

Abstract
During cell division, spindle fibers attach to chromosomes at centromeres. The DNA sequence at regional centromeres is fast evolving with no conserved genetic signature for centromere identity. Instead CENH3, a centromere-specific histone H3 variant, is the epigenetic signature that specifies centromere location across both plant and animal kingdoms. Paradoxically, CENH3 is also adaptively evolving. An ongoing question is whether CENH3 evolution is driven by a functional relationship with the underlying DNA sequence. Here, we demonstrate that despite extensive protein sequence divergence, CENH3 histones from distant species assemble centromeres on the same underlying DNA sequence. We first characterized the organization and diversity of centromere repeats in wild-type Arabidopsis thaliana. We show that A. thaliana CENH3-containing nucleosomes exhibit a strong preference for a unique subset of centromeric repeats. These sequences are largely missing from the genome assemblies and represent the youngest and most homogeneous class of repeats. Next, we tested the evolutionary specificity of this interaction in a background in which the native A. thaliana CENH3 is replaced with CENH3s from distant species. Strikingly, we find that CENH3 from Lepidium oleraceum and Zea mays, although specifying epigenetically weaker centromeres that result in genome elimination upon outcrossing, show a binding pattern on A. thaliana centromere repeats that is indistinguishable from the native CENH3. Our results demonstrate positional stability of a highly diverged CENH3 on independently evolved repeats, suggesting that the sequence specificity of centromeres is determined by a mechanism independent of CENH3.
The single most important locus for stable transmission of genetic material is the centromere, the site where microtubules attach to chromosomes during cell division. A dichotomy exists between centromere function and form: Although the former is strongly conserved, the latter is surprisingly changeable. Centromere architecture ranges from a single localized region on a chromosome (monocentric) to occupying its entire length (holocentric). Species with monocentric chromosomes can either have point centromeres or regional centromeres. Point centromeres are defined by short DNA sequences, whereas regional centromeres contain up to megabases of DNA (Pluta et al. 1995). The DNA sequence at regional centromeres evolves so rapidly and extensively that homology is often undetectable across short evolutionary time scales. Even the core sets of proteins associated with centromeres can vary across eukaryotic taxa (Drinnenberg et al. 2016). Despite diversity, certain dominant themes reoccur in centromere organization. Most eukaryotes have monocentric centromeres embedded in megabase-sized arrays of tandem repeats and/or retrotransposons, bounded by pericentric heterochromatin enriched for repressive histone marks. These specific sequences are not necessary or sufficient for the function of regional centromeres in some contexts (Birchler et al. 2011; McKinley and Cheeseman 2016). The critical feature that distinguishes centromeres from the surrounding pericentromere is the presence of a histone H3 variant, CENH3 (or CENP-A) that localizes exclusively to functional centromeres.
It is generally accepted that the key to establishing a functional centromere is a high local concentration of CENH3 nucleosomes (Allshire and Karpen 2008). The rules governing optimal CENH3 localization, however, are not well understood. Although most eukaryotic centromeres exhibit a strong enrichment for specific repeated sequences, an unresolved question is whether these repeats contribute sequence per se or instead are associated with a unique chromatin environment.
One hypothesis is that CENH3 is coevolving DNA-binding specificity driven by its interactions with underlying centromeric DNA sequence (Henikoff et al. 2001; Malik et al. 2002; Cooper and Henikoff 2004). Unlike canonical histones, the CENH3 protein sequence is hypervariable. In several plant and animal species, signatures of long-term adaptive evolution have been detected, especially in its N-terminal tail domain (Malik and Henikoff 2001; Talbert et al. 2002; Schueler et al. 2010). This concept is also central to the “centromere drive” hypothesis, which reasons that centromeric DNA is evolving selfishly to hijack female meiosis. In turn, this spurs the evolution of either heterochromatin or centromere-associated proteins to suppress this drive (Henikoff et al. 2001; Talbert et al. 2004; Nakano et al. 2008; Malik and Henikoff 2009). Instances of centromeric DNA distorting segregation are well documented, such as Robertsonian translocations in mammals and amplification of a centromere repeat in monkeyflowers (Fishman and Saunders 2008; Chmátal et al. 2014). However, there is no direct evidence addressing whether CENH3 function is adapted or agnostic to centromeric sequence.
From studies of chromosome addition and introgression lines, we know that CENH3 can assemble functional centromeres on DNA repeats of closely related species (Jin et al. 2004; Sanei et al. 2011; Ishii et al. 2015b), and in an extreme case, RNAi depletion of CENH3 in human cells has been partially complemented by the yeast CENH3 homolog (Wieland et al. 2004); however, details of the interaction with non-native DNA remain unknown. In a previous study (Maheshwari et al. 2015), we reported that replacement of A. thaliana CENH3 with CENH3 homologs from distant species resulted in apparently perfect complementation of mitotic and meiotic functions. However, in outcrosses to wild type, chromosomes of the complemented parent missegregate during embryonic growth leading to frequent aneuploidy, haploidy, and death. One explanation for the failure of centromeres specified by divergent CENH3s in crosses might be defective interaction between the endogenous A. thaliana centromeric repeats (CEN180) and the divergent CENH3 resulting in mislocalization to other DNA sequences. Here, we use these genotypes to test whether divergent CENH3s, which evolved in the context of different centromeric DNA sequences, localize differently from native A. thaliana CENH3.
As a prerequisite to answering this question, we first need to decipher the higher-order organization of centromere repeats in A. thaliana. The 180-bp repeat in A. thaliana was shown via DNA hybridization and RFLP mapping to be highly abundant within the genetically mapped A. thaliana centromeres (Maluszynska and Heslop-Harrison 1991; Round et al. 1997; Copenhaver et al. 1999). The gross architecture of all five A. thaliana centromeres was revealed as megabase-sized islands of 180-bp tandem repeats surrounded by flanking DNA rich in retrotransposons and other repetitive sequences (Fransz et al. 2000). CENH3 ChIP followed by Southern blot analysis confirmed the association between this 180-bp repeat sequence and functional A. thaliana centromeres (Nagaki et al. 2003); interestingly, the authors found that only 15% of the total 180-bp repeats were bound by CENH3 and suggested that “only subsets of the 180-bp satellite arrays are involved in centromere function.” Previous studies attempted to characterize variation within the 180-bp repeats by PCR cloning of 180-bp repeats and identifying conserved and variable regions by comparison to a consensus (Heslop-Harrison et al. 1999; Hall et al. 2005). The caveat with comparing PCR clones, beyond that of amplification bias, is that the sequences being compared could be evolving under different functional constraints, such as pericentric or centric repeats. Taken together, although our current view of A. thaliana centromeres is broadly consistent with other regional centromeres, it is still lacking in resolution.
In this study, we exploited the advances in NGS technology to characterize sequence variation of the CEN180 repeats in the context of their physical organization within the A. thaliana centromere. We implement a k-mer-based approach to refine the chromosomal organization of CEN180 variants and interrogate the DNA binding profiles of the related Lepidium oleraceum CENH3 and more distant Zea mays CENH3 in the A. thaliana genome. Our comparative analysis of variant CENH3s binding to the same centromeric sequences casts new light on the effects of CENH3 divergence on centromere specification and haploid induction.
Results
CEN180 repeats cluster into three clades
We were able to identify 2550 CEN180 monomers in the Arabidopsis TAIR10 reference genome assembly. However, because the estimates of centromere size range from 2–4 Mb, the Arabidopsis genome is expected to contain approximately 25–50,000 CEN180 repeats (Copenhaver 2003). The recently released high-coverage long-read Pacific Biosciences (PacBio) assembly of the A. thaliana genome (Kim et al. 2014) incorporates more CEN180 repeats (4780), but still falls significantly short of the estimate. The average pairwise identity between all repeats identified was 73.4%, indicating a significant level of genetic variation. To describe the patterns of diversity within this large population of similar sequences, we used a phylogenetic clustering strategy. We partitioned the repeats into six subpopulations that had the maximum likelihood of evolving from a common ancestral sequence (Fig. 1A). The basis of the clustering becomes obvious upon plotting segregating sites by cluster (Supplemental Fig. S1). Sequences within each cluster share a distinct pattern of SNPs that establish a fingerprint of shared evolutionary history. This grouping of sequences was also largely consistent with a distance matrix-based phylogenetic analysis (Fig. 1B). Overwhelmingly, sequences from the same cluster branched closest to each other. The largest clusters (2, 3, and 6) were also the most diverse and had the greatest branch lengths, whereas the smaller clusters (1, 4, and 5) were more homogeneous. Reducing the clusters to a single representative sequence results in a pairwise branching pattern in which clusters 1 and 4, 2 and 3, and 5 and 6 form monophyletic clades (Fig. 1C). This phylogenetic relationship suggests that the extant repeats are expansions of three ancestral sequences.
Figure 1.
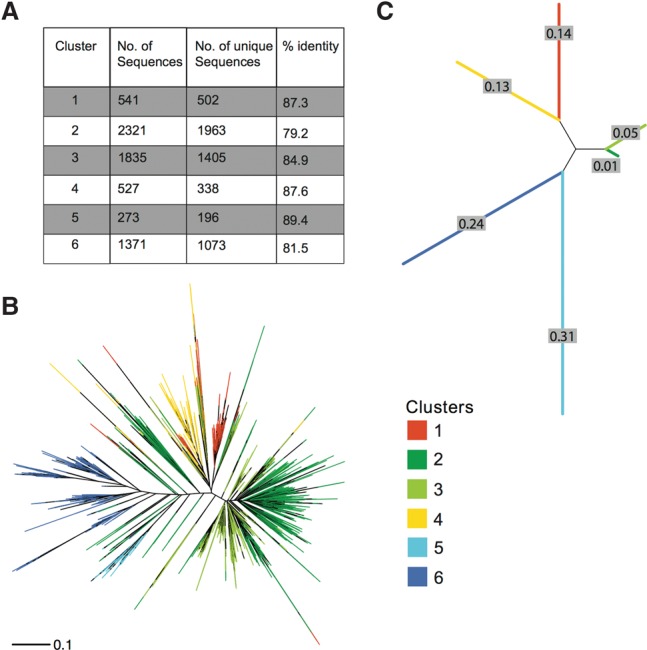
Characterization of genetic diversity within CEN180 repeats. (A) Table summarizing clustering results. Phylogenetic trees constructed using the neighbor-joining method with branches colored by cluster. The trees are drawn to scale, with branch lengths in the units of number of base substitutions per site. (B) Full set of 6868 CEN180 repeats identified from genome assemblies. (C) Consensus sequences of the six clusters.
CEN180 repeats from cluster 4 are dominant in the genome
Next, we asked how well clusters extracted in silico were representative of the genome. Since each cluster is associated with a unique SNP fingerprint, we could define a set of nonoverlapping k-mers from each cluster as “signatures” for that cluster (Supplemental Table S1; Supplemental Fig. S2). In preparation for the forthcoming ChIP-seq analysis, we analyzed the distribution of these “signatures” in the input chromatin, which is effectively an unbiased sampling of the genome occupied by nucleosomes. We performed 2 × 100-bp paired-end sequencing of DNA fragments bound by mononucleosomes generated by micrococcal nuclease (MNase) digestion of nuclei (Supplemental Fig. S3). The additional advantage of this approach is that it allows us to generate overlapping reads that are approximately the length of an actual CEN180 repeat. We observed striking differences in the abundance distribution of CEN180 clusters in the genome assemblies compared to input chromatin (Fig. 2A). Repeats from clusters 1 and 4 are the most abundant in the input chromatin while being the least abundant in both genome assemblies. Conversely, clusters 5 and 6 comprise a negligible fraction of the input in comparison to their abundance in the genome assemblies. We can exclude the possibility that abundance in the input is reflecting the number of signature k-mers associated with that cluster, because clusters 1 and 4 have the smallest set of signature k-mers, whereas clusters 5 and 6 have the largest number of signature k-mers (Supplemental Table S1).
Figure 2.

Distribution of CEN180 clusters in genome assemblies. (A) Stacked proportional bar graph representing relative abundance of each cluster in the TAIR10 reference genome, PacBio contigs, and ChIP input from wild-type Col-0 genetic background. (B) Stacked bar graph illustrating the sequences found adjacent to repeats by cluster. (C) Dot plot of PacBio contig JSAD01000006.1 against itself illustrating four blocks of CEN180 tandem repeats. The colored bar graph running along the x-axis highlights the 219 repeats identified on this contig using the LASTZ search method. (D) Phylogenetic tree of repeats identified in the contig represented in C, with branches colored by cluster.
CEN180 monomers are organized into arrays of similar sequences
To evaluate the robustness of our repeat identification strategy, we looked more closely at PacBio contig JSAD01000006.1, which contained the largest number of cluster 4 repeats (39 of a total of 219, contig length ∼100 kb). Plotting similarity of the PacBio contig against itself by position is an alignment-independent approach that readily highlights regions with tandem repeats (Fig. 2C). Reassuringly, our annotation of CEN180 repeats in this contig concurs entirely with the repeat blocks identified using the dot plot method (Fig. 2C). We can therefore exclude biased sampling from the genome assemblies as a source of discrepancy in cluster abundance. It is notable that repeats from cluster 4 are very similar to one another, inferred from the very short branch lengths of cluster 4 repeats (Fig. 2D). If organization into homogeneous arrays is the norm for cluster 4 repeats, this would be a likely explanation for their reduced representation in the assembled genomes. In contrast, repeats from clusters 2 and 3 are interspersed with one another and monomers within these tandem arrays show a much greater degree of phylogenetic separation. Next, we extracted the sequence neighborhood for all repeats from both the PacBio and TAIR10 assemblies (Fig. 2B). We found that repeats from the same cluster are most frequently adjacent to one another. The exception to this is the frequent embedding of repeats from clusters 5 and 6 in noncentromeric sequences, which could explain their overrepresentation in the genome assemblies (Fig. 2A). Another interesting pattern was that aside from repeats within the same cluster, the next most frequent neighboring sequence was a repeat from a related cluster within the same clade. These results suggest that monomers are organized into arrays of similar sequences with a strong tendency of grouping repeats within a clade.
Evolutionary history visible in the organization of CEN180 repeats
The number of CEN180 repeats that we extracted from the TAIR10 genome assembly ranged from as few as 81 for Chromosome 3 to 995 for Chromosome 5 (Supplemental Table S2). Since no differences in the strength of CEN180 FISH signals across the five centromeres have been reported, we interpret these differences in copy number as variation in the amount of (peri)centromeric sequences included in the genome assembly for each chromosome. To visualize the physical arrangement of the CEN180 repeats, we plotted consecutively identified repeats, colored by cluster, for each chromosome (Fig. 3A). This analysis omits any intervening non-CEN180 sequences; thus, consecutively drawn repeats need not be physically adjacent to one another. Nonetheless, two distinct patterns were observed. First, repeats from clusters 1 and 4 form a core enveloped by repeats from clusters 2 and 3. This arrangement also implies that clusters 2 and 3—the most diverse (79% and 85% pairwise identity) (Fig. 1A) and hence the oldest CEN180 repeats—are pericentric. Second, CEN180 repeats were predominantly arranged in a head-to-tail manner, consistent with unequal crossing over as the model for satellite DNA evolution. In addition, clusters 5 and 6 were revealed as CEN180 variants that had specifically expanded on Chromosome 1 (Fig. 3A). FISH was performed to demonstrate the specificity of the in silico predicted clusters. All centromeres of A. thaliana displayed cluster 4 and CEN180-specific signals, whereas cluster 6 localized to only one chromosome pair (Fig. 3B), which we confirmed was Chromosome 1 using Chr 1–specific BAC FISH (Supplemental Fig. S4).
Figure 3.
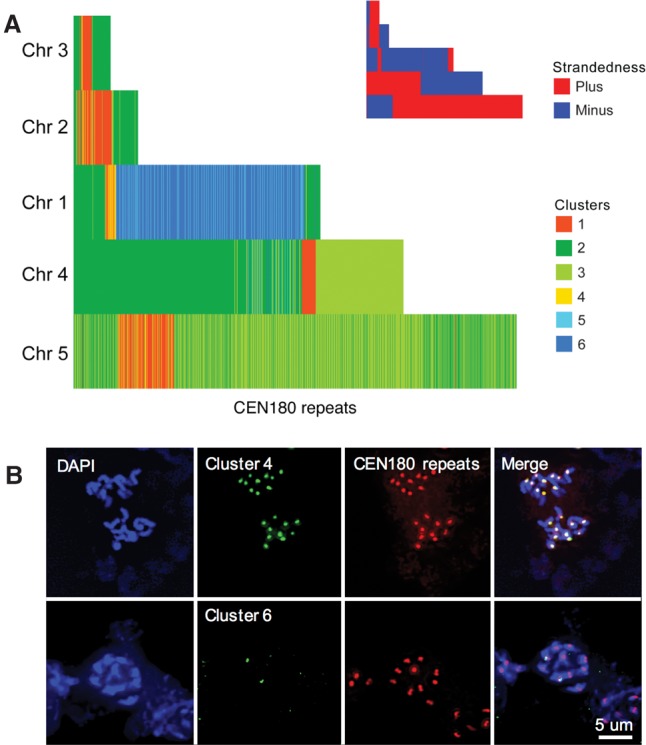
Organization of CEN180 clusters on A. thaliana chromosomes. (A) Representation of consecutive repeats identified on each chromosome. Repeats are colored by cluster, and any intervening sequences are omitted. Inset shows the strandedness of the same. (B) FISH analysis with cluster 4–specific and cluster 6–specific probes. Cluster 4 localized to all chromosomes together with CEN180 repeats. In contrast, the cluster 6–specific probe marks the centromeres of two chromosomes only.
Functional centromeres assemble on cluster 4 repeats regardless of CENH3 species origin
Given the substructure within (peri)centromeric CEN180 repeats, we asked whether CENH3 preferentially associated with repeats from specific clusters. We performed native ChIP against A. thaliana CENH3 (Supplemental Fig. S5) and compared the distribution of CEN180 clusters in the ChIP to abundance in the genome, i.e., the input. We found that A. thaliana CENH3 ChIP was strongly enriched for sequences from cluster 4 and depleted for cluster 3 (Fig. 4B), supporting the phylogenetic partitioning scheme.
Figure 4.

Native and divergent CENH3 exhibit identical binding patterns. (A) Histogram depicting fold enrichment of CENH3 signal over negative control on A. thaliana Chromosome 5, with increasing magnification on the region with maximum signal. Colored bars parallel to the x-axis represent CEN180 repeats, colored by cluster. (B) Distribution of CENH3 ChIP signal across the six CEN180 clusters illustrated in shades of blue. Input in black is shown in comparison and represents abundance in the genome. (C) A. thaliana and Z. mays CENH3s form intermingled subdomains in interphase nuclei of wild-type A. thaliana expressing Z. mays CENH3. Analysis was performed by Structured Illumination Microscopy (SIM): (blue) counterstain with DAPI; (green) A. thaliana CENH3; (red) Z. mays CENH3. Area within the white square is enlarged. (D) Signal intensity of A. thaliana and Z. mays CENH3 measured in eight nuclei. Signal intensities of CENH3s are not significantly different at the 5% level by t-test.
Next, we interrogated localization patterns of non-native CENH3s in A. thaliana nuclei. From our previous study, we know that functional complementation of cenh3 has no effect on vegetative or reproductive phenotypes, except in crosses to wild type (Maheshwari et al. 2015). We had previously shown that centromeres built on L. oleraceum CENH3 are deficient in comparison to wild type, because when outcrossed, they missegregate dramatically. We also tested A. thaliana centromeres assembled with Z. mays CENH3 in crosses to wild type and observed extensive missegregation leading to seed death and aneuploid progeny (Supplemental Table S3). Further, haploid production efficiency was increased proportionally to CENH3 divergence. Up to 19% of the progeny lost the genome of the Z. mays CENH3 expressing parent, whereas in the case of CENH3 from the closer relative L. oleraceum, a maximum of 11% haploids was observed (Maheshwari et al. 2015).
Immunostaining experiments in wild-type A. thaliana expressing both endogenous A. thaliana CENH3 and Z. mays CENH3 show that both CENH3 variants intermingle in centromeric subdomains in equal amounts (Fig. 4C,D). A similar centromere organization was found in species expressing two naturally occurring CENH3 variants (Ishii et al. 2015a; Neumann et al. 2016). To reveal the sequence specificity of these localization patterns, we performed native ChIP against L. oleraceum CENH3 and Z. mays CENH3 in the A. thaliana cenh3 null genetic background. Surprisingly, the genome-wide binding profile of the divergent CENH3s showed high correlation with native A. thaliana CENH3 (Fig. 4A; Supplemental Fig. S6). Moreover, non-native CENH3s preferentially bound cluster 4 repeats, as observed for A. thaliana CENH3. Since the non-native CENH3 transgenes are expressed in the background of an A. thaliana cenh3 null mutation, their conserved localization through two generations, hundreds of mitotic, and two meiotic divisions is not guided by the presence of the native A. thaliana CENH3. More likely, centromere localization is faithfully maintained upon being derived from an epigenetic signal inherited from the heterozygous parent that produced the cenh3− gamete into which the complementing transgene was introduced through floral dip (Maheshwari et al. 2015).
Taken together, our results suggest that even the highly diverged Z. mays CENH3 does not mislocalize in A. thaliana, despite causing dramatic segregation errors in crosses to wild type.
Discussion
In most assembled genomes, the centromere regions are represented by megabase-sized gaps. The long stretches of near-identical tandem repeats make assembly into a linear reference impractical. Currently, even a pixelated view of centromere organization is visible only in a handful of resource-intense genomes with a focus on centromeres enriched for unique sequences (Schueler et al. 2001; Nagaki et al. 2004; Wolfgruber et al. 2009; Miga et al. 2014). On the other hand, given the ease of ChIP-seq, we have catalogs of consensus sequences bound by CENH3 from a wide range of species (Tek et al. 2010; Lee et al. 2011; Gong et al. 2012; Cerutti et al. 2016). However, in these cases, the evolutionary variation and genomic context that characterize the functional centromere sequences remain unknown. Here, we describe the centromere context for A. thaliana, the model plant species, for which insufficient information was available to differentiate centromere core repeats from those at the periphery (Heslop-Harrison et al. 1999; Nagaki et al. 2003; Hall et al. 2005).
We developed a strategy that is easily generalizable to the analysis of repeats from any species with available genomic scaffolds. In brief, we extracted CEN180 variants from all available A. thaliana genomic sequences, partitioned the variants into clusters with shared segregating sites, and then probed for sequence fingerprints of each cluster in CENH3 ChIP-seq data sets. The fact that experimental FISH data supports the predicted chromosome-specificity of the cluster 6 repeats provides strong validation for our computational approach (Fig. 3B; Supplemental Fig. S4).
Our analysis of variation in CEN180 repeats reveals layers of sequences that have undergone independent evolutionary trajectories. This is consistent with differences in methylation profiles observed between 180-bp repeats cloned from CENH3 ChIP versus those present in the flanking pericentromere (Zhang et al. 2008a). Clusters 2 and 3 at the CEN array edges have accumulated mutations, whereas clusters 1 and 4 at the centromere core remain homogeneous, a pattern consistent with the unequal crossing over model for repeat evolution (Smith 1976). We also observed an underrepresentation of cluster 4 sequences in both the TAIR10 and PacBio assemblies (Kim et al. 2014), which is again in agreement with homogeneous arrays of cluster 4 occupying centromere gaps (Lin et al. 1999). Strikingly, A. thaliana CENH3 preferentially bound cluster 4 repeats (Fig. 4B). Sequence-specific enrichment for the youngest and most homogeneous repeat is exactly what has been shown for functional human centromeric chromatin (Henikoff et al. 2015). This finding underscores the conservation in centromere organization across kingdoms, despite apparent changes in its molecular building blocks.
After defining the functional A. thaliana centromeres in this study, we carried out an experiment to test whether sequence divergence in CENH3 can affect centromere location. Remarkably, we find that the binding pattern of divergent CENH3s on the A. thaliana genome is indistinguishable from endogenous A. thaliana CENH3 (Fig. 4). Although this result may not be so surprising in the case of a close relative such as L. oleraceum CENH3, it is remarkable for Z. mays CENH3. The association between CENH3 nucleosomes and maize DNA is variable, as multiple relocation events have been characterized in the domesticated population (Schneider et al. 2016). There is no sequence similarity between centromeric sequences of the monocot Z. mays and dicot A. thaliana. In addition, Z. mays and A. thaliana CENH3 are unalignable at their N-terminal tail domain and differ at 41 of 97 residues in the histone fold domain. Lastly, given that divergence of Z. mays CENH3 does not prevent it from being loaded into A. thaliana, centromeric chromatin suggests that the interaction between the putative plant CENH3 chaperone and its target lacks the degree of specificity observed in Drosophila (Rosin and Mellone 2016).
Our results argue against direct coevolution between CENH3 protein sequence and centromeric DNA sequence (Malik et al. 2002; Cooper and Henikoff 2004). The original centromere drive model hypothesized that CENH3 was evolving to suppress female meiotic drive by changing its DNA binding preference to equalize driving and nondriving allelic centromeres (Henikoff et al. 2001). Given that even extreme changes in CENH3 protein show no detectable effect on centromere positioning, it seems unlikely that evolutionary divergence in CENH3 is an adaptation to mitigate centromere drive through altered DNA-binding specificity. Alternatively, other centromere-associated protein(s) may provide DNA-sequence specificity and evolve to suppress centromere drive (Talbert et al. 2004; Malik and Henikoff 2009). Adaptive evolution of CENH3 could in turn be driven by its interaction with these proteins, for example CENP-C, or instead be driven by modulation of its centromere deposition efficiency through its loading factor as proposed for Drosophila (Rosin and Mellone 2016).
Notwithstanding the complementation of essential functions, data from this study and previous research (Maheshwari et al. 2015) show that centromeres built on non-native CENH3s missegregate during early embryogenesis in crosses to wild type. Clearly, the non-native CENH3 proteins are lacking a species-specific adaptation that the endogenous A. thaliana CENH3 has evolved. Our results indicate that CENH3 binds epigenetically defined centromere domains regardless of sequence context. The remarkable positional stability of divergent CENH3 leaves us even more curious as to why these centromeres are weaker when competing with wild-type centromeres. Identifying this functional deficiency could help in decrypting the evolutionary driver(s) of centromere diversification. Given the parallels between plant and human centromeres, the answer to this question will undoubtedly be of broad significance.
Methods
Plant strains
A. thaliana CENH3 ChIP was performed in wild-type Col-0 background; L. oleraceum CENH3 ChIP in A. thaliana cenh3 null mutant expressing transgenic L. oleraceum CENH3 (T1 family, 19); and Z. mays CENH3 ChIP in A. thaliana cenh3-1 null mutant expressing transgenic Z. mays CENH3 (T1 family, 17). For each CENH3 ChIP, a negative control for antibody specificity was performed in a genetic background that does not express the epitope recognized by that antibody: For the anti-A. thaliana CENH3 antibody, we used cenh3-1/cenh3-1 L. oleraceum CENH3; for anti-L. oleraceum CENH3 antibody, we used cenh3-1/cenh3-1 Z. mays CENH3; and for anti-Z. mays CENH3 antibody, we used wild-type Col-0 (Supplemental Fig. S5).
Antibodies
We used published anti-A. thaliana CENH3 and anti-Z. mays CENH3 antibodies for ChIP (Talbert et al. 2002; Zhong et al. 2002) as well as anti-grass CENH3 for immunostaining (Sanei et al. 2011) experiments. The anti-L. oleraceum CENH3 antibody is an affinity-purified rabbit polyclonal antibody made by LifeTein against the peptide RTKRFASRPQRPRNQTDTTVPC.
Native chromatin immunoprecipitation
Nuclei were isolated from finely ground frozen seedlings using the protocol from Zhou et al. (2005). Crude nuclei were digested with 0.5 gel units/µL Micrococcal Nuclease (NEB) for 6 min at 37°C and stopped by 50 mM EDTA. This digestion mixture was centrifuged at 13,400g for 10 min at 4°C, and the supernatant was collected as the S1 chromatin fraction. Nucleosomes were further extracted by resuspending the pellet in a high-salt buffer (10 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM MgCl2, 2mM EDTA, 0.1% Triton X-100, and Roche protease inhibitor cocktail) to recover the S2 chromatin fraction. For each ChIP, 350 µL of the combined S1 and S2 soluble chromatin was diluted to a final volume of 1 mL in ChIP dilution buffer (20 mM Tris-HCl pH 8.0, 39 mM NaCl, 5 mM EDTA) and incubated with 2–3 µg of antibody overnight at 4°C. The next day, immunoprecipitation was performed using magnetic protein A/G beads (Pierce) following the manufacturer's guidelines. Immunoprecipitated complexes were washed six times in buffers with increasing salt concentration (50 mM Tris-HCl pH 8.0, 10 mM EDTA, and 75 mM/125 mM/175 mM NaCl). After washing, beads were resuspended in TE buffer followed by RNase and Proteinase K treatment to release DNA from the immunoprecipitated nucleosomes. The DNA was isolated using standard SPRI bead-based method.
Illumina sequencing and data processing
Library construction was performed using the KAPA Hyper Prep kit as described in the manual (https://www.kapabiosystems.com/), with the only exception that five PCR cycles post-adaptor ligation libraries were size-selected for mononucleosomes, i.e., 200–400 bp using BluePippin (Sage Science) (Supplemental Fig. S3). qPCR was performed to determine the additional PCR cycles needed for optimal library concentration. Paired-end 100 bp Illumina HiSeq sequencing was performed at the University of California, Davis, DNA technologies core facility. Demultiplexing, adaptor, and quality trimming of the raw reads was performed using the Allprep script (https://github.com/Comai-Lab/allprep). Paired reads were merged using SeqPrep (https://github.com/jstjohn/SeqPrep) with parameters –q 30 (quality) and –L 35 (minimum merged pair length). Read pairs that did not merge were discarded.
Cluster analysis
LASTZ (http://www.bx.psu.edu/~rsharris/lastz/) (Harris 2007) was used to identify CEN180 repeats within the assembled TAIR10 genome and PacBio contigs (Kim et al. 2014). We tested several CEN180 sequences as queries and chose to move forward with LASTZ results that identified the maximum number of repeats and in which a majority of the adjacent repeats were 0 bp apart, i.e., in tandem. The repeats were further filtered to exclude sequences with >14 gapped positions and lengths outside the 165- to 185-bp range. This step pruned 325 sequences, resulting in a total of 7005 repeats (4547 and 2458 from PacBio contigs and TAIR10, respectively).
The 7005 repeats were aligned using Clustal Omega (Sievers et al. 2011) with default parameters, and the alignment was refined by removing sequences that generated rare indels. The final multiple sequence alignment included 6868 sequences and was 204 bp in length. Clustering was performed on this alignment using find.best() function in phyclust (Chen 2011), an R package (R Core Team 2016) developed for exploring population structure of DNA sequence data using a phylogenetic approach. We evaluated a range of clusters (2–10) and using the clustergram visualization approach, found that six clusters was an optimal solution (data not shown).
We assigned ChIP-seq reads to clusters using a k-mer-based strategy. We first generated sets of signature k-mers for each cluster, i.e., high-frequency k-mers that are unique to that cluster. We arbitrarily required the k-mer to be shared by at least 100 repeats within a cluster and tested k-mers in a range of sizes: 20, 25, and 30 bp (Supplemental Table S1). If a read contained signature k-mers, we annotated it as a CEN180 sequence. If k-mers from a read were restricted to a single cluster, we assigned the read to that cluster. We found that the specificity of cluster assignment increased with k-mer size (Supplemental Fig. S2) and therefore chose 30-bp as the default k-mer length.
ChIP-seq read-mapping and peak-calling
Merged paired-end reads were mapped to the TAIR10 reference using the aln and samse algorithms of BWA (Li and Durbin 2009) and up to 10 alignments per read were saved. We performed peak calling using MACS2 (Zhang et al. 2008b) with the additional parameters “--nomodel --extsize 165 --keep-dup all -B” and used the appropriate CENH3 ChIP negative controls as input. We evaluated the quality of the ChIP experiments using the modENCODE recommended metric of FRiP, i.e., fraction of reads in peaks, and calculated correlation between CENH3 ChIP experiments using the R/Bioconductor DiffBind package (Supplemental Table S4; Supplemental Fig. S6; Ross-Innes et al. 2012; R Core Team 2016).
Phylogenetic analysis
Tree construction and rendering was done in R (R Core Team 2016) using the package “APE” (Paradis et al. 2004).
Cytological analysis of centromeres
A. thaliana flower buds were fixed for 5 d at room temperature with 3:1 (V/V) ethanol/glacial acetic acid und used for slide preparation and fluorescence in situ hybridization as described in Schubert et al. (2012). FISH probes: cluster 4 (FAM-5′-CTCATATGGACTTTGGCTACACCAT-3′), cluster 6 (Cy3-5′-TTAGCGGATTTGTAGTCAAATATGACTAGA-3′), and CEN180 repeats (Cy5-5′-GCTTTGAGAAGCAAGAAGAAGG-3′). DNA of the A. thaliana Chromosome 1–specific BACs (F23M19, F12K21, F2J6, and T7O23) were labeled by nick translation with Alexa Fluor-488-dUTP. For immunostaining, the L. oleraceum CENH3 antibody was directly labeled with Alexa Fluor 488 NHS ester (Succinimidyl Ester). Indirect immunostaining and imaging was performed as described in Ishii et al. (2015a). Localization analysis for A. thaliana and Z. mays CENH3 was performed with sequential indirect immunostaining, using anti-Grass CENH3 antibody (1:1000 dilution) to recognize Z. mays CENH3 and directly labeled anti-L. oleraceum CENH3 antibody (1:200 dilution) to recognize A. thaliana CENH3 (Supplemental Fig. S7). To analyze the substructures and spatial arrangement of immunosignals beyond the classical Abbe/Raleigh limit (super-resolution), spatial Structured Illumination Microscopy (3D-SIM) was applied using a Plan-Apochromat 63×/1.4 oil objective of an Elyra PS.1 microscope system and the software ZEN (Carl Zeiss GmbH). The Imaris 8.0 (Bitplane) software was used to measure the intensity of A. thaliana and Z. mays CENH3 in SIM image stacks.
Data access
Raw FASTQ data as well as processed peak and bigWig files from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE88907. Scripts and details of the analysis methods are available in the Supplemental Material and can also be accessed at http://doi.org/10.5281/zenodo.160186 and https://zenodo.org/record/180498 (Allprep).
Supplementary Material
Acknowledgments
We thank Dr. Steve Henikoff and Dr. Kelly Dawe for the generous gift of antibodies; Dr. Steve Henikoff, Siva Kasinathan, Dr. Dan Barbash, and Dr. Greg Smaldone for insightful discussions and critical reading of the manuscript; Marwa Zafarullah's assistance for scoring the Z. mays CENH3 crosses; and Dr. Veit Schubert for performing super high resolution microscopy. This work was funded by the Gordon and Betty Moore Foundation through grant GBMF 2550.03 to the Life Sciences Research Foundation (to S.M.) and by the HHMI and Gordon and Betty Moore Foundation grant GBMF3068 (to L.C.).
Author contributions: S.M., C.T.B., A.H., and L.C. conceived and designed the experiments; S.M. and T.I. performed the experiments; S.M., T.I., and C.T.B. analyzed the data; and S.M., A.H., and L.C. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.214619.116.
References
- Allshire RC, Karpen GH. 2008. Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet 9: 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Gao Z, Sharma A, Presting GG, Han F. 2011. Epigenetic aspects of centromere function in plants. Curr Opin Plant Biol 14: 217–222. [DOI] [PubMed] [Google Scholar]
- Cerutti F, Gamba R, Mazzagatti A, Piras FM, Cappelletti E, Belloni E, Nergadze SG, Raimondi E, Giulotto E. 2016. The major horse satellite DNA family is associated with centromere competence. Mol Cytogenet 9: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WC. 2011. “Overlapping codon model, phylogenetic clustering, and alternative partial expectation conditional maximization algorithm.” PhD thesis, Iowa State University, Ames, IA. [Google Scholar]
- Chmátal L, Gabriel SI, Mitsainas GP, Martínez-Vargas J, Ventura J, Searle JB, Schultz RM, Lampson MA. 2014. Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution in mice. Curr Biol 24: 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JL, Henikoff S. 2004. Adaptive evolution of the histone fold domain in centromeric histones. Mol Biol Evol 21: 1712–1718. [DOI] [PubMed] [Google Scholar]
- Copenhaver GP. 2003. Using Arabidopsis to understand centromere function: progress and prospects. Chromosome Res 11: 255–262. [DOI] [PubMed] [Google Scholar]
- Copenhaver GP, Nickel K, Kuromori T, Benito MI, Kaul S, Lin X, Bevan M, Murphy G, Harris B, Parnell LD, et al. 1999. Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286: 2468–2474. [DOI] [PubMed] [Google Scholar]
- Drinnenberg IA, Henikoff S, Malik HS. 2016. Evolutionary turnover of kinetochore proteins: a ship of theseus? Trends Cell Biol 26: 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman L, Saunders A. 2008. Centromere-associated female meiotic drive entails male fitness costs in monkeyflowers. Science 322: 1559–1562. [DOI] [PubMed] [Google Scholar]
- Fransz PF, Armstrong S, de Jong JH, Parnell LD, van Drunen C, Dean C, Zabel P, Bisseling T, Jones GH. 2000. Integrated cytogenetic map of chromosome arm 4S of A. thaliana: structural organization of heterochromatic knob and centromere region. Cell 100: 367–376. [DOI] [PubMed] [Google Scholar]
- Gong Z, Wu Y, Koblizkova A, Torres GA, Wang K, Iovene M, Neumann P, Zhang W, Novak P, Buell CR, et al. 2012. Repeatless and repeat-based centromeres in potato: implications for centromere evolution. Plant Cell 24: 3559–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SE, Luo S, Hall AE, Preuss D. 2005. Differential rates of local and global homogenization in centromere satellites from Arabidopsis relatives. Genetics 170: 1913–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, Pennsylvania State University, State College, PA. [Google Scholar]
- Henikoff S, Ahmad K, Malik HS. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293: 1098–1102. [DOI] [PubMed] [Google Scholar]
- Henikoff JG, Thakur J, Kasinathan S, Henikoff S. 2015. A unique chromatin complex occupies young α-satellite arrays of human centromeres. Sci Adv 1: e1400234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslop-Harrison JS, Murata M, Ogura Y, Schwarzacher T, Motoyoshi F. 1999. Polymorphisms and genomic organization of repetitive DNA from centromeric regions of Arabidopsis chromosomes. Plant Cell 11: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Karimi-Ashtiyani R, Banaei-Moghaddam AM, Schubert V, Fuchs J, Houben A. 2015a. The differential loading of two barley CENH3 variants into distinct centromeric substructures is cell type- and development-specific. Chromosome Res 23: 277–284. [DOI] [PubMed] [Google Scholar]
- Ishii T, Sunamura N, Matsumoto A, Eltayeb AE, Tsujimoto H. 2015b. Preferential recruitment of the maternal centromere-specific histone H3 (CENH3) in oat (Avena sativa L.) × pearl millet (Pennisetum glaucum L.) hybrid embryos. Chromosome Res 23: 709–718. [DOI] [PubMed] [Google Scholar]
- Jin W, Melo JR, Nagaki K, Talbert PB, Henikoff S, Dawe RK, Jiang J. 2004. Maize centromeres: organization and functional adaptation in the genetic background of oat. Plant Cell 16: 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KE, Peluso P, Baybayan P, Yeadon PJ, Yu C, Fisher W, Chin CS, Rapicavoli NA, Rank DR, Li J, et al. 2014. Long-read, whole genome shotgun sequence data for five model organisms. Sci Data 1: 140045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HR, Hayden KE, Willard HF. 2011. Organization and molecular evolution of CENP-A–associated satellite DNA families in a basal primate genome. Genome Biol Evol 3: 1136–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Kaul S, Rounsley S, Shea TP, Benito MI, Town CD, Fujii CY, Mason T, Bowman CL, Barnstead M, et al. 1999. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature 402: 761–768. [DOI] [PubMed] [Google Scholar]
- Maheshwari S, Tan EH, West A, Franklin FC, Comai L, Chan SW. 2015. Naturally occurring differences in CENH3 affect chromosome segregation in zygotic mitosis of hybrids. PLoS Genet 11: e1004970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. 2001. Adaptive evolution of Cid, a centromere-specific histone in Drosophila. Genetics 157: 1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. 2009. Major evolutionary transitions in centromere complexity. Cell 138: 1067–1082. [DOI] [PubMed] [Google Scholar]
- Malik HS, Vermaak D, Henikoff S. 2002. Recurrent evolution of DNA-binding motifs in the Drosophila centromeric histone. Proc Natl Acad Sci 99: 1449–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maluszynska J, Heslop-Harrison JS. 1991. Localization of tandemly repeated DNA sequences in Arabidopsis thaliana. Plant J 1: 159–166. [Google Scholar]
- McKinley KL, Cheeseman IM. 2016. The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol 17: 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga KH, Newton Y, Jain M, Altemose N, Willard HF, Kent WJ. 2014. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res 24: 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaki K, Talbert PB, Zhong CX, Dawe RK, Henikoff S, Jiang J. 2003. Chromatin immunoprecipitation reveals that the 180-bp satellite repeat is the key functional DNA element of Arabidopsis thaliana centromeres. Genetics 163: 1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaki K, Cheng Z, Ouyang S, Talbert PB, Kim M, Jones KM, Henikoff S, Buell CR, Jiang J. 2004. Sequencing of a rice centromere uncovers active genes. Nat Genet 36: 138–145. [DOI] [PubMed] [Google Scholar]
- Nakano M, Cardinale S, Noskov VN, Gassmann R, Vagnarelli P, Kandels-Lewis S, Larionov V, Earnshaw WC, Masumoto H. 2008. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev Cell 14: 507–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Schubert V, Fukova I, Manning JE, Houben A, Macas J. 2016. Epigenetic histone marks of extended meta-polycentric centromeres of Lathyrus and Pisum chromosomes. Front Plant Sci 7: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20: 289–290. [DOI] [PubMed] [Google Scholar]
- Pluta AF, Mackay AM, Ainsztein AM, Goldberg IG, Earnshaw WC. 1995. The centromere: hub of chromosomal activities. Science 270: 1591–1594. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Rosin L, Mellone BG. 2016. Co-evolving CENP-A and CAL1 domains mediate centromeric CENP-A deposition across Drosophila species. Dev Cell 37: 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. 2012. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round EK, Flowers SK, Richards EJ. 1997. Arabidopsis thaliana centromere regions: genetic map positions and repetitive DNA structure. Genome Res 7: 1045–1053. [DOI] [PubMed] [Google Scholar]
- Sanei M, Pickering R, Kumke K, Nasuda S, Houben A. 2011. Loss of centromeric histone H3 (CENH3) from centromeres precedes uniparental chromosome elimination in interspecific barley hybrids. Proc Natl Acad Sci 108: 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider KL, Xie Z, Wolfgruber TK, Presting GG. 2016. Inbreeding drives maize centromere evolution. Proc Natl Acad Sci 113: E987–E996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert V, Berr A, Meister A. 2012. Interphase chromatin organisation in Arabidopsis nuclei: constraints versus randomness. Chromosoma 121: 369–387. [DOI] [PubMed] [Google Scholar]
- Schueler MG, Higgins AW, Rudd MK, Gustashaw K, Willard HF. 2001. Genomic and genetic definition of a functional human centromere. Science 294: 109–115. [DOI] [PubMed] [Google Scholar]
- Schueler MG, Swanson W, Thomas PJ, Green ED. 2010. Adaptive evolution of foundation kinetochore proteins in primates. Mol Biol Evol 27: 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GP. 1976. Evolution of repeated DNA sequences by unequal crossover. Science 191: 528–535. [DOI] [PubMed] [Google Scholar]
- Talbert PB, Masuelli R, Tyagi AP, Comai L, Henikoff S. 2002. Centromeric localization and adaptive evolution of an Arabidopsis histone H3 variant. Plant Cell 14: 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Bryson TD, Henikoff S. 2004. Adaptive evolution of centromere proteins in plants and animals. J Biol 3: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tek AL, Kashihara K, Murata M, Nagaki K. 2010. Functional centromeres in soybean include two distinct tandem repeats and a retrotransposon. Chromosome Res 18: 337–347. [DOI] [PubMed] [Google Scholar]
- Wieland G, Orthaus S, Ohndorf S, Diekmann S, Hemmerich P. 2004. Functional complementation of human centromere protein A (CENP-A) by Cse4p from Saccharomyces cerevisiae. Mol Cell Biol 24: 6620–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgruber TK, Sharma A, Schneider KL, Albert PS, Koo DH, Shi J, Gao Z, Han F, Lee H, Xu R, et al. 2009. Maize centromere structure and evolution: Sequence analysis of centromeres 2 and 5 reveals dynamic loci shaped primarily by retrotransposons. PLoS Genet 5: e1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Lee HR, Koo DH, Jiang J. 2008a. Epigenetic modification of centromeric chromatin: hypomethylation of DNA sequences in the CENH3-associated chromatin in Arabidopsis thaliana and maize. Plant Cell 20: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008b. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong CX, Marshall JB, Topp C, Mroczek R, Kato A, Nagaki K, Birchler JA, Jiang J, Dawe RK. 2002. Centromeric retroelements and satellites interact with maize kinetochore protein CENH3. Plant Cell 14: 2825–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Zhang L, Duan J, Miki B, Wu K. 2005. HISTONE DEACETYLASE19 is involved in jasmonic acid and ethylene signaling of pathogen response in Arabidopsis. Plant Cell 17: 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.