

Abstract
Background
Dupuytren's disease (DD) is a debilitating fibro-proliferative disorder of the hand characterized by the appearance of fibrotic lesions (nodules and cords) leading to flexion contractures of the fingers and loss of hand function. Although the molecular mechanism of DD is unknown, it has been suggested that transforming growth factor-β2 (TGF-β2) may play an important role in the underlying patho-physiology of the disease. The purpose of this study was to further explore this hypothesis by examining the effects of TGF-β2 on primary cell cultures derived from patient-matched disease and normal palmar fascia tissue using a three-dimensional collagen contraction assay.
Methods
Fibroblast-populated collagen lattice (FPCL) contraction assays using primary cell cultures derived from diseased and control fascia of the same DD patients were studied in response to exogenous TGF-β2 and neutralizing anti-TGF-β2 antibodies.
Results
Contraction of the FPCLs occurred significantly faster and to a greater extent in disease cells compared to control cells. The addition of TGF-β2 enhanced the rate and degree of collagen contraction in a dose-dependent fashion for both control and diseased cells. Neutralizing anti-TGF-β2 antibodies abolished exogenous TGF-β2 stimulated collagen contraction, but did not inhibit the enhanced basal collagen contraction activity of disease FPCL cultures.
Conclusions
Although exogenous TGF-β2 stimulated both disease and control FPCL contraction, neutralizing anti-TGF-β2 antibodies did not affect the elevated basal collagen contraction activity of disease FPCLs, suggesting that the differences in the collagen contraction activity of control and disease FPCL cultures are not due to differences in the levels of endogenous TGF-β2 activity.
Background
Dupuytren's disease (DD) is a fibro-proliferative disorder of the palmar fascia (PF) characterized by the formation of fibrous nodules and cords [1]. The disease results in digital contractures, leading to loss of hand function. Surgical excision of the diseased PF is currently the principal form of management since the lack of a clear etiology has precluded the development of other effective and rational forms of treatment.
Since Baron Guillaume Dupuytren's classical description of the disease in 1831, multiple clinical associations have been described, however, no clear molecular mechanism for the disease has been established [2]. Histochemical studies of DD have demonstrated the presence of myofibroblasts [3], increased production of type III collagen [4-7], and alterations in other extra-cellular matrix proteins including various fibronectin isoforms [8-14]. These biological features are characteristic of abnormal growth factor regulation, specifically fibrogenic cytokines such as transforming growth factor-beta (TGF-β). Several studies have documented TGF-β expression in DD palmar fascia using RT-PCR [15], in-situ hybridization [16], and immunohistochemistry [16-18], while others have shown that TGF-β can stimulate cell proliferation [18-20] and promote myofibroblast differentiation in vitro [21]. As a result of these and other studies it has been suggested that an aberrant TGF-β activity may be involved in the pathogenesis of DD.
In this study, we chose to focus on TGF-β2 and its effects on collagen contraction in vitro using a three-dimensional fibroblast populated collagen lattice (FPCL) contraction assay and DD patient-matched disease and control primary cell cultures. Previous reports have examined the role of TGF-β in DD by comparing disease fibroblasts to 'control' fibroblasts obtained from transverse carpal ligament material obtained from patients undergoing carpal tunnel release (CTR). By contrast, the control fibroblast cultures used in this study were established from unaffected PF tissue from the same patient, thus providing us with unique patient- and tissue- matched control cultures. The observed phenotypic differences between patient/tissue-matched control and disease FPCL cultures, specifically elevated collagen contraction activity, and β-catenin and fibronectin (Fn) expression in disease cells [22-24], raises the intriguing possibility that pro-fibrotic factors, such as TGF-β2, may be regulating these disease-associated events in vitro, since TGF-βs are known to promote fibroblast mediated collagen contraction [21,25,26] and up-regulate collagen, Fn and β-catenin [20,27-30]. As described in detail below, we have found that exogenous TGF-β2 could significantly stimulate 'normal' and disease FPCL contraction in a dose-dependent manner. While neutralizing anti-TGF-β2 antibodies completely blocked exogenous TGF-β2 stimulated FPCL contraction they had no effect on the enhanced basal collagen contraction activity of disease FPCL cultures.
Methods
Patient samples and primary cell cultures
Our study protocol was cleared through the UWO Ethics Committee for Research Involving Human Subjects. Areas of diseased fascia and uninvolved normal (control) PF tissue were collected during surgery. DD explant cultures were initially cultured in starter media consisting of α-MEM (Gibco, Invitrogen Corporation) supplemented with 20% fetal bovine serum (FBS, Clontech Laboratories, Palo Alto, CA), and antibiotics (Penicillin G and streptomycin sulfate) and fungizone (Gibco, Invitrogen Corporation) as previously described [23]. Established primary culture lines were maintained in α-MEM + 10% FBS + antibiotics + fungizone. Culture flasks were incubated at 37°C in a humidified chamber with 5% CO2. Medium was changed every 4–5 days and the cells sub-cultured using 0.05% Trypsin-EDTA (Gibco, Invitrogen Corporation, Grand Island, NY) when confluent.
Fibroblast Populated Collagen Lattice (FPCL) contraction assay
Collagen contraction was carried out using patient-matched disease (D) and control (C) primary cultures (passages 2 – 6) established from patient-matched DD lesions and uninvolved palmar fascia (control). Collagen lattices were prepared by mixing cell suspensions with a neutralized solution of collagen type I matrix (8 parts Vitrogen100 collagen type I, 2.9 mg/ml, Collagen Corp, Santa Clara, CA, USA + 1 part 10 × α-MEM + 1 part HEPES buffer, pH 9.0). The cell-collagen concentrations were adjusted with sterile phosphate buffered solution (PBS) to attain a final collagen concentration of 2.0 mg/ml and a final cell concentration of 8.6 × 104 cells/ml of matrix. The cell-collagen mixture was then aliquoted into 24 well culture dishes (0.5 ml/well) that were pre-treated with a PBS solution containing 2% (w/v) bovine serum albumin (BSA). Following FPCL polymerization (1 hr, 37°C) culture medium (0.5 ml) consisting of α-MEM + 10% fetal bovine serum (FBS) was added atop each lattice. After 2 days of culture the attached FPCLs were mechanically released from the sides of the culture plates. Digital images of the contracting FPCL were captured at various time points over a 5-day assay period using a conventional flatbed scanner. Collagen lattice areas were then quantified using the Image J program [31]. Each assay was done in quadruplicate.
TGF-β2 and neutralizing anti-TGF-β2 antibody treatments
Commercially available human recombinant TGF-β2 (expressed in NSO murine myeloma cells) was acid activated in a solution of 4N HCl + 0.1% (w/v) BSA according to the manufacturer's instructions (Product # T 2815, Sigma, St. Louis, MO). Acid activated human recombinant TGF-β2 was then aliquoted and frozen for extended storage at -70°C. The indicated concentrations of activated human recombinant TGF-β2 were added to complete culture media immediately following FPCL polymerization. Neutralizing anti-TGF-β2 antibodies (R&D Systems, Minneapolis, MN) were added to complete culture media either immediately following FPCL polymerization or added to the cell suspension prior to mixing with the neutralized Vitrogen collagen type I solution. Control FPCL cultures were treated with appropriate carrier solutions.
Cell proliferation assay
FPCL cultures were assayed using a commercially available CellTiter 96® Aqueous One Solution cell proliferation assay according to the manufacturer's instructions (Promega, Madison, WI, USA). This cell proliferation assay is a colorimetric method that uses a tetrazolium compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) in combination with a stable electron coupling reagent PES (phenazine ethosulfate). This produces a chemically stable MTS tetrazolium compound that can be bio-reduced by cells to form a soluble colored formazan product [32,33]. Briefly, cells from patient-matched primary cultures were cultured as stressed-relaxed FPCLs as described above. Each primary cell line was plated as quadruplicate FPCL cultures for each of the indicated time points with 0.5 ml of culture media ± TGF-β2 (1 ng/ml) atop each FPCL. At each of the designated contracting time points 100μl of CellTiter 96® Aqueous One solution reagent was added to the FPCL cultures and incubated for 3 hours (37°C, 5% CO2 atmosphere, humidified chamber). FPCL media was then collected and aliquoted into a 96-well culture plate for absorbance reading at 450 and 650 nm (background reference λ) using a 96-well BIO-RAD microplate reader. Media was also collected from FPCLs containing no cells (negative control) for background absorbance readings. The assay was conducted over 5 days during the course of FPCL contraction. A standard curve was also generated to calculate relative cell numbers per FPCL (range of 4 × 103–105 cells/FPCL).
Statistical analysis
Student two-tailed t test was used to compare data between two groups. Values were expressed as mean ± standard deviation of the mean (SDM). P values < 0.05 were considered statistically significant.
Results
TGF-β2 stimulates FPCL contraction
We first examined the basal collagen contraction activity of three independent patient-matched early passage control and disease primary FPCL cultures. As shown in Figure 1, disease FPCL cultures contracted collagen faster and to a greater degree when compared to patient-matched control FPCL cultures. This is in agreement with previous results that show distinct phenotypic differences between control and disease primary DD cultures [22,24], including enhanced collagen contraction by disease FPCL cultures [23]. Next, we explored the role of exogenous TGF-β2 on FPCL contraction. Initial experiments showed a typical dose-dependent response for TGF-β2 stimulated FPCL contraction (Fig. 2). Because maximum collagen contraction was achieved in response to 1 ng/ml of TGF-β2, all subsequent experiments used this dose. As shown in Figure 3a, we observed enhanced contraction rates and total collagen contraction for both control and disease cells treated with exogenous (1 ng/ml) TGF-β2. This enhanced collagen contraction activity observed in disease FPCL cultures was not due to differences in cell proliferation/viability between control or disease FPCL cultures. In fact, TGF-β2 appears to exert a significant pro-apoptotic effect on relaxed disease FPCL cultures that is absent in the 'control' FPCL cultures (Fig 3b). Thus, the amount of collagen contraction exerted by disease FPCL cultures is more pronounced if one also considers the changes in cell viability over the course of FPCL contraction.
Figure 1.
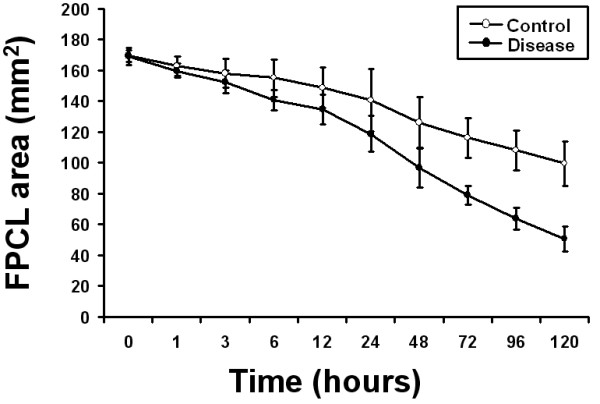
Collagen contraction of primary control and disease FPCL cultures. Digital images of contracting FPCL cultures were captured and analyzed using Image J software to quantify collagen contraction. The mean FPCL surface area (mm2) was plotted over time with each data point representing the mean ± the standard deviation of the mean (SDM) of three independent patient-matched cell lines. Experiments were repeated in quadruplicate.
Figure 2.
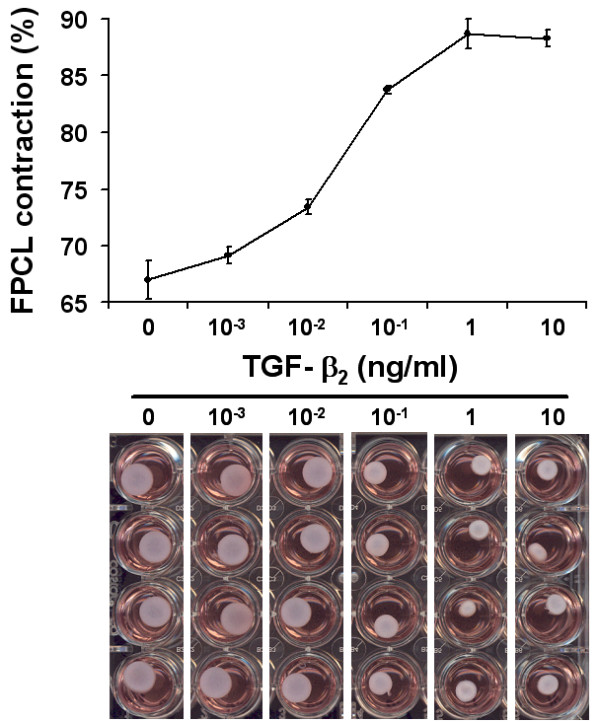
TGF-β2 dose response. The dose response curve (upper panel) shows the accumulated FPCL contraction (%) over 5 days. The degree of TGF-β2 stimulated contraction was measured over a large concentration range (>5 orders of magnitude). The plotted data shows a typical dose-response relationship, with maximal response elicited by 1 ng/ml of TGF-β2. Representative images of contracting FPCL after 5 days are shown for increasing concentrations of TGF-β2 (lower panel).
Figure 3.
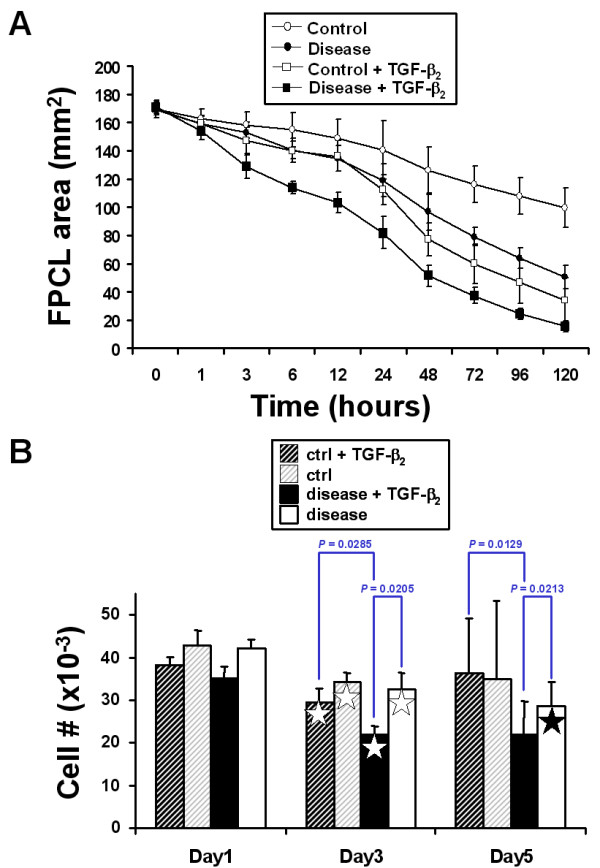
FPCL contraction in response to exogenous TGF-β2. Contracting FPCL cultures (± 1 ng/ml TGF-β2) were analyzed using Image J software to quantify collagen contraction. (A) The plotted data points represent the mean surface area ± SDM for three independent patient-matched primary FPCL cultures. Experiments were repeated in quadruplicate. (B) Cell proliferation/viability assays were performed on contracting FPCL cultures. The plotted bar graph represents the mean cell number ± SDM for the indicated time points for one representative patient-matched disease and control primary culture. Significant differences between groups are indicated by the P-values. The stars denote significance differences (P < 0.05) between the same treatment groups of different time points (white star – Day 1 vs. Day 3, black star – Day 1 vs. Day 5).
Neutralizing anti-TGF-β2 antibodies block TGF-β2 stimulated FPCL contraction but do not alter basal FPCL contraction
Exogenous TGF-β2 stimulated collagen contraction in both control and disease FPCL cultures was inhibited in a dose-dependent manner by neutralizing anti-TGF-β2 antibodies. As shown in Figure 4, lower concentrations of neutralizing antibodies (100 ng/ml) partially inhibited FPCL contraction, while higher concentrations of neutralizing antibodies (1000 ng/ml) completely inhibited TGF-β2 stimulated FPCL contraction, thus confirming the ligand-dependent nature of this response in vitro. We also examined the effect of neutralizing anti-TGF-β2 antibodies on the basal collagen contraction activity of primary disease and control FPCL cultures. As shown in Figure 5, high concentrations (1000 ng/ml) of neutralizing anti-TGF-β2 antibodies had no effect on the basal levels of collagen contraction observed in either control or disease FPCL cultures, suggesting that there is little or no endogenous active TGF-β2 that could account for the FPCL contraction in vitro. To preclude that more localized interactions between endogenously produced TGF-β2 and its cell-surface receptors may account for the observed increase in disease FPCL contraction, we also pre-incubated the cells with neutralizing anti-TGF-β2 antibodies prior to forming FPCL. Regardless, the blocking antibodies had no effect on the basal levels of FPCL contraction (data not shown).
Figure 4.
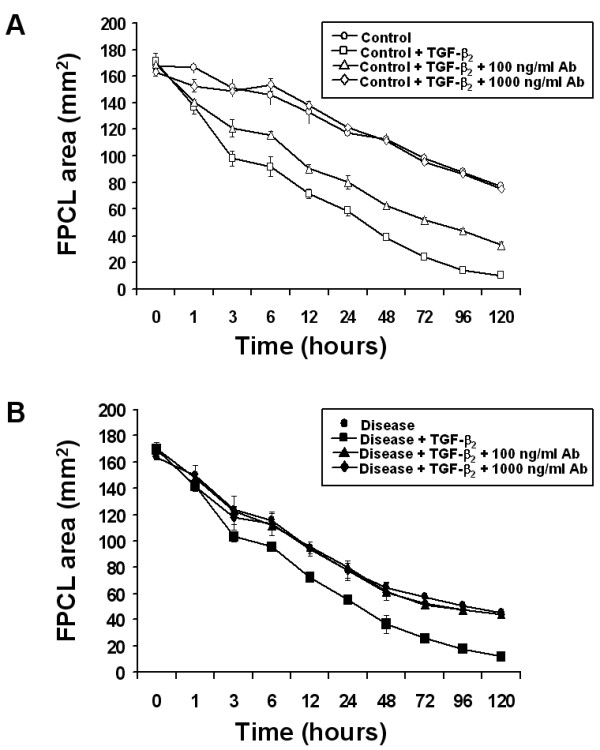
Neutralizing anti-TGF-β2 antibodies block TGF-β2 stimulated FPCL contraction. (A) Control and (B) disease FPCL cultures (± 1 ng/ml TGF-β2) were treated with the indicated concentrations of neutralizing anti-TGF-β2 antibodies. FPCL contraction was analyzed and plotted as the mean surface area ± SDM for quadruplicate cultures per treatment.
Figure 5.
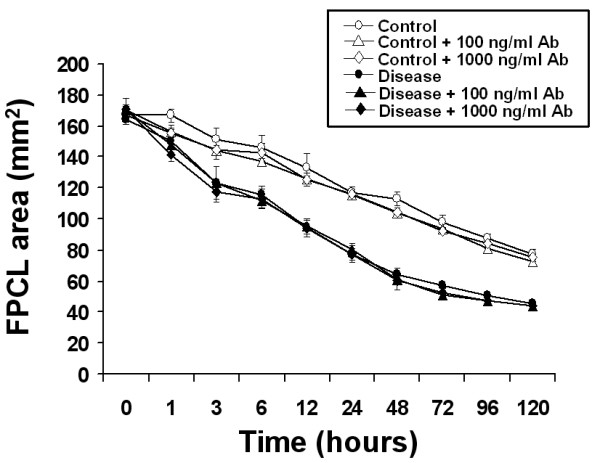
Neutralizing anti-TGF-β2 antibodies do not inhibit basal collagen contraction activity of control or disease FPCL cultures. Disease and control FPCL cultures were incubated in the absence or presence of the indicated concentrations of neutralizing anti-TGF-β2 antibodies. Measurements of the contracting FPCL were plotted as the mean surface area ± SDM for quadruplicate cultures per treatment.
Discussion
Members of the TGF-β family are potent fibrogenic factors that play an important role in the patho-physiology of numerous fibro-proliferative disorders, including DD [34]. The study presented here focused on the effect of TGF-β2 on collagen contraction using primary FPCL cultures derived from patient-matched disease and control unaffected PF tissue. Here, we found that disease FPCL cultures contracted collagen faster and to a greater degree than patient-matched control FPCL cultures. Although neutralizing anti-TGF-β2 antibodies effectively blocked exogenous TGF-β2 stimulated FPCL contraction for both control and disease cultures, the same neutralizing antibodies had no effect on the basal collagen contraction activity of either disease or control FPCL cultures, suggesting that enhanced disease FPCL contraction is not due to elevated levels of endogenous active TGF-β2. These results, however, do not exclude the possibility that there may be differences in the levels of latent TGF-β2 produced by these cell cultures that may be subsequently activated in vivo. Recent work by Kuhn et. al. (2002) reported reduced DD FPCL contraction in response to tamoxifen, which was associated with a decreased TGF-β2 production [35]. However, the TGF-β2 assayed in this study required an acid-activation step, suggesting that TGF-β2 produced by these cells is largely in its latent, non-activate form. Thus, it is possible that TGF-β2 produced by DD fibroblasts and/or other resident cell types in vivo may be important to disease cord contraction, provided appropriate regulators of latent TGF-β activation are also present and active. However, it does not explain the enhanced basal FPCL contraction rates of disease cell cultures, suggesting that other signalling factors may be regulating this disease cell function in vitro. It is interesting to note that both TGF-β and β-catenin signalling pathways show some degree of 'cross-talk' [29,30,36-38], suggesting that β-catenin plays an important role in TGF-β signalling. The degree and significance of signalling 'cross-talk' between β-catenin and TGF-β in the context of DD is unknown and needs further examination.
Although previous DD studies have used different 'control' primary fibroblast as 'disease-free' controls, namely fibroblasts derived from transverse carpal ligament material obtained from patients undergoing carpal tunnel release (CTR), they have the disadvantage of being of different anatomical origin than PF derived 'control' fibroblast cultures. This is an important consideration given that fibroblasts exhibit functional heterogeneity depending on their origin [39-41]. For example, human fibroblasts that express the cell surface antigen Thy-1 are capable of TGF-β1 stimulated myofibroblast differentiation, while Thy-1 negative fibroblasts appear to be only capable of lipofibroblast differentiation [42]. Phenotypic differences have also been attributed to fibroblasts of different dermal origins [43]. For example, Chipev and colleagues showed that TGF-β1 had a pro-apoptotic effect on non-palmoplantar (keloid) fibroblasts and an anti-apoptotic effect on palmoplantar fibroblasts. Similar to what we have observed for detached or contracting DD FPCL cultures, they showed that TGF-β1 treated (in the presence of serum) keloid FPCL cultures underwent the most extensive apoptosis response upon mechanical release compared to dermal fibroblasts from different body sites [43]. Perhaps the similar myofibroblast phenotype attributed to both DD and keloid fibroblast cultures also dictates a similar apoptotic fate to relaxed FPCL cultures in response to TGF-βs. Although the loss of tension in these cultures triggered a mostly uniform loss in cell viability across all groups, unlike disease FPCL cultures TGF-β2 did not appear to have any significant pro-apoptotic effect on relaxed 'control' FPCL cultures. In light of these and other findings, it appears that the patient-matched control fibroblast cultures employed in these studies may truly represent a suitable 'control' phenotype, with the added advantage of having the same PF origins as the disease cell cultures. While this does not exclude the possibility that the control cultures may harbour some residual disease cells, this is not supported by the distinct phenotypic differences we have observed between these two types of patient-matched PF cultures in the current and previous studies. In these earlier studies, we reported elevated levels of β-catenin and fibronectin isoforms in disease FPCL cultures [22-24], as well as enhanced disease FPCL contraction rates which we have subsequently confirmed in these studies. This together with the distinctive pro-apoptotic affects of TGF-β2 on disease FPCL cultures described here, further support the notion that the patient/tissue-matched 'control' cultures have a non-disease phenotype that is suitable for these types of investigations.
Although the 'synthetic' myofibroblast features of the disease cells described in previous studies are known to be stimulated by TGF-β [20,21,25-30,35], our results suggests that endogenous TGF-β2 does not play a role in regulating these phenotypic differences in vitro. Nevertheless, aberrant expression of various TGF-β signalling components have been previously shown to trigger this 'synthetic' myofibroblast phenotype in other fibro-proliferative disorders, specifically keloids and burn hypertrophic scarring [34,44,45], that can to some extent be inhibited by neutralizing anti-TGF-β2 antibodies [46,47]. Hopefully future studies will unravel the extent of these phenotypic differences between these patient/tissue-matched control and disease FPCL cultures with respect to pro-fibrotic factors like TGF-β2, tension and other important intersecting signaling pathways.
Conclusions
Primary disease FPCL cultures contract collagen faster and to a greater extent than control PF-matched FPCL cultures. While neutralizing anti-TGF-β2 antibodies can block exogenous TGF-β2 stimulated collagen contraction for both control and disease FPCL cultures, it had no effect on the basal contraction rates of either control or disease FPCL cultures. We, therefore, conclude that the enhanced collagen contraction activity of disease FPCL cultures is not due to differences in the levels of endogenous active TGF-β2.
List of abbreviations
DD, Dupuytren's disease; PF, palmar fascia; CTR, carpal tunnel release; TGF-β, transforming growth factor-beta; FPCL, fibroblast populated collagen matrix; PBS, phosphate buffered saline; RT, room temperature; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; α-MEM, alpha-minimal essential medium; FBS, fetal bovine serum; Fn, fibronectin; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt; PES, phenazine ethosulfate; RT-PCR, reverse transcriptase polymerase chain reaction; SDM, standard deviation of the mean.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RT and YW carried out the experiments. RT drafted the manuscript. JCH and BSG designed and coordinated the research, and reviewed and edited the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
This study was supported by grants from the Canadian Institutes of Health Research, Plastic Surgery Education Fund, American Society for Surgery of the Hand, and the Lawson Health Research Institute's Internal Research Fund.
Contributor Information
Raymond Tse, Email: rtse@uwo.ca.
Jeffrey Howard, Email: jhoward@lri.sjhc.london.on.ca.
Yan Wu, Email: yan.wu@sjhc.london.on.ca.
Bing Siang Gan, Email: bsgan@lri.sjhc.london.on.ca.
References
- Dupuytren G. Permanent retraction of the fingers, produced by an affection of the palmar fascia. Lancet. 1834;2:222–225. [Google Scholar]
- Burge P. Genetics of Dupuytren's disease. Hand Clin. 1999;15:63–71. [PubMed] [Google Scholar]
- Gabbiani G, Majno G. Dupuytren's contracture: fibroblast contraction? An ultrastructural study. Am J Pathol. 1972;66:131–146. [PMC free article] [PubMed] [Google Scholar]
- Bailey AJ, Sims TJ, Gabbiani G, Bazin S, LeLous M. Collagen of Dupuytren's disease. Clin Sci Mol Med. 1977;53:499–502. doi: 10.1042/cs0530499. [DOI] [PubMed] [Google Scholar]
- Menzel EJ, Piza H, Zielinski C, Endler AT, Steffen C, Millesi H. Collagen types and anticollagen-antibodies in Dupuytren's disease. Hand. 1979;11:243–248. doi: 10.1016/s0072-968x(79)80045-5. [DOI] [PubMed] [Google Scholar]
- Bazin S, Le Lous M, Duance VC, Sims TJ, Bailey AJ, Gabbiani G, D'Andiran G, Pizzolato G, Browski A, Nicoletis C, Delaunay A. Biochemistry and histology of the connective tissue of Dupuytren's disease lesions. Eur J Clin Invest. 1980;10:9–16. doi: 10.1111/j.1365-2362.1980.tb00003.x. [DOI] [PubMed] [Google Scholar]
- Gelberman RH, Amiel D, Rudolph RM, Vance RM. Dupuytren's contracture. An electron microscopic, biochemical, and clinical correlative study. J Bone Joint Surg Am. 1980;62:425–432. [PubMed] [Google Scholar]
- Brickley-Parsons D, Glimcher MJ, Smith RJ, Albin R, Adams JP. Biochemical changes in the collagen of the palmar fascia in patients with Dupuytren's disease. J Bone Joint Surg [Am] 1981;63:787–797. [PubMed] [Google Scholar]
- Tomasek JJ, Schultz RJ, Episalla CW, Newman SA. The cytoskeleton and extracellular matrix of the Dupuytren's disease "myofibroblast": an immunofluorescence study of a nonmuscle cell type. J Hand Surg [Am] 1986;11:365–371. doi: 10.1016/s0363-5023(86)80143-5. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Schultz RJ, Haaksma CJ. Extracellular matrix-cytoskeletal connections at the surface of the specialized contractile fibroblast (myofibroblast) in Dupuytren disease. J Bone Joint Surg [Am] 1987;69:1400–1407. [PubMed] [Google Scholar]
- Tomasek JJ, Haaksma CJ. Fibronectin filaments and actin microfilaments are organized into a fibronexus in Dupuytren's diseased tissue. Anat Rec. 1991;230:175–182. doi: 10.1002/ar.1092300205. [DOI] [PubMed] [Google Scholar]
- Berndt A, Kosmehl H, Katenkamp D, Tauchmann V. Appearance of the myofibroblastic phenotype in Dupuytren's disease is associated with a fibronectin, laminin, collagen type IV and tenascin extracellular matrix. Pathobiology. 1994;62:55–58. doi: 10.1159/000163879. [DOI] [PubMed] [Google Scholar]
- Halliday NL, Rayan GM, Zardi L, Tomasek JJ. Distribution of ED-A and ED-B containing fibronectin isoforms in Dupuytren's disease. J Hand Surg [Am] 1994;19:428–434. doi: 10.1016/0363-5023(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Magro G, Lanzafame S, Micali G. Co-ordinate expression of alpha 5 beta 1 integrin and fibronectin in Dupuytren's disease. Acta Histochem. 1995;97:229–233. doi: 10.1016/S0065-1281(11)80184-4. [DOI] [PubMed] [Google Scholar]
- Baird KS, Crossan JF, Ralston SH. Abnormal growth factor and cytokine expression in Dupuytren's contracture. J Clin Pathol. 1993;46:425–428. doi: 10.1136/jcp.46.5.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt A, Kosmehl H, Mandel U, Gabler U, Luo X, Celeda D, Zardi L, Katenkamp D. TGF beta and bFGF synthesis and localization in Dupuytren's disease (nodular palmar fibromatosis) relative to cellular activity, myofibroblast phenotype and oncofetal variants of fibronectin. Histochem J. 1995;27:1014–1020. doi: 10.1007/BF00175577. [DOI] [PubMed] [Google Scholar]
- Zamora RL, Heights R, Kraemer BA, Erlich HP, Groner JP. Presence of growth factors in palmar and plantar fibromatoses. J Hand Surg [Am] 1994;19:435–441. doi: 10.1016/0363-5023(94)90058-2. [DOI] [PubMed] [Google Scholar]
- Badalamente MA, Sampson SP, Hurst LC, Dowd A, Miyasaka K. The role of transforming growth factor beta in Dupuytren's disease. J Hand Surg [Am] 1996;21:210–215. doi: 10.1016/S0363-5023(96)80102-X. [DOI] [PubMed] [Google Scholar]
- Kloen P, Jennings CL, Gebhardt MC, Springfield DS, Mankin HJ. Transforming growth factor-beta: possible roles in Dupuytren's contracture. J Hand Surg [Am] 1995;20:101–108. doi: 10.1016/S0363-5023(05)80067-X. [DOI] [PubMed] [Google Scholar]
- Kuhn MA, Payne WG, Kierney PC, Pu LL, Smith PD, Siegler K, Ko F, Wang X, Robson MC. Cytokine manipulation of explanted Dupuytren's affected human palmar fascia. Int J Surg Investig. 2001;2:443–456. [PubMed] [Google Scholar]
- Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257:180–189. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- Varallo VM, Gan BS, Seney S, Ross DC, Roth JH, Richards RS, McFarlane RM, Alman B, Howard JC. Beta-catenin expression in Dupuytren's disease: potential role for cell-matrix interactions in modulating beta-catenin levels in vivo and in vitro. Oncogene. 2003;22:3680–3684. doi: 10.1038/sj.onc.1206415. [DOI] [PubMed] [Google Scholar]
- Howard JC, Varallo VM, Ross DC, Roth JH, Faber KJ, Alman B, Gan BS. Elevated levels of beta-catenin and fibronectin in three-dimensional collagen cultures of Dupuytren's disease cells are regulated by tension in vitro. BMC Musculoskelet Disord. 2003;4:16. doi: 10.1186/1471-2474-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JC, Varallo VM, Ross DC, Faber KJ, Roth JH, Seney S, Gan BS. Elevated expression of wound healing associated proteins Hsp-47 and fibronectin in Dupuytren's contracture. J Surg Res. 2004;117:232–238. doi: 10.1016/j.jss.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Montesano R, Orci L. Transforming growth factor beta stimulates collagen-matrix contraction by fibroblasts: implications for wound healing. Proc Natl Acad Sci U S A. 1988;85:4894–4897. doi: 10.1073/pnas.85.13.4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukamizu H, Grinnell F. Spatial organization of extracellular matrix and fibroblast activity: effects of serum, transforming growth factor beta, and fibronectin. Exp Cell Res. 1990;190:276–282. doi: 10.1016/0014-4827(90)90197-i. [DOI] [PubMed] [Google Scholar]
- Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- Ignotz RA, Endo T, Massague J. Regulation of fibronectin and type I collagen mRNA levels by transforming growth factor-beta. J Biol Chem. 1987;262:6443–6446. [PubMed] [Google Scholar]
- Wang H, Chakrabarty S. Requirement of protein kinase Calpha, extracellular matrix remodeling, and cell-matrix interaction for transforming growth factorbeta- regulated expression of E-cadherin and catenins. J Cell Physiol. 2001;187:188–195. doi: 10.1002/jcp.1068. [DOI] [PubMed] [Google Scholar]
- Cheon SS, Nadesan P, Poon R, Alman BA. Growth factors regulate beta-catenin-mediated TCF-dependent transcriptional activation in fibroblasts during the proliferative phase of wound healing. Exp Cell Res. 2004;293:267–274. doi: 10.1016/j.yexcr.2003.09.029. [DOI] [PubMed] [Google Scholar]
- ScionCorporation http://www.scioncorp.com
- Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- Berridge MV, Tan AS. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys. 1993;303:474–482. doi: 10.1006/abbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- Kuhn MA, Wang X, Payne WG, Ko F, Robson MC. Tamoxifen Decreases Fibroblast Function and Downregulates TGF(beta2) in Dupuytren's Affected Palmar Fascia. J Surg Res. 2002;103:146–152. doi: 10.1006/jsre.2001.6350. [DOI] [PubMed] [Google Scholar]
- Letamendia A, Labbe E, Attisano L. Transcriptional regulation by Smads: crosstalk between the TGF-beta and Wnt pathways. J Bone Joint Surg Am. 2001;83-A Suppl 1:S31–9. [PubMed] [Google Scholar]
- Furuhashi M, Yagi K, Yamamoto H, Furukawa Y, Shimada S, Nakamura Y, Kikuchi A, Miyazono K, Kato M. Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol Cell Biol. 2001;21:5132–5141. doi: 10.1128/MCB.21.15.5132-5141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian YC, Phillips AO. Interaction between the transforming growth factor-beta type II receptor/Smad pathway and beta-catenin during transforming growth factor-beta1-mediated adherens junction disassembly. Am J Pathol. 2002;160:1619–1628. doi: 10.1016/s0002-9440(10)61109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider EL, Mitsui Y, Au KS, Shorr SS. Tissue-specific differences in cultured human diploid fibroblasts. Exp Cell Res. 1977;108:1–6. doi: 10.1016/s0014-4827(77)80002-5. [DOI] [PubMed] [Google Scholar]
- Bordin S, Page RC, Narayanan AS. Heterogeneity of normal human diploid fibroblasts: isolation and characterization of one phenotype. Science. 1984;223:171–173. doi: 10.1126/science.6691142. [DOI] [PubMed] [Google Scholar]
- Sorrell JM, Caplan AI. Fibroblast heterogeneity: more than skin deep. J Cell Sci. 2004;117:667–675. doi: 10.1242/jcs.01005. [DOI] [PubMed] [Google Scholar]
- Koumas L, Smith TJ, Feldon S, Blumberg N, Phipps RP. Thy-1 expression in human fibroblast subsets defines myofibroblastic or lipofibroblastic phenotypes. Am J Pathol. 2003;163:1291–1300. doi: 10.1016/S0002-9440(10)63488-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipev CC, Simon M. Phenotypic differences between dermal fibroblasts from different body sites determine their responses to tension and TGFbeta1. BMC Dermatol. 2002;2:13. doi: 10.1186/1471-5945-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TY, Chin GS, Kim WJ, Chau D, Gittes GK, Longaker MT. Expression of transforming growth factor beta 1, 2, and 3 proteins in keloids. Ann Plast Surg. 1999;43:179–184. [PubMed] [Google Scholar]
- Chin GS, Liu W, Peled Z, Lee TY, Steinbrech DS, Hsu M, Longaker MT. Differential expression of transforming growth factor-beta receptors I and II and activation of Smad 3 in keloid fibroblasts. Plast Reconstr Surg. 2001;108:423–429. doi: 10.1097/00006534-200108000-00022. [DOI] [PubMed] [Google Scholar]
- Wang X, Smith P, Pu LL, Kim YJ, Ko F, Robson MC. Exogenous transforming growth factor beta(2) modulates collagen I and collagen III synthesis in proliferative scar xenografts in nude rats. J Surg Res. 1999;87:194–200. doi: 10.1006/jsre.1999.5757. [DOI] [PubMed] [Google Scholar]
- Smith P, Mosiello G, Deluca L, Ko F, Maggi S, Robson MC. TGF-beta2 activates proliferative scar fibroblasts. J Surg Res. 1999;82:319–323. doi: 10.1006/jsre.1999.5580. [DOI] [PubMed] [Google Scholar]