

Abstract
Disability or death due to intracerebral hemorrhage (ICH) is attributed to blood lysis, liberation of iron, and consequent oxidative stress. Iron chelators bind to free iron and prevent neuronal death induced by oxidative stress and disability due to ICH, but the mechanisms for this effect remain unclear. We show that the hypoxia-inducible factor prolyl hydroxylase domain (HIF-PHD) family of iron-dependent, oxygen-sensing enzymes are effectors of iron chelation. Molecular reduction of the three HIF-PHD enzyme isoforms in the mouse striatum improved functional recovery after ICH. A low-molecular-weight hydroxyquinoline inhibitor of the HIF-PHD enzymes, adaptaquin, reduced neuronal death and behavioral deficits after ICH in several rodent models without affecting total iron or zinc distribution in the brain. Unexpectedly, protection from oxidative death in vitro or from ICH in vivo by adaptaquin was associated with suppression of activity of the prodeath factor ATF4 rather than activation of an HIF-dependent prosurvival pathway. Together, these findings demonstrate that brain-specific inactivation of the HIF-PHD metalloenzymes with the blood-brain barrier-permeable inhibitor adaptaquin can improve functional outcomes after ICH in several rodent models.
INTRODUCTION
Intracerebral hemorrhage (ICH) occurs in a host of neurological conditions across the human life span, including stroke, traumatic brain injury, brain tumors, arteriovenous malformations, amyloid angiopathy, anticoagulant use, and sickle cell disease (1). ICH is associated with significant morbidity and mortality that results from both primary and secondary mechanisms. Therapies targeted toward the primary injury have had limited success, which has led to a focus on secondary injury mechanisms (2). Blood breakdown products after ICH appear to be an important source of secondary brain injury. In particular, functional impairments are associated with red blood cell lysis, the release of heme, and increases in redox-active iron. In this state, “free” iron is able to interact with peroxide to generate highly reactive hydroxyl radicals through Fenton chemistry, resulting in oxidative damage to lipids, proteins, and DNA in diverse cell types (3, 4). There is indirect support for the hypothesis that Fenton reactions give rise to dysfunction after ICH. This support derives primarily from observations of the beneficial effects of low-molecular-weight iron chelators in some rodent and pig models of ICH (1), although these results have not been reproduced in all laboratories where iron chelators have been evaluated for ICH treatment (5).
A challenge in the therapeutic use of iron chelators is to reduce pathological iron accumulation without disrupting physiological iron-dependent functions. Iron is essential for several physiological processes, including mitochondrial function, cell signaling, cell division, and myelination (6). Nonselective metal chelators may prevent injury, but they may also have dose-limiting toxicities through nonspecific effects on iron-dependent processes in the cell (7). These observations have led us to try to identify iron-dependent metalloenzymes that are inhibited by iron chelators and are integral to promoting neuronal survival. These metalloenzymes may represent more specific targets for preventing secondary injury after ICH (8).
One class of metalloenzymes that has been implicated in neuronal survival both in vitro and in vivo is a family of oxygen sensors known as the hypoxia-inducible factor prolyl hydroxylase domain enzymes (HIF-PHDs) (9). Catalysis by the HIF-PHDs (oxygen-, 2-oxoglutarate-, and iron-dependent dioxygenases) destabilizes the transcriptional activator HIF-1a under normoxia (10). Iron chelators are known to inhibit the HIF-PHDs under normoxia, thereby inhibiting oxygen-dependent hydroxylation of HIF, its recruitment by the von Hippel-Lindau protein component of an E3 ubiquitin ligase complex, and its consequent proteasomal degradation. Iron chelators have also been shown to stabilize HIF-1a and activate a suite of putative adaptive genes at concentrations that protect neurons from oxidative death (11–14). Here, we used molecular and pharmacological tools to show that HIF-PHD inhibition may be neuroprotective after ICH in vitro and in vivo. Our mechanistic studies suggest an HIF-independent pathway of protection through HIF-PHD inhibition that leads to suppression of activating transcription factor 4 (ATF4) prodeath pathways.
RESULTS
Inhibition of iron-dependent HIF-PHDs protects neurons from hemin-induced toxicity in vitro
Secondary injury from ICH has been attributed, in part, to hemin, a breakdown product of hemoglobin from lysed blood (15, 16). Hemin induces lipid peroxidation and mitochondrial dysfunction to initiate death in endothelial cells (17). To investigate the mechanisms of neuronal toxicity to hemin in vitro, we exposed primary rat cortical neurons, immortalized hippocampal neuroblasts (HT22 cells), or immortalized striatal neuroblasts (Q7 cells) to hemin for 24 hours. As expected, we found a dose-dependent loss in viability in all three cell types as measured by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] reduction or the Live/Dead viability assay (fig. S1, A and B). Treatment of primary neurons with hemin at the median lethal dose (50 mM) results in a time-dependent increase in heme oxygenase expression and iron content, which suggests that hemin is taken up into neurons and metabolized (fig. S1, C and D).
To examine whether iron-dependent HIF-PHDs can be modulated to protect neurons from hemin-induced toxicity, we examined chemically diverse inhibitors of the HIF-PHDs including deferoxamine (DFO), ciclopirox (CPO), and dihydroxybenzoic acid (DHB). The cotreatment of primary rat neurons or mouse neuronal cell lines with hemin and these structurally diverse HIF-PHD inhibitors inhibited hemin-induced neuronal death in all three cell types (Fig. 1, A to K). Previous studies from our laboratory have established that both DFO and DHB can inhibit HIF-PHDs and stabilize HIFs (Fig. 1L), but only DFO functions as an iron chelator (11). These findings suggested that PHD inhibition, rather than metal chelation, is the on-target mechanism of DFO in protecting against hemin toxicity. To test this hypothesis using a chemical-biological approach, we compared the protective effects of CPO, an HIF-PHD inhibitor, and a CPO analog (5342) with similar metal binding affinity to CPO but no PHD inhibitory activity (fig. S2). As expected, only the CPO compound with PHD inhibitory activity was neuroprotective (Fig. 1, M and O to R). Moreover, neuroprotection by CPO did not correlate with a significant reduction in total cellular iron concentration, as measured by inductively coupled plasma optical emission spectroscopy (ICP-OES) (Fig. 1N). Together, these findings suggest that selective inhibition of the HIF-PHDs, and not free iron chelation, is the mechanism for protection from hemin-induced death in an in vitro model of ICH-induced secondary injury.
Fig. 1.
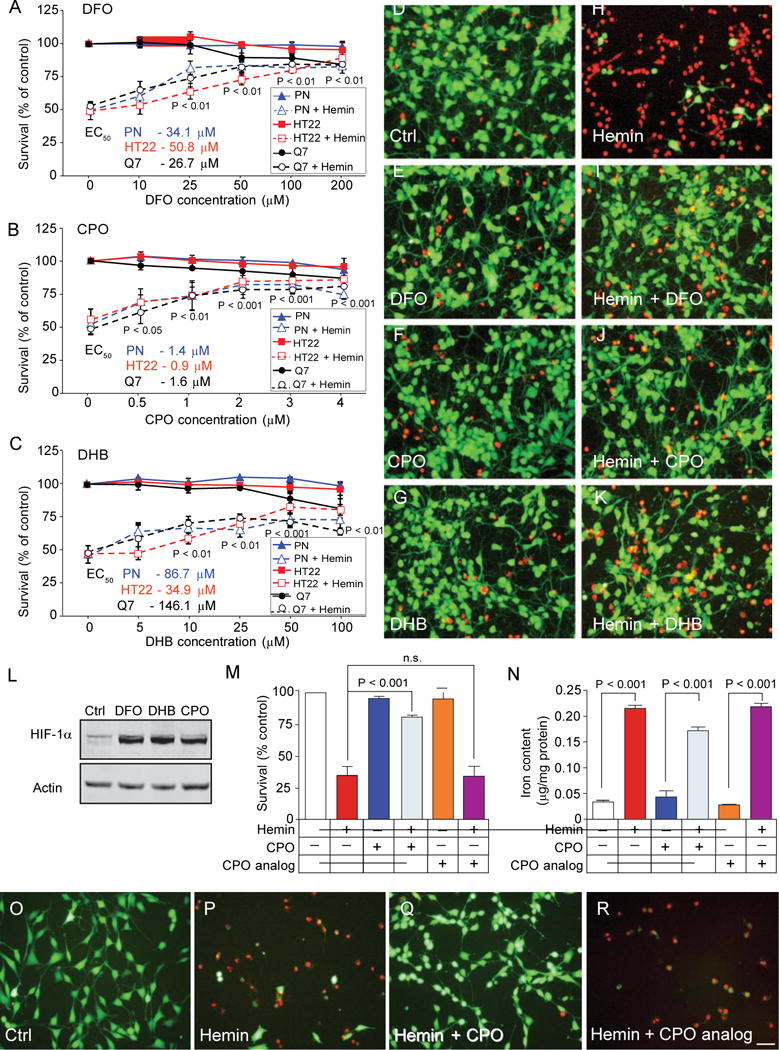
HIF-PHD inhibitors stabilize HIF-1a and abrogate hemin- induced toxicity in rat primary neurons or immortalized neuroblasts independent of iron chelation. (A) DFO, an iron chelator/HIF-PHD inhibitor, blocks hemin-induced toxicity in rat primary cortical neurons (PN; median effective concentration (EC50), 34.1 mM], mouse hippocampal neuroblasts (HT22; EC50, 50.8 mM), and immortalized mouse striatal neuroblasts (Q7; EC50, 26.7 mM) as measured by MTT assay. (B) CPO, an HIF-PHD inhibitor distinct from DFO, abrogates hemin-induced toxicity in primary cortical neurons (EC50, 1.4 mM), hippocampal neuroblasts (EC50 0.9 mM), and immortalized striatal neuroblasts (EC50, 1.6 mM). (C) DHB, a distinct HIF-PHD inhibitor that does not bind to iron (11), abrogates hemin-induced toxicity in primary cortical neurons (EC50, 34.9 mM), hippocampal neuroblasts (EC50, 86.7 mM), and immortalized striatal neuroblasts (EC50, 146.1 mM). (D to K) Representative images of live [calcein-AM, green] and dead (ethidium homodimer, red) primary cortical neurons treated with (D) saline, (E) DFO (100 mM), (F) CPO (3 mM), (G) DHB (100 mM), (H) hemin (50 mM), (I) hemin (50 mM) + DFO (100 mM), (J) hemin (50 mM) + CPO (3 mM), or (K) hemin (50 mM) + DHB (100 mM). Scale bar, 100 mm. (A to C) Significance was determined by two-way analysis of variance (ANOVA) followed by Bonferroni’s comparison test, from three independent experiments. (L) DFO, DHB, and CPO stabilize HIF-1a protein in mouse primary neurons. (M) CPO (3 mM) prevents hemin-induced neuronal death, but a CPO analog (3 mM, 5342; fig. S2) without PHD inhibitory activity does not. n.s., not significant. (N) Protection by CPO is not associated with significant reductions in total iron as measured by ICP-OES. (O to R) Live/Dead staining images of cells treated with (O) saline, (P) hemin (50 mM), (Q) hemin (50 mM) + CPO (1 mM), or (R) hemin (50 mM) + CPO analog (3 mM) in mouse hippocampal neuroblasts. Scale bar, 100 mm. One-way ANOVA followed by Dunnett’s comparison test, from triplicates (M and N) All graphs show the means ± SEM. Immunoblot data are representative of three experiments.
Regional reduction of oxygen-sensing PHDs in the mouse brain improves outcomes after striatal ICH
Although DFO, DHB, and CPO target the HIF-PHDs and prevent hemin-induced toxicity, HIF-PHDs belong to a superfamily of more than 60 2-oxoglutarate-dependent dioxygenases (18). Some members of this superfamily [for example, ten-eleven translocation (TET) DNA demethylases] are inhibited by inhibitors of the HIF-PHDs and could be responsible for the protection we observed. To investigate whether molecular reduction of the oxygen-sensing PHDs is sufficient to improve outcomes after ICH, we conditionally reduced PHD1, PHD2, and PHD3 expression simultaneously in the mouse striatum using dual injections of adeno-associated virus expressing Cre recombinase (AAV8-Cre) in adult mice with homozygous floxed alleles (PHD1/2/3fox/flox) of all three PHDs (Fig. 2A). We chose to reduce all three HIF-PHD isoforms simultaneously because prior studies in the liver have shown that triple knockout is required to stabilize HIFs optimally and drive erythropoietin (EPO) synthesis (19). We focused on the striatum because this is the location where ICH often occurs. We used a floxed-tdTomato reporter to verify that dual injections of AAV8-Cre induced recombination mediolaterally in the striatum, the location corresponding to the center of the hemorrhagic stroke that we induced in these mice (Fig. 2, B to D). We confirmed that injections of AAV8-Cre, but not AAV8-GFP (adeno-associated virus expressing GFP), in floxed mice 2 weeks before the induction of ICH resulted in a significant reduction in mRNA expression for each of the HIF-PHDs (Fig. 2E; PHD1, P = 0.0049; PHD2, P = 0.0010; PHD3, P = 0.0001) and a significant induction of the canonical HIF target genes vascular endothelial growth factor (VEGF) and EPO (Fig. 2F). Because VEGF is produced primarily in neurons mediated by HIF-1a and EPO primarily in glial cells mediated by HIF-2a, these data suggested that AAV8 injections transduced both neurons and glia (20). We induced ICH in wild-type and PHD-deficient mice using collagenase, an enzyme that induces hemorrhage through disruption of the basal lamina of the blood vessels. The resulting hemorrhage fills the striatum and leads to cell death and persistent behavioral deficits. Consistent with our in vitro studies, using small-molecule inhibitors of the HIF-PHDs, the molecular reduction of the three HIF-PHDs resulted in improved somatosensory function 3 and 7 days after the injury (Fig. 2, G and H). Notably, this behavioral improvement occurred in the absence of changes in edema formation or hematoma size, which suggests that the salutary effects of HIF-PHD inactivation occurred downstream of the basal lamina disruption and striatal hemorrhage (fig. S3, A and B).
Fig. 2.
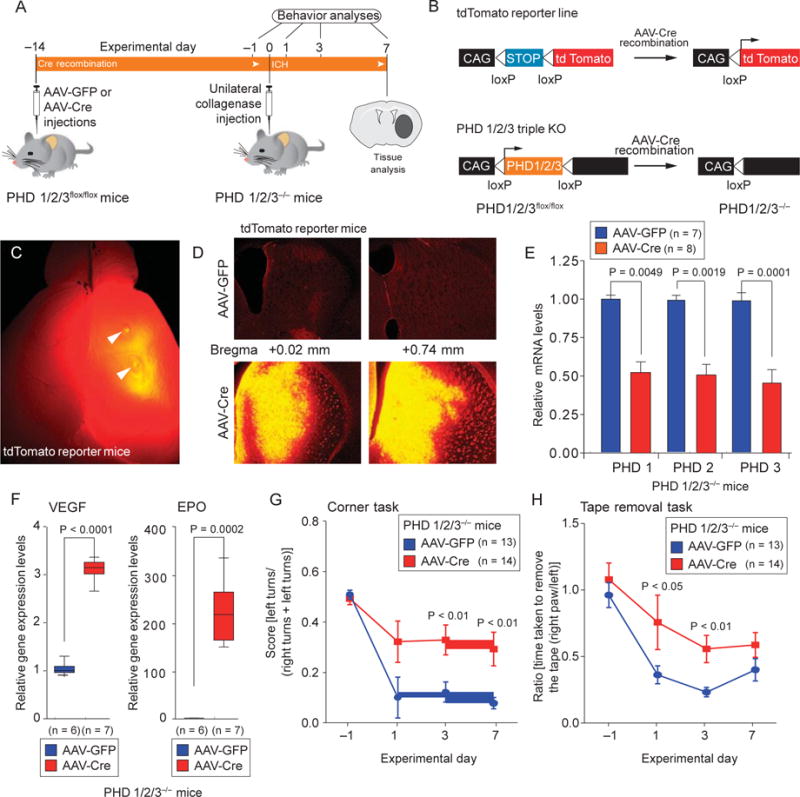
Molecular reduction of HIF-PHD isoforms in the mouse striatum enhances functional recovery after ICH. (A and B) Scheme for validating AAV8-Cre activity and selective deletion of PHD1, PHD2, and PHD3. KO, knockout. (C) Effective recombination by injection of AAV8-Cre into the mouse striatum was verified using a tdTomato floxed reporter. (D) Coronal sections of the tdTomato mouse brain revealed that tdTomato reporter expression was highest mediolaterally at coordinates corresponding to subsequent hemorrhagic stroke. Scale bars, 1 mm (C); 100 mm (D) (E) Quantitative PCR confirmed reduction of PHD1, PHD2, and PHD3 expression in the striatum of AAV8-Cre– injected mice but not in the striatum of AAV8-GFP–injected mice (E). (F) Quantitative PCR confirmed that reduction of striatal PHD expression led to increases in the HIF- dependent genes encoding VEGF and EPO in the mouse striatum. (G and H) Conditional reduction of HIF-PHDs enhances functional recovery in mice after ICH, as shown by two behavioral tasks: the corner task (G) and the tape removal task (H). Significance was determined by two-tailed t test (E and F) or two-way ANOVA with Bonferroni’s post hoc test (G and H). All graphs show the means ± SEM
Identification of a brain-penetrant HIF PHD inhibitor using an HIF reporter mouse
Molecular inactivation studies that involve the HIF-PHDs provide an important in vivo proof of concept for the potential of these oxygensensing enzymes as therapeutic targets for ICH. However, because most ICHs are spontaneous in those not known to be at high risk, it is difficult to imagine a practical prophylactic strategy for this illness. A small molecule that targets the HIF-PHDs would be ideal to facilitate the inhibition of the target after injury. To identify HIF-PHD inhibitors for testing in ICH models, we evaluated whether known HIF-PHD inhibitors could penetrate the blood-brain barrier to act in the central nervous system (CNS). To monitor HIF-PHD activity dynamically, we used mice engineered to ubiquitously express the oxygen degradation domain (ODD) of HIF-1a fused to firefly luciferase (fig. S4A). The ODD domain contains proline residues 402 and 564, which are hydroxylated by the HIF-PHDs to degrade HIF-1 or ODD-luciferase. These mice have been used to track HIF-PHD activity in diverse organs in vivo using bioluminescence imaging (21). Unexpectedly, canonical HIF-PHD inhibitors failed to increase ODD-luciferase activity in the brain (fig. S4, B and C). The failure of these agents to inhibit HIF-PHDs in the brain was not due to a lack of intrinsic inhibitory activity of these drugs because all inhibitors induced ODD-luciferase activity in cell-based assays.
To overcome these unexpected negative results, we turned our attention to an 8-hydroxyquinoline inhibitor of the HIF-PHDs. We have named this molecule adaptaquin for its ability to inhibit the HIF-PHDs and activate adaptive responses to hypoxia in a cell-based assay (22) (Figs. 3A and 4B). Adaptaquin inhibited purified, recombinant PHD2 as shown by either mass spectrometry-based (Fig. 3B) or antibody-based hydroxylation assays (fig. S5A). Nondenaturing mass spectrometry was used to confirm that adaptaquin can bind directly to PHD2 (fig. S5, B to E).
Fig. 3.
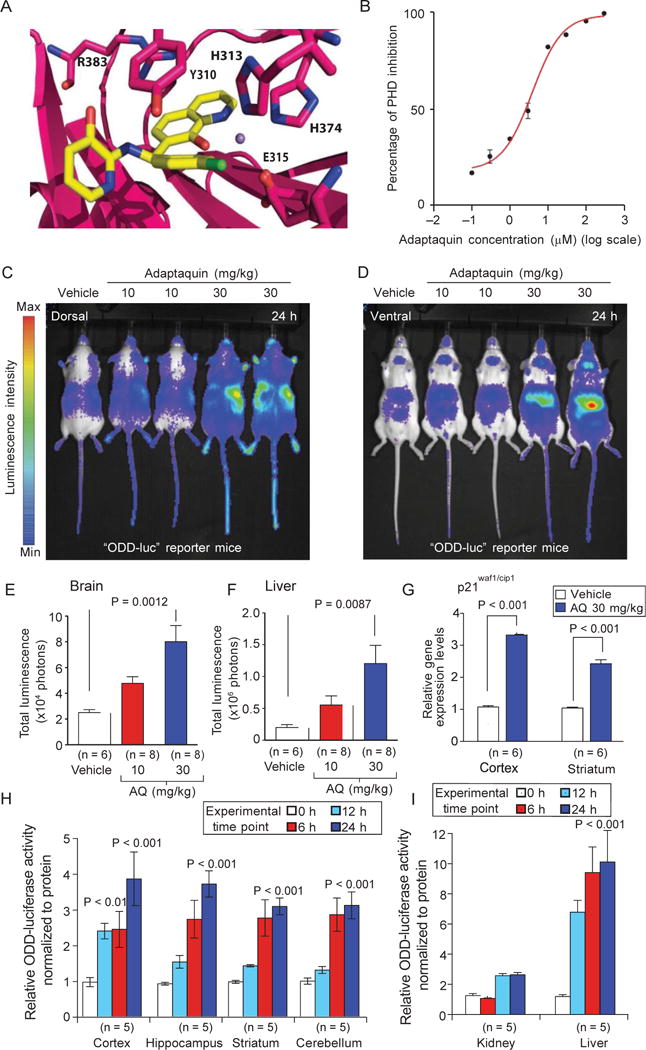
Adaptaquin is an HIF-PHD inhibitor that can penetrate the mouse CNS. (A) In silico modeling predicts that adaptaquin (yellow) fits into the active site of HIF-PHDs. (B) Effect of adaptaquin on hydroxylation of a synthetic HIF peptide by recombinant HIF- PHD2, as assayed by mass spectrometry. (C and D) Adaptaquin dose-dependently inhibits HIF-PHD activity in the mouse brain as monitored by in vivo bioluminescence imaging from dorsal (C) and ventral (D) views. (E and F) Quantitative luciferase activity measurements (from pseudocolored bioluminescence) show that adaptaquin (AQ) increased the stability of ODD-luciferase in the brain and liver. (G) Increased ODD-luciferase activity in the brain was associated with increased transcription of p21waf1/cip1, a gene induced by neuronal HIF-PHD inhibition. (H) Analysis of lysates from several mouse brain regions showed an increase in ODD-luciferase reporter activity at 0, 6, 12, and 24 hours after intraperitoneal injection of adaptaquin (30 mg/kg). (I) Similar results were seen in the kidney and liver. Significance was determined by one-way ANOVA followed by Dunnett’s comparison test for vehicle compared to adaptaquin (10 or 30 mg/kg) treatment (E and F) or by two-way ANOVA with Bonferroni’s post hoc test (G to I). All graphs shows the means ± SEM.
Fig. 4.
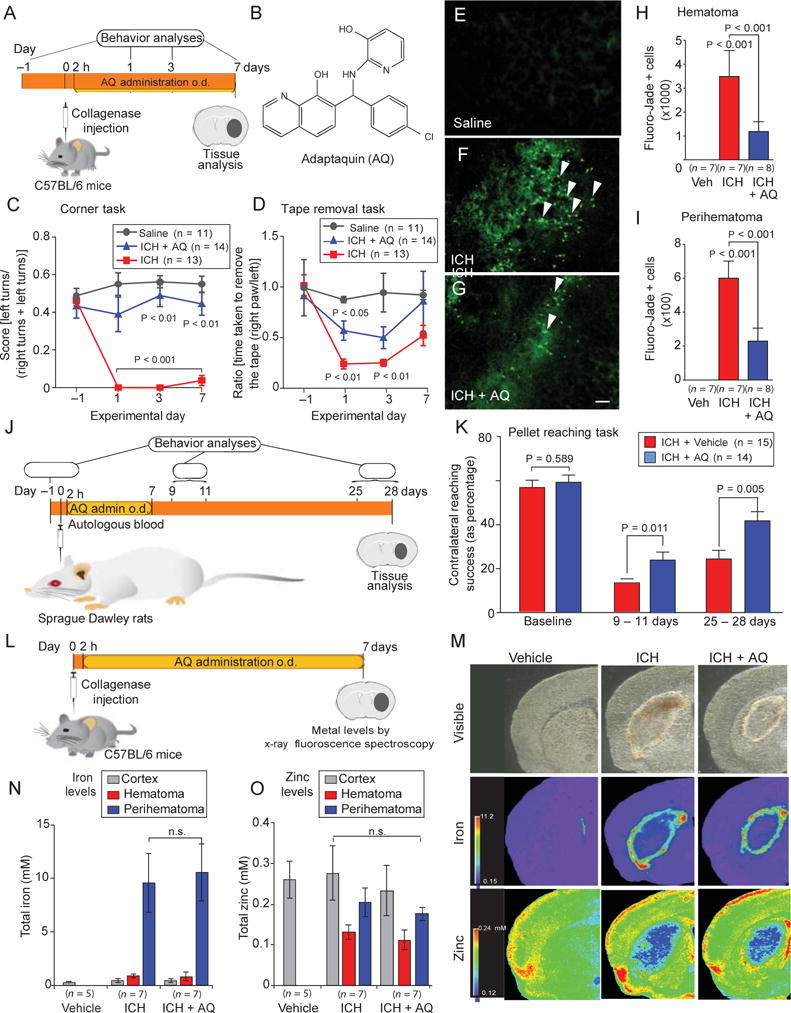
Adaptaquin delivered after injury reduces cell death and enhances functional recovery in rodent models of ICH. (A) Experimental design for delivery of adaptaquin after ICH in mice. (B) Adaptaquin chemical structure. o.d., once daily. (C and D) Adaptaquin improved behavioral deficits associated with ICH in mice undertaking two sensory tasks: the corner task (C) and the tape removal task (D). (E to G) Adaptaquin reduced neuronal degeneration as monitored by Fluoro-Jade staining (green) in the perihematomal regions of the mouse brain. Scale bar, 100 μm. White arrows highlight degenerating neurons in the ICH group (F); this was reduced by adaptaquin treatment (G). (H and I) Stereological counting of Fluoro-Jade–positive neurons confirmed that adaptaquin treatment decreased the number of degenerating neurons in the hematoma (H) and perihematoma (I) regions. Veh, vehicle. (J) Diagram illustrating adaptaquin (30 mg/kg intraperitoneally) treatment in the rat autologous blood infusion model of ICH. (K) Adaptaquin improved single-pellet reaching in rats for up to 1 month after ICH. (L) Protocol for evaluating the total concentration of iron and zinc in the mouse brain after ICH and treatment with either vehicle or adaptaquin (30 mg/kg intraperitoneally). (M) Pseudocolored brain coronal sections from mice treated with collagenase to induce ICH. (N and O) Seven days after adaptaquin treatment, total iron (N) and zinc (O) concentrations were unchanged in the mouse CNS. Significance was determined by one-way ANOVA followed by Dunnett’s comparison test for vehicle or adaptaquin treatment after ICH (H to K) or by two-way ANOVA with Bonferroni’s post hoc test (C and D). All graphs show the means ± SEM.
In contrast to HIF-PHD inhibitors such as DFO and DHB, adaptaquin (30 mg/kg) could significantly increase ODD-luciferase activity as monitored by bioluminescence imaging in the mouse brain (Fig. 3, C and E; P = 0.0012) and mouse liver (Fig. 3, D and F; P = 0.0087). To verify that the observed changes reflected changes in ODD-luciferase levels in the CNS parenchyma rather than changes in skin or dural ODD-luciferase, we measured ODD-luciferase activity in distinct mouse brain regions. As expected, we found increases in activity in response to adaptaquin treatment in the cortex, hippocampus, striatum, and cerebellum (Fig. 3H) and in the kidney and liver (Fig. 3I). Stabilization of ODD-luciferase levels in the brain correlated with increased mRNA expression of p21waf1/cip1, a gene regulated by HIF-PHD inhibition in neurons (Fig. 3G). These results suggest that adaptaquin (30 mg/kg) penetrates the blood-brain barrier, resulting in inhibition of the oxygen-sensing HIF-PHDs and activation of HIF-dependent gene expression.
Adaptaquin reduces neuronal death and improves functional recovery in different rodent models of ICH
To determine whether HIF-PHD inhibition after ICH can improve behavioral outcomes, we administered adaptaquin (30 mg/kg) intraperitoneally once a day for 7 days starting 2 hours after the unilateral injection of collagenase into the mouse striatum (Fig. 4A). Measurements of hematoma size 24 hours after collagenase injection verified that adaptaquin does not inhibit collagenase activity (fig. S6, A to C). Adaptaquin-treated mice, however, showed decreased edema 7 days after collagenase injection, likely because adaptaquin, in contrast to the conditional deletion of HIF-PHD isoforms, is available to target vascular and immune cells (fig. S7B). Mice with striatal hemorrhage showed a preference for ipsilateral turns because of deficits in the weight-balancing movements of the limbs contralateral to the injury, as well as spatial neglect (23). This preference was normalized in adaptaquin-treated mice as measured by the corner turn task (Fig. 4C; P < 0.01). Another behavior (tape removal task), which represents a form of sensory neglect, improved significantly in adaptaquin-treated mice 1 and 3 days after ICH (Fig. 4D; P < 0.05). Adaptaquin-induced behavioral improvements were associated with a reduction in the number of degenerating neurons in perihematomal and hematomal areas of the mouse striatum (Fig. 4, E to I; P < 0.001).
To determine whether adaptaquin could ameliorate outcomes in a different model of ICH, we tested adaptaquin (30 mg/kg) in a rat model of autologous blood infusion into the striatum (Fig. 4J). Autologous blood infusion causes a narrower lesion in the striatum than collagenase but results in motor impairments beyond 30 days (24). As in mice, adaptaquin was administered to rats 2 hours after the autologous blood infusion and then daily for 7 days. Success in the single-pellet reaching task (a test where rats reach through a narrow slot for a food pellet that is placed on a shelf that is attached to the front wall of the test chamber, Fig. 4K) was analyzed by repeated measures ANOVA. The task is sensitive to forepaw impairments after ICH. Compared to sham, ICH impaired the percent of pellets successfully retrieved upon reaching at early (9 to 11 days) and later (25 to 28 days) times after injury (P < 0.001). A planned comparison showed that there were no group differences at baseline (P = 0.589), as expected. However, adaptaquin treatment did improve pellet reaching success at the early times of 9 to 11 days (P = 0.011) and late assessment time periods of 25 to 28 days (P = 0.005). These improvements in motor recovery were not due to the effects of adaptaquin on core body temperature (fig. S8). Together, the rodent data demonstrate that adaptaquin can effectively improve recovery of sensorimotor or motor outcomes after ICH in collagenase or autologous blood rodent models.
Adaptaquin protection is independent of HIF and bulk iron chelation
These studies support HIF-PHDs as targets for therapy after ICH, but they do not identify mechanisms by which adaptaquin abrogates neuronal death. A prediction of our model is that adaptaquin enhances functional recovery without changing total iron in the brain. We monitored total iron concentrations in brain sections from vehicle- and adaptaquin-treated mice at 7 days after ICH (Fig. 4L). Despite improvements in outcomes in the adaptaquin-treated mice (Fig. 4, C and D), there were no differences in the apparent distribution of total iron (Fig. 4, M and N) and zinc (Fig. 4O) in the brain as measured by x-ray fluorescence microscopy. These data are consistent with a model in which specific iron-dependent metalloenzymes, the HIF-PHDs, are inhibited by adaptaquin to prevent neuronal damage after ICH.
The canonical target for HIF-PHD inhibition is the stabilization of HIF-1a, which leads to the induction of an adaptive response to hypoxia (25). To assess whether HIF-1a (or HIF-2a) is required for the protective effects of HIF-PHD inhibition, we examined the effect of selective HIF deletion on protection from hemin toxicity induced by chemically diverse HIF-PHD inhibitors in hippocampal neuroblasts. The reduction of the HIF-1a protein (fig. S9A) and associated target genes, which was accomplished using a previously validated small interfering RNA (26), failed to influence hemin-induced toxicity (fig. S9C) or protection by structurally diverse HIF-PHD inhibitors (fig. S9, E to G). Similar results were obtained with a previously characterized short hairpin RNA to HIF-2a (fig. S9, B, D, and H to J). Together, these results suggest that HIF-PHD inhibition can protect against hemin-induced neuronal death independent of HIF-1 or HIF-2 activation.
Adaptaquin inhibits ATF4-regulated prodeath gene expression
To probe the mechanism of adaptaquin-induced neuroprotection using an unbiased approach, we used microarray analysis with a model of oxidative stress in neurons induced by glutamate or its analog, homocysteate (HCA). Like hemin-induced toxicity (fig. S10, A and B), oxidative glutamate toxicity is abrogated by adaptaquin (Fig. 5, A to C). However, oxidative glutamate toxicity has a wider temporal window before neurons commit to die at 18 hours (as opposed to 4 to 6 hours in hemin-induced cell death). The large window between injury stimulus and death commitment has facilitated the analysis of primary events involved in oxidative death and the mechanism of action of neuroprotectants. We performed detailed dose-response and therapeutic window studies with adaptaquin in mouse cortical neurons exposed to the glutamate analog HCA. We found that adaptaquin afforded complete protection against oxidative death at 1 mM when added up to 16 hours after HCA treatment (Fig. 5, A to C). To probe the mechanism of this potent, delayed protection, we used microarrays to characterize the gene expression profiles of neuronal cultures exposed to HCA for 14 hours compared with vehicle alone and nonprotective and protective doses of adaptaquin (Fig. 5A).
Fig. 5.
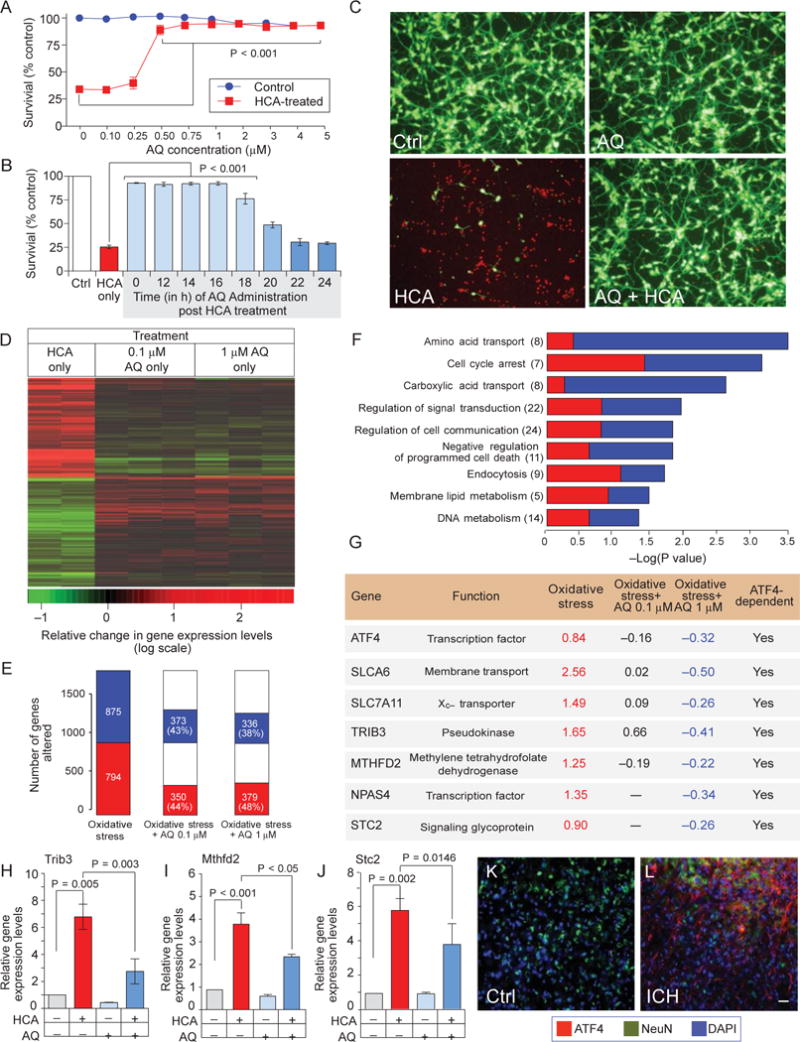
Adaptaquin prevents neuronal death by suppressing ATF4- mediated expression of prodeath genes. (A) Adaptaquin dose- dependently protects mouse primary cortical neurons after treatment with the glutamate analog HCA. (B) Adaptaquin (1 mM) provides complete protection for 16 hours after HCA treatment. (C) Adaptaquin- mediated protection from HCA-induced oxidative stress as shown by Live/Dead imaging, Scale bar, 100mm. (D) Heat map showing the top 1000 differentially expressed genes. Shades of red represent up-regulation and shades of green represent down-regulation of gene expression. A subset of genes dysregulated by HCA treatment are corrected after treatment with adaptaquin. (E) Proportion of oxidative stress–related genes (left) whose expression was altered to more normal levels after treatment with 0.1 mM (middle) and 1 mM (right) adaptaquin. Overrepresented GO gene categories whose expression was altered by adaptaquin. (F) Overrepresented categories included genes involved in amino acid transport and regulation of programmed cell death. (G) The table shows the microarray data for ATF4-regulated genes induced by HCA and down-regulated by protective, but not by nonprotective, concentrations of adaptaquin. Values were transformed to log2 scale. (H to J) Quantitative PCR confirmed that the ATF4 target genes Trib3 (H), MTHFD2 (I), and STC2 (J) were induced by oxidative stress, and this induction was reduced by adaptaquin (1 mM). (K and L) Up-regulation of ATF4 immunostaining 7 days after ICH. ATF4 expression is shown in red, neurons are indicated by NeuN expression (green), and nuclei are stained with 4′,6-diamidino-2-phenylindole (blue). Significance was determined by two-way ANOVA followed by Bonferroni’s comparison test from three independent experiments (A) or by one-way ANOVA followed by Dunnett’s comparison test from triplicates (B and H to J). All graphs show the means ± SEM.
Glutamate analog (HCA) treatment in mouse cortical neurons induced robust gene expression changes, significantly overlapping with those we reported previously (27). This robust signature included overrepresentation of genes implicated in amino acid transport activity, negative regulation of apoptosis, and mitochondria (Fig. 5F). Consistent with these findings, we found that adaptaquin inhibited mitochondrial fission, ATP (adenosine triphosphate) loss, mitochondrial depolarization, and mitochondrial reactive oxygen species production induced by glutamate (fig. S11, A to G). Moreover, protective doses of adaptaquin significantly reversed gene changes induced by the glutamate analog HCA (larger than would be expected by chance; P =1 × 10−27, hypergeometric test). Notably, both nonprotective (0.1 mM) and protective (1 mM) concentrations of adaptaquin caused changes in a similar set of genes (Fig. 5E), although the protective doses showed a greater magnitude of change than nonprotective doses (Fig. 5G).
Our molecular knockdown data suggested that adaptaquin is working through HIF-independent pathways. To determine whether gene changes induced by protective doses of adaptaquin reflected HIF-regulated genes, we compared the transcriptional response of 1 mM adaptaquin (374 unique genes; P < 0.005) with a list of 356 canonical HIF-1 targets, compiled previously (28). Only nine probes were shared between the two lists (P = 0.03, hypergeometric test), suggesting that HIF-related targets are not effectors of the adaptaquin response. The Gene Ontology (GO) categories of adaptaquin-related gene expression changes in HCA-treated neurons (Fig. 5F) included genes involved in apoptosis and amino acid transfer and genes induced by oxidative stress. Because we had previously observed dysregulation of these genes in neurons from ATF4-null mice, we formally compared the transcriptional response to 1 mM adaptaquin (374 unique genes) with a list of 189 probes (179 unique genes) differentially expressed in ATF4 knockout mouse neurons versus neurons from wild-type mice (27). Twenty-two probes were shared with a highly significant, nonrandom overlap (P = 5× 10−14; hypergeometric test). Strikingly, 17 of 22 probes (77%, including ATF4 itself) were changed by adaptaquin in the same direction as in ATF4 knockout neurons, suggesting that the beneficial effect of the adaptaquin treatment in oxidatively stressed cells is mediated through inhibition of ATF4-dependent genes (Fig. 5G).
Protective, but not nonprotective, doses of adaptaquin reduced the expression of a host of ATF4 target genes induced by oxidative stress including tribbles homolog 3 (Trib3), methylenetetrahydrofolate reductase (MTHFD2), the transporter (SLC7A11), and stanniocalcin-2 homeric glycoprotein (STC2) (Fig. 5G). The expression of these genes and cell death are significantly reduced when ATF4 is molecularly deleted (27). Quantitative polymerase chain reaction (PCR) confirmed that adaptaquin significantly reduced the expression of ATF4-dependent genes including Trib3, MTHFD2, and STC2 (Fig. 5, H to J; Tnb3, P = 0.003; MTHFD2, P < 0.05; STC2, P = 0.0146). Moreover, examination of mouse brains after ICH showed increases in ATF4 protein in the perihematomal region, demonstrating induction of this transcription factor after ICH, although at 7 days after injury, a minority of ATF4 staining appeared to be neuronal (Fig. 5, K and L).
Among the genes inhibited by adaptaquin, Trib3, a pseudokinase negative regulator of Akt/CREB (adenosine 3′,5′-monophosphate response element-binding protein), can induce neuronal death and is ATF4-regulated (27, 29–32). To determine if adaptaquin-mediated suppression of Trib3 required the ATF4 binding site in the Trib3 promoter, we investigated the effect of adaptaquin and its analogs on a Trib3 promoter-reporter construct transfected into mouse hippocampal neuroblasts. As expected, we found that HCA increased reporter activity and that activation was dependent on the ATF4 binding site (Fig. 6, A and B). Only protective (1 mM), but not nonprotective (0.1 mM), doses of adapta-quin suppressed the induction of the Trib3 reporter due to HCA-induced oxidative stress. Activation of the Trib3 reporter was occluded by the absence of the ATF4 binding site. An isosteric analog of adaptaquin, which has no cellular HIF-PHD inhibitory activity (22), failed to repress oxidative stress induction of the reporter (Fig. 6, A and B). These findings establish that only protective concentrations of adaptaquin, with known HIF- PHD inhibitory activity (22), suppress oxidative stress-induced Trib3 expression through an ATF4 binding site. Chromatin immunoprecipitation (ChIP) with an ATF4 antibody showed that adaptaquin’s ability to reduce the expression of Trib3 was associated with significantly decreased occupancy of ATF4 at the Trib3 promoter (Fig. 6C).
Fig. 6.
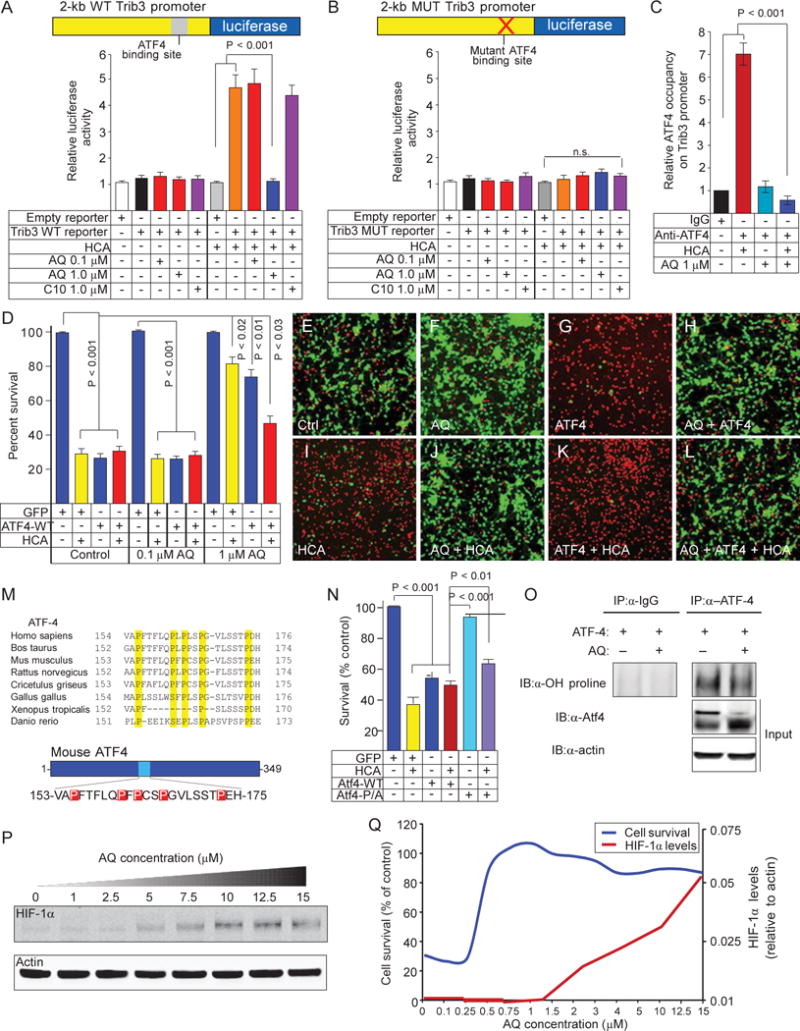
Adaptaquin inhibits ATF4 DNA binding, ATF4 transcriptional activity, and ATF4 prodeath properties in neurons. (A) HCA-induced oxidative stress up-regulated the promoter of the ATF4 target gene Trib3; this was blocked by adaptaquin (1 mM). Treatment with nonprotective concentrations of adaptaquin (0.1 mM) or an inactive oxyquinoline analog (compound 10, 1 mM) did not impede oxidative stress– induced activation of the Trib3 promoter-luciferase reporter construct. WT, wild type. (B) Mutation of the ATF4-binding site occludes oxidative stress inducibility and sensitivity to adaptaquin (1 mM). MUT, mutant. (C) ChIP studies showed that HCA-mediated oxidative stress increased ATF4 occupancy on the Trib3 promoter, which was blocked by protective concentrations of adaptaquin (1 mM). IgG, immunoglobulin G. (D to L) MTT assay (D) or Live/Dead staining (E to L) demonstrated that overexpression of ATF4 was sufficient to induce neuronal death in the absence of HCA, whereas protective concentrations of adaptaquin (1 mM) mitigated death by ATF4 overexpression or HCA and ATF4 overexpression. (M) Proline residues mutated to alanine residues in the mouse ATF4- 5P/A mutant construct are highlighted in red. (N) ATF4- 5P/A mutant does not induce cell death. (O) Adaptaquin inhibited hydroxylation of ATF4. (P) In mouse neurons, treatment with adaptaquin dose-dependently stabilized HIF-1a. (Q) Adaptaquin stabilization of HIF-1a (red line) occurs at concentrations higher than those required for full protection from oxidative death (red line). Significance was determined by two-way ANOVA followed by Bonferroni’s comparison test from three independent experiments (A, B, D, and N) or by one-way ANOVA followed by Dunnett’s comparison test from triplicates (C). All graphs show the means ± SEM.
The reduction in ATF4 binding and activity at the Trib3 promoter suggested the possibility that adaptaquin is working directly on ATF4 through the action of the HIF-PHDs to modulate prodeath gene expression Consistent with this model, we found that adaptaquin, in contrast to the neuroprotective antioxidant N-acetylcysteine, failed to inhibit oxidative stress-induced ATF4 protein expression (fig. S12, A and B). This observation suggested that adaptaquin acts on ATF4 directly to modulate cell death (fig. S12C). To address this possibility, we expressed ATF4 in mouse primary cortical neurons using an adenoviral vector. As expected, ATF4, but not the DNA binding mutant or GFP control, induced substantial cell death (Fig. 6, D and G). Treatment with adaptaquin (1 mM) significantly reduced cell death induced by ATF4 overexpression in the absence or presence of HCA-induced oxidative stress (Fig. 6, D to L; P < 0.03). Concentrations of adaptaquin (0.1 mM) not protective in the HCA model had no effect on ATF4-induced death. In the context of our promoter-reporter (Fig. 6, A and B) and ChIP results (Fig. 6C), these data suggest that adaptaquin can modulate ATF4 directly to affect its ability to induce death.
Recent studies have shown that ATF4 can interact directly with some HIF-PHD isoforms and that ATF4 has phylogenetically conserved prolines (P156, P162, P164, P167, and P174) (33) (Fig. 6M). Mutations of these prolines to alanines modulated ATF4 in HeLa cancer cells (33), but the role of these mutations in neurons has not been explored. Our model predicted that proline-to-alanine mutations should prevent proline hydroxylation and reduce ATF4-dependent cell death. Accordingly, we mutated all five conserved prolines to alanine [5 × P to A (5P/A)]. Consistent with a model in which adaptaquin inhibits the hydroxylation of proline directly on ATF4 to reduce the prodeath effects of this transcription factor, the 5P/A mutant of ATF4, which could nullify proline hydroxylation, was less effective in inducing cortical neuronal death than the wild-type ATF4 protein. The 5P/A mutant also was able to reduce death induced by HCA alone, suggesting a dominant negative function for this mutant (Fig. 6N). Moreover, protective concentrations of adaptaquin (1 mM) reduced hydroxylation of ATF4 (Fig. 6O). Together, these findings suggest that adaptaquin acts to reduce ATF4 transcriptional activity, possibly by reducing proline hydroxylation of ATF4, thereby preventing this transcription factor from being recruited to the promoters of a cassette of death-associated genes including Trib3 in response to oxidative stress. Notably, we found that the lowest dose of adaptaquin required to protect all neurons from oxidative stress was five- to sevenfold lower than those concentrations required to drive HIF stability, further uncoupling the effects of adaptaquin from HIF and its target genes (Fig. 6, P and Q).
Our model predicts that the wild-type ATF4 should drive putative prodeath gene expression in cortical neurons, but the ATF4-P/A, which cannot be hydroxylated by HIF-PHDs at five conserved prolines, should not. We therefore compared the effect of forced expression of wild-type ATF4 to that of forced expression of ATF4-5P/A mutant in mouse cortical neuronal cultures (Fig. 7A). We found that wild-type ATF4 significantly up-regulated mRNAs for Trib3, CHOP, ATF3, MTHFD2, and STC2 (Fig. 7, C to G). ChIP studies confirmed that this increase in Trib3 mRNA induced by wild-type ATF4 was associated with occupancy of ATF4 at the Trib3 promoter (Fig. 7B). As predicted from our model, the ATF4-5P/A mutant did not occupy the Trib3 promoter and did not drive the expression of any ATF4-dependent genes.
Fig. 7.
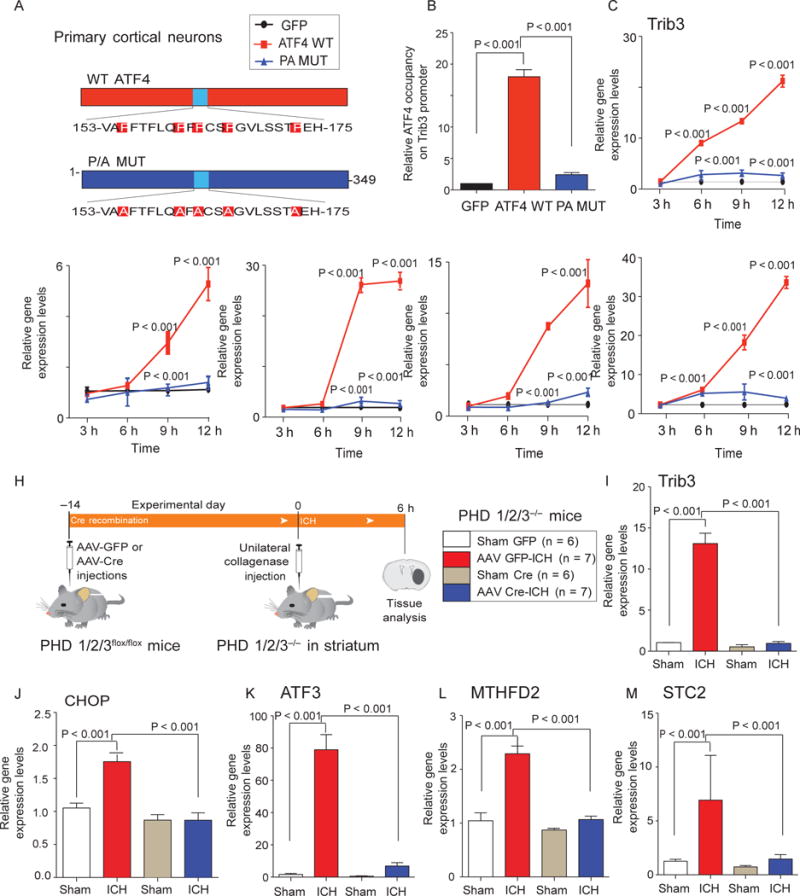
Reduction of HIF-PHD isoforms in the mouse striatum reduces ICH-induced ATF4-dependent gene expression. (A to G) Expression of WT ATF4 (red), but not of ATF4-5P/A mutant (blue), through adenoviral infection of primary cortical neurons leads to time-dependent induction of Trib3 (C), CHOP (D), ATF3 (E), MTHFD2 (F), and STC2 (G). ChIP studies (B) correlate increases in Trib3 gene expression induced by WT ATF4 with occupancy at the Trib3 promoter. (H) Diagram of experimental plan to determine whether reduction of HIF-PHD isoforms can abrogate ATF4-dependent gene expression in vivo. (I to M) Effects of molecular reduction of HIF-PHD isoforms on ICH-induced expression of Trib3 (I), CHOP (J), ATF3 (K), MTHFD2 (L), and STC2 (M). Significance was determined by two-way ANOVA followed by Bonferroni’s comparison test from three independent experiments (B to G) or by one-way ANOVA followed by Dunnett’s comparison test (I to M). All graphs show the means ± SEM.
Together, our findings are consistent with a model in which HIF- PHD inhibition protects neurons by reduced hydroxylation of ATF4 and diminished transactivation of established ATF4-dependent genes. To evaluate this model in vivo, we conditionally deleted HIF-PHD isoforms (as validated in Fig. 2) and studied the effect of this manipulation on a cassette of ATF4-dependent genes (Fig. 7H). We found that molecular HIF-PHD reduction significantly reduced ICH-induced changes in established ATF4-regulated genes (Fig. 7, I to M). To evaluate the effects of adaptaquin delivered after injury on ICH-induced ATF4-dependent gene expression, we focused our attention on Trib3 mRNA and protein expression (Fig. 8A). As expected, we found that adaptaquin completely suppressed ICH-induced Trib3 mRNA and protein, an ATF4 target gene (Fig. 8, C and D). Complete suppression of Trib3 expression occurred with only partial reduction of ICH-induced ATF4 protein induction (Fig. 8B). These data support a model whereby HIF-PHD inhibition suppresses ICH-induced ATF4-mediated prodeath gene expression through modulation of ATF4 transcriptional activity and not by inhibiting upstream oxidative stress. Indeed, adaptaquin had no effect on one established marker of oxidative stress, 3-nitrotyrosine, after ICH (fig. S14).
Fig. 8.
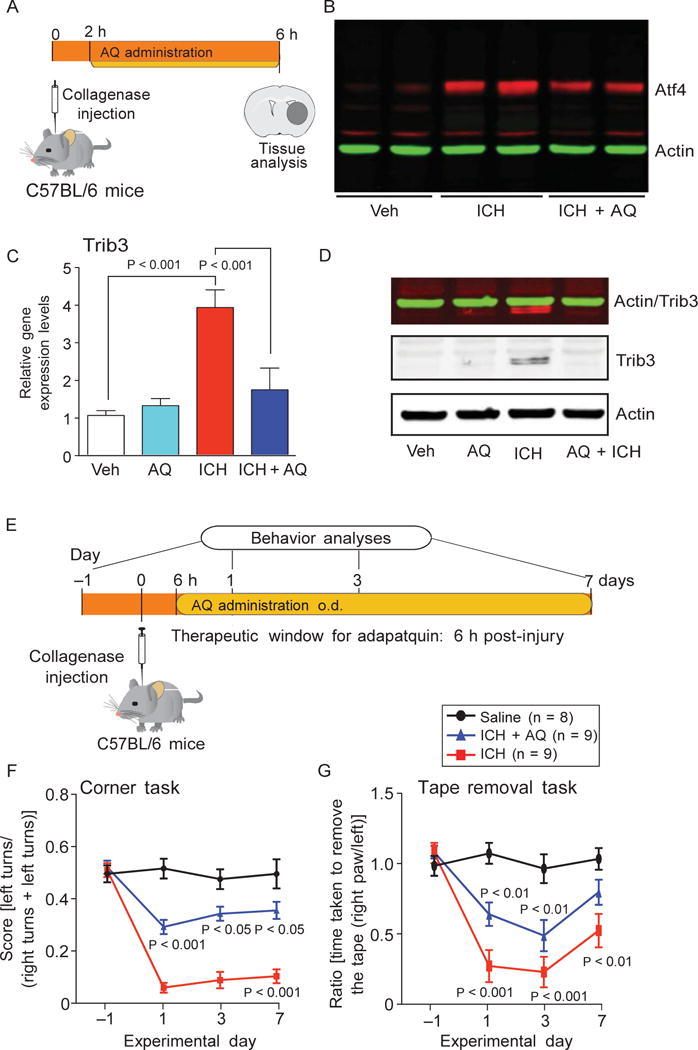
Adaptaquin inhibition of ATF4- dependent gene expression defines a therapeutic window after ICH. (A) Schematic of experimental plan to examine the effect of adaptaquin on ICH-induced Trib3 expression. (B) Immunoblots of ATF4 from nuclear extracts of the mouse striatum in distinct treatment groups. (C) Quantitative PCR of Trib3 message. (D) Immunoblots of Trib3 protein. (E to G) We examined the effect of adaptaquin (30 mg/kg) delivered 6 hours after collagenase injection and then daily for 7 days on ICH-induced spatial neglect in the corner task (F) or sensory neglect in the tape removal task (G). Significance was determined by oneway ANOVA followed by Dunnett’s comparison test (C) or by two-way ANOVA with Bonferroni’s post hoc test (F and G). All graphs show the means ± SEM
Because ATF4-dependent prodeath genes are expressed at 6 hours (Figs. 7 and 8) after collagenase-induced ICH, we predicted a therapeutic window for adaptaquin of 6 hours. As expected, we found that treatment of mice 6 hours after collagenase-induced ICH with adaptaquin significantly improved behavioral outcomes on tasks testing for spatial and sensory neglect compared to vehicle-treated mice (Fig. 8, E to G). Unilateral impairments in sensory and spatial orientation are associated with striatal hemorrhage in mice. Stroke therapeutics targeting ischemic stroke in humans can be delivered within 3 hours of stroke onset; thus, a therapeutic window of 6 hours for adaptaquin in ICH rodent models may be clinically relevant.
DISCUSSION
ICH is a stroke subtype with growing prevalence, high mortality, and significant morbidity (1). Here, we show that the HIF-PHD inhibitor adaptaquin can engage its target in the mouse CNS to reduce cell death in a mouse model of ICH (Fig. 4, E to I) and to enhance functional recovery in two distinct rodent models of brain hemorrhage, when administered after the onset of stroke (Fig. 4, C and D). In the rat autologous blood model of ICH, this functional recovery was observed up to 1 month after injury (Fig. 4K). Functional recovery after subacute treatment with adaptaquin was phenocopied by molecular reduction of three HIF-PHD isoforms in the striatum of mice, supporting the notion that adaptaquin acts through HIF-PHD inhibition and not through an off-target effect (Figs. 2 and 3). Indeed, adaptaquin, at doses required to protect cortical neurons from oxidative death, failed to alter global histone acetylation, histone methylation, or 12-lipoxygenase activity (table S1). Histone deacetylases are zinc-dependent metalloproteins (fig. S13, A and B) that, when inhibited, can increase global acetylation and protect from oxidative death (34). Jumonji domain histone demethylases and TET DNA demethylases can affect histone methylation and DNA methylation, respectively, and contain an iron-, oxygen-, and 2-oxoglutarate-dependent dioxygenase domain similar to that found in the HIF prolyl hydroxylases (fig. S15, A to D). 12-Lipoxygenase is an iron-dependent dioxygenase known to be required for HCA-induced death in neurons in vitro and after stroke in vivo (35) (fig. S13E and table S1). The absence of changes in the activity of these enzymes or markers of this enzyme activity by adaptaquin demonstrates its selectivity for inhibiting iron-dependent HIF prolyl hydroxylases and not other zinc- or iron-dependent enzymes that could have been responsible for adaptaquin-mediated neuroprotection in vitro and in vivo.
Adaptaquin belongs to the 8-hydroxyquinoline chemical family. The 8-hydroxyquinolines have metal chelating ability and are efficacious in Alzheimer’s disease (AD) animal models in vivo (36) and in yeast proteotoxicity models of chronic neurodegeneration in vitro (37). Indeed, 8-hydroxyquinoline analogs are in phase 2 testing for patients with AD or Huntington’s disease, supporting the safety of this chemical backbone in human applications (38). Several lines of evidence suggest that adaptaquin is an 8-hydroxyquinoline that inhibits HIF-PHDs selectively, rather than affecting the global movement of iron in cells to abrogate ICH-induced toxicity. First, structurally diverse HIF-PHD inhibitors, some of which do not bind to metals (for example, DHB; Fig. 1, A to L), were protective against hemin-induced toxicity of neurons in vitro. Second, the structure-activity relationship of some of these inhibitors (for example, CPO) for inhibiting neuronal death correlated with HIF-PHD inhibitory activity and not with affinity for metals, such as iron (Fig. 1, M to R). Third, molecular reduction of metalloenzyme HIF-PHD isoforms before hemorrhage enhanced functional recovery (Fig. 2, G and H). Fourth, x-ray fluorescence microscopy failed to demonstrate a bulk movement of total iron or total zinc out of the brain after treatment with protective doses of adaptaquin in vivo (Fig. 4, L to O). Together, these studies favor a model in which iron-coordinating HIF-PHD inhibitors can protect neurons from brain hemorrhage through inhibition of a specific metalloenzyme rather than through bulk chelation of free iron.
It has been proposed that protection by HIF-PHD inhibitors in stroke would be largely HIF-dependent and mediated through the induction of angiogenic and neurogenic growth factors, such as EPO and VEGF, although data supporting other models exist for other organs (39). We performed transcriptomics along with careful dose-finding studies to exclude HIF-dependent pathways of neuroprotection in an in vitro model of oxidative stress and focused on the prodeath ATF4-dependent pathway (Figs. 5 and 6). The effects of PHD inhibition on oxidative stress-induced prodeath gene expression are reminiscent of the role of PHD3 in regulating the coactivation of hypoxia-induced gene expression by pyruvate kinase M2 (40). However, we found that the HIF-PHD inhibitor adaptaquin can mediate neuroprotection at doses significantly lower than those required to stabilize HIFs (Fig. 6, P and Q) or activate HIF targets. These in vitro findings are consistent with an HIF-independent pathway of neuroprotection by HIF-PHDs. Despite our findings that adaptaquin actually inhibits the expression of some HIF-dependent genes after ICH (fig. S16, C and D), we cannot formally exclude the possibility that adaptaquin-mediated HIF stabilization is necessary for the cell survival effects and functional recovery we observed after ICH in vivo.
The ability of adaptaquin to enhance functional recovery when delivered after ICH in several rodent models tested in distinct laboratories suggests that our findings are robust and reproducible (41). The safety of other oxyquinolines in humans provides confidence that adaptaquin could meet essential safety tests required for its evaluation in humans with brain hemorrhage, a condition of growing prevalence and morbidity.
MATERIALS AND METHODS
Study design
The aim of the study was to use chemical and molecular tools to evaluate whether HIF-PHDs are a target for protection in vitro and in vivo and to fully characterize the specificity and therapeutic utility of a potent and selective inhibitor of HIF-PHDs, which we call adaptaquin. Our initial studies characterized an in vitro model of hemorrhagic stroke and showed that the targets of neuroprotective drugs with the ability to chelate bulk iron, such as DFO and CPO, are the HIF-PHDs. Because chemical inhibitors of HIF-PHDs can have off-target effects, we conditionally reduced all three isoforms in the mouse striatum using PHD1/2/3 triple floxed mice and an AAV8-Cre injected into two sites in the mediolateral striatum. Reduction of each isoform by 50% led to significant induction of VEGF and EPO, known HIF target genes, and improved functional recovery. We showed that adaptaquin penetrates the brain to inhibit the HIF-PHDs using in vivo bioluminescence imaging, and demonstrated that adaptaquin can bind to recombinant HIF-PHD2 and inhibit its activity in a test tube. We then showed that concentrations required to inhibit HIF-PHD activity enhanced functional recovery in two distinct rodent models of hemorrhagic stroke. Our mechanistic studies confirmed that adaptaquin’s salutary effects did not correlate with changes in total iron or zinc in the brain. These findings were consistent with a model in which adaptaquin acts on specific metalloenzymes, the HIF-PHDs, to mediate neuroprotection.
The sample sizes chosen for the drug treatment were adequately powered to observe the effects on the basis of past experience and studies of this type conducted by others. Some animals were excluded because of failure to reach the training criteria and mortality. Exclusions for training were based on scores as routinely done. Mice and rats were randomized to sham or ICH groups. The identity of the animals that received vehicle or adaptaquin was masked to surgeons who performed the ICH. The identity was revealed after the data were collected. We made every effort to minimize the discomfort and pain of the animals. Because HIF transcription factors are stabilized by inhibition of the HIF-PHDs, we used in vitro models to dissect the mechanism. We molecularly reduced HIF-1a or HIF-2a using RNA interference approaches previously characterized in our laboratory. Unexpectedly, deletion of either HIF-1a or HIF-2a neither reduced protection by HIF-PHD inhibitors nor enhanced cell death induced by hemin, a product of blood lysis. We probed the mechanism using an in vitro model of oxidative death through unbiased transcriptomics and found that adaptaquin’s effects were associated with direct modification of ATF4, and not HIF, leading to decreased occupancy at putative prodeath gene promoters. Furthermore, we confirmed in vivo that molecular and pharmacological inhibition of HIF-PHDs by adaptaquin significantly reduced ICH-induced changes in established ATF4-regulated genes.
Animal models
All mice and procedures were approved by the Institutional Animal Care and Use Committee of the Weill Cornell Medical College and were in accordance with the guidelines established by the National Institutes of Health (NIH) and ARRIVE (Animal Research: Reporting of In Vivo Experiments). All mice and rats were housed in a pathogen-free facility on a 12-hour light/dark cycle and, unless otherwise stated, provided ad libitum access to food and water. PHD1/2/3flox/flox mice were mated on a mixed C57BL/6-FVB genetic background. PHD1flox/flox, PHD2flox/flox, and PHD3flox/flox heterozygous mice were then intercrossed to generate PHD1/2/3flox/flox mice. Initially, PHD1/2flox/flox, PHD2/3flox/flox, and PHD1/3flox/flox mice were generated and then intercrossed to generate PHD1/2/3flox/flox. At least three different breeders were used to generate each floxed genotype, and the phenotypes were checked among the litters of all breeders. Only male mice were used for the experiments and litter- mates were used as controls. The genotypes of the PHD1/2/3flox/flox mice were determined with PCR using tail clip DNA as described previously (42). For in vivo PHD inhibition studies, male and female homozygous FVB.129S6-Gt(ROSA)26Sortm1(HIF-1A/luc)Kael/J mice were purchased from The Jackson Laboratory and bred in our colony. All rat study protocols followed the Canadian Council of Animal Care Guidelines and were approved by the Biosciences Animal Care and Use Committee at the University of Alberta. Forty-four male Sprague-Dawley rats (~300 to 450 g, ~8 to 12 weeks old) from the University of Alberta breeding colony were obtained and housed in a temperature- and humidity-controlled room (lights on at 7 a.m., off at 7 p.m.). They had ad libitum access to water and food (Purina rodent chow), except as noted below. Rats were group-housed before surgery and individually after surgery. After acclimatizing to the vivarium, rats were placed on food restriction to limit body weight to 90% of their free-feeding level adjusted for natural gains with age.
Stereotaxic administration of adeno-associated viral vectors into the striatum
To delete PHD1, PHD2, and PHD3 in the striatum specifically by AAV8-Cre-mediated recombination, AAV8-Cre (Vector Biolabs) was sterotaxically injected in the striatum of male PHD1/2/3flox/flox mice. For each mouse, 1.5 ml of AAV8-Cre [1 × 1013 genomic copies (GCs)] was injected into the right striatum at a flow rate of 0.120 ml/min using a nanomite syringe pump (Harvard Apparatus) through a Hamilton syringe. By contrast, control mice received 1.5-ml injections (1 × 1013 GCs) of AAV8-eGFP (Vector Biolabs). All mice received two injections at the following stereotaxic coordinates (all positions are relative to bregma): injection 1: posterior, 0.10; lateral, −0.15; dorsoventral, −0.35; injection 2: posterior, 0.03; lateral, −0.25; dorsoventral, −0.33. The needle was left in place for 5 min after the injection was complete and withdrawn at a rate of 1 mm/min. All mice injected with either AAV8-Cre or AAV8-GFP were subjected to collagenase-induced ICH 14 days later (following the ICH procedure described below). To confirm that the stereotaxic injections enabled Cre-mediated recombination specifically in the striatum, AAV8-Cre was injected into a mouse strain carrying a tdTomato reporter, B6.Cg-Gt(ROSA) 26Sortm9(CAG-tdTomato), using dual injections at the same stereotaxic coordinates used for PHD1/2/3 deletion. Proper postoperative care was taken until the animals recovered completely.
In vivo bioluminescence imaging
For in vivo PHD inhibition studies, male and female homozygous FVB.129S6-Gt(ROSA)26Sortm1(HIF-1A/luc)Kael/J mice (The Jackson Laboratory) were bred in our colony. The male FVB.129S6-Gt(ROSA)26Sortm1(HIF-1A/luc)Kael/J mice (21) were placed in the In Vivo Imaging System (IVIS; PerkinElmer) induction chamber and anesthetized with isoflurane (3 to 4% with an oxygen flow of 1 liter/min). The mice were individually removed from the induction chamber and given an intraperitoneal injection of luciferin (150 mg/kg; Promega) suspended in sterile saline (Invitrogen). After a 10-min incubation period, the mice were placed on the imaging platform of the IVIS Spectrum imaging station supplied with isoflurane at 1.5% with an oxygen flow of 1 liter/min during the imaging procedure. White light and luciferase activity images were obtained at 30-s intervals for 5 min. After imaging, the mice were removed from the imaging stage and were allowed to recover in a heated cage. Images were analyzed to quantify luminescence in either the brain or liver using Living Image software (PerkinElmer).
Collagenase-induced mouse model of ICH
Male C57BL/6 mice (8 to 10 weeks of age; Harlan Laboratories) or PHD1/2/3flox/flox mice (10 to 12 weeks of age) were anesthetized with isoflurane (2 to 5%) and placed on a stereotaxic frame. During the procedure, the animal’s body temperature was maintained at 37°C with a homeothermic blanket. With a nanomite syringe pump (Harvard Apparatus) and a Hamilton syringe, 1 ml of collagenase (0.075 IU; Sigma) was infused into the right striatum at a flow rate of 0.120 ml/min. Relative to the bregma point, the stereotaxic coordinates of the injection were as follows: lateral, −0.20; anteroposterior, 0.62; and dorsoventral, −0.40. In control animals, 1 ml of saline was infused. The treatment group received adaptaquin (30 mg/kg intraperitoneally) once a day for 7 days starting at 2 hours after collagenase infusion. The control groups received vehicle alone. The animals were randomized to sham or ICH groups. The identity of the mice that received vehicle or adaptaquin was masked to surgeons who performed the ICH. The identity was revealed after the data collected. Proper postoperative care was taken until the animals recovered completely (43–49).
Autologous blood-induced ICH rat model
Adult male Sprague-Dawley rats were anesthetized with isoflurane (at about 2% in a 60:40 mixture of N2O/O2). Anesthetized rats were placed in a stereotaxic frame, and their core temperature was kept at 37°C. A hole was drilled in the skull at the level of bregma point and 3.5 mm lateral on the side contralateral to the preferred paw in the reaching task (see below). A 1-ml syringe with a 26-gauge needle containing blood drawn from the tail artery was lowered 6.5 mm below the surface of the skull, and 100 ml of autologous blood was infused over 10 min into the striatum. The needle was slowly withdrawn 5 min after the infusion was completed. Treatment with adaptaquin (30 mg/kg intraperitoneally once a day for 7 days) commenced at 2 hours after the autologous infusion was completed. Rats were housed in groups before surgery but housed individually after surgery and placed on food restriction to limit their body weight to 90% of their free-feeding level adjusted for natural gains with age.
Behavioral analysis
The corner task assessed the integrated sensorimotor function in both stimulation of vibrissae (sensory neglect) and rearing (motor response) (50). Mice were placed between two cardboard pieces forming a corner with a 30° angle. While maintaining the 30° angle, the boards were gradually moved toward the mouse until the mouse approached the corner, reared upward, and turned 180° to face the open end. The direction (left or right) in which the mouse turned around was recorded for each trial. Ten trials were performed for each mouse. The adhesive tape removal task in mice was performed as previously described. Briefly, adhesive tape was placed on the planter region of the forward paw (right and left) of mice. The time from which the tape was applied to when the mouse successfully removed it was recorded for each paw. A maximum of 300 s for each paw was allowed.
Single-pellet reaching task
Sprague-Dawley rats were trained over 4 weeks in the single-pellet reaching task (60 cm long × 14 cm wide × 35 cm high), where they were placed on a shelf in front of a narrow vertical slot (1 cm wide) to obtain food reward pellets (45 mg; Bio-Serv). The dominant forelimb was determined during the first training day, and the rats were then trained to reach with that dominant limb (20 trials per day, 5 days per week, 4 weeks). The percentage success was determined, and rats that did not obtain at least 40% success over the last 5 days of training were excluded from the data set analysis. At the end of training, the rats were returned to ad libitum feeding.
Primary cortical neuronal cultures
Primary cortical neurons were obtained from either embryonic (E15) CD1 mice or embryonic (E17) Sprague-Dawley rats. Briefly, cortices were dissected, homogenized, and plated in minimum essential medium containing 10% fetal bovine serum (FBS), 5% horse serum, and 1% penicillin/streptomycin in 96-well plates, 6-well plates, or 10-cm dishes. Neurons were maintained at 37°C with 5% CO2. All experiments were started at 24 hours after plating.
Cultured cell lines
HT22 murine hippocampal cells were cultured at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM), 10% FBS, and puromycin (4 mg/ml; Sigma). Striatal Q7 cells were maintained at 33°C with 5% CO2 in DMEM containing 25 mM D-glucose, 1 mM L-glutamine, 10% FBS, 1 mM sodium pyruvate, and Geneticin (400 mg/ml; Invitrogen).
In vitro ICH model
Cell death was induced in primary cortical neurons, hippocampal HT22 cells, and striatal Q7 cells by treatment with hemin (10 to 250 mM; Sigma). For the neuroprotection studies, cells were treated with 50 mM hemin in the presence of DFO (10 to 200 mM), CPO (0.5 to 4 mM), DHB (5 to 100 mM), the CPO analog 5342 (0.5 to 4 mM), and adaptaquin (0.25 to 10 mM). Cell viability was analyzed 24 hours after treatment. Cells were rinsed with warm phosphate-buffered saline (PBS) and assessed by MTT assay. The fidelity of MTT assays in measuring viability was verified by calcein-AM/ethidium homodimer-1 staining (Live/Dead assay, Molecular Probes) following the manufacturer’s instructions.
In vitro model of oxidative stress–induced neuronal death Primary cortical neurons, HT22 cells, and striatal Q7 cells were exposed to 5 mM glutamate or its structural analog, HCA (5 mM), to induce oxidative stress—induced cell death. Twenty-four hours after treatment with either glutamate or HCA, the cells were rinsed with warm PBS, and cell viability was assessed by MTT assay (Life Technologies) and calcein-AM/ethidium homodimer-1 staining (Live/Dead assay, Molecular Probes).
Microarray analysis
Total RNA was extracted from cultured embryonic (E15) CD1 mouse primary cortical neurons treated with HCA (5 mM) and either protective or nonprotective doses of adaptaquin. Three replicates were run for each treatment group. RNA quantity was assessed using a NanoDrop spectrophotometer (NanoDrop Technologies), and quality was assessed with the Agilent Bioanalyzer (Agilent Technologies). Total RNA (200 ng) was amplified, biotinylated, and hybridized on Illumina microarray chips. Slides were scanned using the Illumina BeadStation, and the signal was extracted using Illumina BeadStudio software. Raw data were analyzed using Bioconductor packages. Data quality was assessed by inspection of the interarray Pearson’s correlation. Overall data coherence was determined by examining clustering based on top variant genes. Contrast analysis of differential expression was performed using the LIMMA package. After linear model fitting, a Bayesian estimate of differential expression was calculated, and the threshold for statistical significance was set at P = 0.005. Data analysis was aimed at assessing the effect of drug treatment on control cells and cells treated with HCA. GO analysis was performed using DAVID (Database for Annotation, Visualization, and Integrated Discovery). Pathway analysis was performed by using the functional analysis annotation tool in the Ingenuity pathways analysis software (Ingenuity Systems).
Adenoviral overexpression
Constructions of expression plasmids for wild-type ATF4 and the dominant-negative ATF4DRK proteins (provided by J. Alam, Ochsner Foundation, New Orleans, LA) have been previously described. The expression plasmid for the mutant ATF4-5P/A protein (in which proline residues 156, 162, 164, 167, and 174 were changed to alanine) was generated by ViraQuest. The expression cassette for each ATF4 protein construct was cloned into the ad5 pVQ-K-NpA shuttle vector to generate the adenoviruses. ViraQuest performed virus generation, amplification, and sequencing. Primary neuronal cultures were transduced with adenoviruses at a multiplicity of infection of 80 in serum-free medium for 1 hour. Then, the medium was restored and cotreated with adaptaquin. Twelve hours after treatment, cell survival was determined by MTT assay and Live/Dead assay (Invitrogen).
Immunoprecipitation assays
Nuclear and cytoplasmic proteins were obtained using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology) in the presence of the protease inhibitors phenylmethylsulfonyl fluoride (PMSF), N-carbobenzyloxy-L-leucyl-L-leucyl-L-leucinal (MG132), and sodium orthovanadate according to the manufacturer’s protocol. Nuclear proteins (200 mg) were diluted in 50 mM tris-HCl (pH 7.4) in the presence of protease inhibitors (Sigma). Proteins were then precleared with agarose beads (Santa Cruz Biotechnology) for 30 min and were immunoprecipitated with 5 mg of rabbit ATF4 antibody (ABE387, Millipore) overnight.
Immunoblot analyses
Nuclear and cytoplasmic proteins were obtained using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology) in the presence of the protease inhibitors PMSF, MG132, and sodium orthovanadate according to the manufacturer’s protocol. For determination of acetyl histone H4 protein levels, cells were lysed in RIPA-B [1% Triton X-100, 1% SDS, 50 mM tris-Cl (pH 7.4), 500 mM NaCl, and 1 mM EDTA] buffer before being diluted with Laemmli buffer and boiled. All samples were electrophoresed under reducing conditions on NuPAGE Novex 4–12% bis-tris polyacrylamide gels (Invitrogen) before being transferred onto a nitrocellulose membrane (Bio-Rad). For detection of specific proteins, the following primary antibodies and dilutions were used: ATF4 (L. A. Greene, Columbia University; 1:1000), HIF- 1a (NB100-105, Novus Biologicals; 1:1000), Trib3 (ST1032, Millipore; 1:1000), histone H4 (05-858R, Millipore; 1:2000), acetyl histone H4 (06-866, Millipore; 1:2000), H3K4me2 (A300-060A, Bethyl Laboratories; 1:1000), hydroxy proline (3434S, Cell Signaling; 1:1,000), and b-actin (AC-74, Sigma-Aldrich; 1:10,000). Primary antibodies were visualized by 1-hour incubation at room temperature with either IRDye- 680 or IRDye-800 secondary antibodies (LI-COR Biosciences; 1: 10,000) and were used at 1:10,000 dilution. All proteins were detected using an Odyssey infrared imaging system (LI-COR Biosciences).
Quantitative real-time PCR
Total RNA was prepared using the NucleoSpin RNA II kit (MACHEREY- NAGEL) according to the manufacturer’s protocol. Duplex real-time PCR reactions were performed with gene expression assays using 6- carboxyfluorescein-labeled probes (Applied Biosystems) for EPO (Mm 00433126_m1), VEGF (Mm 01281449_m1), p21waf1/ap1 (Mm 00432448_m1), Trib3 (Mm 00454879_m1), MTHFD2 (Mm 00485276_m1), and STC2 (Mm 00441560_m1). All expression levels were normalized to b-actin gene expression levels, which were determined with a VIC-labeled probe (Applied Biosystems). All experiments were performed using a 7500 Real-Time PCR System (Applied Biosystems).
Chromatin immunoprecipitation
The ChIP assays were performed with the EZ-Magna ChIP assay kit (Millipore) following the manufacturer’s instructions. Briefly, primary cortical cells were cross-linked with 1% formaldehyde at 37°C for 7 min and then sonicated using the Bioruptor (Diagenode). Genomic targets were immunoprecipitated with 5 mg of rabbit ATF4 antibody (ABE387, Millipore). Quantitation of immunoprecipitated genomic DNA regions was performed with real-time PCR using the SYBR Green Master Mix (Applied Biosystems) on a 7500 Real-Time PCR System (Applied Biosystems) and the following primers for the Trib3 promoter: 5′-GGCTCGGATCCCCGCCCGGCTGATGTA-3′ and 5′-GAAGGTGAGGGCGGAGCTCACTCGGCGA-3′.
Statistical analyses
Data are reported as means ± SEM of multiple individual experiments each carried out in triplicate. Unless stated otherwise, the statistical analysis was carried out with GraphPad Prism 5. A two-tailed t test was used if two groups were compared, a one-way ANOVA with Bonferroni’s multiple comparisons post hoc test if more than two groups were compared, and a two-way ANOVA with Bonferroni’s post hoc test if two independent variables were compared.
Supplementary Material
Fig. S1. Hemin, the reactive ferric protoporphyrin IX group of hemoglobin, induces concentration-dependent death in multiple cell types.
Fig. S2. CPO or a CPO analog (5342) differentially inhibit HIF-PHD activity, although they have a similar affinity for iron.
Fig. S3. Conditional reduction of HIF-PHD1, HIF-PHD2, and HIF-PHD3 in the striatum does not affect ICH-induced hematoma size or brain edema.
Fig. S4. Canonical HIF-PHD inhibitors DFO, CPO, and DHB do not stabilize ODD-luciferase in brains of mice.
Fig. S5. Adaptaquin binds to and inhibits recombinant PHD2.
Fig. S6. Adaptaquin does not inhibit collagenase activity in vivo.
Fig. S7. Adaptaquin significantly reduced ICH-induced edema.
Fig. S8. Adaptaquin’s beneficial effects on ICH outcomes are not associated with changes in core body temperature.
Fig. S9. Reduction of HIF-1 a or HIF-2 a by RNA interference does not potentiate hemin-induced toxicity or affect the ability of chemically diverse HIF-PHD inhibitors to prevent hemin toxicity.
Fig. S10. Hemin-induced neuronal death is abrogated by the HIF-PHD inhibitor adaptaquin.
Fig. S11. Adaptaquin inhibits glutamate-induced mitochondrial dysfunction.
Fig. S12. Adaptaquin is distinct from the classical antioxidant N-acetylcysteine in abrogating ATF4-mediated death.
Fig. S13. Adaptaquin does not affect global histone acetylation or methylation or inhibit 12- lipoxygenase activities.
Fig. S14. Adaptaquin does not affect 3-nitrotyrosine, a biomarker of oxidative stress.
Fig. S15. Adaptaquin does not affect TET enzyme activity, a subfamily of the iron-, 2-oxoglutarate-, and oxygen-dependent dioxygenases that demethylate DNA, at concentrations where it completely abrogates neuronal death.
Fig. S16. Adaptaquin protection is not associated with induction of HIF-dependent genes VEGF and EPO in the striatum of mice.
Table S1. Inability of adaptaquin to influence likely off-target enzymes at concentrations where it fully protects neurons.
Acknowledgments
We thank H. Aleyasin, M. Basso, N. Smirnova, T. Ma, H. Pinto, D. Willis, T. Holland, and F. Gao for technical assistance. We thank M. Shelanski and L. A. Greene (Columbia University) for the gift of the ATF4 antibody, and M. MacDonald for the gift of the Q7 cell line. We thank A.-H. Lee and T. McGraw (Cornell), H. Kornblum and T. Carmichael (Univesity of California, Los Angeles), and E. Lo and K. van Leyen (Massachusetts General Hospital) for the constructive comments on the manuscript. Portions of this work were performed at Beamline X27A, National Synchrotron Light Source (NSLS), Brookhaven National Laboratory. X27A is supported in part by the Geosciences Division of the U.S. Department of Energy (DE-FG02-92ER14244 to the Center for Advanced Radiation Sources of the University of Chicago) and the Department of Environmental Sciences of Brookhaven National Laboratory. The use of the NSLS was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-98CH10886. Funding: This work was funded with support from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the Burke Foundation, the Sperling Center for Hemorrhagic Stroke Recovery at the Burke Medical Research Institute, New York State Department of Health Center for Research Excellence in Spinal Cord Injury (grant DOHC019772), NIH (grant P01 NIA AG014930, project 1, to R.R.R.). G.C. thanks the National Institute of Neurological Disorders and Stroke Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691). C.J.S. thanks the Welcome Trust and the British Heart Foundation for funding Cancer Research, UK. C.C. thanks the Neuroscience PhD program of the University of Marburg. Author contributions: S.S.K. and R.R.R. conceived and designed the research. S.S.K. performed most experiments with the help of I.A., S.J.K., M.W.B., S.F.S., and I.G. R.J. and F.C. carried out and analyzed studies with the in vivo rat autologous blood model of ICH. C.C.T., T.-L.Y., M.D., and C.J.S. performed and analyzed the mass spectrometry-based hydroxylation assays, AlphaScreen, and nondenaturing mass spectrometric studies. M.W.B. and R. V.T. carried out and analyzed metal concentrations by x-ray fluorescence microscopy. D.W.K. measured metal concentrations by ICP-OES. S.N. and C.C. carried out and analyzed the mitochondrial assays. D.C. and F.R.M. carried out and analyzed the macrophage assay. G.X., T.S., R.F.K., and LM. helped with the in vivo ICH models and behavioral outcomes. T.R.H. carried out and analyzed the lipoxygenase assays. G.-H.F. and J.Z. provided the PHD1flox/flox, PHD2flox/flox, and tdTomato reporter mice. G.C. performed microarray analysis and analyzed the data. J.W.C. and S.C. helped with the preparation of the figures and manuscript. R.R.R. wrote the manuscript with help from S.S.K. All authors discussed the results and commented on the manuscript. Competing interests: Cornell University owns and has licensed the patent on the use of adaptaquin described in this study entitled “Prolylhydroxylase/ATF4 inhibitors for treating neural cell injury’ (US2014/070052). R.R.R., S. S.K., and I.G. are named as co-inventors. C.J.S. was a cofounder of ReOX, a company that seeks to exploit knowledge of the hypoxia response for therapeutic benefit. Data and materials availability: Microarray data have been deposited in the National Center for Biotechnology Information and are available at the Gene Expression Omnibus under accession no. GSE75048.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/8/328/328ra29/DC1
Materials and Methods
REFERENCES AND NOTES
- 1.Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurol ME, Greenberg SM. Management of intracerebral hemorrhage. Curr Atheroscler Rep. 2008;10:324–331. doi: 10.1007/s11883-008-0050-y. [DOI] [PubMed] [Google Scholar]
- 3.Greenough MA, Camakaris J, Bush AI. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int. 2013;62:540–555. doi: 10.1016/j.neuint.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 4.Andersen HH, Johnsen KB, Moos T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell Mol Life Sci. 2014;71:1607–1622. doi: 10.1007/s00018-013-1509-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warkentin LM, Auriat AM, Wowk S, Colbourne F. Failure of deferoxamine, an iron chelator, to improve outcome after collagenase-induced intracerebral hemorrhage in rats. Brain Res. 2010;1309:95–103. doi: 10.1016/j.brainres.2009.10.058. [DOI] [PubMed] [Google Scholar]
- 6.Lee DL, Strathmann FG, Gelein R, Walton J, Mayer-Proschel M. Iron deficiency disrupts axon maturation of the developing auditory nerve. J Neurosci. 2012;32:5010–5015. doi: 10.1523/JNEUROSCI.0526-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karuppagounder SS, Ratan RR. Hypoxia-inducible factor prolyl hydroxylase inhibition: Robust new target or another big bust for stroke therapeutics? J Cereb Blood Flow Metab. 2012;32:1347–1361. doi: 10.1038/jcbfm.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Youdim MBH, Kupershmidt L, Amit T, Weinreb O. Promises of novel multi-target neuroprotective and neurorestorative drugs for Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(Suppl 1):S132–S136. doi: 10.1016/S1353-8020(13)70032-4. [DOI] [PubMed] [Google Scholar]
- 9.Ozer A, Bruick RK. Non-heme dioxygenases: Cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3:144–153. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- 10.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, Patton SM, Connor JR, Cherny RA, Volitakis I, Bush AI, Langsetmo I, Seeley T, Gunzler V, Ratan RR. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 2009;29:8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunze R, Zhou W, Veltkamp R, Wielockx B, Breier G, Marti HH. Neuron-specific prolyl-4- hydroxylase domain 2 knockout reduces brain injury after transient cerebral ischemia. Stroke. 2012;43:2748–2756. doi: 10.1161/STROKEAHA.112.669598. [DOI] [PubMed] [Google Scholar]
- 14.Chen RL, Nagel S, Papadakis M, Bishop T, Pollard P, Ratcliffe PJ, Pugh CW, Buchan AM. Roles of individual prolyl-4-hydroxylase isoforms in the first 24 hours following transient focal cerebral ischaemia: Insights from genetically modified mice. J Physiol. 2012;590:4079–4091. doi: 10.1113/jphysiol.2012.232884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dang TN, Robinson SR, Dringen R, Bishop GM. Uptake, metabolism and toxicity of hemin in cultured neurons. Neurochem Intl. 2011;58:804–811. doi: 10.1016/j.neuint.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Chen-Roetling J, Cai Y, Lu X, Regan RF. Hemin uptake and release by neurons and glia. Free Rad Res. 2014;48:200–205. doi: 10.3109/10715762.2013.859386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higdon AN, Benavides GA, Chacko BK, Ouyang X, Johnson MS, Landar A, Zhang J, Darley-Usmar VM. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am J Physiol Heart Circ Physiol. 2012;302:H1394–H1409. doi: 10.1152/ajpheart.00584.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aragonés J, Fraisl P, Baes M, Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell Metab. 2009;9:11–22. doi: 10.1016/j.cmet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Minamishima YA, Kaelin WG., Jr Reactivation of hepatic EPO synthesis in mice after PHD loss. Science. 2010;329:407. doi: 10.1126/science.1192811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chavez JC, Baranova O, Lin J, Pichiule P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J Neurosci. 2006;26:9471–9481. doi: 10.1523/JNEUROSCI.2838-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saftan M, Kim WY, O’Connell F, Flippin L, Günzler V, Horner JW, DePinho RA, Kaelin WG., Jr Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: Assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci USA. 2006;103:105–110. doi: 10.1073/pnas.0509459103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smirnova NA, Rakhman I, Moroz N, Basso M, Payappilly J, Kazakov S, Hernandez-Guzman F, Gaisina IN, Kozikowski AP, Ratan RR, Gazaryan IG. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem Biol. 2010;17:380–391. doi: 10.1016/j.chembiol.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hua Y, Schallert T, Keep RF, Wu J, Hoff JT, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33:2478–2484. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- 24.MacLellan CL, Paquette R, Colbourne F. A critical appraisal of experimental intracerebral hemorrhage research. J Cereb Blood Flow Metab. 2012;32:612–627. doi: 10.1038/jcbfm.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Semenza GL. HIF-1, O2, and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 26.Aminova LR, Chavez JC, Lee J, Ryu H, Kung A, LaManna JC, Ratan RR. Prosurvival and prodeath effects of hypoxia-inducible factor-1a stabilization in a murine hippocampal cell line. J Biol Chem. 2005;280:3996–4003. doi: 10.1074/jbc.M409223200. [DOI] [PubMed] [Google Scholar]
- 27.Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117:e207–e217. doi: 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 30.Zareen N, Biswas SC, Greene LA. A feed-forward loop involving Trib3, Akt and FoxO mediates death of NGF-deprived neurons. Cell Death Differ. 2013;20:1719–1730. doi: 10.1038/cdd.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, Mayes PA, Wise DR, Thompson CB, Maris JM, Hogarty MD, Simon MC. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell. 2012;22:631–644. doi: 10.1016/j.ccr.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liew CW, Bochenski J, Kawamori D, Hu J, Leech CA, Wanic K, Malecki M, Warram JH, Qi L, Krolewski AS, Kulkarni RN. The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse b cells. J Clin Invest. 2010;120:2876–2888. doi: 10.1172/JCI36849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Köditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, Myllyharju J, Wenger RH, Katschinski DM. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–3617. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- 34.Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci USA. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Leyen K, Holman TR, Maloney DJ. The potential of 12/15-lipoxygenase inhibitors in stroke therapy. Future Med Chem. 2014;6:1853–1855. doi: 10.4155/fmc.14.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Ab. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 37.Tardiff DF, Tucci ML, Caldwell KA, Caldwell GA, Lindquist S. Different 8-hydroxyquinolines protect models of TDP-43 protein, a-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J Biol Chem. 2012;287:4107–4120. doi: 10.1074/jbc.M111.308668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H, Ingelsson M, Masters CL, Tanzi RE, Cummings JL, Herd CM, Bush AI. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J Alz Dis. 2010;20:509–516. doi: 10.3233/JAD-2010-1390. [DOI] [PubMed] [Google Scholar]
- 39.Wong BW, Kuchnio A, Bruning U, Carmeliet P. Emerging novel functions of the oxygen-sensing prolyl hydroxylase domain enzymes. Trends Biochem Sci. 2013;38:3–11. doi: 10.1016/j.tibs.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, Crystal RG, Darnell RB, Ferrante RJ, Fillit H, Finkelstein R, Fisher M, Gendelman HE, Golub RM, Goudreau JL, Gross RA, Gubilz AK, Hesterlee SE, Howells DW, Huguenard J, Kelner K, Koroshetz W, Krainc D, Lazic SE, Levine MS, Macleod MR, McCall JM, Moxley RT, III, Narasimhan K, Noble LJ, Perrin S, Porter JD, Steward O, Unger E, Utz U, Silberberg SD. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–191. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor a levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rynkowski MA, Kim GH, Komotar RJ, Otten ML, Ducruet AF, Zacharia BE, Kellner CP, Hahn DK, Merkow MB, Garrett MC, Starke RM, Cho BM, Sosunov SA, Connolly ES. A mouse model of intracerebral hemorrhage using autologous blood infusion. Nat Protoc. 2008;3:122–128. doi: 10.1038/nprot.2007.513. [DOI] [PubMed] [Google Scholar]
- 44.Grossetete M, Rosenberg GA. Tissue inhibitor of matrix metalloproteinases-3 (TIMP-3) lacks involvement in bacterial collagenase-induced intracerebral hemorrhage in mouse. Acta Neurochir Suppl. 2008;105:89–93. doi: 10.1007/978-3-211-09469-3_18. [DOI] [PubMed] [Google Scholar]
- 45.Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Rad Biol Med. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Zhuang H, Doré S. Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiol Dis. 2006;22:473–476. doi: 10.1016/j.nbd.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 48.Krafft PR, McBnde DW, Lekrc T, Rolland WB, Mansell CE, Ma Q, Tang J, Zhang JH. Correlation between subacute sensorimotor deficits and brain edema in two mouse models of intracerebral hemorrhage. Behav Brain Res. 2014;264:151–160. doi: 10.1016/j.bbr.2014.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC, Aronowski J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007;38:3280–3286. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- 50.Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- 51.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IKH, Li XS, Woon ECY, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TDW, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron- specific inactivation of the hypoxia inducible factor 1a increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Hemin, the reactive ferric protoporphyrin IX group of hemoglobin, induces concentration-dependent death in multiple cell types.
Fig. S2. CPO or a CPO analog (5342) differentially inhibit HIF-PHD activity, although they have a similar affinity for iron.
Fig. S3. Conditional reduction of HIF-PHD1, HIF-PHD2, and HIF-PHD3 in the striatum does not affect ICH-induced hematoma size or brain edema.
Fig. S4. Canonical HIF-PHD inhibitors DFO, CPO, and DHB do not stabilize ODD-luciferase in brains of mice.
Fig. S5. Adaptaquin binds to and inhibits recombinant PHD2.
Fig. S6. Adaptaquin does not inhibit collagenase activity in vivo.
Fig. S7. Adaptaquin significantly reduced ICH-induced edema.
Fig. S8. Adaptaquin’s beneficial effects on ICH outcomes are not associated with changes in core body temperature.
Fig. S9. Reduction of HIF-1 a or HIF-2 a by RNA interference does not potentiate hemin-induced toxicity or affect the ability of chemically diverse HIF-PHD inhibitors to prevent hemin toxicity.
Fig. S10. Hemin-induced neuronal death is abrogated by the HIF-PHD inhibitor adaptaquin.
Fig. S11. Adaptaquin inhibits glutamate-induced mitochondrial dysfunction.
Fig. S12. Adaptaquin is distinct from the classical antioxidant N-acetylcysteine in abrogating ATF4-mediated death.
Fig. S13. Adaptaquin does not affect global histone acetylation or methylation or inhibit 12- lipoxygenase activities.
Fig. S14. Adaptaquin does not affect 3-nitrotyrosine, a biomarker of oxidative stress.
Fig. S15. Adaptaquin does not affect TET enzyme activity, a subfamily of the iron-, 2-oxoglutarate-, and oxygen-dependent dioxygenases that demethylate DNA, at concentrations where it completely abrogates neuronal death.
Fig. S16. Adaptaquin protection is not associated with induction of HIF-dependent genes VEGF and EPO in the striatum of mice.
Table S1. Inability of adaptaquin to influence likely off-target enzymes at concentrations where it fully protects neurons.