

Abstract
Toxoplasma kinase ROP18 is a key molecule responsible for the virulence of Toxoplasma gondii; however, the mechanisms by which ROP18 exerts parasite virulence via interaction with host proteins remain limited to a small number of identified substrates. To identify a broader array of ROP18 substrates, we successfully purified bioactive mature ROP18 and used it to probe a human proteome array. Sixty eight new putative host targets were identified. Functional annotation analysis suggested that these proteins have a variety of functions, including metabolic process, kinase activity and phosphorylation, cell growth, apoptosis and cell death, and immunity, indicating a pleiotropic role of ROP18 kinase. Among these proteins, four candidates, p53, p38, UBE2N, and Smad1, were further validated. We demonstrated that ROP18 targets p53, p38, UBE2N, and Smad1 for degradation. Importantly, we demonstrated that ROP18 phosphorylates Smad1 Ser-187 to trigger its proteasome-dependent degradation. Further functional characterization of the substrates of ROP18 may enhance understanding of the pathogenesis of Toxoplasma infection and provide new therapeutic targets. Similar strategies could be used to identify novel host targets for other microbial kinases functioning at the pathogen-host interface.
Toxoplasma gondii is a widespread pathogen parasite that infects a broad range of warm-blooded animals, including humans, causing severe toxoplasmosis in congenitally infected newborns and immunocompromised individuals (1, 2). Despite progress in the clinical management of this disease, important limitations remain, such as the lack of a commercial vaccine and imperfect drugs for toxoplasmic encephalitis (3, 4). Therefore, the discovery of novel targets and effective therapeutic strategies against toxoplasmosis is of acute importance.
During infection, T. gondii delivers numerous effectors into the parasitophorous vacuole and the host cytoplasm to co-opt the host cell for survival and replication (5). Among these effectors, ROP18 is suggested to be a key determinant responsible for the virulence of T. gondii (6, 7). ROP18 is a member of the ROP2 subfamily of protein kinase, which is secreted from apical organelle rhoptries into the host cell and localizes to the parasitophorous vacuole membranes (PVMs)1 during invasion (8). ROP18 is a Ser/Thr protein kinase, which mediates parasite virulence through targeting and phosphorylating host proteins (9).
In recent years, several substrates have been identified for ROP18 (10–13). ROP18 has been reported to phosphorylate the immunity-related GTPases, Irga6 and Irgb6, preventing their accumulation at PVMs and the clearance of parasites at an early stage after infection (10, 11). Given the absence of an immunity-related GTPase system in humans, it is likely that additional host defense systems may be targeted by ROP18. Using a yeast two-hybrid screening, Yamamoto et al. (12) have identified another target of ROP18, the host endoplasmic reticulum-bound transcription factor ATF6β. ROP18 phosphorylates ATF6β and triggers its proteasomal degradation, and thus it impairs the ATF6β-dependent immune responses at a later stage after infection (12). More recently, a study showed that via phosphorylation, ROP18 mediates ubiquitin-dependent degradation of p65, leading to suppression of the NF-κB pathway to limit expression of proinflammatory cytokines (13). Although Cheng et al. (14) have identified several ROP18-interacting human proteins using a yeast two-hybrid technology, it remains unclear whether these proteins are substrates of ROP18. Overall, these studies have provided important insights into the basis of ROP18-mediated parasite virulence; however, the full scope of ROP18's contribution to virulence remains poorly understood. Identification of other ROP18 substrates is needed to more completely understand the mechanisms of ROP18-mediated augmentation of the disease.
Functional proteome array technology has been established as a powerful tool for proteome-wide identification of substrates for a variety of post-translational modification enzymes, such as protein kinases, acetyltransferases, and ubiquitin E3 ligases (15–17). For example, functional human protein microarrays have been used to successfully identify substrates of >370 human kinases (18–24). In this study, we utilized a human proteome array (i.e. HuProt), composed of >16,000 human proteins, to globally identify host cell proteins that could be phosphorylated by ROP18 kinase, and we found multiple potential host cellular substrates involved in a wide range of functions. Four of these, p53, p38, UBE2N, and Smad1, were selected for further in vitro and in vivo characterization.
MATERIALS AND METHODS
Cells and Parasites
All parasites were maintained by serial passage in human foreskin fibroblasts (HFFs) as described previously (25). Tachyzoites of the RH strain and ROP18 knockout (ROP18-KO) RH strain of T. gondii were kindly provided by Professor Vern B. Carruthers (University of Michigan School of Medicine). Tachyzoites of the T. gondii ROP18-Ty1 (overexpression of Ty1-tagged ROP18) RH strain were kindly provided by Professor Jian Du (Anhui Medical University, Anhui, China). The HFF, HeLa, and 293T cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (DMEM, high glucose, and pyruvate from Gibco) containing 100 units/ml penicillin and 100 μg/ml streptomycin (Gibco) and 10% fetal bovine serum (Gibco).
Reagents
Monoclonal antibody against the V5 epitope tag (catalog no. MCA1360GA) was purchased from AbDSerotec (Kidlington, Oxford, UK). Antibodies against Ty1 (catalog no. SAB4800032), FLAG (catalog no. F7425), and GFP (catalog no. G1544) tags were purchased from Sigma-Aldrich. Antibodies against UBE2N (catalog no. ab109286) and phospho-Smad1 (Ser-187) (catalog no. ab73211) were obtained from Abcam (Cambridge, MA). Antibodies against GAPDH (14C10) (catalog no. 2118S), p53 (1C12) (catalog no. 2524), p38 MAPK (D13E1) XP (catalog no. 8690), and Smad1 (D59D7) (catalog no. 6944) were obtained from Cell Signaling Technology (Beverly, MA). Antibody against SAG1 was kindly provided by Professor Ho-Woo Nam (College of Medicine, The Catholic University of Korea, Seoul, Korea) (26, 27). Horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies were purchased from Sigma-Aldrich. Protease inhibitor mixture (catalog no. CW2200S) and phosphatase inhibitor mixture (catalog no. CW2383S) were purchased from CWbiotech (Beijing, China). Alexa Fluor®488 goat anti-mouse IgG (catalog no. A11001) and Alexa Fluor®555 goat anti-rabbit IgG (catalog no. A21428) were obtained from Life Technologies, Inc. Recombinant human EGFR protein was purchased from Thermo Fisher Scientific (Waltham, MA). MG132 (catalog no. 474790) was from Millipore (Billerica, MA).
Generation of Constructs
The ROP18 gene (GenBankTM accession no. JX045330) was PCR-amplified from genomic DNA of T. gondii RH strain parasites with the forward primer 5′-ACCGTCGTCCGAATGGGTTTAG-3′ and the reverse primer 5′-CAATAATGCCATCCGTCGAGAG-3′. The fragment of ROP18 lacking the N-terminal signal peptide and prodomain (ROP18Δ83, amino acids 83–539, numbering based on the second ATG codon) (28) was further amplified with the primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAAGGGCTCAACACCGGG-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTTATTCTGTGTGGAGATGTTC-3′. Underlines indicate the recombination sites attB1 and attB2, respectively. GST-ROP18Δ83 and V5-ROP18Δ83 plasmids were constructed by utilizing the Gateway cloning system (Invitrogen). Briefly, the PCR product was cloned into pDONR201 vector through BR recombination reaction to construct the entry clone (pDONR201-ROP18Δ83). After confirming by restriction enzyme digestion and sequencing, ROP18Δ83 was subcloned from the entry clone into the pEGH-A vector that produces GST-His6 fusion protein (GST-ROP18Δ83) or pcDNA3.1/nV5-DEST vector that produces V5-tagged protein (V5-ROP18Δ83) through LR recombination reaction. The kinase-dead ROP18 mutant (D394A) (ROP18Δ83-KD) was generated using the QuikChange® Lightning Multi Site-directed mutagenesis kit (Agilent Technologies) with the primer 5′-ATTGTGCATACGGCTATCAAACCGGCG-3′ using the pDONR201-ROP18Δ83 plasmid as a template. Then ROP18Δ83-KD was subcloned from the entry clone into pEGH-A vector to construct GST-ROP18Δ83-KD or pcDNA3.1/nV5-DEST to construct V5-ROP18Δ83-KD, through LR recombination reaction. Total RNA was extracted from 293T cells by TRIZOL reagent (Invitrogen), and the cDNA was synthesized by Moloney murine leukemia virus reverse transcription kit (Promega). The human TP53 cDNA (GenBankTM accession no. NM_000546) was amplified by PCR with the following primers, 5′-CGGGGATCCATGGAGGAGCCGCAGTCAGA-3′ and 5′-CGCGAATTCTCAGTCTGAGTCAGGCCCTTC-3′, and cloned into pCMV-Tag2B vector to produce a FLAG-tagged protein (FLAG-p53) with BamHI and EcoRI enzyme digestion sites (underlined). The human SMAD1 cDNA (GenBankTM accession no. NM_001003688) was amplified with primers 5′-CGGGGATCCATGAATGTGACAAGTTTA-3′ and 5′-CGCAAGCTTTTAAGATACAGATGAAATAG-3′ and cloned into pCMV-Tag2B vector to produce a FLAG-tagged protein (FLAG-Smad1) or bacterial expression vector pET-30a to produce a His-tagged protein (His-Smad1), using the BamHI and HindIII digestion sites (underlined). Mutations in SMAD1 were induced using the QuikChange® Lightning Multi Site-directed mutagenesis kit using the following primers: S187A, 5′-CCACCCGTTTCCTCACGCTCCCAATAGCAGTTA-3′; S195A, 5′-GCAGTTACCCAAACGCTCCTGGGAGCAGC-3′; S206A, 5′-CACCTACCCTCACGCTCCCACCAGCTC-3′; and S214A, 5′-ACCAGCTCAGACCCAGGAGCCCCTTTCCAGATG-3′. The ROP18Δ27-GFP plasmid was kindly provided by Professor Jian Du (Anhui Medical University, Anhui, China) (13).
Protein Expression and Purification
Protein expression and purification were performed as described previously (29). Briefly, GST-ROP18Δ83 and GST-ROP18Δ83-KD recombinant constructs were transformed into yeast host strain Y258. Yeast was cultured in SC-URA/glucose liquid medium at 30 °C with shaking to saturation. The saturated cultures were diluted 1:125 into SC-URA/raffinose liquid medium, and the cultures were incubated at 30 °C with shaking. When the A600 of cultures reached 0.7–0.9, the cultures were induced with 2% galactose for 4 h at 30 °C. Then the yeast cells were harvested and lysed with zirconia beads (0.5 mm diameter from BSP, Germany) in cold lysis buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm EGTA, 10% glycerol, 0.1% Triton X-100, and 0.1% β-mercaptoethanol, 1 mm PMSF, and protease mixture inhibitors). GST fusion proteins were bound to glutathione-Sepharose (Amersham Biosciences) for 1 h at 4 °C and washed four times for 15 min each time with wash I buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm EGTA, 0.1% Triton X-100, 0.1% β-mercaptoethanol, 0.5 mm PMSF, and protease mixture inhibitors) and two times with wash II buffer (50 mm HEPES, pH 7.4, 100 mm NaCl, 1 mm EGTA, 10% glycerol, 0.1% β-mercaptoethanol, and 0.5 mm PMSF) and eluted in elution buffer (100 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10 mm MgCl2, 20 mm glutathione, and 20% glycerol). Eluates were collected and stored at −80 °C for further experiments.
Dot Blot Kinase Assay
A dot blot kinase assay was performed as described previously with slight modifications (30). Briefly, 10 μl of GST-ROP18Δ83 or GST-ROP18Δ83-KD protein solution was added to a 96-well plate with 10 μl of kinase assay buffer containing 2 μCi of [γ-32P]ATP and incubated for 30 min at 30 °C. Then 5 μl of mixture was dotted onto the nitrocellulose membrane. After air-drying, the membrane was washed three times with TBST buffer (100 mm Tris-HCl, pH 7.5, 0.9% NaCl, and 0.1% Tween 20). After air-drying again, the membrane was exposed to the X-ray film.
Protein Array Phosphorylation Assays
The HuProt array was fabricated by spotting down 16,368 unique full-length human recombinant proteins in duplicate along with several control proteins provided by CDI Laboratories, Inc. (31, 32). The protocol for phosphorylation assays on HuProt arrays was modified from that of previous publications (23, 24). Briefly, HuProt arrays were preincubated in Superblock (Pierce) containing 0.2% BSA and 1% Triton X-100 for 1 h at room temperature. Purified GST-ROP18Δ83 and GST-ROP18Δ83-KD recombinant proteins were diluted with kinase buffer (50 mm Tris-HCl, pH 7.4, 20 mm HEPES, pH 7.5, 100 mm NaCl, 10 mm MgCl2, 1 mm DTT, 1 mm EGTA, and 20 mm cold ATP) and [γ-32P]ATP (33.3 nm final concentration). Duplicate HuProt arrays were incubated with the kinase mixture in a humidity chamber for 30 min at 30 °C. Slides were subjected to three 10-min washes with TBST buffer (25 mm Tris-HCl, pH 7.4, 3 mm KCl, 150 mm NaCl, 0.05% Tween 20) and three 10-min washes with 10 mm Tris-HCl, pH 7.4, and 0.5% SDS, rinsed briefly with doubly distilled water, and spun dry. Control slides were incubated with kinase buffer without kinase and processed in parallel. The slides were exposed to X-ray films that were scanned at 1,800 pixels/inch and analyzed using GenePix 3.0 (Molecular Devices). The experiments were performed in duplicate, and only those that were reproducible were scored as positives.
Protein Microarray Data Analysis
Following chip scanning, background correction was performed to obtain the signal intensity value for each spot using the ratio of foreground to background signals. Then, between-array normalization was performed. To identify positive hits on the microarrays, we estimated the standard deviation from the signal intensity distribution, as described in detail previously (33). Cutoff values of 2.0 and 2.5 standard deviations above the mean were chosen in this study. For proteins printed in duplicate, the ones with both spots meeting our defined cutoff were defined as positive hits. Finally, because it is impossible to distinguish signals arising from auto-phosphorylation events and trans-phosphorylation events, we conducted negative control experiments during each set of assays to identify proteins that underwent auto-phosphorylation on the protein microarrays. The proteins that exhibited auto-phosphorylation activity in the negative control experiments were excluded from the substrate list for further analysis. These proteins were obtained using cutoff values of 2.0 standard deviations above the mean. Background correction and between-array normalization were conducted using limma package (34).
Gene Ontology and Functional Protein Analysis
We calculated the occurrence frequency of each GO term or KEGG pathway within foreground (e.g. ROP18 substrates) and background gene groups (e.g. the whole human genome), respectively. Then we used a hypergeometric model to evaluate the statistical significance of comparison between foreground and background, followed by multiple-test Bonferroni correction (for GO) or false discovery rate correction (for KEGG pathway). A GO term or KEGG pathway was defined as an enriched one if its frequency in the foreground was larger than that in the background and its corrected p value was less than 0.05.
Because many detailed GO terms were found enriched for ROP18 substrates, we merged them into five general groups as follows: metabolic process, kinase activity/phosphorylation, response to growth factor, apoptotic process/cell death, and immune system process. We merged associated GO terms into these five categories and then re-calculated the occurrence frequencies for the foreground and background gene groups, respectively, and we obtained corresponding p values based on hypergeometric distribution.
Similarity Comparison with ROP18
We used the database, PhosphoNetworks, to investigate phosphorylation substrates of 289 kinases. The hypergeometric probability of finding a certain number of common targets between kinase i and ROP18 is given in Equation 1,
| (Eq. 1) |
where S0 is the total number of unique substrates in the database; Ki is the number of substrates of kinase i; n is the number of ROP18 substrates detected in the database; and m is the number of common targets between ROP18 and kinase i.
In Vitro Infection
Cells were infected by tachyzoites of RH strain, as described previously (35) with slight modifications. Briefly, cells were plated in 12-well plates 24 h before infection. Fresh tachyzoites of the RH strain were added to plates at a density of 5.0 × 105 per well. After 5 h of infection, uninvaded parasites were washed with pre-warmed PBS and refilled with fresh medium. At 36 h post-infection, total cell lysates were harvested for further experiments.
Transfection
Transfections were carried out using LipoFiterTM reagent (HANBio, Shanghai, China) as instructed by the manufacturer. 293T cells (2 × 105 per well) were seeded into 12-well plates coated with poly-l-lysine (Sangon Biotech, Shanghai, China). 24 h later, cells were incubated with plasmids mixed with LipoFiterTM reagent in DMEM basic medium for 6 h. Then the DMEM basic medium was replaced with complete DMEM. Cells were harvested at the indicated time points and used for further experiments.
Immunofluorescence Staining
Immunofluorescence staining was performed as described previously with slight modifications (25, 28). Briefly, HeLa monolayers grown on coverslips were transfected with V5-ROP18Δ83 alone or together with FLAG-p53. At 24 h post-transfection, cells were infected with tachyzoites of T. gondii. 24 h after infection, cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.02% digitonin in PBS for 10 min (26). After blocking with 3% BSA in PBS for 30 min, cells were incubated with primary anti-V5 and anti-FLAG (or anti-p38 MAPK) antibodies at 4 °C overnight followed by incubation with Alexa Fluor®488 goat anti-mouse IgG and Alexa Fluor®555 goat anti-rabbit IgG. Coverslips were mounted with ProLong® Gold antifade reagent containing DAPI (Life Technologies, Inc.) that was used for DNA visualization. Slides were viewed under an inverted fluorescence microscope (OLYMPUS 1X71) with appropriate filters.
GST Pulldown Assay
GST pulldown was performed as described previously (36). GST-ROP18Δ83 and GST tag proteins were purified and conjugated to glutathione-Sepharose beads. 293T cells were harvested and lysed in PBS buffer containing 1.0% Nonidet P-40, 0.5% deoxycholic acid sodium, and protease inhibitor mixtures. After incubation on ice for 25 min, the lysate was centrifuged at 13,000 × g for 25 min at 4 °C, and the supernatant was collected. The supernatant was buffer exchanged with PBS, pH 7.4, containing protease inhibitor mixtures using the Amicon® Ultra 10K device (Millipore) and then incubated with GST-ROP18Δ83-conjugated or GST-conjugated beads for 4 h at 4 °C. After washing three times with PBS containing 0.1% Triton X-100, beads were eluted in elution buffer (100 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10 mm MgCl2, 20 mm glutathione, and 20% glycerol). Eluates were boiled for 5 min with Laemmli buffer and analyzed by Western blotting.
Coimmunoprecipitation
Immunoprecipitation assay was performed using the coimmunoprecipitation kit (Thermo Scientific Pierce) according to the manufacturer's instructions. Briefly, cells were lysed in IP lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, and 5% glycerol) on ice for 25 min and centrifuged at 13,000 × g for 25 min at 4 °C. Corresponding primary antibodies were covalently coupled onto the AminoLink Plus Coupling Resin. Cell lysates were incubated with the coupled resin for 4 h at room temperature. The resin was washed once with IP lysis buffer and two times with conditioning buffer and eluted with elution buffer provided in the kit. Eluates were analyzed by Western blotting with the indicated antibodies.
Western Blotting
Cells were harvested and lysed in lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm EDTA, and 1% Na3VO4) supplemented with phosphatase inhibitor mixture on ice for 20 min. Total cell lysates were resolved by SDS-PAGE and transferred to PVDF membranes (Millipore). The membranes were blocked for 1 h in 5% non-fat dry milk in TBST buffer (100 mm Tris-HCl, pH 7.5, 0.9% NaCl, and 0.1% Tween 20) and incubated with primary antibodies diluted in 3% BSA at 4 °C overnight. Then the membranes were incubated with HRP-conjugated secondary antibodies and detected with ECL Plus (Millipore).
In Vitro Phosphorylation Assay
The His-tagged full-length Smad1 was expressed in Escherichia coli BL21 (DE3) strain and purified by using Ni2+-Sepharose beads (Qiagen) as described previously (37). The GST-tagged ROP18Δ83 and ROP18Δ83-KD recombinant proteins were purified as described above. For the in vitro phosphorylation assay, GST-ROP18Δ83 or GST-ROP18Δ83-KD recombinant proteins were incubated with purified recombinant His-Smad1 in kinase buffer (50 mm Tris-HCl, pH 7.4, 20 mm HEPES, pH 7.5, 100 mm NaCl, 10 mm MgCl2, 1 mm DTT, 1 mm EGTA, and 20 μm ATP) with 0.5 μCi of [γ-32P]ATP. The reaction mixtures were incubated at 30 °C for 30 min and terminated by adding SDS-PAGE sample buffer and heating immediately at 95 °C for 5 min. Proteins were analyzed by SDS-PAGE, and the gel was exposed to X-ray films after dried.
RESULTS
Expression and Purification of ROP18 Recombinant Proteins
To ensure purification of sufficient amounts of active ROP18 kinase proteins to carry out phosphorylation reactions on the HuProt arrays, we employed the budding yeast as an expression and purification system, as it has been successfully used to purify >400 active human and viral kinases in our previous studies (18, 22). The GST-tagged ROP18Δ83 and the kinase-dead ROP18 mutant (D394A) (ROP18Δ83-KD), a negative control, were expressed in yeast cells and purified (Fig. 1A). As judged by Coomassie staining, both GST-tagged ROP18Δ83 and ROP18Δ83-KD proteins showed a single band at the expected molecular weights, indicating that both proteins were successfully purified with high purity (Fig. 1B). To examine whether the purified ROP18Δ83 retained its kinase activity, we employed auto-phosphorylation reactions using [γ-32P]ATP as a labeling reagent. Commercially available EGFR kinase proteins were used as a positive control. Both ROP18Δ83 and EGFR kinases showed auto-phosphorylation activity, whereas ROP18Δ83-KD did not, as demonstrated by the dot blot kinase assay (Fig. 1C). Together, these results confirmed that the purified ROP18 kinase was active and validated the kinase-dead mutant as an inactive negative control.
Fig. 1.
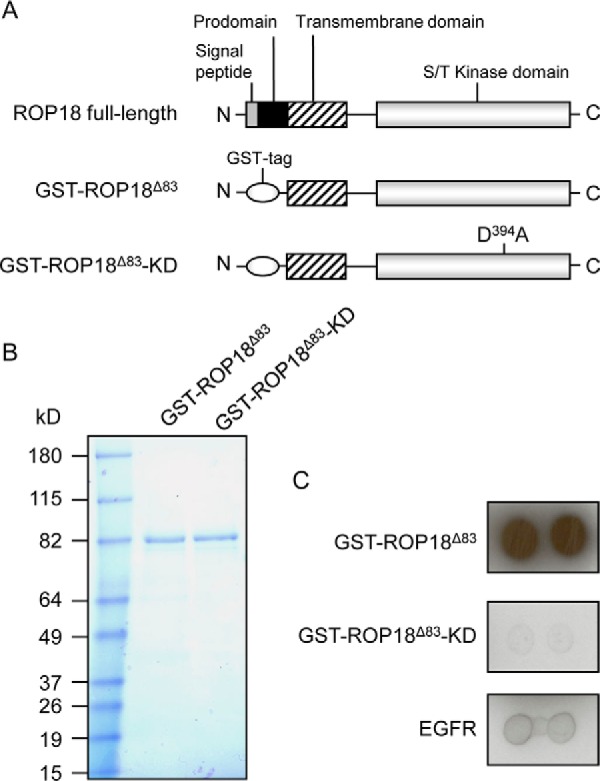
Purification of active ROP18 kinase proteins. A, schematic illustration of the GST-tagged ROP18 constructs GST-ROP18Δ83 and GST-ROP18Δ83-KD. ROP18Δ83 lacks the signal peptide and the prodomain, and the kinase-dead mutant ROP18Δ83-KD is a D394A-mutated version of ROP18Δ83. B, GST-ROP18Δ83 and GST-ROP18Δ83-KD proteins were expressed in yeast and purified using glutathione beads. The purified proteins were separated by SDS-PAGE and stained with Coomassie Brilliant Blue. C, purified GST-ROP18Δ83, GST-ROP18Δ83-KD, and EGFR (positive control) were incubated with kinase assay buffer containing [γ-32P]ATP. Proteins were then dotted on nitrocellulose membrane, followed by autoradiography.
Identification of ROP18 Substrates in Vitro
To better understand the contribution of ROP18 to parasite virulence via its kinase activity, we set out to systematically identify the ROP18 substrates using the HuProt arrays (version II) that contain 16,368 unique, full-length human proteins that were individually purified (Fig. 2, A and B) (31, 32). Purified GST-tagged ROP18Δ83 and ROP18Δ83-KD protein were separately incubated with a pre-blocked HuProt array in a generic kinase reaction buffer containing [γ-32P]ATP. After the kinase reactions, the HuProt arrays were washed under denaturing conditions to ensure removal of the added kinases and unincorporated [γ-32P]ATP. The phosphorylation signals on the HuProt arrays were detected by exposure to X-ray film (Fig. 2C). Phosphorylated proteins on each array were identified using an algorithm that measures the relative signal intensity of each spot at cutoff values of 2.0 and 2.5 standard deviations (S.D.) above the background signals (18). Proteins that showed phosphorylation signals on the array incubated with ROP18Δ83-KD were omitted from the ROP18Δ83 hit list, because the signals were likely produced by auto-phosphorylation reactions. This approach has resulted in the identification of 172 and 68 potential substrates of ROP18 kinase at the cutoff values of 2.0 and 2.5 S.D., respectively (Table I and supplemental Tables S1 and S2).
Fig. 2.

Identification of ROP18 kinase substrates using HuProt arrays. A, overall scheme to identify the ROP18 kinase substrates. Purified GST-ROP18Δ83 and GST-ROP18Δ83-KD proteins were assayed on HuProt arrays in kinase buffer containing [γ-32P]ATP. After the kinase reaction, protein microarrays were washed to remove added proteins and unincorporated [γ-32P]ATP followed by exposure to X-ray films. The films were then scanned, and bioinformatics analysis and partial validation were performed. B, representative image of the HuProt array. Individually purified human proteins tagged with GST were spotted in duplicate on a single slide, and the immobilized human proteins were visualized by probing with anti-GST antibody. C, representative photographs of X-ray films. After the kinase reactions on the HuProt arrays, the phosphorylation signals were detected by exposure of the slides to a piece of X-ray film. Developed X-ray film was then scanned, and the enlarged images of p53, MAPK11 (i.e. p38), UBE2N, and Smad1 protein dots are shown.
Table I. Putative ROP18 substrates identified on the HuProt arrays.
68 proteins were identified as putative ROP18 substrates under the condition of 2.5 S.D.
| Protein | Protein (continued) | Protein (continued) |
|---|---|---|
| ANUBL1 | MAP3K5 | RBBP5 |
| BCL11A | MAPK10 | REM1 |
| C18orf25 | MAPK11 | RFK |
| C4orf19 | MAPK9 | RFX4 |
| CLK2 | MOSPD3 | RNF25 |
| CNBP | NADK | SFRS12 |
| CSNK1G3 | NAP1L1 | SMAD1 |
| CSRP3 | NME2 | SMYD2 |
| CYB5B | NPM2 | SRPX2 |
| DAPK2 | OSBPL6 | SSPN |
| DIXDC1 | PACSIN2 | SUPT6H |
| FAM5B | PARN | TBC1D16 |
| FLT1 | PAX9 | THUMPD3 |
| FN3K | PCDHGC3 | TP53 |
| GSK3B | PDPK1 | TSSC4 |
| HARS | PELI3 | UBE2N |
| HDGF2 | PLEK2 | WDR45 |
| HNRPK | PRAC | XRCC4 |
| IRF1 | PRKAR1A | ZBTB22 |
| KIF3C | PRKCA | ZMYM3 |
| KLF11 | PRKRA | ZNF16 |
| KPNA2 | PSMD7 | ZNF641 |
| MAGEB4 | RAB43 |
Bioinformatics Analyses of ROP18-Substrate Relationships
We were well aware that the 68 substrates identified using the above in vitro kinase reactions might not reflect in vivo kinase-substrate relationships. To improve the likelihood of revealing ROP18's in vivo function, we decided to take advantage of the previous proteomics studies by integrating those existing large datasets with ours.
Because a common means for a pathogen to hijack the cellular pathways is by targeting multiple components of the same host signaling pathways (20), we first performed the STRING analysis (STRING.org), which integrates known protein-protein interaction (PPI) information, against the 68 in vitro substrates of ROP18, to determine whether there is any significant PPI network among these substrates (Fig. 3A) (38). As expected, 32 of the 68 substrates of ROP18 formed a tightly connected PPI network with 54 edges (i.e. known PPI relationships). Interestingly, p53 (i.e. TP53) is identified as the central hub located in the middle of the PPI network with 20 edges. Overall, 31 of the 68 substrates were also identified as either the kinases (Fig. 3A, circled in red) or kinase substrates (Fig. 3A, with asterisk) in our previous study, in which an activity-based kinase-substrate relationship (KSR) network was established (18, 39). Interestingly, 8 of the 31 ROP18 substrates were protein kinase themselves, whereas only 105 of the total 1,967 substrates also phosphorylated others in the KSR networks. This phenomenon is more pronounced in the PPI network formed by the 32 ROP18 substrates, 11 are known protein kinases and 21 are found in the KSR networks. Note that the majority of the KSR was determined by performing phosphorylation reactions on an early version of the human protein microarrays, each composed of ∼4,200 unique proteins (18, 39). Taken together, these analyses indicate a tendency of ROP18 to act as a master kinase that regulates many host kinases and signaling components (p = 1.2E-4).
Fig. 3.

Bioinformatics analysis of the identified in vitro substrates of ROP18. A, integration of known PPI relationships revealed that 32 of the 68 identified substrates of ROP18 form a highly connected PPI network. TP53 is located as the most connected hub in the center of the PPI network. B, GO analysis demonstrated that 68 ROP18 targets (left panel), especially 32 of them with annotated PPI relationships (right panel), are significantly enriched in some biological functions, e.g. metabolic process, kinase activity/phosphorylation, response to growth factor, apoptotic process/cell death, and immune system process. C, ROP18 targets are over-represented in some signaling pathways. Examples of the insulin signaling and MAPK signaling pathways are shown. Identified ROP18 substrates are indicated by circles with GO terms annotated in different colors using the same code as A. D, using the M3 algorithm, ROP18 was found to specifically recognize an acidic phospho-motif, which is most similar to CSNK2A1. E, comparison of identified substrates using the protein microarray approach revealed 14 human kinases that share a significant portion of targets as ROP18. p values of the statistical significance of each human kinase are shown in the bottom panel.
Second, we performed Gene Ontology (GO) analysis to determine possible roles of ROP18 in pathogen-host interactions. We found that the 68 ROP18 substrates were significantly enriched in following biological functions: metabolic process (p = 1.2E-5); kinase activity or phosphorylation (p = 5.6E-6); response to growth factor (p = 5.0E-5); apoptotic process or cell death (p = 6.1E-4), and immune system process (p = 2.9E-4) (Fig. 3B). When focusing on the 32 substrates that form a highly connected PPI network, the p values of the corresponding GO terms were further improved by at least 1.7 logs (right panel, Fig. 3B). This analysis suggests that ROP18 is likely to play a pleiotropic role as a master kinase that is capable of regulating multiple signaling pathways in parallel.
Indeed, ROP18 substrates were found enriched in several specific signaling pathways, such as the insulin signaling and mitogen-activated protein kinase (MAPK) signaling pathways. For instance, ROP18 was found to phosphorylate multiple kinases, including MAPK9, MAPK10, PDPK1, GSK3β, and PRKAR1A, which are the downstream components of the insulin signaling pathway (p = 8.3E-4) with multiple physiological outcomes, such as protein synthesis, glycogensis, and antilipolysis (upper panel, Fig. 3C). Similarly, ROP18 seems to regulate two parallel MAPK signaling pathways (p = 1.2E-3) by targeting MAPKKK (i.e. MAP3K5) and p38 (i.e. MAPK11) and by targeting JNK2/3 (i.e. MAPK9 and MAPK10), which converge at p53 that leads to transcription activation of the p53 target genes (lower panel, Fig. 3C).
To better understand how ROP18 recognizes its substrates, we next employed a previously reported M3 algorithm, which integrates the phosphorylation sites identified with mass spectrometry approaches with ROP18's substrates, and we determined the phosphorylation motif logo of ROP18 as (S/T)XXXXXDE (Fig. 3D) (18, 39). After searching the KSR database, we found that the acidic phospho-motif recognized by ROP18 is most similar to that of the catalytic α subunit of casein kinase 2 (i.e. CSNK2A1), which is well known to be involved in many cellular processes, including apoptosis. Like ROP18, CSNK2A1 was also identified as a master kinase (p = 0.025), which tends to phosphorylate many other kinases.
Because a common theme utilized by pathogenic kinases is to imitate the host counterparts, we directly compared the substrate similarity of ROP18 with the 289 human kinases in the PhosphoNetworks database. We observed that 14 human kinases showed a significant similarity (p < 0.01) with ROP18 in terms of substrate specificity (Fig. 3E). Moreover, four of these 14 kinases, including NIM1, PRKX, PRKCH, and GSK3β, have a strong tendency to phosphorylate other kinases. This is significant (p < 0.05) because only 57 of the 289 tested kinases showed a similar behavior. This observation suggests again that ROP18 is likely to mimic human master kinases rather than general kinases. Of the 14 kinases, STK3 showed the lowest p value, at least 10-fold lower than the second best one. STK3 (also known as MST2) is a proapoptotic serine/threonine kinase and plays a critical role in the Hippo-YAP signaling pathway (40). In response to proapoptotic molecules, STK3 is activated by caspase cleavage of the auto-inhibitory domain, and the cleaved N-terminal portion of STK3 goes to the nucleus and forms the active kinase dimer. A recent study by Wu et al. (41) suggested that ROP18 might manipulate the host mitochondrial apoptosis pathways. They observed that overexpression of T. gondii ROP18 significantly suppressed 293T cell apoptosis induced by actinomycin D and maintained mitochondrial membrane potential and integrity. Interestingly, STK3 recognizes a very different phospho-motif (i.e. HXRXX(S/T), where X is any amino acid) than ROP18, suggesting that ROP18 phosphorylates different Ser/Thr residues of these shared substrates, which might lead to different physiological outcomes of the substrates. Two other kinases, RPS6KA6 and PRKX, were found to recognize a very similar acidic phospho-motif as ROP18. Although not much is known about PRKX functions in cells, RPS6KA6 (i.e. RSK4) is well characterized for its role in the neurotrophin signaling (42). RSK4 acts as a downstream kinase of ERK1/2 that activates cell differentiation and cell survival processes. Therefore, it is plausible that ROP18 hijacks RSK4's role by phosphorylating the same downstream substrates of RSK4.
ROP18-mediated p53 Degradation via Phosphorylation
On the basis of the study by Wu et al. (41) and the above bioinformatics analyses, we suspected that ROP18 might play an important role in anti-apoptosis. Intriguingly, p53 was identified as a novel substrate of ROP18 in our in vitro kinase assays and is located at the central hub position in the PPI network among the 32 ROP18 substrates (Fig. 3A). p53 is a master transcription factor and is known to respond to diverse cellular stresses to regulate expression of target genes, thereby inducing cell cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism (43). p53 is also a well known tumor suppressor involved in immunity (44). It has been reported that infection from T. gondii induces endogenous p53 degradation; however, the exact molecular mechanism remains unknown (45, 46). Therefore, we decided to validate the relationship between ROP18 and p53 and determine the impact of ROP18 phosphorylation on p53.
To do so, immunofluorescence staining was performed. The results showed that p53 was colocalized with ROP18 at the PVMs in host cells (Fig. 4A), indicating that ROP18 and p53 might form a protein complex. To access whether ROP18 interacts with p53, we performed a GST pulldown assay with recombinant GST-ROP18Δ83 protein. The results showed that GST-ROP18Δ83, but not the GST tag, was able to pull down p53 (Fig. 4B). Moreover, we performed coimmunoprecipitation assay and detected an interaction between ROP18 and p53 (Fig. 4C). We then investigated the influence of ROP18 on p53 expression. We observed that overexpression of ROP18Δ83 but not ROP18Δ83-KD significantly decreased the protein level of p53 (Fig. 4D), indicating that the kinase activity is essential for ROP18-mediated p53 degradation. In addition, ROP18 decreased the protein levels of p53 in a dose-dependent manner (Fig. 4E). To further demonstrate the specificity of ROP18-dependent p53 degradation, we cotransfected the FLAG-tagged p53 construct with different amounts of the V5-tagged ROP18Δ83 constructs to 293T cells. We found that overexpression of ROP18Δ83 caused dose-dependent degradation of the exogenous p53 protein (Fig. 4F). To investigate the impact of ROP18 on p53 expression in vivo, we infected 293T cells with wild-type (WT) RH or ROP18-Ty1 RH strain. Consistent with the previous studies (45, 46), we found that in 293T cells infection with WT RH strain led to a significant degradation of p53 protein. Additionally, infection of 293T cells with ROP18-Ty1 RH strain further extended the degradation (Fig. 4G). Taken together, these results demonstrated that ROP18 relies on its kinase activity to inhibit apoptosis in T. gondii-infected cells via targeting p53 for degradation.
Fig. 4.

ROP18 interacts with p53 and promotes its degradation. A, HeLa cells were transfected with V5-ROP18Δ83 and FLAG-p53 followed by infection with tachyzoites of RH strain for 24 h, and then immunofluorescence staining was performed with mouse monoclonal anti-V5 (green) and rabbit polyclonal anti-FLAG (red) antibodies. DAPI (blue) was used to visualize cell nuclei. Phase, bright field; bar, 5 μm. B, GST pulldown assay was performed using GST-ROP18Δ83 protein purified from yeast and lysates of 293T cells overexpressing FLAG-p53. IB, immunoblot. C, 293T cells were transfected with ROP18Δ27-GFP expression plasmid, and coimmunoprecipitation was performed with anti-GFP, followed by immunoblotting with the indicated antibodies. D, 293T cells were transfected with V5-ROP18Δ83 or V5-ROP18Δ83-KD plasmids for 36 h, and the total cell lysates were subjected to Western blotting with the indicated antibodies. E, lysates of 293T cells transiently transfected with the indicated amounts of V5-ROP18Δ83 plasmids were detected by Western blotting with the indicated antibodies. F, 293T cells were cotransfected with the indicated amounts of V5-ROP18Δ83 and 1 μg of FLAG-p53 plasmids. 36 h after transfection, total cell lysates were analyzed by Western blotting. G, lysates of 293T cells infected with wild-type (WT) RH and ROP18-Ty1 RH strains were detected by Western blotting with the indicated antibodies. SAG1 and GAPDH were used as loading controls.
ROP18-mediated p38 Degradation via Phosphorylation
MAPK signaling has been reported to be involved in the infection of T. gondii (47, 48). Intriguingly, our bioinformatics analyses suggested that ROP18 might regulate the MAPK pathways via targeting p38 (e.g. MAPK11). The p38 MAPKs are known to be activated by cytokines, LPS, and environmental stress and to be implicated in several processes, including cell survival, apoptosis, and immune function (49). The p38 MAPK pathway has been related to T. gondii invasion (50, 51). To confirm the association between ROP18 and p38, immunofluorescence staining was performed. The results revealed a colocalization of p38 and ROP18 at the PVMs (Fig. 5A), indicating that ROP18 might form a complex with p38. To confirm this assumption, we performed a GST pulldown assay. The results showed that GST-ROP18Δ83 was able to pull down p38 (Fig. 5B), suggesting that ROP18 interacts with p38. We next investigated the potential impact of ROP18 on p38 in cells. We found that overexpression of ROP18Δ83 rather than ROP18Δ83-KD significantly decreased the expression level of p38 protein (Fig. 5C), indicating that the kinase activity is required for ROP18-mediated p38 degradation. To further determine whether ROP18 affects the stability of p38 in vivo, 293T cells were infected with WT RH or ROP18-Ty1 RH strain. The results showed that infection with WT RH strain significantly reduced the p38 protein level as compared with that in un-infected cells, and infection with ROP18-Ty1 RH strain resulted in a more significant reduction of p38 protein level (Fig. 5D). Together, these results suggested that ROP18 targets p38 for degradation via phosphorylation.
Fig. 5.

ROP18 interacts with p38 and promotes its degradation. A, HeLa cells transiently expressing V5-ROP18Δ83 were infected with tachyzoites of RH strain. 24 h after infection, immunofluorescence staining was performed with mouse monoclonal anti-V5 (green) and rabbit monoclonal anti-p38 (red) antibodies. DAPI (blue) was used to visualize cell nuclei. Phase, bright field; bar, 5 μm. B, GST pulldown assay was performed using GST-ROP18Δ83 protein and 293T cell lysates. IB, immunoblot. C, 293T cells were transfected with V5-ROP18Δ83 or V5-ROP18Δ83-KD plasmids for 36 h, and then total cell lysates were subjected to Western blotting analysis. D, 293T cells were infected without or with wild-type (WT) RH or ROP18-Ty1 RH strain for 36 h. Cellular lysates were separated by SDS-PAGE and immunoblotted with the indicated antibodies. SAG1 and GAPDH were used as loading controls.
ROP18-mediated UBE2N Degradation via Phosphorylation
Du et al. (13) reported that ROP18 inactivates the NF-κB pathway via phosphorylation and degradation of p65. Interestingly, we identified another upstream regulator of NF-κB pathway, UBE2N (52, 53), as a putative substrate of ROP18. UBE2N is also found as a hub in the PPI network that is directly connected to p53, PDPK1, PELI3, ZFAND4, and NME2 (Fig. 3A). UBE2N (also known as Ubc13) is a unique E2 ubiquitin-conjugating enzyme and plays an important role in host inflammatory responses (54). For example, a secreted Shigella effector, OspI, was reported to specifically target Ubc13 and deamidates Gln-100 to a glutamic acid residue, leading to the disruption of TRAF6-catalyzed poly-ubiquitylation. This results in suppressing the diacylglycerol → CBM → TRAF6→ NF-κB signaling pathway and dampens the host inflammatory responses (55). To validate the relevance of ROP18 and UBE2N, we expressed V5-ROP18Δ83 or V5-ROP18Δ83-KD in 293T cells. Western blotting analysis showed that overexpression of ROP18Δ83 but not ROP18Δ83-KD led to a significant reduction in the protein level of UBE2N (Fig. 6A), suggesting that the kinase activity of ROP18 plays an important role in ROP18-mediated UBE2N degradation. We then investigated whether ROP18 affects UBE2N expression in vivo. The data showed that, although infection with the WT RH strain significantly decreased the UBE2N expression relative to non-infection, infection with the ROP18-Ty1 RH strain resulted in a more significant decrease in the protein level of UBE2N (Fig. 6B). Together, these results suggested that UBE2N is a ROP18-targeting host protein.
Fig. 6.

ROP18 promotes degradation of UBE2N. A, 293T cells were transfected with V5-ROP18Δ83 or V5-ROP18Δ83-KD plasmids for 36 h, and then total cell lysates were subjected to Western blotting analysis. IB, immunoblot. B, 293T cells were infected without or with wild-type (WT) RH or ROP18-Ty1 RH strain for 36 h. Cell lysates were separated by SDS-PAGE and immunoblotted with the indicated antibodies. SAG1 and GAPDH were used as loading controls.
ROP18-mediated Smad1 Proteasomal Degradation via Phosphorylation
Interference of the TGF-β signaling pathway has been shown to be highly associated with Toxoplasma infection (56). Accordingly, Smad1, one of the family members that act downstream of receptors of TGF-β family members (57), was identified as a substrate of ROP18 kinase in this study. To determine whether ROP18 interacted with Smad1, we performed the coimmunoprecipitation assay and observed that ROP18 could readily immunoprecipitate Smad1 in cotransfected cells (Fig. 7A). We then investigated the influence of ROP18 on Smad1 expression. The results showed that expression of ROP18Δ83 but not ROP18Δ83-KD markedly reduced the protein level of Smad1 (Fig. 7B) in a dose-dependent manner (Fig. 7C). Interestingly, the protein level of Smad1 was restored in the presence of a proteasome inhibitor, MG132 (Fig. 7D), suggesting that ROP18 phosphorylation-induced Smad1 degradation was mediated by the proteasome. To confirm that Smad1 could be phosphorylated by ROP18, we carried out an in vitro kinase assay with purified ROP18Δ83 and ROP18Δ83-KD proteins. The results clearly showed that GST-ROP18Δ83, but not GST-ROP18Δ83-KD, strongly phosphorylated the His6-tagged Smad1 protein (Fig. 7E).
Fig. 7.

ROP18 interacts with Smad1 and triggers its proteasomal degradation via phosphorylation. A, 293T cells were transfected with ROP18Δ27-GFP expression plasmid, and coimmunoprecipitation was performed with anti-GFP, followed by immunoblotting with the indicated antibodies. IB, immunoblot. B, 293T cells were transfected with V5-ROP18Δ83 or V5-ROP18Δ83-KD and/or FLAG-Smad1 plasmids for 36 h, and then total cell lysates were analyzed by Western blotting with the indicated antibodies. C, 293T cells transiently cotransfected with the indicated amounts of V5-ROP18Δ83 or V5-ROP18Δ83-KD and FLAG-Smad1 plasmids and cell lysates were detected by Western blotting. D, 293T cells were transfected with V5-ROP18Δ83 and/or FLAG-Smad1 plasmids in the absence or presence of 10 μm MG132 for 24 h, and lysates were detected by Western blotting. E, in vitro kinase assay was performed to detected His-Smad1 phosphorylation with purified GST-ROP18Δ83 and GST-RO18Δ83-KD proteins. F, 293T cells were cotransfected with FLAG-Smad1 mutants (M1, S187A, S195A, S206A, and S214A; M2, S187A, S195A, and S206A; M3, S206A; M4, S187A) and V5-ROP18Δ83 for 48 h, and the protein levels of Smad1 mutants were detected by Western blotting. G, 293T cells were transfected with V5-ROP18Δ83 or V5-ROP18Δ83-KD and/or FLAG-Smad1 for 48 h, and Smad1 phosphorylation was detected by Western blotting with phospho-Smad1 (Ser-187) antibody. H, HFF cells were infected with wild-type (WT) RH, ROP18-KO RH, or ROP18-Ty1 RH strain, and Smad1 phosphorylation was detected by Western blotting with phospho-Smad1 (Ser-187) antibody. SAG1 and GAPDH were used as loading controls.
To further investigate the molecular basis of ROP18-mediated Smad1 degradation, we sought to determine which residue(s) in Smad1 could be phosphorylated by ROP18. In previous studies, phosphorylation in the linker region of Smad1 has been shown to trigger its degradation (58, 59). The observation that ROP18 also induced Smad1 proteasomal degradation prompted us to test the hypothesis that ROP18 might utilize a similar mechanism to regulate Smad1 protein levels. Thus, we performed site-directed mutagenesis at Ser-187, Ser-195, Ser-206, and Ser-214 in the linker region, which were identified as the phosphorylation sites in previous studies (59). By testing different combinations of mutations at these Ser residues, we found that Ser-187 to Ala mutation blocked the reduction of Smad1 protein levels induced by ROP18 (Fig. 7F), suggesting that Ser-187 phosphorylation of Smad1 by ROP18 was responsible for its degradation. To verify that Smad1 Ser-187 was phosphorylated by ROP18, we examined the phosphorylation status of Smad1 Ser-187 by performing immunoblotting analysis with a commercial phospho-specific antibody against Smad1 Ser-187 in transfected cells. The results showed that, compared with the control vector, transfection of ROP18Δ83 significantly increased the phosphorylation level of Smad1 Ser-187, especially considering that the Smad1 protein was strongly reduced by ROP18Δ83 (Fig. 7G). In contrast, no significant difference was observed upon transfection of ROP18Δ83-KD (Fig. 7G). To further determine whether ROP18 phosphorylates Smad1 at Ser-187 in vivo, HFF cells were infected with the WT RH, ROP18-KO RH, or ROP18-Ty1 RH strain. Immunoblotting analysis showed that as compared with non-infection, infection with the WT RH and ROP18-Ty1 RH strain resulted in increased phosphorylation levels of Smad1 Ser-187 accompanied by decreased Smad1 protein levels, whereas infection with ROP18-KO RH strain did not alter the phosphorylation status of Smad1 at Ser-187 and had no effect on Smad1 protein levels (Fig. 7H). These results suggested that ROP18 phosphorylates Smad1 Ser-187 to facilitate its degradation.
DISCUSSION
In this study, we performed phosphorylation reactions on a HuProt array to globally screen for host substrates of ROP18 kinase, which is a key determinant of parasite virulence. A number of novel putative host targets involved in various cellular functions were identified. Among these proteins, four candidates, p53, p38, UBE2N, and Smad1, were further validated as ROP18 substrates.
A key to the success of this study was the purification of active ROP18 kinase proteins from the budding yeast. Although the bacterial systems have been more popular for protein expression, they suffer from a lack of proper protein post-translational modifications, and overexpression of foreign proteins in bacteria often resulted in an insoluble or inactive form of the desired proteins (28, 60). Yeast cells combine the advantages of being single cells, such as fast growth and easy genetic manipulation, as well as eukaryotic features, including a secretory pathway leading to correct protein processing and post-translational modifications (61). Indeed, recombinant ROP18 proteins currently purified from E. coli are only suitable for immunity or structural analysis, requiring little or no activity (60, 62). A truncated ROP18 protein that corresponds to the kinase catalytic domain was successfully expressed in E. coli, but the recombinant protein had to be refolded to obtain bioactivity. The refolded recombinant protein was rather unstable and could not be obtained in sufficient amounts (28). To our knowledge, we have provided the first demonstration that by using a yeast system, the mature ROP18 protein with high kinase activity and satisfactory quantity was successfully purified. This finding paves a way for oncoming in-depth studies, such as crystal structure analysis of phosphorylated ROP18, in vitro kinase assays, and substrate screening.
Identification of key host targets of ROP18 is crucial for understanding the pathogenesis of T. gondii. So far the discovery of ROP18 substrates mainly relies on the yeast two-hybrid method, in which interactions are typically detected in the nucleus, thus restricting the types of interaction that can be detected. In addition, the two-hybrid approach detects protein-protein interaction but not the protein bioactivity (29). Protein microarray is a powerful tool for comprehensive screens of protein functions, which has been applied for global analysis of protein modified by ubiquitylation, SUMOylation, acetylation, and S-nitrosylation (17). Additionally, protein microarray has been expanded to identify kinase substrates (18–24). Whereas creating protein microarrays requires a specialized system, the arrays can be stored and saved for later use, providing a faster and cheaper detection tool for high-throughput studies. In this study, using the human protein microarray, we globally determined the substrates of ROP18 kinase and identified a total of 68 potential host targets.
T. gondii strictly relies on host cellular metabolism to obtain the energy and nutrients for its replication and survival (63). But how T. gondii modulates the metabolism of its host to create a more hospitable metabolic niche remains vague. Transcriptomics analysis has shown that infection of T. gondii results in alteration of protein expression in key metabolic pathways, as characterized by the down-regulation of mitochondrial respiratory chain and the metabolism of fatty acids, lipids, and xenobiotics (64). Interestingly, in this study, we found that the majority of ROP18 substrates are enriched in the metabolic process, suggesting an important role of ROP18 in manipulating the host cellular metabolism. In particular, ROP18 seems to regulate the host insulin signaling, which modulates the metabolism of glucose, lipids, proteins, and energy (65, 66), as several putative substrates take part in this pathway. T. gondii preferentially utilizes host glucose as a source for the synthesis of ATP, nucleic acid, proteins, and lipids and intracellular replication (67). Thus, ROP18-mediated insulin pathway might be beneficial for the metabolism of T. gondii.
Induction of apoptosis is a common host response to infection. Therefore, pathogens including T. gondii have developed strategies to hamper host cell apoptosis. Consistent with this notion, ROP18 has been shown to inhibit host cell apoptosis with a not yet known mechanism. Previous studies reported that T. gondii infection alters host MAPK signaling pathway (47, 48), which regulates multiple cellular processes, including apoptosis (68). Intriguingly, we identified several MAPK pathway effectors, including MAP3K5, MAPK9, MAPK10, MAPK11, PRKCA, and p53, as the putative ROP18 substrates. In addition, three substrates, PDPK1, GSK3β, and p53, are implicated in the PI3K/AKT pathway that are important for maintaining the cellular anti-apoptotic state (69). Moreover, host proteins FLT1 (70) and PRKRA (71) involved in apoptosis and cell growth were also identified as the ROP18 targets. Overall, our data suggest that ROP18 might regulate host cell apoptosis through multiple host signaling pathways and targets.
Host immunity limits the parasite growth. In turn, T. gondii interferes with the host immune-related signaling, enabling the parasite to evade the host immune response. ROP18 has been reported to modulate the NF-κB signaling pathway that is critical in regulating inflammation and innate immune responses to T. gondii infection (13). In agreement with this idea, host proteins UBE2N (72, 73) and RNF25 (74), which take part in NF-κB pathway, were identified as the ROP18 substrates, indicating that these proteins might be required for ROP18-mediated NF-κB signaling regulation. IFN-γ secretion is crucial for controlling T. gondii infection (75). T. gondii has been reported to down-regulate the expression of IRF1 (76, 77), a transcription factor that regulates most of the other half of IFN-γ-responsive genes (78). Intriguingly, we identified IRF1 as a potential substrate of ROP18, implying that T. gondii probably regulates IRF1/IFN-γ expression via ROP18 kinase.
Phosphorylation by protein kinases is a common way to regulate the complex signaling networks. Interestingly, we identified at least eight host kinases as ROP18 substrates, including CLK2, CSNK1G3, PDPK1, PRKCA, GSK3β, MAPK10, MAPK11, and MAPK9. All of these kinases are key regulators involved in multiple biological processes, including cell cycle, apoptosis, cell proliferation, immunity, and metabolism, to name a few (Fig. 3A). Bioinformatics analyses based on our dataset now suggest that ROP18 may serve as a master kinase modulating multiple host signaling pathways. Notably, ATF6β (12) and p65 (13) that have been reported to be the substrates of ROP18 were not identified in our assays. ATF6β was absent in the human ORF collection used to express the proteins for HuProt array fabrication, whereas p65 was available in our collection but its protein level was not detected on the HuProt, presumably due to failure in its purification (29). Although the possibility of false positives or false negatives due to incorrect folding or an insufficient amount of some proteins on the array cannot be excluded (29), our follow-up validation of four substrates, p53, p38, UBE2N, and Smad1, confirmed the efficacy of the screening.
The central position of p53 occupied in the PPI network formed by ROP18 substrates indicates that p53 might be a key host target required for ROP18-dependent parasite virulence. Studies have reported that infection of T. gondii induces degradation of endogenous p53, although the mechanism is not clearly understood (45, 46). In this study, we found that p53 is a host target of ROP18 kinase, and overexpression of ROP18 induced p53 degradation. Combined with previous studies, our results indicate that T. gondii induces p53 degradation via active ROP18 kinase. ROP18 has been reported to inhibit the apoptosis of infected cells, thereby providing a stable environment for parasitic proliferation (41). Considering the important role of p53 in apoptosis, it is possible that ROP18 mediates p53 degradation and thus suppresses host cell apoptosis. In addition to its function in apoptosis, p53 also plays a critical role in innate immunity (43, 44). It has been reported that down-regulation of p53 results in impaired expression of genes involved in interferon or toll-like receptor signaling during viral infection (79, 80). Gadd45α is a downstream target of p53 (43). The deficiency of Gadd45α in mice results in impaired Th1 development due to the lack of p38 activation, IL-12, and CD40 production in dendritic cells and diminished Th1 immune response to T. gondii antigen (81). Therefore, it is also likely that during pathogen infection, ROP18 binds to and degrades p53, leading to down-regulation of Gadd45α expression, which in turn results in reduction of IL-12 and CD40 production, thereby conferring an immune-privileged microenvironment for parasite proliferation, but such a mechanism remains to be experimentally demonstrated.
The p38 MAPK pathways controlling gene expression and early immune response contribute to the infection and replication of T. gondii (47, 48). It has been reported that parasite invasion triggers p38 activation and drives IL-12 production through an association with TAB1 (51). Braun et al. (82) also reported that during Toxoplasma infection the dense granule protein GRA24 binds to p38 and promotes p38 auto-phosphorylation and nuclear translocation, which results in up-regulation of the transcription factors Egr-1 and c-Fos and the synthesis of cytokines IL-12 and MCP1. Parasite infection activates the p38 MAPK pathway; however, this activation is rapidly decreased thereafter (50), implying a feedback mechanism subverting the host p38 MAPK pathway may exist. Interestingly, in this study, we identified p38 as a host target of ROP18. Moreover, we found that ROP18 interacted with p38 and triggered its degradation. Our results indicate that ROP18 might be a key negative modulator of the p38 MAPK pathway that disrupts the autonomous host cell protecting early immune response.
Ubiquitylation is an important process modulating protein destruction. It has been reported that ROP18 induced ubiquitin-dependent degradation of host protein (13) indicates that ROP18 is involved in ubiquitylation-related signaling. Here, we demonstrated that UBE2N, a member of the E2 ubiquitin-conjugating enzyme family (52), is a host target of ROP18. Previous studies have shown that UBE2N is essential for the immune signaling. In thymocytes, UBE2N regulates NF-κB, MAPK, and TAK1 (TGF-β-activated kinase 1) signaling pathways and thus regulates T cell receptor-mediated signaling and innate immunity (52, 53, 73, 83). Conjunction of TRAF6 with UBE2N is necessary for the activation of TNF receptor-associated factor (TRAF)-dependent signal transduction pathways (NF-κB, JNK, and p38 MAPK) and inflammatory responses (73, 84). Given the critical role of UBE2N in innate and inflammatory responses, disruption of UBE2N by ROP18 may result in impaired immune responses, thus facilitating the intracellular growth of T. gondii. Further studies are required to validate this hypothesis.
TGF-β signaling, which functions in cell proliferation, apoptosis, embryonic development, angiogenesis, and immunity, plays important roles in controlling Toxoplasma infection (56, 85). TGF-β1 is an important cytokine that has a dual role in the regulation of immune response against T. gondii infection as follows: induction of immune responses by development of Th17 lymphocytes and mucosal immunity and suppression of immune responses by inducing T regulatory cells, which restrict Th17 development, and inhibiting pro-inflammatory cytokine production, such as TNF-α (85). Smad1 is one of the members of the intracellular effectors of TGF-β signaling, which has important roles in cell proliferation, inflammation, and T cell differentiation (86–88). Smad1 is a mediator of bone morphogenetic protein signals, but it can also be activated by TGF-β1 (89). In this study, we identified for the first time that Smad1 is a target of ROP18, but its function and mechanisms underlying the parasite virulence remain to be defined. It has been reported that manipulation of host cell targets by T. gondii contributes to its proliferation (90). Considering the important role of Smad1 in cell proliferation, we determined whether targeting Smad1 affects T. gondii intercellular growth; however, the results showed that silencing Smad1 in HFF cells was not obviously beneficial for T. gondii replication (data no shown), implying that Smad1 might mediate parasite virulence via a mechanism that is distinct from regulating the parasite proliferation. Studies have been reported that Ids, the downstream targets of Smad1 (91), are pivotal regulators of lymphocyte differentiation and proliferation, particularly the CD8+ effector and memory T cells and CD4+ T cells (92–95). Our data showed that ROP18 interacted with Smad1 and prompted its proteasomal degradation. Therefore, it is possible that after parasite infection, ROP18 impairs the T cell development and function by down-regulating Smad1 and its downstream effectors Ids, and thus it facilitates an escape of T. gondii from the host immune system, but this hypothesis remains to be verified in the parasite-infected Smad1 mutant mouse model.
In summary, we report for the first time that with the use of a human proteome array, the substrates of T. gondii ROP18 kinase were systemically identified. Moreover, four important proteins, p53, p38, UBE2N, and Smad1 were validated as the host targets. Notably, we demonstrated that ROP18 phosphorylates Smad1 Ser-187 to promote its proteasome-dependent degradation. Functional analysis of the novel substrates of ROP18 kinase will be of benefit for understanding Toxoplasma virulence regulatory mechanisms mediated by ROP18 and providing potential targets for therapeutic intervention. The approaches utilized in this study may offer a new strategy for discovering novel host targets during host-pathogen interactions.
Supplementary Material
Acknowledgments
We thank Dr. Jianfei Hu (The Johns Hopkins University) and Zhihao Zhang (Sungkyunkwan University) for their technical assistance. We thank Professor Vern B. Carruthers (University of Michigan Medical School) for many helpful comments.
Footnotes
Author Contributions: X.Z., H.Z., and W.L. designed the research and wrote the manuscript; Z.Y., Y.H., T.H., and H.R. performed the experiments; J.W. and Y.L. performed the bioinformatics analysis; X.G., J.Y., and A.P. analyzed the data; Z.X. and J.Q. provided technical assistances; all authors have read and approved the manuscript.
* This work was supported in part by National Natural Science Foundation of China Grants 81328014 and 31602040, Natural Science Foundation of Guangdong Province Grant 2015A030313105, Science and Technology Planning Project of Guangdong Province Grants 2014A020214002, 2013B021800243, and 2015A020210048, and National Institutes of Health Grant 1RO1GM111514-02 (to H.Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PVM
- parasitophorous vacuole membrane
- HuProt
- human proteome array
- HFF
- human foreskin fibroblast
- KSR
- kinase-substrate relationship
- MAPK
- mitogen activated protein kinase
- GO
- Gene Ontology
- TAK1
- TGF-β-activated kinase 1
- PPI
- protein-protein interaction
- EGFR
- EGF receptor.
REFERENCES
- 1. Billi P., Della Strada M., Pierro A., Semprini S., Tommasini N., and Sambri V. (2016) Three years retrospective analysis of the incidence of Toxoplasma gondii infection in pregnant women living in the Greater Romagna Area (North Eastern Italy). Clin. Microbiol. Infect. 22, 572. [DOI] [PubMed] [Google Scholar]
- 2. Shen G., Wang X., Sun H., and Gao Y. (2016) Seroprevalence of Toxoplasma gondii infection among HIV/AIDS patients in Eastern China. Korean J. Parasitol. 54, 93–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dubey J. P. (2008) The history of Toxoplasma gondii–the first 100 years. J. Eukaryot. Microbiol. 55, 467–475 [DOI] [PubMed] [Google Scholar]
- 4. Boothroyd J. C. (2009) Toxoplasma gondii: 25 years and 25 major advances for the field. Int. J. Parasitol. 39, 935–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boothroyd J. C., and Dubremetz J. F. (2008) Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat. Rev. Microbiol. 6, 79–88 [DOI] [PubMed] [Google Scholar]
- 6. Saeij J. P., Boyle J. P., Coller S., Taylor S., Sibley L. D., Brooke-Powell E. T., Ajioka J. W., and Boothroyd J. C. (2006) Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314, 1780–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor S., Barragan A., Su C., Fux B., Fentress S. J., Tang K., Beatty W. L., Hajj H. E., Jerome M., Behnke M. S., White M., Wootton J. C., and Sibley L. D. (2006) A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314, 1776–1780 [DOI] [PubMed] [Google Scholar]
- 8. Bradley P. J., Ward C., Cheng S. J., Alexander D. L., Coller S., Coombs G. H., Dunn J. D., Ferguson D. J., Sanderson S. J., Wastling J. M., and Boothroyd J. C. (2005) Proteomic analysis of rhoptry organelles reveals many novel constituents for host-parasite interactions in Toxoplasma gondii. J. Biol. Chem. 280, 34245–34258 [DOI] [PubMed] [Google Scholar]
- 9. Sinai A. P. (2007) The Toxoplasma kinase ROP18: an active member of a degenerate family. PLoS Pathog. 3, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steinfeldt T., Konen-Waisman S., Tong L., Pawlowski N., Lamkemeyer T., Sibley L. D., Hunn J. P., and Howard J. C. (2010) Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 8, e1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fentress S. J., Behnke M. S., Dunay I. R., Mashayekhi M., Rommereim L. M., Fox B. A., Bzik D. J., Taylor G. A., Turk B. E., Lichti C. F., Townsend R. R., Qiu W., Hui R., Beatty W. L., and Sibley L. D. (2010) Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8, 484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamamoto M., Ma J. S., Mueller C., Kamiyama N., Saiga H., Kubo E., Kimura T., Okamoto T., Okuyama M., Kayama H., Nagamune K., Takashima S., Matsuura Y., Soldati-Favre D., and Takeda K. (2011) ATF6β is a host cellular target of the Toxoplasma gondii virulence factor ROP18. J. Exp. Med. 208, 1533–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du J., An R., Chen L., Shen Y., Chen Y., Cheng L., Jiang Z., Zhang A., Yu L., Chu D., Shen Y., Luo Q., Chen H., Wan L., Li M., et al. (2014) Toxoplasma gondii virulence factor ROP18 inhibits the host NF-κB pathway by promoting p65 degradation. J. Biol. Chem. 289, 12578–12592 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Cheng L., Chen Y., Chen L., Shen Y., Shen J., An R., Luo Q., and Du J. (2012) Interactions between the ROP18 kinase and host cell proteins that aid in the parasitism of Toxoplasma gondii. Acta Trop. 122, 255–260 [DOI] [PubMed] [Google Scholar]
- 15. Lin Y. Y., Lu J. Y., Zhang J., Walter W., Dang W., Wan J., Tao S. C., Qian J., Zhao Y., Boeke J. D., Berger S. L., and Zhu H. (2009) Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136, 1073–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu J. Y., Lin Y. Y., Qian J., Tao S. C., Zhu J., Pickart C., and Zhu H. (2008) Functional dissection of a HECT ubiquitin E3 ligase. Mol. Cell. Proteomics 7, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sutandy F. X., Qian J., Chen C. S., and Zhu H. (2013) Overview of protein microarrays. Curr. Protoc. Protein Sci. Chapter 27, Unit 27.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Newman R. H., Hu J., Rho H. S., Xie Z., Woodard C., Neiswinger J., Cooper C., Shirley M., Clark H. M., Hu S., Hwang W., Jeong J. S., Wu G., Lin J., Gao X., et al. (2013) Construction of human activity-based phosphorylation networks. Mol. Syst. Biol. 9, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu J., Rho H. S., Newman R. H., Hwang W., Neiswinger J., Zhu H., Zhang J., and Qian J. (2014) Global analysis of phosphorylation networks in humans. Biochim. Biophys. Acta 1844, 224–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Woodard C. L., Goodwin C. R., Wan J., Xia S., Newman R., Hu J., Zhang J., Hayward S. D., Qian J., Laterra J., and Zhu H. (2013) Profiling the dynamics of a human phosphorylome reveals new components in HGF/c-Met signaling. PLoS ONE 8, e72671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woodard C., Shamay M., Liao G., Zhu J., Ng A. N., Li R., Newman R., Rho H. S., Hu J., Wan J., Qian J., Zhu H., and Hayward S. D. (2012) Phosphorylation of the chromatin binding domain of KSHV LANA. PLoS Pathog. 8, e1002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li R., Zhu J., Xie Z., Liao G., Liu J., Chen M. R., Hu S., Woodard C., Lin J., Taverna S. D., Desai P., Ambinder R. F., Hayward G. S., Qian J., Zhu H., and Hayward S. D. (2011) Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10, 390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woodard C., Liao G., Goodwin C. R., Hu J., Xie Z., Dos Reis T. F., Newman R., Rho H., Qian J., Zhu H., and Hayward S. D. (2015) A screen for extracellular signal-regulated kinase-primed glycogen synthase kinase 3 substrates identifies the p53 inhibitor iASPP. J. Virol. 89, 9232–9241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu J., Liao G., Shan L., Zhang J., Chen M. R., Hayward G. S., Hayward S. D., Desai P., and Zhu H. (2009) Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J. Virol. 83, 5219–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fentress S. J., Steinfeldt T., Howard J. C., and Sibley L. D. (2012) The arginine-rich N-terminal domain of ROP18 is necessary for vacuole targeting and virulence of Toxoplasma gondii. Cell. Microbiol. 14, 1921–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sohn W. M., and Nam H. W. (1999) Western blotting analysis of stray cat sera against Toxoplasma gondii and the diagnostic availability of monoclonal antibodies in sandwich-ELISA. Korean J. Parasitol. 37, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Son E. S., and Nam H. W. (2001) Detection and characterization of excretory/secretory proteins from Toxoplasma gondii by monoclonal antibodies. Korean J. Parasitol. 39, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. El Hajj H., Lebrun M., Arold S. T., Vial H., Labesse G., and Dubremetz J. F. (2007) ROP18 is a rhoptry kinase controlling the intracellular proliferation of Toxoplasma gondii. PLoS Pathog. 3, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu H., Bilgin M., Bangham R., Hall D., Casamayor A., Bertone P., Lan N., Jansen R., Bidlingmaier S., Houfek T., Mitchell T., Miller P., Dean R. A., Gerstein M., and Snyder M. (2001) Global analysis of protein activities using proteome chips. Science 293, 2101–2105 [DOI] [PubMed] [Google Scholar]
- 30. Glover C. V., and Allis C. D. (1991) Enzyme activity dot blots for assaying protein kinases. Methods Enzymol. 200, 85–90 [DOI] [PubMed] [Google Scholar]
- 31. Lee Y. I., Giovinazzo D., Kang H. C., Lee Y., Jeong J. S., Doulias P. T., Xie Z., Hu J., Ghasemi M., Ischiropoulos H., Qian J., Zhu H., Blackshaw S., Dawson V. L., and Dawson T. M. (2014) Protein microarray characterization of the S-nitrosoproteome. Mol. Cell. Proteomics 13, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jeong J. S., Jiang L., Albino E., Marrero J., Rho H. S., Hu J., Hu S., Vera C., Bayron-Poueymiroy D., Rivera-Pacheco Z. A., Ramos L., Torres-Castro C., Qian J., Bonaventura J., Boeke J. D., Yap W. Y., Pino I., Eichinger D. J., Zhu H., and Blackshaw S. (2012) Rapid identification of monospecific monoclonal antibodies using a human proteome microarray. Mol. Cell. Proteomics 11, O111.016253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu S., Xie Z., Onishi A., Yu X., Jiang L., Lin J., Rho H. S., Woodard C., Wang H., Jeong J. S., Long S., He X., Wade H., Blackshaw S., Qian J., and Zhu H. (2009) Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 139, 610–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang Z., Ahn H. J., Park Y. H., and Nam H. W. (2016) Afatinib reduces STAT6 signaling of host ARPE-19 cells infected with Toxoplasma gondii. Korean J. Parasitol. 54, 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Einarson M. B., Pugacheva E. N., and Orlinick J. R. (2007) Identification of protein-protein interactions with glutathione-S-transferase (GST) fusion proteins. CSH Protoc. 2007, pdb.top11 [DOI] [PubMed] [Google Scholar]
- 37. Zhou X. W., Mudannayake M., Green M., Gigena M. S., Wang G., Shen R. F., and Rogers T. B. (2007) Proteomic studies of PP2A-B56γ1 phosphatase complexes reveal phosphorylation-regulated partners in cardiac local signaling. J. Proteome Res. 6, 3433–3442 [DOI] [PubMed] [Google Scholar]
- 38. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu J., Rho H. S., Newman R. H., Zhang J., Zhu H., and Qian J. (2014) PhosphoNetworks: a database for human phosphorylation networks. Bioinformatics 30, 141–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoa L., Kulaberoglu Y., Gundogdu R., Cook D., Mavis M., Gomez M., Gomez V., and Hergovich A. (2016) The characterisation of LATS2 kinase regulation in Hippo-YAP signalling. Cell. Signal. 28, 488–497 [DOI] [PubMed] [Google Scholar]
- 41. Wu L., Wang X., Li Y., Liu Y., Su D., Fu T., Guo F., Gu L., Jiang X., Chen S., and Cao J. (2016) Toxoplasma gondii ROP18: potential to manipulate host cell mitochondrial apoptosis. Parasitol. Res. 115, 2415–2422 [DOI] [PubMed] [Google Scholar]
- 42. Sapkota G. P., Cummings L., Newell F. S., Armstrong C., Bain J., Frodin M., Grauert M., Hoffmann M., Schnapp G., Steegmaier M., Cohen P., and Alessi D. R. (2007) BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 401, 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kruse J. P., and Gu W. (2009) Modes of p53 regulation. Cell 137, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miciak J., and Bunz F. (2016) Long story short: p53 mediates innate immunity. Biochim. Biophys. Acta 1865, 220–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brunet J., Pfaff A. W., Abidi A., Unoki M., Nakamura Y., Guinard M., Klein J. P., Candolfi E., and Mousli M. (2008) Toxoplasma gondii exploits UHRF1 and induces host cell cycle arrest at G2 to enable its proliferation. Cell. Microbiol. 10, 908–920 [DOI] [PubMed] [Google Scholar]
- 46. Bougdour A., Durandau E., Brenier-Pinchart M. P., Ortet P., Barakat M., Kieffer S., Curt-Varesano A., Curt-Bertini R. L., Bastien O., Coute Y., Pelloux H., and Hakimi M. A. (2013) Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe 13, 489–500 [DOI] [PubMed] [Google Scholar]
- 47. Kim J. Y., Ahn M. H., Song H. O., Choi J. H., Ryu J. S., Min D. Y., and Cho M. H. (2006) Involvement of MAPK activation in chemokine or COX-2 productions by Toxoplasma gondii. Korean J. Parasitol. 44, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quan J. H., Chu J. Q., Kwon J., Choi I. W., Ismail H. A., Zhou W., Cha G. H., Zhou Y., Yuk J. M., Jo E. K., and Lee Y. H. (2015) Intracellular networks of the PI3K/AKT and MAPK pathways for regulating Toxoplasma gondii-induced IL-23 and IL-12 production in human THP-1 cells. PLoS ONE 10, e0141550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zarubin T., and Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18 [DOI] [PubMed] [Google Scholar]
- 50. Valère A., Garnotel R., Villena I., Guenounou M., Pinon J. M., and Aubert D. (2003) Activation of the cellular mitogen-activated protein kinase pathways ERK, P38 and JNK during Toxoplasma gondii invasion. Parasite 10, 59–64 [DOI] [PubMed] [Google Scholar]
- 51. Kim L., Del Rio L., Butcher B. A., Mogensen T. H., Paludan S. R., Flavell R. A., and Denkers E. Y. (2005) p38 MAPK autophosphorylation drives macrophage IL-12 production during intracellular infection. J. Immunol. 174, 4178–4184 [DOI] [PubMed] [Google Scholar]
- 52. Yamamoto M., Okamoto T., Takeda K., Sato S., Sanjo H., Uematsu S., Saitoh T., Yamamoto N., Sakurai H., Ishii K. J., Yamaoka S., Kawai T., Matsuura Y., Takeuchi O., and Akira S. (2006) Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat. Immunol. 7, 962–970 [DOI] [PubMed] [Google Scholar]
- 53. Chen J., Hao L., Li C., Ye B., Du Y., Zhang H., Long B., Zhu P., Liu B., Yang L., Li P., Tian Y., and Fan Z. (2015) The endoplasmic reticulum adaptor protein ERAdP initiates NK cell activation via the Ubc13-mediated NF-κB pathway. J. Immunol. 194, 1292–1303 [DOI] [PubMed] [Google Scholar]
- 54. Cheng J., Fan Y. H., Xu X., Zhang H., Dou J., Tang Y., Zhong X., Rojas Y., Yu Y., Zhao Y., Vasudevan S. A., Zhang H., Nuchtern J. G., Kim E. S., Chen X., Lu F., and Yang J. (2014) A small-molecule inhibitor of UBE2N induces neuroblastoma cell death via activation of p53 and JNK pathways. Cell Death Dis. 5, e1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sanada T., Kim M., Mimuro H., Suzuki M., Ogawa M., Oyama A., Ashida H., Kobayashi T., Koyama T., Nagai S., Shibata Y., Gohda J., Inoue J., Mizushima T., and Sasakawa C. (2012) The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 483, 623–626 [DOI] [PubMed] [Google Scholar]
- 56. Clemente A. M., Severini C., Castronovo G., Tanturli M., Perissi E., Cozzolino F., and Torcia M. G. (2014) Effects of soluble extracts from Leishmania infantum promastigotes, Toxoplasma gondii tachyzoites on TGF-beta mediated pathways in activated CD4+ T lymphocytes. Microbes Infect. 16, 778–787 [DOI] [PubMed] [Google Scholar]
- 57. Massagué J. (1998) TGF-β signal transduction. Annu. Rev. Biochem. 67, 753–791 [DOI] [PubMed] [Google Scholar]
- 58. Fuentealba L. C., Eivers E., Ikeda A., Hurtado C., Kuroda H., Pera E. M., and De Robertis E. M. (2007) Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131, 980–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sapkota G., Alarcón C., Spagnoli F. M., Brivanlou A. H., and Massagué J. (2007) Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol. Cell 25, 441–454 [DOI] [PubMed] [Google Scholar]
- 60. Lim D., Gold D. A., Julien L., Rosowski E. E., Niedelman W., Yaffe M. B., and Saeij J. P. (2013) Structure of the Toxoplasma gondii ROP18 kinase domain reveals a second ligand binding pocket required for acute virulence. J. Biol. Chem. 288, 34968–34980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mattanovich D., Branduardi P., Dato L., Gasser B., Sauer M., and Porro D. (2012) Recombinant protein production in yeasts. Methods Mol. Biol. 824, 329–358 [DOI] [PubMed] [Google Scholar]
- 62. Qu D., Han J., and Du A. (2013) Enhancement of protective immune response to recombinant Toxoplasma gondii ROP18 antigen by ginsenoside Re. Exp. Parasitol. 135, 234–239 [DOI] [PubMed] [Google Scholar]
- 63. Zhou C. X., Zhou D. H., Elsheikha H. M., Zhao Y., Suo X., and Zhu X. Q. (2016) Metabolomic profiling of mice serum during toxoplasmosis progression using liquid chromatography-mass spectrometry. Sci. Rep. 6, 19557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. He J. J., Ma J., Elsheikha H. M., Song H. Q., Zhou D. H., and Zhu X. Q. (2016) Proteomic profiling of mouse liver following acute Toxoplasma gondii infection. PLoS ONE 11, e0152022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Choi E., Zhang X., Xing C., and Yu H. (2016) Mitotic checkpoint regulators control insulin signaling and metabolic homeostasis. Cell 166, 567–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Titchenell P. M., Quinn W. J., Lu M., Chu Q., Lu W., Li C., Chen H., Monks B. R., Chen J., Rabinowitz J. D., and Birnbaum M. J. (2016) Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab. 23, 1154–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. MacRae J. I., Sheiner L., Nahid A., Tonkin C., Striepen B., and McConville M. J. (2012) Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of Toxoplasma gondii. Cell Host Microbe 12, 682–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wu K., Yang Y., Liu D., Qi Y., Zhang C., Zhao J., and Zhao S. (2016) Activation of PPARγ suppresses proliferation and induces apoptosis of esophageal cancer cells by inhibiting TLR4-dependent MAPK pathway. Oncotarget 7, 44572–44582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen S., Fisher R. C., Signs S., Molina L. A., Shenoy A. K., Lopez M. C., Baker H. V., Koomen J. M., Chen Y., Gittleman H., Barnholtz-Sloan J., Berg A., Appelman H. D., and Huang E. H. (2016) Inhibition of PI3K/Akt/mTOR signaling in PI3KR2-overexpressing colon cancer stem cells reduces tumor growth due to apoptosis. Oncotarget 10.18632/oncotarget.9919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Koch S., Tugues S., Li X., Gualandi L., and Claesson-Welsh L. (2011) Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 437, 169–183 [DOI] [PubMed] [Google Scholar]
- 71. Peters G. A., Li S., and Sen G. C. (2006) Phosphorylation of specific serine residues in the PKR activation domain of PACT is essential for its ability to mediate apoptosis. J. Biol. Chem. 281, 35129–35136 [DOI] [PubMed] [Google Scholar]
- 72. Ye Y., and Rape M. (2009) Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lamothe B., Besse A., Campos A. D., Webster W. K., Wu H., and Darnay B. G. (2007) Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of IκB kinase activation. J. Biol. Chem. 282, 4102–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Asamitsu K., Tetsuka T., Kanazawa S., and Okamoto T. (2003) RING finger protein AO7 supports NF-κB-mediated transcription by interacting with the transactivation domain of the p65 subunit. J. Biol. Chem. 278, 26879–26887 [DOI] [PubMed] [Google Scholar]
- 75. Sa Q., Ochiai E., Tiwari A., Perkins S., Mullins J., Gehman M., Huckle W., Eyestone W. H., Saunders T. L., Shelton B. J., and Suzuki Y. (2015) Cutting edge: IFN-γ produced by brain-resident cells is crucial to control cerebral infection with Toxoplasma gondii. J. Immunol. 195, 796–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lang C., Algner M., Beinert N., Gross U., and Lüder C. G. (2006) Diverse mechanisms employed by Toxoplasma gondii to inhibit IFN-γ-induced major histocompatibility complex class II gene expression. Microbes Infect. 8, 1994–2005 [DOI] [PubMed] [Google Scholar]
- 77. Lüder C. G., Walter W., Beuerle B., Maeurer M. J., and Gross U. (2001) Toxoplasma gondii down-regulates MHC class II gene expression and antigen presentation by murine macrophages via interference with nuclear translocation of STAT1α. Eur. J. Immunol. 31, 1475–1484 [DOI] [PubMed] [Google Scholar]
- 78. Ramsauer K., Farlik M., Zupkovitz G., Seiser C., Kröger A., Hauser H., and Decker T. (2007) Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-γ-inducible gbp2 gene. Proc. Natl. Acad. Sci. U.S.A. 104, 2849–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yuan L., Chen Z., Song S., Wang S., Tian C., Xing G., Chen X., Xiao Z. X., He F., and Zhang L. (2015) p53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. J. Biol. Chem. 290, 3172–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Heinrich A., Haarmann H., Zahradnik S., Frenzel K., Schreiber F., Klassert T. E., Heyl K. A., Endres A. S., Schmidtke M., Hofmann J., and Slevogt H. (2016) Moraxella catarrhalis decreases antiviral innate immune responses by down-regulation of TLR3 via inhibition of p53 in human bronchial epithelial cells. FASEB J. 30, 2426–2434 [DOI] [PubMed] [Google Scholar]
- 81. Jirmanova L., Jankovic D., Fornace A. J. Jr, and Ashwell J. D. (2007) Gadd45α regulates p38-dependent dendritic cell cytokine production and Th1 differentiation. J. Immunol. 178, 4153–4158 [DOI] [PubMed] [Google Scholar]
- 82. Braun L., Brenier-Pinchart M. P., Yogavel M., Curt-Varesano A., Curt-Bertini R. L., Hussain T., Kieffer-Jaquinod S., Coute Y., Pelloux H., Tardieux I., Sharma A., Belrhali H., Bougdour A., and Hakimi M. A. (2013) A Toxoplasma dense granule protein, GRA24, modulates the early immune response to infection by promoting a direct and sustained host p38 MAPK activation. J. Exp. Med. 210, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., and Chen Z. J. (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 [DOI] [PubMed] [Google Scholar]
- 84. Fukushima T., Matsuzawa S., Kress C. L., Bruey J. M., Krajewska M., Lefebvre S., Zapata J. M., Ronai Z., and Reed J. C. (2007) Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc. Natl. Acad. Sci. U.S.A. 104, 6371–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zare-Bidaki M., Assar S., Hakimi H., Abdollahi S. H., Nosratabadi R., Kennedy D., and Arababadi M. K. (2016) TGF-βin toxoplasmosis: friend or foe? Cytokine 86, 29–35 [DOI] [PubMed] [Google Scholar]
- 86. Liu H., Jia D., Li A., Chau J., He D., Ruan X., Liu F., Li J., He L., and Li B. (2013) p53 regulates neural stem cell proliferation and differentiation via BMP-Smad1 signaling and Id1. Stem Cells Dev. 22, 913–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Besson-Fournier C., Gineste A., Latour C., Gourbeyre O., Meynard D., Martin P., Oswald E., Coppin H., and Roth M. P. (2016) Hepcidin upregulation by inflammation is independent of Smad1/5/8 signaling by activin B. Blood, blood-2016-10-748541 [DOI] [PubMed] [Google Scholar]
- 88. Gandhi R., Kumar D., Burns E. J., Nadeau M., Dake B., Laroni A., Kozoriz D., Weiner H. L., and Quintana F. J. (2010) Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat. Immunol. 11, 846–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nurgazieva D., Mickley A., Moganti K., Ming W., Ovsyi I., Popova A., Sachindra, Awad K., Wang N., Bieback K., Goerdt S., Kzhyshkowska J., and Gratchev A. (2015) TGF-beta1, but not bone morphogenetic proteins, activates Smad1/5 pathway in primary human macrophages and induces expression of proatherogenic genes. J. Immunol. 194, 709–718 [DOI] [PubMed] [Google Scholar]
- 90. Zhou W., Quan J. H., Lee Y. H., Shin D. W., and Cha G. H. (2013) Toxoplasma gondii proliferation require down-regulation of host Nox4 expression via activation of PI3 kinase/Akt signaling pathway. PLoS ONE 8, e66306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hai Y., Sun M., Niu M., Yuan Q., Guo Y., Li Z., and He Z. (2015) BMP4 promotes human Sertoli cell proliferation via Smad1/5 and ID2/3 pathway and its abnormality is associated with azoospermia. Discov. Med. 19, 311–325 [PubMed] [Google Scholar]
- 92. Shaw L. A., Bélanger S., Omilusik K. D., Cho S., Scott-Browne J. P., Nance J. P., Goulding J., Lasorella A., Lu L. F., Crotty S., and Goldrath A. W. (2016) Id2 reinforces TH1 differentiation and inhibits E2A to repress TFH differentiation. Nat. Immunol. 17, 834–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lin Y. Y., Jones-Mason M. E., Inoue M., Lasorella A., Iavarone A., Li Q. J., Shinohara M. L., and Zhuang Y. (2012) Transcriptional regulator Id2 is required for the CD4 T cell immune response in the development of experimental autoimmune encephalomyelitis. J. Immunol. 189, 1400–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Miyazaki M., Miyazaki K., Chen S., Itoi M., Miller M., Lu L. F., Varki N., Chang A. N., Broide D. H., and Murre C. (2014) Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat. Immunol. 15, 767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yang C. Y., Best J. A., Knell J., Yang E., Sheridan A. D., Jesionek A. K., Li H. S., Rivera R. R., Lind K. C., D'Cruz L. M., Watowich S. S., Murre C., and Goldrath A. W. (2011) The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat. Immunol. 12, 1221–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.