinstead et al. 2016).
Although ample evidence points to the dynamic nature of enhancers and TF activity, how these events come into play following a physiological signal (e.g., hormones, nutritional status) in intact tissues is unexplored. The hepatic response to fasting is an optimal process to study TF dynamics in a physiologically relevant system for several reasons. First, the upstream hormonal and metabolic signals affecting the fasting response are well-defined (Corssmit et al. 2001). Second, the fasting response is heavily regulated at the transcriptional level. TFs from various TF families and classes were documented to regulate fasting-related genes: forkhead box proteins (FOXO1, FOXO3, FOXO4, FOXO6, FOXA2); cAMP responsive element binding proteins (CREB1, CREBH); CCAAT enhancer binding proteins (CEBPA, CEBPB); and nuclear receptors (glucocorticoid receptor [GR], peroxisome proliferator activated receptor alpha [PPARA], thyroid receptor, hepatocyte nuclear factor 4 alpha, retinoic acid receptor, estrogen-related receptor) (for review, see Goldstein and Hager 2015). Third, fuel production during fasting is highly dynamic and is temporally organized. The process of gluconeogenesis (i.e., de novo synthesis of glucose) plays a more prominent role in early fasting, whereas ketogenesis (i.e., the production of ketone bodies from fatty-acid oxidation-derived acetyl CoA) becomes the major fuel-producing pathway as fasting continues (Cahill 2006). Thus, during fasting transcriptional regulation occurs that dynamically adjusts to changes in metabolic needs (Bahar Halpern et al. 2015).
The TFs involved in regulating the fasting response have been studied mostly with a focus on a single TF's role. A systematic view of the TF network controlling the fasting response and the dynamics of this network is lacking. Thus, identity of the principal TFs that drive the fasting response, the crosstalk between them, and how they cooperate during fasting is largely unknown. Finally, the regulatory mechanism driving the temporal organization of fuel production (gluconeogenesis first with ketogenesis increasing later) is unexplored.
Results
Fasting massively reorganizes hepatic chromatin accessibility to expose ‘fasting-induced enhancers’
To globally evaluate the changes in chromatin landscape following fasting and to map fasting-responsive enhancers in mouse liver, we assessed chromatin accessibility using DNase-seq. We found substantial changes in chromatin accessibility for 3955 DHS sites (Fig. 1A,B), mostly occurring at intergenic regions (Supplemental Fig. S1A). Specifically, there were 1481 fasting-induced DHS sites and 2474 fasting-repressed DHS sites following 24 h of food deprivation (Fig. 1B). We examined whether DNase I hypersensitivity patterns are correlated with changes in H3K27 acetylation, a histone modification marking active enhancers (Creyghton et al. 2010). We plotted all fasting-responsive H3K27ac sites that overlap with DHS sites; fasting-induced H3K27ac sites are shown in red, and fasting-repressed H3K27ac sites are shown in blue. Nearly all fasting-induced H3K27ac sites overlapped with fasting-induced DHS; the same pattern was observed for fasting-repressed sites (Fig. 1B). To quantitatively measure a possible correlation between both enhancer features in terms of signal strength, we plotted the overlapping fasting-induced DHS/H3K27ac sites and found the two enhancer marks to be well correlated (Supplemental Fig. S1B). Moreover, during fasting, H3K27ac is higher in fasting-induced DHS sites compared to fasting-repressed DHS sites (Fig. 1C,D). Such broad changes in accessibility suggest extensive alterations in TF occupancy and binding dynamics at these sites during fasting, possibly leading to a highly modulated transcriptional program.
Figure 1.

Fasting substantially affects the hepatic chromatin landscape, exposing “fasting-induced enhancers.” (A) Genome browser tracks of the Cyp4a14 locus depicting increases in DNase I hypersensitivity, H3K27 acetylation, and RNA levels upon fasting (24 h; three animals were assayed in each group). (B) Volcano plot of all hepatic DNase I hypersensitive sites. DHS sites within the shaded areas show a significant change in accessibility upon fasting (twofold or greater, adjusted P-value ≤0.05). DHS sites overlapping with fasting-induced H3K27ac sites are depicted by a red X. These DHS sites tend themselves to be enhanced by fasting (mostly to the right). DHS sites overlapping with fasting-repressed H3K27ac sites are depicted by a blue X. These DHS sites tend themselves to be decreased by fasting (mostly to the left). (C,D) DHS (C) and H3K27ac (D) ChIP-seq tag density at fasting-induced enhancers in the fed and fasted states. (E) The number of DHS sites in the vicinity of fasting-induced genes (n = 1055) or unresponsive genes (random set, n = 1055) was plotted as a function of distance from gene transcriptional start site (TSS; plotted in 10-kb bins).
To examine the effect of these alterations in chromatin landscape on gene expression, we measured the changes in the hepatic transcriptome following fasting using RNA-seq (Supplemental Table S1). We found that fasting-induced DHS sites are proximally located to fasting-induced genes (Fig. 1E; Supplemental Table S2), and the DHS signal next to fasting-induced genes is increased following fasting compared to fasting-repressed genes (Supplemental Fig. S1C). Collectively, the data reveal a network of thousands of loci in mouse liver that contain classic enhancer features (DHS, H3K27ac, and proximity to regulated genes). Chromatin accessibility and histone marks are dynamically altered in these loci in response to fasting. Therefore, the loci with increased accessibility were termed “fasting-induced enhancers,” and the reciprocal loci were termed “fasting-repressed enhancers” (Fig. 1B, shaded areas).
Footprint depth of specific TFs, the accessibility proximal to them, and their binding at fasting-induced enhancers increase upon fasting
Having mapped the enhancer regions involved in the fasting response, we sought to identify TFs that regulate fasting-related transcription. Many TFs are known to regulate the fasting response. However, it is not clear which factors play a major role in the response and which factors have a supporting role (Goldstein and Hager 2015). Thus, instead of focusing on TFs from prior knowledge, we utilized an unbiased method to identify fasting-related TFs. We measured two parameters indicative of TF function: footprint depth at TF motifs and motif-flanking chromatin accessibility. An increase in footprint depth following fasting suggests increased TF binding, leading to increased protection from DNase I digestion (Gusmao et al. 2016), whereas increased flanking accessibility suggests TF-mediated opening of chromatin (Fig. 2A; Sherwood et al. 2014).
Figure 2.

An unbiased method for detecting differences in footprinting depth and motif-flanking accessibility reveals the key TFs that bind at fasting-induced enhancers following fasting. (A,B) The number of DNase I cuts (i.e., “cut count”) is collected at and around a motif (±100 bp from motif center) in all motif occurrences within DHS sites. This cut count profile is then compared between the fed and fasted conditions, and a delta value is given for each motif (x-axis in the scatter plot). Then, a log ratio between the observed and expected (due to DNase I cut bias) cut counts at the motif is calculated. This ratio is normalized to provide a reliable footprint depth value that is unaffected by surrounding hypersensitivity (Methods). The delta value between footprint depth in the fasted and fed conditions is then given for each motif (y-axis in the scatter plot). The bigger the size of the circle marking the motif, the deeper the footprint is in the fasted state. Cut count data for B were pooled from three replicates. When single replicates were used, the observed pattern was similar (Supplemental Fig. S1D). (C) Individual normalized footprint depth aggregate plots for the CEBP, CRE, and GRE motifs. Footprint depth is illustrated with horizontal dashed lines: (red) fasted; (blue) fed. (D) Scatter plot depicting changes in footprint depth and hypersensitivity of fasting-related motifs in total liver DHS sites compared to fasting-induced enhancers. (E) Extent of CEBPB, CREB1, and GR binding (measured by ChIP-seq tag density) at fasting-responsive enhancers in liver following fasting (24 h).
The difference between the fed and fasted conditions in terms of flanking accessibility and footprint depth for all known TF motifs in all DHS sites was determined (Fig. 2B). As expected, the footprint depth (y-axis) of most TF motifs as well as the flanking accessibility/hypersensitivity around them (x-axis) did not change following fasting, and the majority of TF motifs accumulated around the origin. However, four groups of outliers were observed (for statistical characterization of outliers, see Supplemental Methods; Supplemental Table S3). A group comprised of CEBP and CEBP-like motifs showed increased footprint depth upon fasting, with no increase in accessibility (“Group A”). A second group containing GRE and GRE-like motifs showed increased accessibility upon fasting with no change in footprint depth (“Group B”). A third group comprised of CRE/Jun/ATF motifs (all are highly similar) showed an increase both in footprint depth and accessibility upon fasting (“Group C”). Individual plots depicting changes in footprint depth (normalized to changes in flanking accessibility) at representative motifs from Groups A–C are shown in Figure 2C. The CEBP and CRE motifs show deeper footprints in the fasted condition, whereas the GRE motif shows no apparent footprint as previously reported (Sung et al. 2014, 2016).
Last, a group of STAT motifs showed a reciprocal pattern to Group C, whereby both footprint depth and accessibility were decreased upon fasting (“Group D”) (Fig. 2B). The decrease in both footprint depth and flanking accessibility around STAT motifs suggests a role for STAT proteins in repressing fasting-related pathways and is consistent with a recent report of STAT3-dependent suppression of gluconeogenesis (Nie et al. 2009).
We next explored the possibility that footprint depth and flanking accessibility are more prominently increased at fasting-induced enhancers compared to total DHS sites. Indeed, at each relevant motif, the footprint became deeper and the surrounding more accessible at fasting-induced enhancers compared to total DHS sites (Fig. 2D). An increase in these parameters suggests amplified binding of associated TFs at those motifs upon fasting. To explore this hypothesis, we performed ChIP-seq following fasting of the three major TFs known to bind these motifs in liver and to be involved in the fasting response—CREB1, GR, and CEBPB. We compared TF binding at fasting-altered enhancers in the fed and fasted states. Binding of all three TFs was significantly enriched at fasting-induced enhancers compared to fasting-repressed enhancers. Importantly, the three factors showed markedly increased binding at these enhancers following fasting compared with the fed state (Fig. 2E). In addition to increased binding of CREB1 at fasting-induced enhancers, we performed ChIP-seq for the fasting-activated form of CREB1 (Ser133-phosphorylated CREB1, or pCREB1) (Supplemental Fig. S2A). Similarly to CREB1, we found pCREB1 to bind favorably at fasting-induced enhancers during fasting (Supplemental Fig. S2B).
To examine whether the increases in footprint depth and flanking accessibility are indeed directly associated with TF binding, we partitioned genome-wide motif occurrences to TF-bound motifs and unbound motifs (in which a ChIP-seq peak was not called). Flanking accessibility as well as footprint depth at the relevant motifs increased in TF-bound motifs compared to total motifs. In contrast, all unbound motifs presented only marginal increases in the two parameters following fasting, and most unbound motifs were not identified as outliers according to our analysis (Supplemental Fig. S2C; Supplemental Table S3).
In line with the findings in Figure 2, de novo motif analysis revealed that the most highly enriched TF binding motif in fasting-repressed enhancers was STAT. Accordingly, the most highly enriched TF binding motifs in fasting-induced enhancers were CEBP, CREB1, and GR, together with the binding motif for PPARA (Supplemental Fig. S2D). The lack of a footprint at PPAR motifs and only mild changes in accessibility around the motif (Fig. 2B; Supplemental Fig. S2E) explains its absence from the four outlier groups described in Figure 2B. Nonetheless, PPARA did show increased binding at fasting-induced enhancers compared to fasting-repressed enhancers (Supplemental Fig. S2F). Taken together, these data indicate that a particular, context-dependent set of motifs show a deeper footprint in response to fasting, which is associated with increased TF binding. However, some TFs do not leave a detectable footprint but still bind specific regions in a context-dependent manner (i.e., during fasting), leading to chromatin opening at their vicinity. These findings reveal a complex transcriptional response to fasting comprised of several key TFs, raising the possibility of spatial and/or temporal crosstalk between these fasting-related TFs.
Gene regulation during fasting is governed by two TF modules
CREB1 and GR are activated by the major fasting-related hormones glucagon and corticosterone, respectively, whereas PPARA is activated by fatty acids, whose levels increase in hepatocytes during fasting. CEBPB is mostly considered a critical factor, without which the fasting response is significantly impaired, but which is constitutively active in contrast to the other three TFs (Goldstein and Hager 2015). Thus, downstream analyses aiming at deciphering the possible crosstalk between the key TFs would focus on the three signal-activated TFs. We first evaluated gene expression following these fasting-related signals. We treated primary mouse hepatocytes with combinations of glucagon, corticosterone, and a PPARA agonist (WY-14643) and examined fasting-induced genes from our RNA-seq analysis related to gluconeogenesis, FAO, and ketogenesis (Supplemental Table S1). In the event in which one TF cooperates with another TF in increasing transcription, we would expect a synergistic effect, i.e., an increase in transcription in the combined treatment that is greater than the sum of each treatment alone. In contrast, when two TFs independently regulate transcription, the combined treatment should amount only to the sum of two single treatments. Glucagon was able to induce known gluconeogenic genes; this induction was synergized by corticosterone but was either unaffected (Ppargc1a) or even slightly reduced (Tat, Pck1) by PPARA. In contrast, GR and PPARA had a synergistic effect on FAO/ketogenic genes with no substantial effect on these targets by glucagon (Fig. 3A; Supplemental Fig. S3A).
Figure 3.

The transcriptional response to fasting in liver is comprised of two TF modules. (A) Nascent RNA levels of fasting-induced genes in primary hepatocytes following a 3-h treatment with different combinations of glucagon (gluc), corticosterone (cort), and WY-14643 (wy). (B) Time course of fasting-induced genes shows an early induction of gluconeogenic genes and a later induction of FAO/ketogenic genes in liver. (C) Glucose production in primary hepatocytes following treatment with different combinations of glucagon, corticosterone, and WY-14643. (D) Nascent RNA levels of Ppara following corticosterone treatment (3 h) in primary hepatocytes. (E) Nascent RNA levels of Pdk4 in primary hepatocytes following 1, 3, and 8 h of treatment with different combinations of WY-14643 and corticosterone: (*) statistical significance (P ≤ 0.05) compared to a nontreated sample (NT) in each time point. (F) Time course of serum corticosterone and β-hydroxybutyrate levels during fasting: (*) statistical significance (P ≤ 0.05) compared to control values (fed). (n.s.) Not significant.
To evaluate these two expression patterns in mice, animals were fasted for varying durations, and gene expression levels were examined. Induction of gluconeogenic genes was evident as soon as 4 h following food removal in line with early activation of gluconeogenesis (Cahill 2006). In contrast, expression of FAO/ketogenic genes only increased at prolonged fasting (24 h) when ketones are produced (Fig. 3B,F). To correlate these gene expression patterns with fuel production, we measured glucose produced by primary hepatocytes in the presence of fasting-related signals. As expected, glucagon increased glucose production. Corticosterone was not able to increase glucose production alone but significantly augmented it in the presence of glucagon. Activating PPARA had no effect on glucose production under any circumstance (Fig. 3C). These observations are in line with the above findings showing the involvement of GR and CREB1, but not PPARA, in a gluconeogenic gene program. These data suggest that the transcriptional response to fasting is governed by two TF modules. One module is mediated by CREB1-GR crosstalk, resulting in synergistic expression of gluconeogenic genes at short fasting periods. The other module, brought about by GR-PPARA crosstalk, leads to increased FAO/ketogenic gene expression as fasting persists and gluconeogenic precursors are depleted.
One possible route by which the GR-PPARA module operates is through a “TF cascade,” namely, one TF increases the level of the second TF thereby indirectly augmenting its activity. This option seemed plausible due to GR being able to induce the expression of the gene encoding PPARA in primary hepatocytes (Fig. 3D), in agreement with previous studies (Lemberger et al. 1994; Steineger et al. 1994). Furthermore, GR bound at a fasting-induced enhancer within the Ppara gene body upon food removal, suggesting direct regulation of Ppara by GR during fasting (Supplemental Fig. S3B). Evidence to support the TF cascade model came from a time-course experiment wherein the transcript levels of FAO/ketogenic genes were measured. Nascent transcripts rather than mature RNA levels were measured to reflect transcriptional activation and not post-transcriptional events (to achieve this, we amplified regions spanning intron–exon junctions). We determined that the levels of FAO/ketogenic genes increase as early as 1 h of PPARA activation. At this time point, cotreatment with corticosterone did not augment induction. However, following 3 h of cotreatment, transcript levels increased significantly, whereas the levels of transcript in the single treatment condition began to decrease. This pattern was even more pronounced after 8 h of treatment, and in several examined genes, it was only apparent at that time point (Fig. 3E; Supplemental Fig. S3C). The expression pattern identified here is consistent with a TF cascade model in which GR gradually affects PPARA activity only at later time points, after PPARA gene induction and protein synthesis have been initiated. Although the primary induction in the single treatment wanes quickly, the continuous increase in PPARA in the presence of corticosterone prolongs the effect of WY-14643. However, we cannot exclude that some of the increased PPARA activity is due to a corticosterone-dependent increase in endogenous PPARA ligands.
The timeline of this TF cascade is strengthened by evidence from measurements in fasted mice. Corticosterone levels started to increase at midterm fasting (10 h), consistent with previous reports (Champy et al. 2004). Conversely, the main ketone body, β-hydroxybutyrate, was not increased until 24 h of fasting accompanied by a further increase in corticosterone and induction of PPARA transcript levels (Fig. 3F; Supplemental Table S1). Thus, corticosterone levels rose several hours prior to ketogenesis and prior to the increase in the levels of PPARA, the major ketogenic TF. We then evaluated enhancer activity using H3K27ac ChIP-seq through the course of fasting. The increase in H3K27ac signal at fasting-induced enhancers harboring a PPARA motif became more apparent and significant at long-term fasting (24 h) compared with midterm fasting (10 h), suggesting increased enhancer activity in prolonged fasting (Supplemental Fig. S3D). Accordingly, the increase in H3K27ac signal next to PPARA target genes markedly increased 24 h following food removal, with only modest increases at 10 h of fasting (Supplemental Fig. S3E). Taken together, the findings presented in Figure 3 delineate the progression of the transcriptional response to fasting from a CREB1-GR-based gluconeogenic program to a GR-PPARA FAO/ketogenic program.
GR assists the loading of CREB1 at multiple genomic loci to augment gene expression
To examine the modus operandi of the CREB1-GR module, we performed a time-course experiment measuring the nascent transcript levels of CREB1-GR synergistic genes following glucagon alone or in combination with corticosterone. The rapid gene induction (1 h following glucagon treatment) resembled the pattern observed in the PPARA target gene in the single treatment (Fig. 3E). However, in stark contrast to the delayed synergistic effect seen in the GR-PPARA module, we observed a rapid synergistic effect between glucagon and corticosterone. Transcript levels synergistically rose 1 h following treatment. In some cases, transcript levels began to decrease already by 3 h (Fig. 4A; Supplemental Fig. S4A). This observation suggests that CREB1 and GR directly cooperate to regulate transcription. To examine the nature of that cooperation, we performed ChIP-seq of CREB1 in primary hepatocytes following short (1 h) single and dual treatments. As expected, glucagon-dependent CREB1 binding events were observed next to Tat, a CREB1-regulated gene (Fig. 4B). Remarkably, cotreatment with corticosterone (which leads to synergized Tat expression as shown in Fig. 3A) led to a major increase in CREB1 binding upstream of Tat. Moreover, these sites (as well as others) became more accessible following fasting, suggesting similar events take place in the liver during fasting (Fig. 4B; Supplemental Fig. S4B). Similarly to gene expression and glucose production patterns, corticosterone did not significantly affect CREB1 binding in the absence of glucagon (Supplemental Fig. S4C). To determine whether this is a global phenomenon, we performed several genome-wide analyses. First, comparing the total number of CREB1 binding sites between the single and dual treatments revealed that the dual treatment doubled CREB1 binding events (3452 sites in glucagon-treated cells vs. 7081 sites in the dual treatment group) (Fig. 4C). In addition to the absolute number of sites, cotreatment led to a twofold increase in CREB1 binding intensity at 39% of sites, whereas only a negligible 0.3% of sites showed decreased CREB1 binding (Fig. 4D). Furthermore, corticosterone-increased CREB1 sites generally show more CREB1 binding than corticosterone-independent sites, affirming the augmenting effect of corticosterone on CREB1 binding (Fig. 4E,F).
Figure 4.

Corticosterone increases the number of CREB1 binding sites and CREB1 binding intensity. (A) Nascent RNA levels of Ppargc1a in primary hepatocytes following 1, 3, and 8 h of treatment with different combinations of glucagon (gluc) and corticosterone (cort): (*) statistical significance (P ≤ 0.05) compared to a nontreated sample (NT) in each time point. (B) Genome browser tracks of the Tat locus depicting increases in CREB1 binding following glucagon treatment (1 h) and cotreatment with corticosterone (1 h) as well as increased accessibility following either fasting or GR activation by dexamethasone (dex). Adrenalectomized mice were treated with dex (1 mg/kg) for 1 h, DNase-seq data were generated and described in Grøntved et al. (2013). (C,D) Cotreatment with glucagon and corticosterone leads to an increase in the number (C) and intensity (D) of CREB1 binding sites. (E,F) Corticosterone-increased CREB1 binding sites show more CREB1 binding than corticosterone-unaffected sites (measured by ChIP-seq tag density). (**) Statistical significance (P ≤ 0.0001).
Notably, some CREB1-bound loci were unaltered by corticosterone. In accordance, glucagon-induced gluconeogenic genes adjacent to these sites were unaltered by corticosterone (Supplemental Fig. S5A–C). This suggests that GR affects glucagon-dependent transcription only on a subset of enhancers, where it assists CREB1 loading, and does not indiscriminately augment CREB1 DNA binding activity. To address that possibility, we performed ChIP-seq of GR in primary hepatocytes. Indeed, GR colocalized with CREB1 at corticosterone-increased CREB1 sites at the Tat locus (Fig. 4B). Globally, GR binding was significantly increased in the vicinity of the 3678 CREB1 sites augmented by corticosterone (i.e., the sites unique to the dual treatment). This was evident both in terms of binding intensity (Fig. 5A,B) and in the number of GR binding events adjacent to CREB1 sites (Fig. 5C). We also found that in liver, corticosterone-increased CREB1 sites are opened following GR activation by dexamethasone (a synthetic GR agonist) more readily than unaffected sites as measured by DNase-seq (Figs. 4B, 5D,E). Linking this effect of GR to fasting, we observed that fasting-induced enhancers also become more accessible by dexamethasone (Fig. 5F,G).
Figure 5.

GR binds at corticosterone-increased binding sites and increases their accessibility. (A,B) Corticosterone-increased CREB1 binding sites show more GR binding than corticosterone-unaffected sites (measured by ChIP-seq tag density). (**) Statistical significance (P ≤ 0.0001). (C) The number of GR peaks in the vicinity of cort-unaffected or cort-increased CREB1 peaks was plotted as a function of distance from CREB1 peak center (plotted in 50-bp bins). (D,E) Corticosterone-increased CREB1 binding sites show more DNase I hypersensitivity than corticosterone-unaffected sites in livers from adrenalectomized mice treated with dexamethasone (dex, 1 mg/kg) for 1 h, DNase-seq data were generated and described in Grøntved et al. (2013) (measured by DNase-seq tag density). (**) Statistical significance (P ≤ 0.0001). (F,G) GR increases DNase I hypersensitivity at fasting-induced enhancers (measured by DNase-seq tag density). Adrenalectomized mice were treated with dexamethasone (dex, 1 mg/kg) for 1 h, DNase-seq data were generated and described in Grøntved et al. (2013). (**) Statistical significance (P ≤ 0.0001).
Furthermore, we isolated corticosterone-increased CREB1 sites with the highest GR ChIP-seq signal (n = 1230) and analyzed them for motif enrichment. In further support of an assisted loading model, the top motif was the CRE (P = 10−161) with the GRE ranking second (P = 10−152) compared to either total CREB1 binding sites (n = 7130), where CRE was the top motif (P = 10−642) and GRE only ranking 24th (P = 10−78), or to a random set of enhancers (n = 1230) where GRE was not enriched altogether. These observations further attest to GR's role in opening fasting-induced enhancers and facilitating CREB1 binding and are in agreement with the finding that among all known motifs, the highest change in accessibility following fasting was documented around the GR motif (Fig. 2B).
Next, we measured transcript levels in the presence of an inhibitor of GR trans-activation (RU486). The synergistic effect corticosterone has on glucagon-induced genes (Tat, Ppargc1a, Pck1) was completely abolished upon RU486 treatment. Conversely, RU486 did not affect glucagon-induced genes that show no corticosterone-dependent increase in CREB1 binding or synergistic gene expression (G6pc, Nr4a1) (Supplemental Fig. S5D). Thus the synergistic effect of corticosterone is mediated by GR and not mineralocorticoid receptor, a different corticosterone-activated TF that is not inhibited by RU486 (Cadepond et al. 1997).
Collectively, the data suggest a mode of action, whereby GR augments CREB1 binding through increasing enhancer accessibility and allowing synergistic gene expression. This dynamic assisted loading mechanism is likely independent of physical interaction between the two TFs because it is enhancer-selective and does not encompass the entirety of CREB1 binding sites. Moreover, GR-dependent increases in chromatin accessibility are abundant in fasting-induced enhancers, causally linking GR to chromatin rearrangements during fasting.
GR increases the number of bound CREB1 molecules and their DNA residence time
The preceding observations clearly point to a facilitating effect of GR on CREB1 binding, CREB1-dependent gene expression, and glucose production. To examine the real-time dynamics of this relationship, we tracked CREB1 movement at the single-molecule level. We were unable to image single molecules in primary hepatocytes owing to technical reasons (Supplemental Methods). Thus, we utilized HepG2 cells, which are of hepatic origin and preserve many hepatic-related features including glucagon- and corticosterone-induced gene expression profiles. Using Halo-Tag-fused CREB1 and the JF549 dye (which covalently binds to the HaloTag) (Grimm et al. 2015), we identified and tracked single CREB1 molecules using highly inclined laminated optical sheet (HILO) illumination (Tokunaga et al. 2008). Quantitative analysis of large track sets (Supplemental Fig. S6A) revealed that about half of CREB1 molecules are chromatin-bound at a given time. Within that bound population of molecules, we observed a continuum of exponentially distributed DNA residence times. A single exponential model was insufficient to fit the data, whereas a two component model fits the data with high precision (Supplemental Fig. S6B). This indicates that the DNA-bound population of molecules is divided into two mathematically distinguished subpopulations, or two fractions: a short-lived (Tns, fast bound) and a longer-lived (Ts, slow bound) fraction. Previous reports suggest that the slow fraction of molecules represents specific binding events associated with enhancers or promoters, whereas the fast fraction defines nonspecific binding to chromatin or a DNA scanning mechanism (Chen et al. 2014; Morisaki et al. 2014; Swinstead et al. 2016).
To examine a potential effect of GR on the binding properties of CREB1, we cotreated cells with forskolin (a CREB1 agonist) and dexamethasone and tracked single CREB1 molecules in live cells. These agonists are commonly used in liver cell lines to achieve maximal activity of these two TFs (Goldstein et al. 2013). Indeed, forskolin led to CREB1 phosphorylation as well as to induction of CREB1 target genes, whereas dexamethasone led to GR nuclear localization and induction of GR target genes (Supplemental Fig. S6C–E). Both the residence time of CREB1 molecules on DNA and the amount of slow-bound molecules increased upon forskolin treatment. Remarkably, cotreatment with dexamethasone led to a further increase in both DNA residence time and the DNA-bound fraction of CREB1 molecules (Fig. 6A,B; Supplemental Fig. S6A,B; Supplemental Movies S1–S3). These real-time observations portray GR as a facilitating factor to productive CREB1 binding and are in agreement with our findings showing an assisted loading mechanism for the two TFs.
Figure 6.
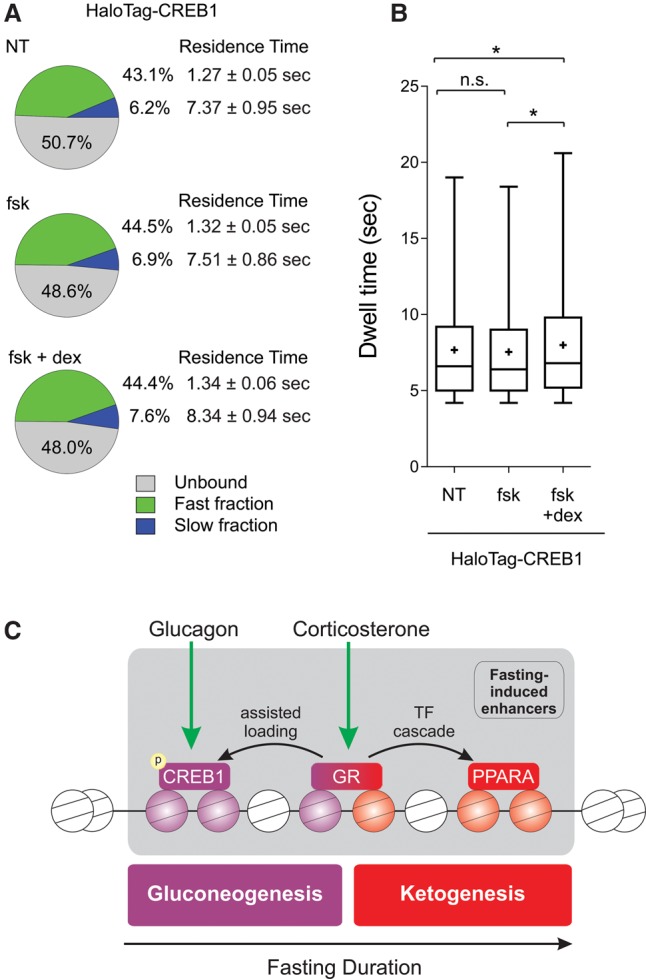
GR increases the number and residence time of CREB1 molecules. (A) Summary of single-molecule tracking data. The percentage of the unbound, fast-bound, and slow-bound fractions as well as each fraction's average residence time is depicted under different treatments: (fsk) forskolin; (dex) dexamethasone. (B) Box plot depicting dwell time distributions of CREB1 molecules under different treatments. (*) Statistical significance (P ≤ 0.05) compared to indicated sample. (n.s.) Not significant. (C) A model for hepatic TF dynamics during fasting. The liver is exposed to glucagon in early fasting, leading to CREB1 activation by phosphorylation. At midterm fasting, GR is activated by increasing levels of corticosterone leading to two trajectories: First, GR assists the loading of CREB1 onto gluconeogenic enhancers resulting in synergized gluconeogenic gene expression and glucose production; and second, GR induces the gene level of PPARA which, as fasting persists, promotes a FAO/ketogenic gene program.
Discussion
Mammals have adapted to periods of nutrient deprivation by activating fuel-producing pathways such as gluconeogenesis and ketogenesis. Both pathways are supported by ATP produced in liver by FAO, which also provides precursors for ketogenesis (Goldstein and Hager 2015). A major conclusion drawn from our observations is the significant contribution of enhancer and TF dynamics to these frequently occurring metabolic events. Upon fasting, a global effect on chromatin landscape was evident with thousands of sites altering their accessibility to TFs, resulting in a prominent effect on the transcriptome. This finding expands beyond the common tendency to focus only on the induction of a few gluconeogenic genes (mainly Pck1 and G6pc) as the main events in fasting-related transcription.
We used several unbiased genomic methodologies to isolate key fasting-related TFs from the chromatin landscape data. We developed a program for detecting differences in TF footprint depth and motif-flanking accessibility. Successful attempts at deriving TF activity from footprint depth (Gusmao et al. 2016) or flanking accessibility (Sherwood et al. 2014) were reported. Our algorithm combines these two attributes to provide a bivariate prediction for TF activity. Using this program, we found motifs with increased protection from DNase I (i.e., deeper footprint) and higher flanking accessibility upon fasting. Footprint detection is a powerful approach because it does not rely on prior knowledge of a specific TF or of a subset of genomic loci. Moreover, this method measures biological properties that are directly indicative of TF function. However, the main pitfall is that not all specifically bound TFs protect motif DNA from DNase I digestion (Sung et al. 2014). Thus, the program we developed also measures changes in motif-flanking accessibility that are thought to result from secondary events following TF binding (e.g., recruitment of chromatin-modifying machinery and histone modifying enzymes) (Voss and Hager 2014). Indeed, using this program, we were able to detect GR, a key fasting-related TF that does not leave a footprint. Combining these two TF-derived parameters provides a motif-wide map of key TFs without relying on prior knowledge of TFs involved in a given pathway.
Additionally, we found enriched motifs within fasting-induced enhancers. Motif enrichment is also a useful approach, but it is weakened by the need to analyze only a subset of genomic loci (e.g., fasting-induced enhancers) rather than use the entire enhancer landscape as was done in the presented algorithm. These two complementary approaches independently detected three key TFs—CEBPB, CREB1, and GR—focusing our attention on them. We also included PPARA (which was only detected through motif enrichment) for further analyses due to its well established role in the hepatic fasting response. These unbiased approaches provided evidence for the involvement of these TFs in the global response to fasting. Attention to these factors is further supported by their strong tendency to bind fasting-induced enhancers and their documented role in certain aspects of the fasting response (Goldstein and Hager 2015).
Using an ex vivo approach to examine fasting-induced genes, we found two transcription-regulating modules dominated by GR-CREB1 crosstalk (in the gluconeogenic module) and GR-PPARA crosstalk (in the FAO/ketogenic module). The FAO/ketogenic module operates through a TF cascade, whereby GR induced the PPARA gene. This leads to a gradual increase in ketogenic enhancer activity and late induction of ketogenic genes, resulting in activation of ketogenesis only in prolonged fasting. In contrast, the gluconeogenic module functions through a rapid and dynamic assisted loading mechanism. Upon activation by corticosterone, GR binds near CREB1 binding sites, making the enhancer chromatin structure more accessible. This results in increased CREB1 binding, synergistic gene induction, and higher glucose production.
Direct physical interaction between TFs is the commonly offered model for synergistic gene expression. Our findings would argue against this model. Synergistic gene expression was only evident in a subset of gluconeogenic genes, and GR-dependent CREB1 assisted loading was only apparent in a subset of CREB1 binding sites. In line with the genomic data, the SMT findings show that activation of GR affected only a fraction of CREB1 molecules rather than shifting the binding kinetics of the entire population of CREB1 molecules. The fact that many CREB1 molecules are unaffected by GR explains the mild (but significant nonetheless) effect of GR on CREB1 real-time dynamics. Taken together, we provide evidence from molecular, biochemical, genomic, and live-cell imaging approaches to describe a dynamic assisted loading mechanism resulting in synergistic gene expression and culminating in increased glucose production.
From a metabolic standpoint, our findings delineate the transcriptional regulatory events underlying temporally organized fuel production during fasting. Glucagon is secreted promptly after the decrease of blood glucose levels, leading to CREB1 activation (Altarejos and Montminy 2011). Upon subsequent secretion of corticosterone at midterm fasting, GR is activated and augments a CREB1-dependent gluconeogenic gene program. Additionally, GR induces PPARA expression and boosts a PPARA-dependent ketogenic gene program that is at play during longer fasting periods (Fig. 6C). In summary, our findings shed light on the intricate regulation of fuel production by hormonal signals and reveal the major role of enhancer and TFs dynamics in homeostatic gene regulation.
Methods
Animals and fasting procedure
Male C57BL/6 mice (8–12 wk old) purchased from Charles River were maintained according to NIH guidelines under 12-h light–dark cycles (lights on at 6 a.m.). In fasting experiments, food was removed at 6 a.m. Excluding fasting time-course experiments, the fed group was fed ad libitum. For collection of liver tissue, mice were sacrificed by cervical dislocation.
For fasting time-course experiments, each group contained at least six mice; error bars in qPCR represent SEM between biological replicates (the SEM is calculated for the normalized average cycle threshold (ct) value of two technical replicates per mouse). Control mice were fasted and then re-fed for 2 h. For collection of liver tissue, mice were sacrificed by cervical dislocation. For collection of blood metabolites/hormones, mice (n = 6, error bars in Fig. 1F represent SD) were anesthetized using isoflurane, and blood was extracted by cardiac puncture. Blood was left to clot at room temperature for 30 min followed by incubation at 4°C overnight. Serum was isolated after centrifugation of 20,000 relative centrifugal force for 1 min at 4°C. Glucose was measured from 2 μL of serum using a OneTouch glucometer (BD). β-hydroxybutyrate was measured from 3 μL of serum using β-hydroxybutyrate assay kit (Sigma, MAK041). Corticosterone was measured from 15 μL of serum using RIA (Ani-Lytics).
Isolation of primary mouse hepatocytes, reagents, and Western blot procedure are described in the Supplemental Methods.
DNase I hypersensitivity assay
DNase-seq was essentially performed as previously described (Grøntved et al. 2012). Briefly, isolated nuclei from fresh livers were treated with 40 units/mL DNase I and DNA fragments of 100–500 bp were purified using sucrose gradients. DNA was precipitated and reconstituted in nuclease free H2O. Each group contained three mice.
Hepatic glucose production
One day following isolation, primary hepatocytes (1.7 × 105 cells/well in 12-well plates) were incubated with “starvation media” (DMEM without glucose, pyruvate, glutamine, or phenol red; Gibco, A14430-01) for 24 h. Then, starvation media containing pyruvate (5 mM) and glutamine (5 mM) was added along with glucagon, corticosterone, and WY-14643 as described in Figure 3C legend. Following 24 h, 40 µL of media was sampled, and glucose was measured using the glucose oxidase colorimetric method according to the manufacturer's instructions (Sigma, GAGO20). The experiment was replicated three independent times. Error bars represent SD of three technical replicates.
Sequencing, initial data analysis, and peak calling
Chromatin immunoprecipitation is described in the Supplemental Methods. Sequence reads (50-mer) were generated for ChIP-seq and DNase-seq experiments on the Illumina HiSeq 2000 and Illumina NextSeq 500 platforms at the Advanced Technology Center (ATC), National Cancer Institute (NCI), and the tags were uniquely aligned to the mouse reference genome (NCBI37/mm9 assembly). Regions of enriched tags known as “hotspots” (i.e., peaks) were determined using the algorithms and methods previously described with minor modification (http://sourceforge.net/projects/dnase2hotspots) (Baek et al. 2012). Replicate concordant hotspots have been calculated between replicates. For each individual replicate, hotspot regions were called with a minimal threshold (Z-score >2), and initial replicate concordant regions were defined as the intersection of hotspots between replicates. Then, the tags from each replicate are combined, and hotspots from the pooled tags are determined using a false discovery rate (FDR) of 0%. Finally, replicate concordant hotspots were defined as the intersection of the FDR-thresholded set of hotspots from the merged data and the initial replicate concordant regions.
Tag density profiles (used for genome browser screenshots)
We constructed a tag density profile of the data by extending each mapped read to the 150-bp length into the 3′ direction relative to that strand and counted the distribution of tag counts over the genome. The scale factor is given by 10 million/the total number of nonmitochondrial reads. By multiplying the scale factor, the normalized tag density profiles were obtained.
Differential DHS and H3K27ac sites
Differentially regulated DHS and H3K27ac sites were identified using DESeq (Anders and Huber 2010) from three biological replicates (fold change ≥2, adjusted P-value ≤0.05) (Supplemental Table S5). Normalization was performed using the standard normalization provided in the DESeq Bioconductor package (version 1.22.1) by calling the estimateSizeFactors and sizeFactors functions, and based on the hypothesis that a majority of genes/sites are not differentially expressed, a scaling factor is computed for each lane, and raw read counts were multiplied by the associated scaling factor.
Analysis of corticosterone-affected CREB1 binding sites
Two replicates of glucagon-treated cells and dual treated cells (glucagon+corticosterone) were used for CREB1 ChIP-seq (peak maximal densities were well correlated between replicates as determined with linear regression: R2 = 0.8217 and 0.8586, respectively). Corticosterone-affected CREB1 sites were determined by a twofold change or higher in the site's maximal density value in the gluc+cort condition compared to the gluc condition (Supplemental Table S4). CREB1 binding sites with maximal tag density lower than 30 were excluded.
Differentially regulated genes
RNA isolation and quantitative real-time PCR (qPCR) are detailed in the Supplemental Methods. Significant change of RNA expression was analyzed by Cufflinks and CuffDiff (Trapnell et al. 2010, 2013) using three replicates (fold change cutoff ≥1.5, Q-value ≤0.05) (Supplemental Table S1).
Scatterplots
DHS/H3K27ac scatterplot
Commonly enriched sites of DHS and H3K27ac were defined as DHS hotspot sites that are located within 1 kb of a H3K27ac site. The values of the x-axis are the maximum tag counts in commonly enriched DHS sites, and the corresponding y-axis values are the maximum tag count of the nearest H3K27ac site. Tag counts were normalized to 10 million reads. The R2 value was calculated by Spearman correlation.
CREB1 binding scatterplot
The fold change (glucagon+cort/glucagon) in maximal tag density is plotted for each CREB1 hotspot.
Footprint depth calculation, flanking accessibility, and the resulting scatterplot are described in the Supplemental Methods.
Volcano plot
The x-axis shows the log2 fold change between the fasted and fed conditions of the tag count of the sites. The y-axis shows the negative log10 transformed adjusted P-values of the t-test. DHS sites overlapping H3K27ac sites are color coded as described in Figure 1B. Overlap parameters: The maximal density of a H3K27ac site is 1 kb or closer to the maximal density of a DHS site.
Motif analysis, genome annotation, and binned genomic distance analysis
Analyses were performed using HOMER (http://homer.salk.edu/homer/) (Heinz et al. 2010). The analysis described in Supplemental Figure S2 is de novo motif analysis, and all enriched motifs are presented. The known motif analysis described in Figure 5 was done on three groups of sites: (1) a group of corticosterone-increased CREB1 sites overlapping with GR binding sites with the highest GR signal (maximal tag density ≥10; n = 1230, “Assisted loading cluster”) (Supplemental Table S4); (2) total CREB1 sites (n = 7130); and (3) a random set of DHS sites (n = 1230). The distance of GR peaks from CREB1 peaks and DHS sites from TSS was also done using HOMER.
Box plots and aggregation plots
Box plots represent sequenced tag density (per bp) around (±100 bp) the center of fasting-altered DHS sites or ChIP-seq peak or around (±50 kb) fasting-altered genes (generated using HOMER). The mean is denoted by a “+” sign. SMT box plots represent the distribution of individual dwell times for each single molecule in the slow long-lived fraction. The distribution is used to calculate the residence time of CREB1. Outliers, representing molecules with residence time over the one-and-a-half times the interquartile range of the box plot, are not shown.
Aggregation plots represent average sequenced tag density (10-bp bins) around (±500 bp) the center of fasting-altered DHS sites or ChIP-seq peak (generated using HOMER).
Single-molecule tracking and the resulting box plot are described in the Supplemental Methods.
Statistics
Asterisks denote statistical significance (P ≤ 0.05) (double asterisks, P ≤ 0.0001) compared to control or to indicated sample as determined by an unpaired, two-tailed t-test (n.s. = not significant). P-value in the SMT dwell time comparison represents a two-sample Kolmogorov Smirnov test defined by the brackets shown above the box plots in Figure 6B.
Published data sets
The GR-dependent DHS raw data were obtained from GEO (GSE46047) (Grøntved et al. 2013). PPARA ChIP-seq raw data were obtained from GEO (GSE35262) (Boergesen et al. 2012).
Study approval
All animal procedures were approved by the Animal Users and Care Committee, the National Cancer Institute, National Institutes of Health.
Data access
All high-throughput sequencing data from this study are summarized in Supplemental Table S6 and have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE72087.
Supplementary Material
Acknowledgments
We thank Lars Grøntved, Tom Johnson, Lyuba Varticovski, Tom Misteli, and Irwin Arias for their comments and help. This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), the National Cancer Institute (NCI), and the Center for Cancer Research (CCR). V.P. was supported, in part, by the Sigrid Jusélius Foundation.
Author contributions: I.G. conceived and performed experiments, performed bioinformatic analyses, and wrote the manuscript. S.B. performed bioinformatic analyses. D.M.P. and V.P. performed SMT experiments and analyses. E.E.S. helped in bioinformatic analyses. G.L.H. helped in experimental design and wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.212175.116.
References
- Altarejos JY, Montminy M. 2011. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 12: 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S, Sung MH, Hager GL. 2012. Quantitative analysis of genome-wide chromatin remodeling. Methods Mol Biol 833: 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar Halpern K, Tanami S, Landen S, Chapal M, Szlak L, Hutzler A, Nizhberg A, Itzkovitz S. 2015. Bursty gene expression in the intact mammalian liver. Mol Cell 58: 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, et al. 2011. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43: 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boergesen M, Pedersen TÅ, Gross B, van Heeringen SJ, Hagenbeek D, Bindesbøll C, Caron S, Lalloyer F, Steffensen KR, Nebb HI, et al. 2012. Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor α in mouse liver reveals extensive sharing of binding sites. Mol Cell Biol 32: 852–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosisio D, Marazzi I, Agresti A, Shimizu N, Bianchi ME, Natoli G. 2006. A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-κB-dependent gene activity. EMBO J 25: 798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadepond F, Ulmann A, Baulieu EE. 1997. RU486 (mifepristone): mechanisms of action and clinical uses. Annu Rev Med 48: 129–156. [DOI] [PubMed] [Google Scholar]
- Cahill GF Jr. 2006. Fuel metabolism in starvation. Annu Rev Nutr 26: 1–22. [DOI] [PubMed] [Google Scholar]
- Champy MF, Selloum M, Piard L, Zeitler V, Caradec C, Chambon P, Auwerx J. 2004. Mouse functional genomics requires standardization of mouse handling and housing conditions. Mamm Genome 15: 768–783. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang Z, Li L, Chen BC, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, et al. 2014. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156: 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corssmit EP, Romijn JA, Sauerwein HP. 2001. Review article: Regulation of glucose production with special attention to nonclassical regulatory mechanisms: a review. Metabolism 50: 742–755. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galas DJ, Schmitz A. 1978. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res 5: 3157–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I, Hager GL. 2015. Transcriptional and chromatin regulation during fasting—the genomic era. Trends Endocrinol Metab 26: 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I, Yizhak K, Madar S, Goldfinger N, Ruppin E, Rotter V. 2013. p53 promotes the expression of gluconeogenesis-related genes and enhances hepatic glucose production. Cancer Metab 1: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. 2015. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 12: 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grøntved L, Bandle R, John S, Baek S, Chung HJ, Liu Y, Aguilera G, Oberholtzer C, Hager GL, Levens D. 2012. Rapid genome-scale mapping of chromatin accessibility in tissue. Epigenetics Chromatin 5: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grøntved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, Aguilera G, Hager GL. 2013. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J 32: 1568–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grøntved L, Waterfall JJ, Kim DW, Baek S, Sung MH, Zhao L, Park JW, Nielsen R, Walker RL, Zhu YJ, et al. 2015. Transcriptional activation by the thyroid hormone receptor through ligand dependent receptor recruitment and chromatin remodeling. Nat Commun 6: 7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusmao EG, Allhoff M, Zenke M, Costa IG. 2016. Analysis of computational footprinting methods for DNase sequencing experiments. Nat Methods 13: 303–309. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, Zhang Z, Sabo PJ, Chen X, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. 2009. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods 6: 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeddin I, Récamier V, Bosanac L, Cissé II, Boudarene L, Dugast-Darzacq C, Proux F, Bénichou O, Voituriez R, Bensaude O, et al. 2014. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. eLife 3: e02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberger T, Staels B, Saladin R, Desvergne B, Auwerx J, Wahli W. 1994. Regulation of the peroxisome proliferator-activated receptor α gene by glucocorticoids. J Biol Chem 269: 24527–24530. [PubMed] [Google Scholar]
- Madsen MS, Siersbæk R, Boergesen M, Nielsen R, Mandrup S. 2014. Peroxisome proliferator-activated receptor γ and C/EBPα synergistically activate key metabolic adipocyte genes by assisted loading. Mol Cell Biol 34: 939–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally JG, Müller WG, Walker D, Wolford RG, Hager GL. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287: 1262–1265. [DOI] [PubMed] [Google Scholar]
- Miranda TB, Voss TC, Sung MH, Baek S, John S, Hawkins M, Grøntved L, Schiltz RL, Hager GL. 2013. Reprogramming of the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res 73: 5130–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T, Müller WG, Golob N, Mazza D, McNally JG. 2014. Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat Commun 5: 4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller F, Stasevich TJ, Mazza D, McNally JG. 2013. Quantifying transcription factor kinetics: at work or at play? Crit Rev Biochem Mol Biol 48: 492–514. [DOI] [PubMed] [Google Scholar]
- Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q. 2009. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol 11: 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. 2013. Latent enhancers activated by stimulation in differentiated cells. Cell 152: 157–171. [DOI] [PubMed] [Google Scholar]
- Sharp ZD, Mancini MG, Hinojos CA, Dai F, Berno V, Szafran AT, Smith KP, Lele TT, Ingber DE, Mancini MA. 2006. Estrogen-receptor-α exchange and chromatin dynamics are ligand- and domain-dependent. J Cell Sci 119: 4101–4116. [DOI] [PubMed] [Google Scholar]
- Sherwood RI, Hashimoto T, O'Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, Gifford DK. 2014. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol 32: 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A. 2014. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 15: 272–286. [DOI] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, Emmett MJ, Briggs ER, Fang B, Everett LJ, et al. 2015. Genetic variation determines PPARγ function and anti-diabetic drug response in vivo. Cell 162: 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626. [DOI] [PubMed] [Google Scholar]
- Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, Johnson TA, Voss TC, Lightman SL, Hager GL. 2009. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol 11: 1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steineger HH, Sørensen HN, Tugwood JD, Skrede S, Spydevold O, Gautvik KM. 1994. Dexamethasone and insulin demonstrate marked and opposite regulation of the steady-state mRNA level of the peroxisomal proliferator-activated receptor (PPAR) in hepatic cells. Hormonal modulation of fatty-acid-induced transcription. Eur J Biochem 225: 967–974. [DOI] [PubMed] [Google Scholar]
- Sung MH, Guertin MJ, Baek S, Hager GL. 2014. DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol Cell 56: 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung MH, Baek S, Hager GL. 2016. Genome-wide footprinting: ready for prime time? Nat Methods 13: 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, Karpova TS, Ball D, Mazza D, Lavis LD, et al. 2016. Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell 165: 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, Sakata-Sogawa K. 2008. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 5: 159–161. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. 2013. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Hager GL. 2014. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 15: 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Schiltz RL, Sung MH, Yen PM, Stamatoyannopoulos JA, Biddie SC, Johnson TA, Miranda TB, John S, Hager GL. 2011. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 146: 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Gates LA, Stashi E, Dasgupta S, Gonzales N, Dean A, Dacso CC, York B, O'Malley BW. 2015. Coactivator-dependent oscillation of chromatin accessibility dictates circadian gene amplitude via REV-ERB loading. Mol Cell 60: 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.