

Abstract
The success of recombinant monoclonal immunoglobulins (IgG) is rooted in their ability to target distinct antigens with high affinity combined with an extraordinarily long serum half-life, typically around 3 weeks. The pharmacokinetics of IgGs is intimately linked to the recycling mechanism of the neonatal Fc receptor (FcRn). For long serum half-life of therapeutic IgGs, the highly pH-dependent interaction with FcRn needs to be balanced to allow efficient FcRn binding and release at slightly acidic pH and physiological pH, respectively. Some IgGs, like the antibody briakinumab has an unusually short half-life of ∼8 days. Here we dissect the molecular origins of excessive FcRn binding in therapeutic IgGs using a combination of hydrogen/deuterium exchange mass spectrometry and FcRn affinity chromatography. We provide experimental evidence for a two-pronged IgG-FcRn binding mechanism involving direct FcRn interactions with both the Fc region and the Fab regions of briakinumab, and correlate the occurrence of excessive FcRn binding to an unusually strong Fab-FcRn interaction.
The interaction between the neonatal Fc receptor (FcRn)1 and IgG is of high importance for effective humoral immunity and of considerable pharmaceutical interest because of its role in regulating IgG pharmacokinetics (1). FcRn binds antibodies in a strict pH-dependent manner with high binding affinity at mild acidic conditions (pH 6.0) and no measurable affinity at physiological pH (pH 7.4). This characteristic pH dependence is crucial for FcRn mediated IgG recycling. In the vascular endothelium FcRn binds IgG in the slightly acidic environment of the endosomes and release IgG to the blood stream at physiological pH thereby circumventing lysosomal degradation of IgG and leading to the extraordinary long half-life of most antibodies of 21 days (2).
Human IgGs consist of two antigen binding Fab regions and a Fc region involved in Fc receptor interactions. FcRn binds to the CH2-CH3 regions of the Fc domain of IgG (3, 4). However, several studies showing differential FcRn binding of recombinant IgGs with identical Fc regions (5, 6) suggest that the interaction could be influenced by the Fab regions. Schlothauer et al. (6) recently demonstrated that intact antibodies can dissociate from FcRn at different pH values than their corresponding Fc fragments strongly suggesting a contribution of the antibody Fab in FcRn binding. However, the role of the Fab regions in FcRn binding is still a matter of discussion in the literature (7–12) and the molecular mechanism is not clear. Limited structural information is available for the complex of full-length IgG and FcRn and much of our knowledge of the binding interface comes from X-ray crystal structures of only the Fc region bound to FcRn (3, 4).
We recently applied a method based on hydrogen/deuterium exchange mass spectrometry (HDX-MS) to detect local structural conformation and dynamics of IgG and FcRn upon interaction (13). HDX-MS monitors the isotopic exchange of hydrogen and deuterium of proteins in solution and reports on the protection of amide hydrogens from intra- and intermolecular hydrogen bonds and to a lesser extent solvent accessibility (14, 15). Our studies (13) revealed structural stabilization of the Fab regions upon FcRn interaction, which could be caused by a conformational link between the Fc and Fab regions or by direct interaction with FcRn.
Here, we perform HDX-MS analyses of a unique set of antibodies with different FcRn binding characteristics to dissect the conformational origins of the involvement of Fab in FcRn binding. The antibodies ustekinumab (Stelara) and briakinumab (Ozespa) targeting the p40 subunit of IL-12 and IL-23 show remarkable differences in terminal half-life with about 20 days for the former and only 8–9 days for the latter (16). Recent studies by Schoch et al. (17) indicates this difference in the elimination phase is FcRn-mediated as briakinumab showed excessive FcRn binding mediated by positively charged amino acids in the variable domain. While the antibodies had similar binding affinity at pH 5.5 (17), they showed large differences in FcRn dissociation pH when analyzed by FcRn affinity chromatography (17) (described in detail in (6)). Ustekinumab dissociated at a pH of ∼7.45 whereas the dissociation pH of briakinumab was 7.85. Mutation of three residues in the light chain (LC) complementarity determining region (CDR) of briakinumab (R27A, R57A and R97A) had a dramatic impact in dissociation pH changing the elution pH to 7.5. Interestingly, the isolated Fc fragments of the antibodies showed identical FcRn dissociation at a pH of about 7.45 (17) indicating a clear role of the briakinumab Fab region in differential FcRn dissociation of the intact IgGs.
EXPERIMENTAL PROCEDURES
Expression and Purification of Monoclonal Antibodies and FcRn
The recombinant human monoclonal IgG1 antibodies ustekinumab (CNTO 1275, Stelara, CAS Registry Number 815610-63-0), briakinumab (ABT 874, J 695, Ozespa, SEQ ID NO 36, PN WO2001014162-A2) and a briakinumab variant (R27A, R57A and R97A) were expressed and purified as described previously (17) and buffered in 50 mm Na phosphate, 50 mm NaCl, pH 6.5 (Roche Diagnostics, Penzberg, Germany). Recombinant human FcRn was expressed in HEK293F cells and purified as described previously (6) and buffered in 50 mm Na phosphate, 50 mm NaCl, pH 6.5 (Roche Diagnostics, Penzberg, Germany).
Hydrogen/Deuterium Exchange Mass Spectrometry
The antibodies ustekinumab, briakinumab and the briakinumab variant (72 pmol/μl) and FcRn (165 pmol/μl) were mixed and diluted in 99.9% D2O 50 mm Na phosphate, 50 mm NaCl, pH 6.5 to a final D2O content of 90% and an Ab and FcRn concentration of 1.2 pmol/μl and 8.5 pmol/μl, respectively. Hydrogen exchange was performed at 22 °C for various time points from 15 s up to 5 h. Fully deuterated samples were prepared by overnight incubation in 6 m deuterated Gnd-HCl at a final D2O content of 90%. Hydrogen exchange was quenched by a 1:1 (v/v) dilution of proteins with ice-cold 6 m Gnd-HCl, 0.5 m TCEP pH 2.3 resulting in a final pH of 2.5. The quenched samples were immediately frozen and stored at −80 °C. The samples were loaded onto a cooled Waters NanoAcquity UPLC system (Waters Corp., Milford, MA) for online pepsin digestion (Pierce, Rockford, IL, USA), desalting (Waters VanGuard C18, 1.7 μm, 2.1 × 5 mm) and reverse phase separation (Waters BEH C18, 1.7 μm, 1 × 100 mm) using a gradient from 8–40% 0.23% formic acid in acetonitrile (pH 2.5) in 7 min at a flow rate of 40 μl/min. Mass analysis was performed with a Waters Synapt G2 HDMS mass spectrometer using positive ion-electrospray and ion mobility. Peptic peptides were identified in by collision induced tandem mass spectrometry using both data dependent and -independent acquisition methods. Deuterium incorporation was determined in DynamX ver. 3.0 (Waters Corp.). The generation of the homology model of briakinumab is described elsewhere(17) and visualized using PyMOL (Schrödinger LLC).
FcRn Affinity Chromatography
FcRn affinity columns were prepared as described previously (6). For analysis of IgG-antigen complexes, briakinumab and ustekinumab were bound to their target interleukin-12 (CellSciences, Canton, MA), by mixing 0.3 nmol IgG with 0.2 nmol IL-12. In addition, a briakinumab-IL-12 complex was assembled using an IL-12 excess by mixing 0.3 nmol mAb with 0.8 nmol IL-12. The mAb-IL12 complexes were incubated for 30 min at RT before FcRn affinity chromatography. The antibodies were eluted by a linear pH gradient from pH 5.5 to 8.8 within 120 min using 20 mm MES, 140 mm NaCl, pH 5.5 and 20 mm TRIS, 140 mm NaCl, pH 8.8 as eluents. To determine the elution pH at particular retention times, samples were collected every 5 min and the pH was measured offline.
RESULTS
Backbone Amide Hydrogens in the Antibody Fab Become Protected from HDX Upon FcRn Binding
To investigate the molecular mechanism of FcRn interaction of these antibodies, we have performed solution phase HDX-MS experiments of briakinumab, the briakinumab variant (R27A, R57A, and R97A) and ustekinumab in the absence and presence of FcRn (supplemental Fig. S1–S6). Our analyses have revealed that several segments of the Fab regions of briakinumab and its variant, particularly the LC CDRs, are stabilized upon FcRn binding (Fig. 1).
Fig. 1.

Conformational stabilization of Fab peptides revealed by HDX-MS. Homology model of briakinumab with a zoomed view of one Fab arm with residues R27, R57 and R97 shown. In the zoomed view, regions with no difference in HDX in unbound and FcRn bound IgG are shown in gray, regions showing reduced HDX upon FcRn binding are shown in red and regions without coverage are shown in white. Deuterium uptake plots show HDX curves of briakinumab (black), briakinumab variant (red) without (bold line) and with FcRn (dashed line). Deuterium incorporation was determined in triplicates in a time range from 15 s to 5 h labeling of unbound or FcRn-bound IgG. The fully deuterated control of briakinumab is shown (gray star) at the last time point. **: p < 0.01, *: p < 0.05 indicate the level of significance between of the deuterium uptake of IgG in the absence and presence of FcRn.
The HDX of LC peptides 13–39, 48/49–73/74, 50–74, 75–86, 88–101, and 92–101 was slowed as a result of the formation/stabilization of hydrogen bonds or/and shielding from solvent, upon FcRn binding. Interestingly, the protected peptides except peptide 75–86 correlates to the regions of the Fab that were previously implicated in the charge dependent interaction with FcRn (17). The difference in HDX in the unbound and FcRn-bound state is comparable for briakinumab and the variant. However, as the HDX of the FcRn-bound briakinumab variant is higher relative to that of briakinumab, the variant is not stabilized to the same level as briakinumab in the presence of FcRn in any of the peptides. The HDX of the variant was increased for all peptides containing the point mutations indicating a destabilization of hydrogen bonds in these regions. Differences in intrinsic exchange rates (18) because of the mutation of arginine to alanine does not cause significant changes in deuterium incorporation that could account for the difference observed between briakinumab and the variant (supplemental Fig. S8). We thus observe that the conformation of the Fab of briakinumab variant is destabilized by mutation. Furthermore, these same regions are not stabilized to the same level upon FcRn binding, as for briakinumab, in good correlation with a reduced FcRn binding of this variant. In the heavy chain (HC), peptides in the region 51–80 also show reduced HDX upon FcRn interaction (supplemental Fig. S1). Interestingly, previous studies (17) showed substitution of the positively charged amino acids (K57A, K64A) in this region impact FcRn binding negatively albeit not to the same level as the briakinumab variant with LC substitutions (R27A, R57A, and R97A) described here. Comparison of the deuterium content of briakinumab and ustekinumab LC peptides is not directly possible as the LC sequence identity is only 43% (supplemental Fig. S8) because of the different types of LCs (lambda for briakinumab and kappa for ustekinumab). Ustekinumab only showed reduced deuterium level upon FcRn binding in one region (LC peptide 50–70) in the variable domain (supplemental Fig. S5) corresponding to our previous results of another IgG with a kappa LC (13).
Differential HDX Protection in the CH2 Region of Briakinumab and Variant Upon FcRn Binding
In the antibody Fc region the HDX-MS data revealed several regions in the antibody CH2 and CH3 domain that become protected from HDX upon binding to FcRn in good overall agreement with the IgG binding interface as revealed by X-ray crystallography of FcRn and the Fc region of IgG (4) (supplemental Fig. S1). Several overlapping peptides in the region 233–250 constituting a small α-helix-loop region in the CH2/CH3 domain interface (Fig. 2) show reduced deuterium uptake when bound to FcRn for both briakinumab and the variant which was also observed previously for an unrelated antibody (13).
Fig. 2.

Differential impact of FcRn binding in the Fc region (CH2) of briakinumab and its variant revealed by HDX-MS. A, Homology model of briakinumab with zoomed view of region 241–250 (red) with the backbone amide hydrogens shown as green spheres. B, Deuterium uptake plot for peptide 241–250 showing HDX curves of briakinumab (black), the briakinumab variant (red) without (bold) and bound to FcRn (dashed). The fully deuterated control for briakinumab is shown (in gray star) at the last time point. **: p < 0.01, *: p < 0.05 indicate significance levels comparing briakinumab and the variant when bound to FcRn. The HDX-MS experiments were performed as described in Fig. 1. C, Difference in HDX of peptide 241–250 at the 1 h time point for briakinumab (gray), the briakinumab variant (red) and ustekinumab (blue).
Strikingly, briakinumab showed more pronounced reduction in HDX upon FcRn binding in this region in comparison to the variant. This suggests that FcRn binding to briakinumab leads to an increased stabilization of this CH2 region relative to the variant with mutations in the LC. Notably, no differences in HDX are observed the CH2 region between unbound briakinumab and the variant, indicating that the amino acid substitution in the LC Fab alone does not result in altered conformational dynamics in this region. The differential HDX between briakinumab and its variant in the CH2 region is thus not associated to structural differences in the Fc region of the unbound states of these two antibodies. Interestingly, the reduction in HDX in CH2 region of the briakinumab variant (residues 241–250) upon FcRn binding is comparable to that of ustekinumab (Fig. 2C). Both these antibodies do not display excessive FcRn binding in contrast to briakinumab. The FcRn affinity of all antibodies at pH 6.5 are in the very low micromolar range (estimated Kd from 0.3–3 μm) with a measured relative affinity normalized to the Kd of ustekinumab of 0.4 and 0.2 for the briakinumab variant and briakinumab, respectively (17). As the labeling is performed with a large molar surplus of FcRn, the small differences in FcRn affinity between the antibodies cannot account for the observed differences in HDX of the FcRn-bound states.
Influence of Antigen Binding on FcRn Dissociation
To investigate the role of the Fab regions on FcRn binding we compared the FcRn interaction of unbound and IL-12 bound ustekinumab and briakinumab by FcRn affinity chromatography (Fig. 3).
Fig. 3.
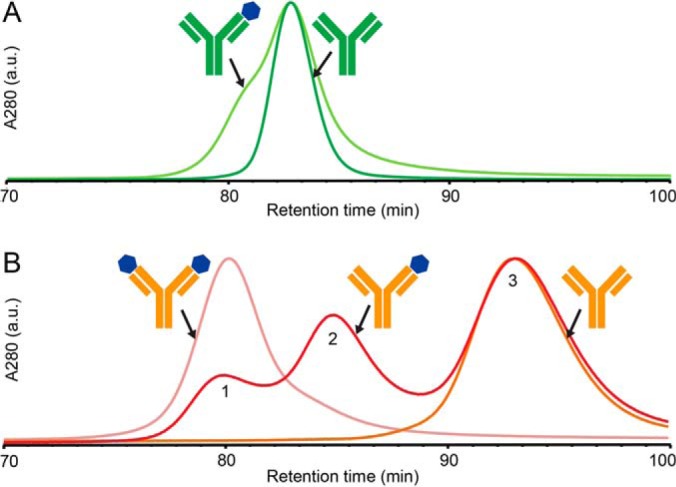
FcRn affinity chromatography of ustekinumab and briakinumab in complex with IL-12. A, ustekinumab (dark green) and ustekinumab-IL-12 complex with excess ustekinumab (light green). B, briakinumab (orange), briakinumab-IL-12 complex with excess briakinumab (red) and with excess IL-12 (light red). Chromatographic peaks are normalized to main peak of ustekinumab in A, and briakinumab-IL-12 complex assembled using IL-12 excess in B. IgGs and IgG-IL-12 complexes were loaded on the FcRn column at pH 5.5 and were eluted with a 2 h pH gradient to pH 8.8.
Target binding of ustekinumab only had a minor effect on FcRn dissociation as IL-12 interactions lead to a reduced retention of approximately 2 mins in comparison to unbound ustekinumab (peak fronting in Fig. 3A). Oppositely, briakinumab showed large target-mediated differences in FcRn dissociation pH. Although unbound briakinumab shows one late eluting peak with a retention time of approx. 93 min. (peak 3, Fig. 3B), the briakinumab-IL-12 complex assembled using an excess of IL-12 eluted as a single peak with a markedly reduced retention time at approx. 80 min (peak 1). The briakinumab-IL-12 complex assembled using an excess of briakinumab show three peaks with vastly different FcRn column retention times (Fig. 3B), corresponding to unbound briakinumab (peak 3), briakinumab bound to one (peak 2) or two IL-12 molecules (peak 1), respectively.
Antigen binding thus leads to a large shift in FcRn dissociation pH for briakinumab whereas the effect on ustekinumab is minor. Antigen bound briakinumab therefore appears to display reduced FcRn binding under elevated pH conditions because of direct steric hindrance of either the Fab-FcRn interaction or the Fc-FcRn interaction. The latter option does, however, not seem plausible as antigen binding to ustekinumab only has a minor impact on the FcRn interaction as would not be expected if IL-12 binding shields the Fc-FcRn interaction. Our results thus strongly support that impaired FcRn binding of IL12-bound briakinumab is a result of a reduced Fab-FcRn interaction, in turn, leading to a dissociation pH comparable to ustekinumab and Fc fragments (17).
DISCUSSION
So how do Fab arm mutations cause differential HDX in the Fc region upon binding to FcRn? We propose a two-step binding mechanism where the briakinumab Fab arms are directly involved in FcRn binding to explain the differences in HDX observed in the Fab and Fc region (Fig. 4).
Fig. 4.
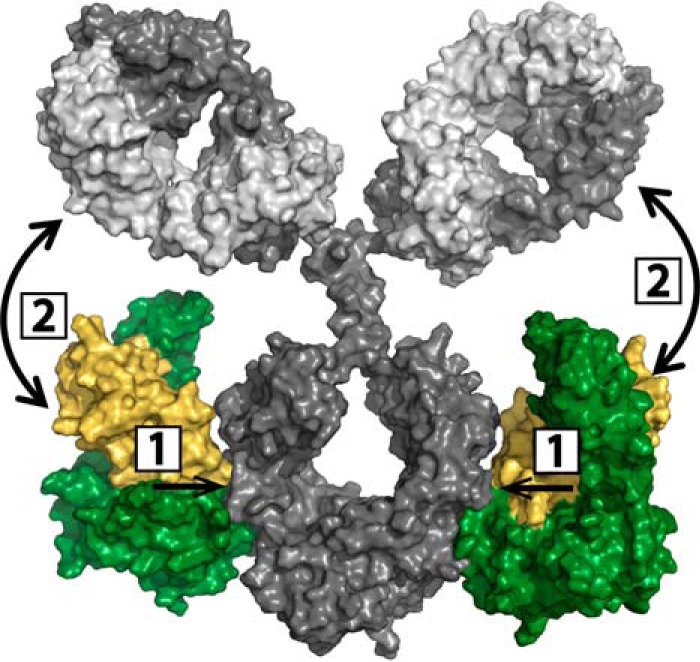
Model for two-pronged IgG-FcRn binding mechanism. Step 1: High affinity interaction of IgG CH2/CH3 domains and FcRn, step 2: Fab-FcRn interaction for briakinumab. The structural model shows the IgG HC (dark gray), LC (light gray), FcRn α-chain (green) and β2-microglobulin (yellow).
In the two-pronged binding mechanism for briakinumab the initial step (Step 1) is the high affinity binding of FcRn to the Fc region (3, 4), which is followed by a direct interaction of Fab regions with FcRn (Step 2, Fig. 4). In support of this we have shown that cleaved Fab arms have no measurable affinity to FcRn by SPR or FcRn chromatography (6). Thus, the Fab-FcRn interaction requires binding of FcRn to Fc, bringing Fab and FcRn in close spatial proximity. Our HDX-MS data shows that the subsequent Fab-FcRn interaction in turn leads to a more stable Fc-FcRn interaction in the 233–250 region, as we see increased stabilization upon FcRn binding for briakinumab relative to the variant for which the Fab-FcRn interaction (Step 2) is weakened (Fig. 1 and 2). The involvement of direct Fab-FcRn interactions following the high affinity Fc-FcRn binding thus leads to a more stable complex in comparison to IgG-FcRn complexes that do not involve strong Fab interactions.
As a consequence of a strong involvement of Fab arms in the FcRn interaction of briakinumab as opposed to ustekinumab, we hypothesized that antigen binding (IL-12) could have a significant effect on the FcRn binding of briakinumab as opposed to ustekinumab. The results from FcRn chromatography revealed a large impact of antigen binding that was unique for briakinumab and not observed for ustekinumab. These observations thus further support the proposed two-step binding mechanism involving a direct interaction of Fc- and subsequently Fab regions with the FcRn (Fig. 4). Interestingly, it appears that both Fab arms of briakinumab can simultaneously interact with cognate FcRn molecules bound on each side of the Fc region as three peaks with distinct FcRn binding are observed for the briakinumab-IL-12 complex. The Fab arms of IgG molecules are inherently very flexible in solution and the required movements for FcRn interaction for both Fab arms is accordance with previously measured Fab arm rotation angles and Fab-Fc and Fab-Fab angle ranges (19). Our data clearly show how a limitation of Fab arm mobility as a consequence of antigen binding can limit the Fab-FcRn interaction. This may explain some of the current discrepancies in the literature on the role of the Fab in FcRn interaction as Fab arm mobility has been limited by interaction with antigen or Fab specific antibodies in some of the used in vitro assays (7, 10).
In summary, our data reveal a direct impact of the Fab region of a therapeutic IgG on FcRn binding and significantly expand current knowledge on a functionally important structural interplay between both the Fc and Fab regions of IgG upon complex formation with FcRn. Based on the results described here we conclude that the involvement of the Fab of briakinumab in FcRn binding occurs by a direct interaction. HDX data on the FcRn binding of another IgG1 (13) have also shown reduced HDX in the Fab region. This strongly suggests that the two-pronged binding mechanism of Fc and Fab to FcRn, uncovered here for briakinumab, is an inherent property of the IgG1 architecture and also occurs in other IgG1 molecules, albeit with a varying strength of the Fab interaction as reported in (5, 6). While the LC of briakinumab strongly influences the FcRn interaction as described here and previously (17), a possible correlation to LC type (kappa or lambda) needs to be investigated further. Our results demonstrate that differences in FcRn binding of therapeutic IgGs can be because of differences in the involvement of the Fc and, in cases such as briakinumab, the Fab regions. Evidently, the contribution of both Fc and Fab regions to FcRn binding can have significant impact on terminal half-life as observed for briakinumab and ultimately efficacy of therapeutic IgGs and should be dissected and analyzed for successful development of therapeutic IgGs with tailored pharmacokinetics.
Supplementary Material
Acknowledgments
We thank Dr. Alex Bujozeck at Roche Innovation Center Penzberg, for making the IgG-FcRn model in figure 4.
Footnotes
Author contributions: P.F.J., A.S., M.H., T.S., T.E., and K.D.R. designed research; P.F.J., A.S., and V.L. performed research; P.F.J., A.S., T.S., T.E., and K.D.R. contributed new reagents or analytic tools; P.F.J., A.S., V.L., and K.D.R. analyzed data; P.F.J., A.S., M.H., T.S., T.E., and K.D.R. wrote the paper.
* This work was supported by the Roche Biologics Technology Evaluation & Development program, the Marie Curie Actions Programme of the EU (Grant no. PCIG09-GA-2001-292414) and the Danish Council for Independent Research Natural Sciences (Steno Grant no.11-104058).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- FcRn
- neonatal Fc receptor
- HDX-MS
- hydrogen/deuterium exchange mass spectrometry
- HC
- heavy chain
- LC
- light chain
- CDR
- complementarity determining region.
REFERENCES
- 1. Waldmann T. A., and Strober W. (1969) Metabolism of immunoglobulins. Prog. Allergy 13, 1–110 [DOI] [PubMed] [Google Scholar]
- 2. Roopenian D. C., and Akilesh S. (2007) FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. 7, 715–725 [DOI] [PubMed] [Google Scholar]
- 3. Burmeister W. P., Huber A. H., and Bjorkman P. J. (1994) Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 372, 379–383 [DOI] [PubMed] [Google Scholar]
- 4. Oganesyan V., Damschroder M. M., Cook K. E., Li Q., Gao C., Wu H., and Dall'acqua W. F. (2014) Structural Insights into Neonatal Fc Receptor-based Recycling Mechanisms. J. Biol. Chem. 289, 7812–7824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang W., Lu P., Fang Y., Hamuro L., Pittman T., Carr B., Hochman J., and Prueksaritanont T. (2011) Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab. Dispos. 39, 1469–1477 [DOI] [PubMed] [Google Scholar]
- 6. Schlothauer T., Rueger P., Stracke J. O., Hertenberger H., Fingas F., Kling L., Emrich T., Drabner G., Seeber S., Auer J., Koch S., and Papadimitriou A. (2013) Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. mAbs 5, 576–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Igawa T., Tsunoda H., Tachibana T., Maeda A., Mimoto F., Moriyama C., Nanami M., Sekimori Y., Nabuchi Y., Aso Y., and Hattori K. (2010) Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng., Design Selection 23, 385–392 [DOI] [PubMed] [Google Scholar]
- 8. Li B., Tesar D., Boswell C. A., Cahaya H. S., Wong A., Zhang J., Meng Y. G., Eigenbrot C., Pantua H., Diao J., Kapadia S. B., Deng R., and Kelley R. F. (2014) Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 6, 1255–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neuber T., Frese K., Jaehrling J., Jager S., Daubert D., Felderer K., Linnemann M., Hohne A., Kaden S., Kolln J., Tiller T., Brocks B., Ostendorp R., and Pabst S. (2014) Characterization and screening of IgG binding to the neonatal Fc receptor. mAbs 6, 928–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdiche Y. N., Yeung Y. A., Chaparro-Riggers J., Barman I., Strop P., Chin S. M., Pham A., Bolton G., McDonough D., Lindquist K., Pons J., and Rajpal A. (2015) The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. mAbs 7, 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu Q., Lee H. Y., Wong P. Y., Jiang G., and Gazzano-Santoro H. (2015) Development and applications of AlphaScreen-based FcRn binding assay to characterize monoclonal antibodies. J. Immunol. Methods 420, 31–37 [DOI] [PubMed] [Google Scholar]
- 12. Souders C. A., Nelson S. C., Wang Y., Crowley A. R., Klempner M. S., and Thomas W. Jr. (2015) A novel in vitro assay to predict neonatal Fc receptor-mediated human IgG half-life. mAbs 7, 912–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jensen P. F., Larraillet V., Schlothauer T., Kettenberger H., Hilger M., and Rand K. D. (2015) Investigating the Interaction between the Neonatal Fc Receptor and Monoclonal Antibody Variants by Hydrogen/Deuterium Exchange Mass Spectrometry. Mol. Cell. Proteomics 14, 148–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Skinner J. J., Lim W. K., Bedard S., Black B. E., and Englander S. W. (2012) Protein dynamics viewed by hydrogen exchange. Protein Sci. 21, 996–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Skinner J. J., Lim W. K., Bedard S., Black B. E., and Englander S. W. (2012) Protein hydrogen exchange: Testing current models. Protein Sci. 21, 987–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weger W. (2010) Current status and new developments in the treatment of psoriasis and psoriatic arthritis with biological agents. Br. J. Pharmacol. 160, 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schoch A., Kettenberger H., Mundigl O., Winter G., Engert J., Heinrich J., and Emrich T. (2015) Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc. Natl. Acad. Sci. USA 112, 5997–6002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bai Y., Milne J. S., Mayne L., and Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sandin S., Öfverstedt L.-G., Wikström A.-C., Wrange Ö., and Skoglund U. (2004) Structure and Flexibility of Individual Immunoglobulin G Molecules in Solution. Structure 12, 409–415 [DOI] [PubMed] [Google Scholar]
- 20. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.