

Abstract
Metagenomics has made accessible an enormous reserve of global biochemical diversity. To tap into this vast resource of novel enzymes, we have screened over one million clones from metagenome DNA libraries derived from sixteen different environments for carboxylesterase activity and identified 714 positive hits. We have validated the esterase activity of 80 selected genes, which belong to 17 different protein families including unknown and cyclase-like proteins. Three metagenomic enzymes exhibited lipase activity, and seven proteins showed polyester depolymerization activity against polylactic acid and polycaprolactone. Detailed biochemical characterization of four new enzymes revealed their substrate preference, whereas their catalytic residues were identified using site-directed mutagenesis. The crystal structure of the metal-ion dependent esterase MGS0169 from the amidohydrolase superfamily revealed a novel active site with a bound unknown ligand. Thus, activity-centered metagenomics has revealed diverse enzymes and novel families of microbial carboxylesterases, whose activity could not have been predicted using bioinformatics tools.
We are living on a “Planet of microbes” with microorganisms and their communities occupying every biological niche and representing the largest part of the global biodiversity. Our present knowledge of microorganisms and their enzymes is based largely on laboratory studies of pure microbial cultures. However, more than 99% of environmental microbes cannot be cultivated in the lab using routine techniques and therefore cannot be studied using classical experimental approaches1,2,3. Metagenomics has emerged as a strategic approach to explore unculturable microbes through the sequencing and analysis of DNA extracted from environmental samples, as well as using such experimental methods as DNA hybridization, gene expression, proteomics, metabolomics, and enzymatic screening4,5,6,7. The remarkable contribution of this approach to global DNA sequencing efforts has been demonstrated by several large scale metagenomic projects including the Sargasso Sea sampling (over one million novel protein encoding genes), the Global Ocean Survey (over six million genes), and human gut microbiome studies (over 3 million genes)8,9,10,11.
Due to the progress in DNA sequencing technology, the number of sequenced genomes and protein sequences in public databases has expanded exponentially. As of July 2015, the UniProtKB/TrEMBL database contained over 50 million sequences (European Bioinfomatics Institute (EBI) website http://www.ebi.ac.uk/). However, it is estimated that the global protein universe of microorganisms exceeds 1012 proteins indicating that we know astonishingly little about microbial proteins and enzymes12,13. Even more, based on conservative estimates, over 50% of the sequences available in the databases have uncertain (general), unknown, or incorrectly annotated functions14. Therefore, the direct experimental determination of protein function or enzyme activity for millions of biochemically uncharacterized proteins or genes of unknown function represents one of the major challenges in postgenomic biology. In addition to sequence similarity-based and comparative genomics methods of gene function prediction, there are several experimental approaches to annotation including analysis of gene or protein interactions, gene expression, gene knockouts, protein localization, and protein structures15,16,17,18,19. However, in most cases, these approaches produce predictions or general annotations of biochemical or cellular function requiring subsequent experimental verification. In contrast, screening of purified proteins or metagenome gene libraries for enzymatic activity represents a direct experimental approach to identify the biochemical function of unknown proteins5,7,20,21,22. The feasibility and merits of general and specific enzymatic assays for screening of purified proteins and metagenome libraries has already been demonstrated for many hydrolases and oxidoreductases, two very broad classes of enzymes5,7,20,21,23.
The metagenomic enzyme screening approach involves directly assaying proteins expressed from environmental DNA in a surrogate host (most often E. coli) for enzymatic activity against a specific chemical substrate24. An alternate approach is to clone environmental DNA fragments into a lambda phage-based system and to screen for enzymatic activities directly on phage plaques25. Enzymatic screening of metagenome libraries provides the possibility to mine for new enzyme activities and discover novel families of enzymes with no sequence similarity to previously characterized proteins. This method has greatly expanded the number of novel enzymes, including over 130 new nitrilases and many cellulases, carboxylesterases, and laccases26,27,28. A recent high-throughput metagenomics project has identified over 27,000 putative carbohydrate-active genes in the cow rumen metagenome and demonstrated the presence of glycosyl hydrolase activity in 51 out of 90 tested proteins29. In addition, metagenomes from several extreme environments have revealed a rich biochemical diversity of enzymes adapted to function under extreme conditions, such as low/high temperatures, low/high pH, and high salt concentrations or high pressure5,30,31. Biochemical and structural characterization of these enzymes has revealed different molecular mechanisms of adaptation to extreme environmental conditions32,33,34. A recent analysis of metagenome screening works published in the last two decades revealed that these studies identified almost 6,000 genes with 70% of them representing carboxylesterases and lipases35. Based on sequence, most known carboxylesterases and lipases belong to the large protein superfamilies of α/β hydrolases and β-lactamases and have been classified into 16 families36,37,38. Since these enzymes are of high interest for applications in biotechnology, a significant number of these proteins have been characterized both structurally and biochemically, mostly esterases from the α/β hydrolase superfamily36,39,40,41.
Here we present the results of enzymatic screening of 16 metagenomic gene libraries from different environments for novel carboxylesterases. We have identified over 700 positive clones, from which 80 selected genes were expressed in E. coli, and their esterase activities were confirmed using additional assays. Four enzymes representing unknown (DUF3089) and hypothetical (MGS0084) proteins, cyclase-like enzymes (PF04199), as well as polyester hydrolyzing and lipolytic enzymes were characterized biochemically including substrate and temperature profiles. The active site residues of new enzymes were identified using site-directed mutagenesis, and the crystal structure of a metal-dependent cyclase-like esterase provided insight into the molecular mechanisms of its activity.
Results and Discussion
Enzymatic screening of metagenome libraries for carboxylesterase activity
To probe the biochemical diversity of carboxylesterases from uncultured microbes of environmental metagenomes, we screened 16 metagenome DNA libraries prepared from different geographic sites including various marine environments, soils, and waste treatment facilities (Table 1, Supplementary Table S1). The environments include moderate to hypersaline (3.8% to 10% NaCl, w/vol) conditions, low to elevated temperatures (3 °C to 50 °C), as well as sites contaminated with petroleum or heavy metals, from public or industrial wastewater sludge digesters (Supplementary Table S1). Overall, we screened over 1 million fosmid and Lambda-ZAP clones (approximately 7,000 Mbp DNA) for the ability to degrade tributyrin, generating a total 714 positive fosmid and Lambda-ZAP clones (Fig. 1, Table 1). Lambda-ZAP clones (208) and 178 fosmids (from the total 506) were sequenced by primer walking or using Illumina HiSeq, respectively. All genes predicted to have hydrolytic enzyme activity were cloned for protein purification. Where no sequence similarity to known esterases was found (two Haven library clones), we subcloned all predicted open reading frames and identified the presence of esterase activity in one hypothetical protein (MGS0084) and one predicted cyclase (MGS0169).
Table 1. Metagenomic libraries used in this work and esterase screening results.
| Metagenome library | Number of clones in library | Clones screened | Positive clones |
Validated esterasesa | |
|---|---|---|---|---|---|
| Identified | Sequenced | ||||
| 1. Anaerobic waste water digester (Evry, France) | 99,840 | 47,616c | 254 | 110 | 28 |
| 2. Composting plant (Liemehna, Germany) | 49,000 | 60,000b | 6 | 6 | 5 |
| 3. Kolguev Island (Barents Sea) | 100,000 | 142,000b | 34 | 34 | 4 |
| 4. Messina harbour (Mediterranean Sea) | 1,000 | 24,000b | 18 | 18 | 8 |
| 5. Messina Int II (Mediterranean Sea) | 10,368 | 5,760c | 208 | 50 | 1 |
| 6. Michle soil (Czech Republic) | 15,000 | 99,900b | 20 | 20 | 3 |
| 7. Milazzo (Mediterranean Sea) | 2,400 | 20,000b | 8 | 8 | 3 |
| 8. MT Haven sunken shipwreck (Ligurian Sea, Italy) | 25,000 | 36,800b | 19 | 19 | 11 |
| 9. Priolo (Gargallo, Italy) | 40,000 | 118,500b | 4 | 4 | 2 |
| 10. Port of Murmansk (Barents Sea) | 100,000 | 108,000b | 43 | 43 | 3 |
| 11. Rimicaris exoculata gill | 150,000 | 21,100b | 5 | 5 | 2 |
| 12. Rimicaris exoculata gut | 350,000 | 137,500b | 4 | 4 | 2 |
| 13. Sobeslav soil (Czech Republic) | 2,500 | 114,000b | 5 | 5 | 2 |
| 14. Tembec Paper Mill (Ontario, Canada) | 100,000 | 53,500b | 1 | 1 | 1 |
| 15. Urania DHAL (Mediterranean Sea) | 100,000 | 90,800b | 41 | 41 | 1 |
| 16. Vulcano Island (Mediterranean Sea) | 3,456 | 1,920c | 44 | 18 | 1 |
| 17. Cycloclasticus sp. 78-ME (genomic library) | 768 | 768c | 27 | 24 | 3 |
| Total | 1,080,628 | 714 | 386 | 80 | |
aEsterase activity of selected proteins was confirmed by subcloning and individual expression in E. coli followed by assays with crude extracts or purified proteins.
bLambda-ZAP libraries, mixed clone pools.
cFosmid libraries.
Figure 1. Agar-based screening of metagenomic libraries and purified metagenomic proteins for esterase and lipase activities.

(a) Fosmid library screening (tributyrin plate); (b) lambda-Zap library screening (tributyrin plate); (c) screening of purified proteins (activity validation; tributyrin plate); (d) screening of purified proteins for lipase activity (olive oil plate). Purified proteins (50 μg, except for MGS0169 and GEN0034, for which 100 μg were used) were spotted onto agar plates containing 1% tributyrin (a,b,c) or 3% olive oil (d) and incubated overnight at 30 °C. Commercial enzymes pig liver esterase (PLE, 5 μg) and Lipase A (LipA, 5 μg) were used as positive controls. A clearing on tributyrin plates indicates esterase activity while fluorescence visualized under UV light (bright yellow spots) on olive oil plates indicates lipase activity.
We confirmed the presence of esterase activity in 80 selected genes using agar plates with 1% tributyrin (Fig. 1, Supplementary Table S2). These enzymes were also tested for the presence of lipase activity, based on the ability to hydrolyze long chain-length lipids (C16, C18), using an olive oil agar plate assay (Fig. 1d). Three enzymes (MGS0084, MGS0156 and GEN0160) out of 80 tested clones were found to have lipase activity (Fig. 1), consistent with previous metagenome screens where low frequency of lipase activity was reported7.
Sequence analysis and enzyme families of identified metagenomic esterases
BLASTp searches of the NCBI database using 80 validated metagenomic esterases as queries indicated that most of these proteins represent genuine metagenomic enzymes with just 11 sequences from known genomes including Alcanivorax borkumensis, Cycloclasticus sp. 78-ME, Marinobacter hydrocarbonoclasticus, Parvibaculum lavamentivorans, and Serratia fonticola (99–100% identity) (Fig. 2, Supplementary Table S2). Sixty-nine remaining esterases showed 28–98% sequence identity to sequences from the NCBI database with most sequences within the range of 50–80% identity. Analysis of phylogenetic distribution of the 80 validated metagenomic esterases revealed that these proteins and their top sequence homologues are present in a broad range of Gram-positive and Gram-negative microorganisms with most proteins found in Proteobacteria (52 proteins), Terrabacteria (11 proteins), and the Fibrobacteres, Chlorobi and Bacteroidetes (FCB) group (10 proteins) (Fig. 2).
Figure 2. Phylogenetic distribution of 80 metagenomic carboxylesterases and their top sequence homologues across the tree of life.
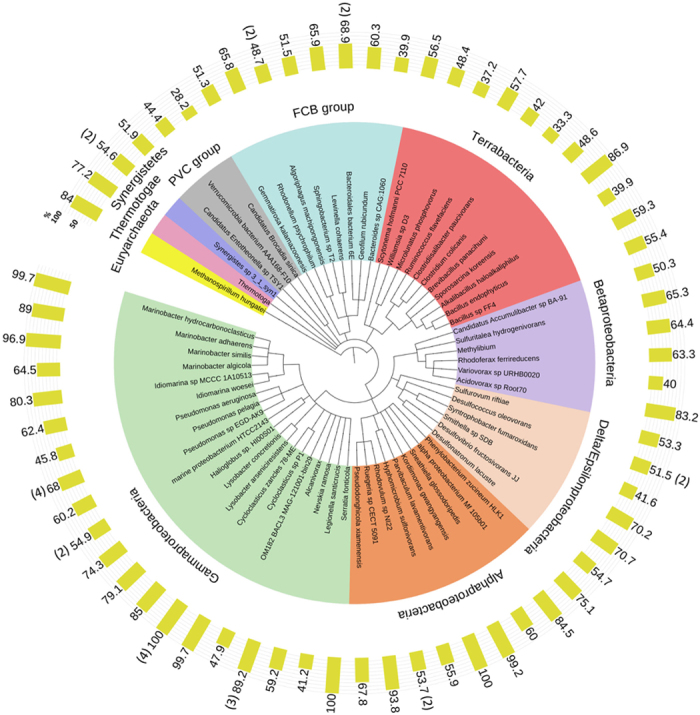
The numbers on yellow bars show sequence identity (%) of metagenomic esterases to protein sequences from indicated organisms. For organisms/genera with more than one metagenome esterase homologue, the means are indicated (with the number of esterases shown in brackets).
Based on sequence analysis, the 80 validated esterases belong to 17 protein families (Fig. 3). A majority of these enzymes are predicted to belong to the α/β hydrolase superfamily (59 proteins), which represents one of the largest groups of structurally related proteins (148 families in the ESTHER database) with diverse catalytic and non-catalytic functions including hydrolases, dehalogenases, haloperoxidases, and hydroxynitrile lyases36,38,42,43. Their catalytic activity depends on the conserved catalytic triad, which consists of a nucleophile (serine, aspartate or cysteine), a histidine and a catalytic acid (aspartate or glutamate). Most α/β hydrolases that we have identified have a conserved Gly-x-Ser-x-Gly catalytic motif and are distributed among eleven different families, with a majority belonging to α/β hydrolase-3, α/β hydrolase-1, and α/β hydrolase-6. The remaining enzymes are distributed among Hydrolase_4, Esterase, Peptidase_S9, COesterase, Chlorophyllase_2, Esterase_phd and DUF676 families, with the exception of two (Fig. 3, Supplementary Fig. S1). Although enzymes MGS0032 and MGS0156 are predicted to belong to the α/β superfamily, they are not associated with known hydrolase families, suggesting that these proteins may belong to new branches.
Figure 3. Protein family classification of 80 carboxylesterases identified by activity screening of metagenomic libraries.

For all genes, the presence of esterase activity was confirmed by individual subcloning and expression in E. coli. Enzymes were classified into protein families using HMMER and BLAST searches of Pfam and COG databases. Polypeptide chain lengths are proportional to size and predicted domains are depicted as boxes. Domains predicted/confirmed to confer esterase activity are shown in yellow. Proteins selected for biochemical characterization in this work are shown in red font.
Two metagenomic esterases, MGS0012 and GEN0034, belong to the DUF3089 family, which appears to be related to α/β hydrolases (Fig. 3, Supplementary Fig. 1). Recently, several members of this family were also isolated from metagenomic libraries and have been shown to exhibit esterase activity44,45,46,47,48,49. Interestingly, MGS0012 shares 99% protein sequence identity with the hypothetical protein WP_026168275 from Kordiimonas gwangyangensis (97.6% at the nucleotide level), which has been shown to have the ability to degrade high-molecular weight polycyclic aromatic hydrocarbons50.
Ten isolated enzymes belong to the esterase family VIII, which includes β-lactamase-like enzymes with promiscuous β-lactam hydrolytic activity responsible for resistance to β-lactam antibiotics. Previously, several metagenomic β-lactamase-like esterases have been characterized revealing high esterase activity against shorter chain p-nitrophenyl esters (C2-C5) and detectable hydrolytic activity against the β-lactamase substrates nitrocefin and cephalosporin31,51. All ten identified β-lactamase-like esterases are serine hydrolases with a conserved Ser-x-x-Lys catalytic motif typical for the class C β-lactamases. Interestingly, the α/β-hydrolase-like esterase GEN0169 has an additional Metallo-β-lactamase domain (PF00753). This is a domain commonly found in class B β-lactamases, a structurally unrelated enzyme family also capable of hydrolyzing β-lactam antibiotics.
The remaining hydrolase-like proteins share sequence similarity with Patatin-like phospholipases (5 proteins), SGNH-hydrolases (3 proteins), and 3-hydroxybutyrate oligomer hydrolase (one protein, PF10605). The unknown protein MGS0084 has only eight homologous sequences in the Uniprot and non-redundant GenBank databases. A protein sequence alignment of MGS0084 with its homologues shows a conserved Gly-His-Ser-His-(Ala/Gly)-Gly motif, which resembles Gly-x-Ser-x-Gly commonly found in α/β hydrolases suggesting that these proteins may represent a new branch of this superfamily (Supplementary Fig. S1).
MGS0169 belongs to the PF04199 family of putative cyclase-like enzymes, which contain a conserved His-x-Gly-Thr-His-x-Asp-x-Pro-x-His motif predicted to form part of the active site. This motif is only partially conserved in MGS0169 and its closest homologues with the two first His residues replaced by Gln (Supplementary Fig. S1). Several cyclase-like proteins from different bacteria have been shown to exhibit metal-dependent amidohydrolase activity against formylkynurenine and isatin52,53,54, but carboxylesterase activity of PF04199 proteins has not been reported before. Thus, enzymatic screens of metagenomic libraries have revealed carboxylesterases from diverse protein families, including several candidates, which could not have been annotated based on sequence analysis.
Biochemical characterization of selected metagenomic esterases
For biochemical and structural characterization of metagenomic esterases, we selected the lipolytic enzymes MGS0084 and GEN0160, as well as the novel esterases MGS0012 (DUF3089) and MGS0169 (a cyclase-like protein). The selected proteins were over-expressed in E. coli and affinity-purified to over 95% homogeneity. The acyl chain length preference of metagenomic esterases was analyzed using 11 model esterase substrates including three α-naphthyl and eight p-nitrophenyl (pNP) esters with different chain lengths (Fig. 4). MGS0012 showed the highest activity with α-naphthyl acetate, MGS0169 against pNP-acetate, whereas MGS0084 exhibited comparable activity against α-naphthyl- and pNP-acetate and propionate (Fig. 4). In contrast, GEN0160 showed a preference to substrates with longer acyl chains with the highest activity against pNP-octanoate α-naphthyl- or pNP-butyrate (C4, GEN0160). This protein showed detectable esterase activity against pNP-palmitate (C16), which is a representative substrate for lipases (Fig. 4). This is in line with the presence of hydrolytic activity of this enzyme toward olive oil (Fig. 1).
Figure 4. Substrate acyl chain length preference of metagenomic esterases.

Esterase activity of purified proteins against p-nitrophenyl esters (pNP-, white bars) or α-naphthyl esters (αN-, gray bars) with different acyl chain lengths. The reaction mixtures contained the indicated substrate (1 mM) and purified proteins: (a) 0.2 μg of MGS0012; (b) 0.2 μg of MGS0084; (c) 0.025 μg of MGS0169 (and 0.1 mM MnCl2); (d) 0.3 μg of GEN0160.
In contrast to the other three proteins, esterase activity of MGS0169 was greatly stimulated by the addition of divalent metal cations (Mn2+ > Mg2+ > Co2+ ≫ Ni2+) (Supplementary Fig. S2). Several biochemically characterized members of the cyclase-like protein family (PF04199) exhibited metal ion dependent amidohydrolase activity against formylkynurenine and isatin52,53,54. MGS0169 also showed detectable metal dependent amidohydrolase activity against isatin (kcat/KM 0.1 × 103 M−1 s−1), but its esterase activity against pNP-acetate was at least three orders of magnitude higher (kcat/KM ~ 0.2 × 106 M−1 s−1) (Fig. 4, Table 2). Previously, the presence of metal ion-stimulated esterase activity was demonstrated in the amidohydrolase proteins from the phosphotriesterase family (PF02126) including Rsp3690 from Rhodobacter sphaeroides and Pmi1525 from Proteus mirabilis55,56. However, these enzymes have different structural folds and active sites. Thus, MGS0169 and homologous proteins from the PF04199 family do indeed represent a novel group of metal-dependent esterases from the amidohydrolase superfamily.
Table 2. Kinetic parameters of purified metagenomic esterases.
| Protein | Variable substrate | KM, mM | kcat, s−1 | kcat/KM, M−1s−1 |
|---|---|---|---|---|
| MGS0012 | α-NAa (C2) | 0.71 ± 0.04 | 51.0 ± 1.0 | 0.7 × 105 |
| β-NA (C2) | 0.41 ± 0.05 | 25.3 ± 1.0 | 0.6 × 105 | |
| α-NP (C3) | 0.11 ± 0.01 | 28.8 ± 0.4 | 2.6 × 105 | |
| α-NB (C4) | 0.10 ± 0.01 | 29.5 ± 0.5 | 3.0 × 105 | |
| pNP-acetate (C2) | 2.12 ± 0.29 | 33.7 ± 2.3 | 0.2 × 105 | |
| pNP-propionate (C3) | 1.62 ± 0.31 | 28.1 ± 2.8 | 0.2 × 105 | |
| pNP-butyrate (C4) | 1.46 ± 0.19 | 16.8 ± 1.2 | 0.1 × 105 | |
| pNP-valerate (C5) | 0.87 ± 0.12 | 12.1 ± 0.8 | 0.1 × 105 | |
| pNP-octanoate (C8) | 0.78 ± 0.09 | 12.3 ± 0.8 | 0.2 × 105 | |
| pNP-decanoate (C10) | 0.23 ± 0.02 | 2.8 ± 0.1 | 0.1 × 105 | |
| MGS0084 | α-NA (C2) | 0.71 ± 0.11 | 39.8 ± 2.9 | 0.6 × 105 |
| α-NP (C3) | 0.43 ± 0.04 | 39.8 ± 2.0 | 0.9 × 105 | |
| pNP-acetate (C2) | 4.1 ± 1.2 | 35.8 ± 8.0 | 0.8 × 104 | |
| pNP-butyrate (C4) | 2.4 ± 0.5 | 7.4 ± 1.1 | 0.3 × 104 | |
| pNP-valerate (C5) | 0.53 ± 0.06 | 1.1 ± 0.1 | 0.2 × 104 | |
| GEN0160 | α-NA (C2) | 1.07 ± 0.04 | 6.1 ± 0.2 | 0.6 × 104 |
| α-NP (C3) | 0.65 ± 0.03 | 7.3 ± 1.7 | 0.1 × 105 | |
| α-NB (C4) | 0.32 ± 0.04 | 10.6 ± 0.5 | 0.3 × 105 | |
| MGS0169 | α-NA (C2) | 0.49 ± 0.11 | 14.9 ± 1.2 | 0.3 × 105 |
| α-NP (C3) | 0.42 ± 0.12 | 14.7 ± 1.4 | 0.4 × 105 | |
| pNP-acetate (C2) | 0.40 ± 0.29 | 71.2 ± 1.7 | 1.8 × 105 | |
| isatin (amidohydrolase activity) | 19.7 ± 4.2 | 1.9 ± 0.2 | 0.1 × 103 |
aα-NA, α-naphthyl acetate; β-NA, β-naphthyl acetate; α-NB, α-naphthyl butyrate; α-NP, α-naphthyl propionate.
The purified metagenomic esterases showed saturation kinetics and high catalytic efficiencies with low KM values toward the tested model esterase substrates (Table 2). The ester substrate profiles of four metagenomic esterases were determined using a library of 89 various monoesters including alkyl and aryl esters (Supplementary Fig. S3, Supplementary Table S3). These proteins showed hydrolytic activity against a broad range of substrates with different substrate preferences. MGS0012, MGS0169 and GEN0160 were most active against phenyl acetate, whereas MGS0084 against vinyl laurate (Supplementary Fig. S3). From these proteins, MGS0012 was found to be the most efficient esterase showing high kcat/KM values toward a broad range of substrates including the medium acyl chain esters (C4-C10) (Table 2).
The esterase activities of metagenomic esterases showed different temperature profiles determined in the range from 5 °C to 70 °C (Supplementary Fig. S4). MGS0084 was most active at 25 °C but retained almost 50% of maximal activity at 5 °C suggesting that it is a cold-adapted enzyme. In contrast, the other metagenomic esterases showed maximal activity at 40 °C and retained less than 15% of maximal activity at 5 °C, which is typical of mesophilic enzymes (Supplementary Fig. S4). The esterases also showed different sensitivities to high salt concentrations with MGS0084 exhibiting strong inhibition by 0.25 M NaCl or KCl (Supplementary Fig. S5). In contrast, the esterase activity of MGS0012 and GEN0160 was slightly stimulated by the addition of salt, and they showed no inhibition even at 2 M or 3 M salt concentration (Supplementary Fig. S5). The metagenomic esterases also showed different sensitivities to solvents (acetonitrile and DMSO) with MGS0012 being the most sensitive enzyme and GEN0160 being the most resistant enzyme (Supplementary Fig. S5). Thus, the characterized metagenomic esterases exhibit different temperature profiles and sensitivities to inhibition by salt and solvents perhaps reflecting differences in native environmental conditions and hosts.
Polyester depolymerization activity of purified metagenomic esterases
Recent studies including our work have demonstrated the presence of hydrolytic activity against polylactic acid (PLA), a biodegradable polyester, in several lipolytic enzymes and carboxylesterases31,57,58. In this work, 26 purified metagenomic esterases were screened for PLA-degrading activity using an agarose plate assay with the emulsified PLA2 (Mw 2 K). These screens revealed the presence of PLA hydrolytic activity in seven enzymes including the lipolytic esterases MGS0084 and MGS0156 (Fig. 5). An agarose-based screening of purified esterases using the emulsified polycaprolactone PCL10 (Mw 10 K), another biodegradable polyester, demonstrated the presence of high PCL10 depolymerization activity in MGS0084, GEN0105, and GEN0160, as well as in MGS0009 and MGS0156 (Fig. 5). The hydrolytic activity of the identified metagenomic esterases against different polyester substrates makes these enzymes attractive candidates for studies toward enzyme-based depolymerization of polyester plastics.
Figure 5. Hydrolytic activity of purified metagenomic esterases against polyester substrates.

Agarose-based polyester depolymerase screens with emulsified PLA2 (a) or polycaprolactone PCL10 (b). Agarose gels (1.5%) contained 0.2% PLA2 or PCL10 emulsified in 50 mM Tris-HCl (pH 8.0) containing 0.01% Plysurf A210G. The wells in agarose gel were loaded with 50 μg of purified proteins including the positive (PlaM4) and negative (PLE) controls. The formation of a clear halo around the wells is attributed to the enzymatic hydrolysis of insoluble polymeric substrates.
Crystal structure of the metal ion dependent esterase MGS0169
The purified seleno-methionine-substituted metagenomic esterases were also submitted to crystallization trials. MGS0169 (21–341 aa) produced diffracting crystals, and its crystal structure was determined at 1.61 Å resolution (Supplementary Table 4). The MGS0169 protomer core has a slightly distorted central β-barrel containing both parallel and anti-parallel β-strands surrounded by ten α-helices, whose fold resembles the swivelling β/α/β domain of metal-dependent α/β hydrolases53,59. The small sub-domain of MGS0169 is comprised two β-strands (β2 and β3) connected by a flexible loop containing a short α-helix with the strands of one protomer forming a four-stranded anti-parallel β-sheet with the two related β-strands of another protomer (Fig. 6). This results in the formation of a tightly packed twisted (~90°) tetramer through the interaction between the β-sheets stabilized by interactions between the surrounding α-helices (Fig. 6). Analysis of the crystal contacts using the quaternary structure prediction server PISA suggests that MGS0159 is likely to form tetramers through multiple interactions between tightly packed monomers burying ~7,000 Å2 of the solvent accessible surface per monomer (~30% of the total solvent accessible surface). The tetrameric organization of MGS0169 is supported by the results of size-exclusion chromatography suggesting a trimeric or tightly packed tetrameric organization (112 kDa, predicted Mw 37 kDa).
Figure 6. Crystal structure of MGS0169.

(a) Overall structure of the MGS0169 protomer (several orientations related by 90° rotation). The ribbon diagram of the core domain is colored gray (helices) and cyan (β-strands). (b) Three views of the MGS0169 tetramer related by 90° rotation with monomers colored gray, cyan, green, and orange. The last view is also shown in a surface presentation to demonstrate the tight packing of monomers. (c) Close-up view of the MGS0159 active site showing the bound unknown ligand (UL). The amino acid side chains and ligand molecule are shown as sticks along a protein ribbon colored gray. In the MGS0169 structure, the second protomer (colored wheat) contributes to the substrate binding site of the first protomer (shown in gray).
A Dali search for MGS0169 structural homologues identified several protein structures as the best matches including the isatin hydrolase IH-b from Labrenzia aggregata (PDB codes 4J0N and 4M8D; Z-score 21.3, rmsd 2.5 Å), the three microbial kynurenine formamidases KynB (PDB codes 4COB, 4COG, and 4CO9; Z-score 19.4–19.9, rmsd 2.1–2.7 Å), and the uncharacterized predicted hydrolase P84132_GEOSE from Geobacillus stearothermophilus (PDB code 3KRV; Z-score 18.6, rmsd 2.9 Å). These proteins share low sequence similarity to MGS0169 (17–22% sequence identity), and the two biochemically characterized enzymes (IH-b and KynB) have metal-dependent amidohydrolase activity against isatin and N-formylkynurenine53,54.
The active site of MGS0169
The location of the MGS0169 active site is indicated by the unknown electron density with a tetrahedral-like geometry located in the narrow cavity formed mainly by α-helices near the end of the central β-barrel (Fig. 6, Supplementary Fig. S2). Based on its shape, this density might represent a molecule of acetyl-phosphate, probably captured by the enzyme from E. coli cells. The bottom part of the ligand is positioned close to the side chains of the conserved Gln127 (5.3 Å), Gln131 (4.3 Å), Asp133 (3.8 Å), and His137 (2.6 Å) (Fig. 6). These residues represent a modified cyclase-like motif Gln-x-x-x-Gln-x-Asp-x-x-x-His found in several cyclase-like proteins (PF04199) including the trans-dienelactone hydrolase from Pseudomonas reinekei MT160 and are likely to be involved in metal ion binding. The bound ligand also interacts with the side chains of the conserved Arg87 (2.9 Å), Glu299 (2.6 Å), and His286 (2.6 Å). The MGS0169 substrate binding site is less conserved with only two residues (Phe84 and His286) identical to the isatin hydrolase substrate binding site (Phe41 and His212, PDB code 4J0N) (Fig. 6).
In the crystal structures of isatin hydrolase IH-b (PDB code 4M8D) and kynurenine formamidase KynB (PDB code 4COB), which contain the classical cyclase-like motif His-x-x-x-His-x-Asp-x-x-x-His, the side chains of the motif residues coordinate one Mn2+ ion (IH-b) or two Zn2+ ions (KynB), which are required for the enzymatic activity of these enzymes53,54. However, no metal ion was found in the corresponding site of the uncharacterized cyclase-like protein MJ0783 from Methanocaldococcus jannaschii (PDB code 2B0A), or in MGS0169. We propose that metal ion binding to MGS0169 was prevented by the presence of the bound acetylphosphate-like ligand, and the active site of the catalytically active MGS0169 contains one or two metal ions (probably Mn2+, based on the MGS0169 metal ion profile), as was suggested for the the trans-dienelactone hydrolase from Pseudomonas reinekei MT161.
The structure of the MGS0169 dimer also revealed that the side chain of Phe117 (and possibly Phe110 and Lys112) from one protomer contributes to the substrate binding site of another protomer and is positioned near the bound ligand (3.7 Å) and side chains of His137 (3.6 Å) and His286 (4.0 Å) (Fig. 6). This suggests that the two active sites of the MGS0169 dimer can allosterically interact through the residues of the composite binding sites, which is in line with the observed range of Hill coefficients of 1.2–1.5. The other highly conserved residues of the MGS0169 active site, which can potentially contribute to substrate binding include Phe84 (4.6 Å to the acetyl-like ligand), Arg87 (2.9 Å to the ligand), His286 (2.5 Å), and Glu299 (2.6 Å). Thus, the active site residues of MGS0169 and other cyclase-like proteins are different from those of non-specific carboxylesterases from the amidohydrolase superfamiliy55,56.
Validation of the catalytic residues of metagenomic esterases using site-directed mutagenesis
The potential active site residues of metagenomic esterases, selected based on their sequence alignments (Supplementary Fig. S1) and the MGS0169 crystal structure (Fig. 6), were verified using site-directed mutagenesis (alanine replacement). The mutant proteins were purified using affinity chromatography, and their enzymatic activity was compared to that of wild type proteins. As shown in Fig. 7, the catalytic triad of the DUF3089 hydrolase MGS0012 includes Ser193, Asp360, and His375, because the corresponding mutant proteins showed a greatly reduced catalytic activity. Low activity was also found in the MGS0012 D232A mutant protein, whereas S131A, S244A, and D247A retained high enzymatic activity (Fig. 7). Similarly, alanine replacement mutagenesis of the unknown protein MGS0084 suggested that it is a novel Ser-dependent hydrolase with the catalytic triad comprising Ser172, Asp300, and His386 (Fig. 7).
Figure 7. Identification of catalytic residues of novel esterases using site-directed mutagenesis: esterase activity of purified mutant proteins.
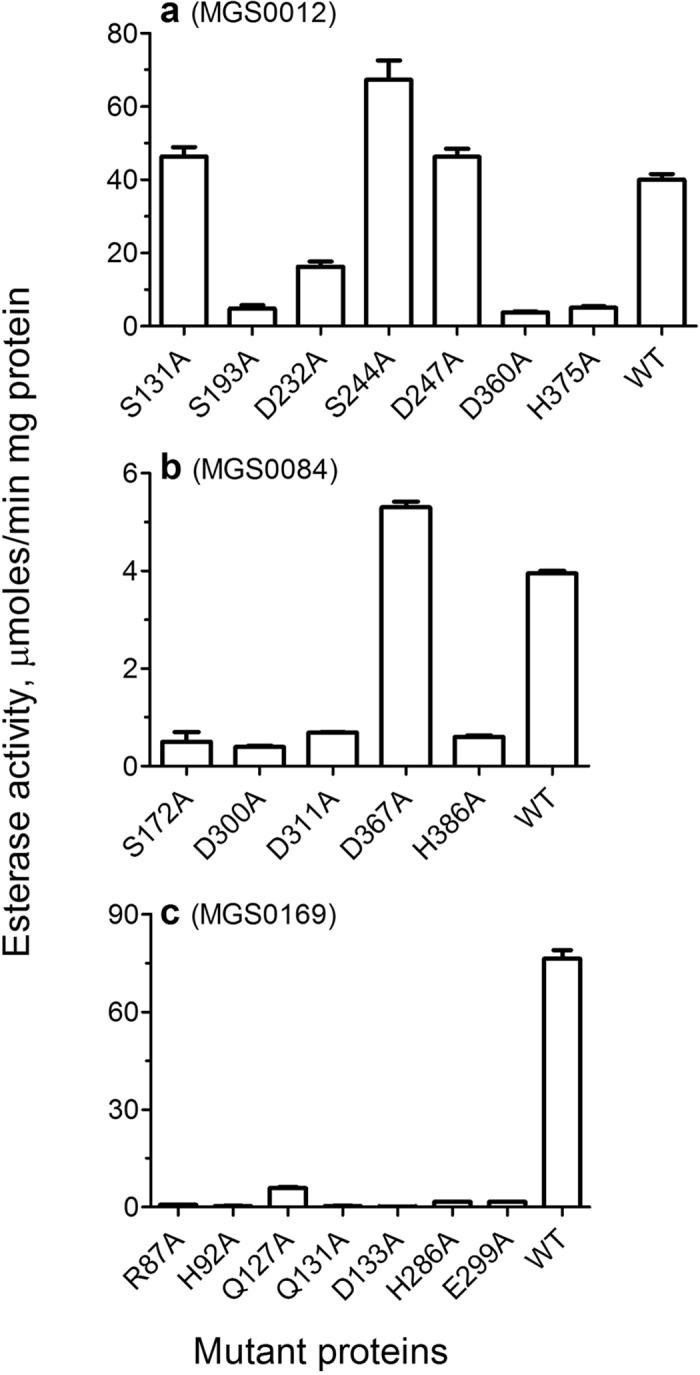
The reaction mixtures contained: (a) 2 mM α-naphthyl acetate and 0.2 μg of MGS0012; (b) 1.5 mM α-naphthyl propionate and 2 μg of MGS0084; (c) 1.5 mM pNP-acetate, 0.1 mM MnCl2, and 0.03 μg of MGS0169.
Alanine replacement mutagenesis of the metal-dependent esterase MGS0169 (from the cyclase-like family) revealed that this enzyme is sensitive to mutations in the active site including the residues of the modified cyclase-like motif Gln127, Gln131, and Asp133 (H137A was insoluble), as well as those involved in substrate binding (Arg87, H286, and Glu299) (Fig. 7). Thus, site-directed mutagenesis of metagenomic esterases revealed that MGS0012 and MGS0084 represent novel Ser-dependent hydrolases, whereas MGS0169 is a novel metal-dependent esterase with the modified cyclase-like motif Gln127-Gln131-Asp133-His137 potentially involved in metal ion binding.
Conclusions
The discovery of new enzymes in environmental bacteria contributes greatly to our fundamental knowledge of protein structure-function relationships, expands the biocatalytic toolbox of enzymes for metabolic engineering and synthetic biology, and improves the quality of gene annotation in public databases, on which bioinformatics tools rely. Enzymatic screening of environmental gene libraries presented in this work revealed a huge sequence and biochemical diversity with identification of 80 esterases from 17 different enzyme families (Fig. 3). These enzymes exhibit diverse substrate preferences, from short to long acyl chain esters with a significant number of enzymes possessing polyesterase activity against polylactic acid and polycaprolactone. The differences in their sensitivities to temperature and salt conditions likely reflect environmental adaptations. Through activity-based screening, we have been able to identify three novel Ser-dependent esterases and present the crystal structure of a new carboxylesterase subfamily within the cyclase-like family of metal ion dependent amidohydrolases. This work contributes a 30% increase in experimentally validated metagenomic esterases (288 enzymes according to a recent analysis35). By combining metagenomic enzyme discovery with protein and metabolic engineering, we may gain access to virtually unlimited diversity of enzyme sequences, with the potential to discover tailor-made enzymes for any biotransformation reaction.
Methods
Metagenome library preparation, gene cloning, and protein purification
Extraction of metagenomic DNA from environmental samples and preparation of fosmid and lambda-ZAP DNA libraries (Supplementary Table S1) were performed as described previously25,62,63,64. Genes were amplified by PCR from purified fosmids or excised lambda-ZAP plasmids and cloned into a modified pET15b vector, containing an N-terminal 6His tag, as described previously65. Tagged genes were overexpressed in E. coli BL21(DE3) and affinity purified using metal-chelate chromatography on Ni-NTA (Qiagen). Site-directed mutagenesis of selected enzymes was performed based on the QuikChange® site-directed mutagenesis kit (Stratagene). All mutations were verified by DNA sequencing, and the mutant proteins were overexpressed and purified like the wild-type.
Enzymatic screening of metagenomic libraries
E. coli fosmid clones were cultured at 37 °C in 384-well microtiter plates, and spotted onto Luria Broth (LB) agar plates containing chloramphenicol (12.5 μg/mL), arabinose (0.001–0.01%), gum Arabic (0.5%), and emulsified tributyrin (1%). Clones were grown overnight at 37 °C, then at 30 °C for 3–4 days. Colonies with clear halos were considered positive for esterase activity, and selected for plasmid extraction. Lambda-ZAP clones were screened as follows. 300 μL of mid-log phase E. coli XL1-Blue MRF’ cells were infected the Lambda-ZAP library added to 4 mL of 0.7% LB agar containing 10 mM MgSO4, 1 mM IPTG, 0.5% gum arabic and 1% of emulsified tributyrin, at 48 °C. The mixture was immediately layered onto LB agar plates containing 1 mM IPTG, at approximately 1,000 plaque forming units per plate, and the plates were incubated at 37 °C. Phage plaques exhibiting a clear halo over 3–4 days were isolated, and plasmids containing the metagenomic segments were extracted from phage DNA according to the manufacturer’s protocol.
To confirm esterase activity in cloned proteins, E. coli expressing the cloned genes were streaked onto LB-agar plates containing 1% tributyrin (as above) or purified enzymes were spotted directly and the plates were incubated at 30 °C or 37 °C. Clones were also checked for lipase activity either by streaking E. coli colonies or spotting 5–10 μg of the purified enzymes onto LB agar plates containing 3% emulsified olive oil and 0.001% Rhodamine B indicator dye, and incubating at 30 °C or 37 °C. Lipase activity was identified under UV light as orange fluorescence66.
Sequencing of metagenomic fragments and bioinformatics analysis
Lambda-ZAP clones were sequenced by primer walking, while fosmids were sequenced as mixed pools using Illumina or Roche 454 platforms (at TCAG, Genome Quebec and Genoscope). Reads were dereplicated and assembled into contigs using the Velvet algorithm67 in Geneious68 version 6.0.6, and contigs were mapped to specific fosmids using Sanger sequenced fosmid end sequences. Contigs were submitted to the MG-RAST69 pipeline for gene annotation. In parallel, open reading frames were predicted using the Glimmer algorithm70, and translated protein sequences were annotated through BLAST searches of UniProt and the non-redundant GenBank protein database71. Genes predicted as esterases, lipases, or hydrolases were selected for recombinant expression in E. coli. Where no such gene was found, smaller esterase positive genetic fragments were identified by subcloning, and all predicted genes were cloned and rescreened.
Proteins with confirmed esterase activity (Supplementary Table S2) were classified into families through sequence analysis using HMMER72 searches against the Pfam database and BLAST searches of the COG database73, with an E-value cut-off of 1E-5 unless otherwise indicated. Where an enzyme had a significant score to more than one protein family, the family with the smaller E-value and/or larger sequence coverage was assigned. Multiple sequence alignments were generated using MUSCLE74. The phylogenetic tree was produced using the NCBI taxonomy of the closest sequence homologues and the PhyloT tree generator (http://phylot.biobyte.de/) and visualized using the iTOL v3 online tool75.
Enzymatic assays with purified proteins
Carboxylesterase activity of purified proteins against p-nitrophenyl (pNP) or α-naphthyl esters of various fatty acids was measured spectrophotometrically as described previously31. The effect of temperature, salts, and solvents on esterase activity of purified proteins against the indicated α-naphthyl substrate was measured using the same protocol. Hydrolytic activity of purified enzymes against a library of 89 ester substrates was determined spectrophotometrically using p-nitrophenol as described previously31. Depolymerization activity of purified enzymes against polylactic acid (PLA) or polycaprolactone (PCL) was determined essentially as described previously31. These assays were performed in agarose plates (1.5%) containing emulsified substrates (poly (DL-lactide), average M.W. 2,000, or PCL10), or in solution (20 mg of PLA10 in 1 ml of 0.4 M Tris-HCl buffer, pH 8.0, 0.01% Plysurf A210G) at 32 °C. For determination of kinetic parameters (KM and kcat), esterase activity was determined over a range of substrate concentrations (0.01–5.0 mM). Kinetic parameters were calculated by non-linear regression analysis of raw data fit (to the Michaelis-Menten or Hill functions) using GraphPad Prism software (version 4.00 for Windows).
Crystallization and structure determination of MGS0169
The selenomethionine substituted MGS0169 (21–341 aa) was crystallized at 22 °C using the sitting-drop vapor diffusion method by mixing 0.5 μl of the purified protein (20 mg/ml) with 0.5 μl of the crystallization solution containing 0.2 M ammonium acetate, 0.1 M Tris-HCl (pH 8.0), and 30% (w/v) PEG 2KMME. The crystals were stabilized by cryoprotection in Paratone-N prior to flash-freezing in liquid nitrogen. Diffraction data were collected at the beamline 19-ID with an ADSC Quantum 315 R detector of the Structural Biology Center, Advanced Photon Source, Argonne National Laboratory76,77. Diffraction data were processed using the HKL3000 suit of programs78, and structural statistics is summarized in Supplementary Table S4. The MGS0169 structure was determined by the single-wavelength anomalous diffraction (SAD) method using phasing, density modification, and initial protein model building as implemented in the HKL3000 software package79,80,81,82,83,84. Several cycles of manual corrections of the model were carried out using the programs COOT85 and REFMAC of the CCP486 and finalized using Phenix87. The final model was refined against all reflections except for 5% randomly selected reflections, which were used for monitoring Rfree. The final refinement statistics are presented in Supplementary Table S4.
Additional Information
Accession codes: The sequences of 80 metagenomic esterases identified and verified in this project have been submitted to GenBank with the accession numbers shown in Supplementary Table S2. The atomic coordinates and structure factors for the MGS0169 structure have been deposited in the RCSB Protein Data Bank with accession code 5IBZ.
How to cite this article: Popovic, A. et al. Activity screening of environmental metagenomic libraries reveals novel carboxylesterase families. Sci. Rep. 7, 44103; doi: 10.1038/srep44103 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We thank all members of the Centre for Structural Proteomics in Toronto for help in conducting these experiments. This work was supported by the Government of Canada through Genome Canada, the Ontario Genomics Institute (2009-OGI-ABC-1405), Ontario Research Fund (ORF-GL2-01-004), and the NSERC Strategic Network grant IBN. Structural results shown in this manuscript are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The work was also supported by European Community project MAMBA (FP7-KBBE-2008-226977), MAGIC-PAH (FP7-KBBE-2009-245226), ULIXES (FP7-KBBE-2010-266473), MicroB3 (FP7-OCEAN.2011-2-287589), KILL-SPILL (FP7-KBBE-2012-312139), EU Horizon 2020 Project INMARE (Contract Nr 634486) and ERA Net IB2 Project MetaCat through UK Biotechnology and Biological Sciences Research Council (BBSRC) Grant BB/M029085/1. This work was further funded by grants BIO2011-25012, PCIN-2014-107 and BIO2014-54494-R from the Spanish Ministry of Economy and Competitiveness, the UK Biotechnology and Biological Sciences Research Council (BBSRC), and the German Federal Ministry of Education and Research (BMBF) within the ERA NET-IB2 program (grant number ERA-IB-14-030). The authors gratefully acknowledge the financial support provided by the European Regional Development Fund (ERDF).
Footnotes
The authors declare no competing financial interests.
Author Contributions M.F., D.L.P., O.V.G., P.N.G., and A.F.Y. conceived and supervised the study. A.P., T.H., M.M.Y., A.J., and A.S. designed the experiments. A.P., T.H., A.T., M.H., G.B., J.G., R.F., A.N.K., T.N.C., B.N., T.S., and V.Y. performed the experiments. D.L.P., M.M.Y., A.J., M.F., O.V.G., A.S., P.N.G., and A.F.Y. supervised and gave advice. T.B., D.L.P., M.M.Y., O.V.G., and P.N.G. contributed reagents and materials. A.P., B.N., D.L.P., A.J., A.S., P.N.G., and A.F.Y. analysed data. A.P., B.N., M.F., P.N.G., and A.F.Y. wrote the manuscript.
References
- Amann R. I., Ludwig W. & Schleifer K. H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59, 143–169 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsvik V., Goksoyr J. & Daae F. L. High diversity in DNA of soil bacteria. Appl Environ Microbiol 56, 782–787 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe M. S. & Giovannoni S. J. The uncultured microbial majority. Annu Rev Microbiol 57, 369–394, doi: 10.1146/annurev.micro.57.030502.090759 (2003). [DOI] [PubMed] [Google Scholar]
- Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68, 669–685 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M., Golyshina O., Beloqui A. & Golyshin P. N. Mining enzymes from extreme environments. Curr Opin Microbiol 10, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
- Vieites J. M., Guazzaroni M. E., Beloqui A., Golyshin P. N. & Ferrer M. Metagenomics approaches in systems microbiology. FEMS Microbiol Rev 33, 236–255 (2009). [DOI] [PubMed] [Google Scholar]
- Uchiyama T. & Miyazaki K. Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr Opin Biotechnol 20, 616–622 (2009). [DOI] [PubMed] [Google Scholar]
- Venter J. C. et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74 (2004). [DOI] [PubMed] [Google Scholar]
- Rusch D. B. et al. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5, e77 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yooseph S. et al. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol 5, e16 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M. Nature of the protein universe. Proc Natl Acad Sci USA 106, 11079–11084 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godzik A. Metagenomics and the protein universe. Curr Opin Struct Biol 21, 398–403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A. et al. The Enzyme Function Initiative. Biochemistry 50, 9950–9962 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky E. M. & Fields S. Protein-protein interactions: methods for detection and analysis. Microbiol Rev 59, 94–123 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P. O. & Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet 21, 33–37 (1999). [DOI] [PubMed] [Google Scholar]
- Galperin M. Y. & Koonin E. V. Who’s your neighbor? New computational approaches for functional genomics. Nat Biotechnol 18, 609–613 (2000). [DOI] [PubMed] [Google Scholar]
- Winzeler E. A. et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 (1999). [DOI] [PubMed] [Google Scholar]
- Christendat D. et al. Structural proteomics of an archaeon. Nat Struct Biol 7, 903–909 (2000). [DOI] [PubMed] [Google Scholar]
- Martzen M. R. et al. A biochemical genomics approach for identifying genes by the activity of their products. Science 286, 1153–1155 (1999). [DOI] [PubMed] [Google Scholar]
- Kuznetsova E. et al. Enzyme genomics: Application of general enzymatic screens to discover new enzymes. FEMS Microbiol Rev 29, 263–279 (2005). [DOI] [PubMed] [Google Scholar]
- Zhu H. & Snyder M. Protein arrays and microarrays. Curr Opin Chem Biol 5, 40–45 (2001). [DOI] [PubMed] [Google Scholar]
- Phizicky E. M. & Grayhack E. J. Proteome-scale analysis of biochemical activity. Crit Rev Biochem Mol Biol 41, 315–327 (2006). [DOI] [PubMed] [Google Scholar]
- Rondon M. R. et al. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 66, 2541–2547 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M., Martinez-Abarca F. & Golyshin P. N. Mining genomes and ‘metagenomes’ for novel catalysts. Curr Opin Biotechnol 16, 588–593 (2005). [DOI] [PubMed] [Google Scholar]
- Robertson D. E. et al. Exploring nitrilase sequence space for enantioselective catalysis. Appl Environ Microbiol 70, 2429–2436 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz P. & Eck J. Metagenomics and industrial applications. Nat Rev Microbiol 3, 510–516 (2005). [DOI] [PubMed] [Google Scholar]
- Beloqui A. et al. Novel polyphenol oxidase mined from a metagenome expression library of bovine rumen: biochemical properties, structural analysis, and phylogenetic relationships. J Biol Chem 281, 22933–22942 (2006). [DOI] [PubMed] [Google Scholar]
- Hess M. et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467 (2011). [DOI] [PubMed] [Google Scholar]
- Alcaide M. et al. Pressure adaptation is linked to thermal adaptation in salt-saturated marine habitats. Environ Microbiol 17, 332–345 (2015). [DOI] [PubMed] [Google Scholar]
- Tchigvintsev A. et al. The environment shapes microbial enzymes: five cold-active and salt-resistant carboxylesterases from marine metagenomes. Appl Microbiol Biotechnol 99, 2165–2178 (2015). [DOI] [PubMed] [Google Scholar]
- Feller G. & Gerday C. Psychrophilic enzymes: hot topics in cold adaptation. Nat Rev Microbiol 1, 200–208 (2003). [DOI] [PubMed] [Google Scholar]
- Olufsen M., Smalas A. O., Moe E. & Brandsdal B. O. Increased flexibility as a strategy for cold adaptation: a comparative molecular dynamics study of cold- and warm-active uracil DNA glycosylase. J Biol Chem 280, 18042–18048 (2005). [DOI] [PubMed] [Google Scholar]
- Siddiqui K. S. & Cavicchioli R. Cold-adapted enzymes. Annu Rev Biochem 75, 403–433 (2006). [DOI] [PubMed] [Google Scholar]
- Ferrer M. et al. Estimating the success of enzyme bioprospecting through metagenomics: current status and future trends. Microb Biotechnol 9, 22–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenfant N. et al. ESTHER, the database of the alpha/beta-hydrolase fold superfamily of proteins: tools to explore diversity of functions. Nucleic Acids Res 41, D423–429 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpigny J. L. & Jaeger K. E. Bacterial lipolytic enzymes: classification and properties. Biochem J 343Pt 1, 177–183 (1999). [PMC free article] [PubMed] [Google Scholar]
- Bornscheuer U. T. Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol Rev 26, 73–81 (2002). [DOI] [PubMed] [Google Scholar]
- Wei Y. et al. Crystal structure of brefeldin A esterase, a bacterial homolog of the mammalian hormone-sensitive lipase. Nat Struct Biol 6, 340–345 (1999). [DOI] [PubMed] [Google Scholar]
- Jaeger K. E., Dijkstra B. W. & Reetz M. T. Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu Rev Microbiol 53, 315–351 (1999). [DOI] [PubMed] [Google Scholar]
- Turner J. M. et al. Biochemical characterization and structural analysis of a highly proficient cocaine esterase. Biochemistry 41, 12297–12307 (2002). [DOI] [PubMed] [Google Scholar]
- Ollis D. L. et al. The alpha/beta hydrolase fold. Protein Eng 5, 197–211 (1992). [DOI] [PubMed] [Google Scholar]
- Nardini M. & Dijkstra B. W. Alpha/beta hydrolase fold enzymes: the family keeps growing. Curr Opin Struct Biol 9, 732–737 (1999). [DOI] [PubMed] [Google Scholar]
- Bayer S., Kunert A., Ballschmiter M. & Greiner-Stoeffele T. Indication for a new lipolytic enzyme family: isolation and characterization of two esterases from a metagenomic library. J Mol Microbiol Biotechnol 18, 181–187 (2010). [DOI] [PubMed] [Google Scholar]
- Fu J. et al. Functional and structural studies of a novel cold-adapted esterase from an Arctic intertidal metagenomic library. Appl Microbiol Biotechnol 97, 3965–3978 (2013). [DOI] [PubMed] [Google Scholar]
- Kim M. K., Kang T. H., Kim J., Kim H. & Yun H. D. Cloning and identification of a new group esterase (Est5S) from noncultured rumen bacterium. J Microbiol Biotechnol 22, 1044–1053 (2012). [DOI] [PubMed] [Google Scholar]
- Lee M. H. et al. A new esterase EstD2 isolated from plant rhizosphere soil metagenome. Appl Microbiol Biotechnol 88, 1125–1134 (2010). [DOI] [PubMed] [Google Scholar]
- Prive F. et al. Isolation and characterization of novel lipases/esterases from a bovine rumen metagenome. Appl Microbiol Biotechnol 99, 5475–5485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. C. et al. Est10: A Novel Alkaline Esterase Isolated from Bovine Rumen Belonging to the New Family XV of Lipolytic Enzymes. PLoS One 10, e0126651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon K. K., Lee H. S., Yang S. H. & Kim S. J. Kordiimonas gwangyangensis gen. nov., sp. nov., a marine bacterium isolated from marine sediments that forms a distinct phyletic lineage (Kordiimonadales ord. nov.) in the ‘Alphaproteobacteria’. Int J Syst Evol Microbiol 55, 2033–2037 (2005). [DOI] [PubMed] [Google Scholar]
- Rashamuse K., Magomani V., Ronneburg T. & Brady D. A novel family VIII carboxylesterase derived from a leachate metagenome library exhibits promiscuous beta-lactamase activity on nitrocefin. Appl Microbiol Biotechnol 83, 491–500 (2009). [DOI] [PubMed] [Google Scholar]
- Kurnasov O. et al. Aerobic tryptophan degradation pathway in bacteria: novel kynurenine formamidase. FEMS Microbiol Lett 227, 219–227 (2003). [DOI] [PubMed] [Google Scholar]
- Bjerregaard-Andersen K. et al. A proton wire and water channel revealed in the crystal structure of isatin hydrolase. J Biol Chem 289, 21351–21359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Saez L., Srikannathasan V., Zoltner M. & Hunter W. N. Structures of bacterial kynurenine formamidase reveal a crowded binuclear zinc catalytic site primed to generate a potent nucleophile. Biochem J 462, 581–589 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang D. F., Kumaran D., Swaminathan S. & Raushel F. M. Structural characterization and function determination of a nonspecific carboxylate esterase from the amidohydrolase superfamily with a promiscuous ability to hydrolyze methylphosphonate esters. Biochemistry 53, 3476–3485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang D. F. et al. Function discovery and structural characterization of a methylphosphonate esterase. Biochemistry 54, 2919–2930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayumi D., Akutsu-Shigeno Y., Uchiyama H., Nomura N. & Nakajima-Kambe T. Identification and characterization of novel poly(DL-lactic acid) depolymerases from metagenome. Appl Microbiol Biotechnol 79, 743–750 (2008). [DOI] [PubMed] [Google Scholar]
- Akutsu-Shigeno Y. et al. Cloning and sequencing of a poly(DL-lactic acid) depolymerase gene from Paenibacillus amylolyticus strain TB-13 and its functional expression in Escherichia coli. Appl Environ Microbiol 69, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzberg O. et al. Swiveling-domain mechanism for enzymatic phosphotransfer between remote reaction sites. Proc Natl Acad Sci USA 93, 2652–2657 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara B. et al. trans-Dienelactone hydrolase from Pseudomonas reinekei MT1, a novel zinc-dependent hydrolase. Biochem Biophys Res Commun 376, 423–428 (2008). [DOI] [PubMed] [Google Scholar]
- Marin M. & Pieper D. H. Novel metal-binding site of Pseudomonas reinekei MT1 trans-dienelactone hydrolase. Biochem Biophys Res Commun 390, 1345–1348 (2009). [DOI] [PubMed] [Google Scholar]
- Alcaide M. et al. Single residues dictate the co-evolution of dual esterases: MCP hydrolases from the alpha/beta hydrolase family. Biochem J 454, 157–166 (2013). [DOI] [PubMed] [Google Scholar]
- Placido A. et al. Diversity of hydrolases from hydrothermal vent sediments of the Levante Bay, Vulcano Island (Aeolian archipelago) identified by activity-based metagenomics and biochemical characterization of new esterases and an arabinopyranosidase. Appl Microbiol Biotechnol 99, 10031–10046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier E. et al. “Candidatus Cloacamonas acidaminovorans”: genome sequence reconstruction provides a first glimpse of a new bacterial division. J Bacteriol 190, 2572–2579 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C. F. et al. Molecular basis of formaldehyde detoxification. Characterization of two S-formylglutathione hydrolases from Escherichia coli, FrmB and YeiG. J Biol Chem 281, 14514–14522 (2006). [DOI] [PubMed] [Google Scholar]
- Kouker G. & Jaeger K. E. Specific and sensitive plate assay for bacterial lipases. Appl Environ Microbiol 53, 211–213 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D. R. & Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass E. M., Wilkening J., Wilke A., Antonopoulos D. & Meyer F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb Protoc 2010, pdb prot5368 (2010). [DOI] [PubMed] [Google Scholar]
- Delcher A. L., Bratke K. A., Powers E. C. & Salzberg S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D. et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D., Clements J. & Eddy S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39, W29–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin M. Y., Makarova K. S., Wolf Y. I. & Koonin E. V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res 43, D261–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I. & Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum G. et al. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat 13, 30–45 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocek B., Mulligan R., Bargassa M., Collart F. & Joachimiak A. Crystal structure of aminopeptidase N from human pathogen Neisseria meningitidis. Proteins 70, 273–279 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W., Cymborowski M., Otwinowski Z. & Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866 (2006). [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
- Terwilliger T. C. & Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr 55, 849–861 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T. SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchrotron Radiat 11, 49–52 (2004). [DOI] [PubMed] [Google Scholar]
- Cowtan K. Fast Fourier feature recognition. Acta Crystallogr D Biol Crystallogr 57, 1435–1444 (2001). [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr 66, 479–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. & Main P. Miscellaneous algorithms for density modification. Acta Crystallogr D Biol Crystallogr 54, 487–493 (1998). [DOI] [PubMed] [Google Scholar]
- Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- Murshudov G. N., Vagin A. A. & Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.