

Abstract
Background
Enterococcus is ubiquitous in nature and is a commensal of both the bovine and human gastrointestinal (GI) tract. It is also associated with clinical infections in humans. Subtherapeutic administration of antibiotics to cattle selects for antibiotic resistant enterococci in the bovine GI tract. Antibiotic resistance genes (ARGs) may be present in enterococci following antibiotic use in cattle. If located on mobile genetic elements (MGEs) their dissemination between Enterococcus species and to pathogenic bacteria may be promoted, reducing the efficacy of antibiotics.
Results
We present a comparative genomic analysis of twenty-one Enterococcus spp. isolated from bovine feces including Enterococcus hirae (n = 10), Enterococcus faecium (n = 3), Enterococcus villorum (n = 2), Enterococcus casseliflavus (n = 2), Enterococcus faecalis (n = 1), Enterococcus durans (n = 1), Enterococcus gallinarum (n = 1) and Enterococcus thailandicus (n = 1). The analysis revealed E. faecium and E. faecalis from bovine feces share features with human clinical isolates, including virulence factors. The Tn917 transposon conferring macrolide-lincosamide-streptogramin B resistance was identified in both E. faecium and E. hirae, suggesting dissemination of ARGs on MGEs may occur in the bovine GI tract. An E. faecium isolate was also identified with two integrative conjugative elements (ICEs) belonging to the Tn916 family of ICE, Tn916 and Tn5801, both conferring tetracycline resistance.
Conclusions
This study confirms the presence of enterococci in the bovine GI tract possessing ARGs on MGEs, but the predominant species in cattle, E. hirae is not commonly associated with infections in humans. Analysis using additional complete genomes of E. faecium from the NCBI database demonstrated differential clustering of commensal and clinical isolates, suggesting that these strains may be specifically adapted to their respective environments.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-017-0962-1) contains supplementary material, which is available to authorized users.
Keywords: Bovine feces, Enterococcus, Comparative genomics
Background
The genus Enterococcus is ubiquitous in nature and can be found in a range of habitats, being associated with soil, plants, fresh and salt water, sewage and the gastrointestinal (GI) tract of animals (including mammals, birds, fish, reptiles and insects) and humans [1]. Although typically a commensal of the human GI tract, enterococci are often associated with a variety of clinical infections including urinary tract infections, hepatobiliary sepsis, endocarditis, surgical wound infections, bacteraemia and neonatal sepsis [2, 3]. Enterococcus faecalis and Enterococcus faecium are the two species responsible for the majority of healthcare-associated enterococcal infections [4]. Difficulties in treating enterococcal infections have emerged due to their ability to readily acquire resistance to many antibiotics, most notably vancomycin. As a result, the ability to successfully treat clinical infections has been reduced [5].
Antibiotic use in livestock production has been correlated with the emergence of antibiotic resistant bacteria. This was first recognised in the 1990s when use of the glycopeptide avoparcin as a subtherapeutic growth promotant led to the emergence of glycopeptide-resistant E. faecium in livestock and poultry [6]. Consumption of meat products contaminated with resistant bacteria was suggested to lead to the transmission of glycopeptide-resistant E. faecium to healthy, non-hospitalised humans. This association demonstrated the transmission of resistant bacteria from animals to humans through the food chain [7, 8]. Consequently, avoparcin was banned as a growth promotant in Europe in 1997 [9]. However, many antibiotics continue to be administered subtherapeutically to livestock in North America. For example, tylosin phosphate, a member of the macrolide family, is administered subtherapeutically to cattle to control liver abscesses. We recently demonstrated that subtherapeutic administration of tylosin phosphate selected for macrolide resistant enterococci in the bovine GI tract [10]. Enterococci have the ability to transfer antibiotic resistance and virulence genes horizontally to other bacteria [11]. The creation of a reservoir of resistant enterococci in the bovine GI tract could promote the dissemination of antibiotic resistance genes (ARGs) to other bacteria, particularly if they are associated with mobile genetic elements (MGEs).
Comparative genomic analysis can be used to identify genes coding for virulence, antibiotic resistance and gene mobility as well as elucidate the evolutionary relationship among bacteria. The number of complete or draft genome sequences available for E. faecalis and E. faecium is 446 and 436, respectively, comprising the bulk of enterococcal genome sequences available (http://www.ncbi.nlm.nih.gov/genome), as several comparative genomic studies of these species have been conducted [12–14]. There are comparatively few draft genome sequences available for other Enterococcus spp. with only 11, 10, 6, 5, 2 and 1 genomes are available for Enterococcus casseliflavus, Enterococcus hirae, Enterococcus durans, Enterococcus gallinarum, Enterococcus villorum and Enterococcus thailandicus, respectively (http://www.ncbi.nlm.nih.gov/genome). Furthermore, there is a poor representation of genomic sequences for enterococci isolated from non-human sources [15].
Previously, we identified a number of enterococci from bovine feces that carried at least one ARG, but only a few isolates carried multiple ARGs [10]. We also identified E. hirae as the principle species of the bovine GI tract, with infrequent isolation of E. faecium and E. faecalis, the species associated with nosocomial infections in humans. In the current study, we selected twenty-one isolates of enterococci originating from bovine feces for whole-genome sequencing and comparative genomic analysis. We hypothesized that E. faecium and E. faecalis would present more genes coding for virulence and antibiotic resistance than other Enterococcus spp. isolated from bovine feces.
Methods
Isolate selection
Twenty-one Enterococcus spp. isolated from bovine feces including E. hirae (n = 10), E. faecium (n = 3), E. villorum (n = 2), E. casseliflavus (n = 2), E. faecalis (n = 1), E. durans (n = 1), E. gallinarum (n = 1) and E. thailandicus (n = 1) were selected for whole genome sequencing (Table 1). These were selected from an archive of isolates collected between 2004 and 2005, which were previously characterized by PFGE and antimicrobial susceptibility testing [10]. At least one representative of each species isolated from bovine feces was selected, and for E. hirae and E. faecium, selection was based on maximizing diversity as measured by PFGE profiles as well as selecting isolates that displayed unique antimicrobial resistance profiles.
Table 1.
Genome characteristics of Enterococcus spp. isolated from bovine feces
| Strain | No. contigs | Size (bp) | %GC | Genes | CDSs | STa |
|---|---|---|---|---|---|---|
| E. hirae 1 | 32 | 2926392 | 36.7 | 2785 | 2712 | – |
| E. hirae 2 | 29 | 2850950 | 36.7 | 2678 | 2631 | – |
| E. hirae 3 | 81 | 3088947 | 36.6 | 2977 | 2906 | – |
| E. hirae 4 | 28 | 3042973 | 36.7 | 2825 | 2753 | – |
| E. hirae 5 | 28 | 2869170 | 36.8 | 2741 | 2670 | – |
| E. hirae 6 | 62 | 2966815 | 36.6 | 2848 | 2777 | – |
| E. hirae 7 | 235 | 2766361 | 37.0 | 2602 | 2535 | – |
| E. hirae 8 | 47 | 2922437 | 36.7 | 2801 | 2730 | – |
| E. hirae 9 | 47 | 3178271 | 36.6 | 2971 | 2899 | – |
| E. hirae 10 | 71 | 3018341 | 36.6 | 2885 | 2814 | – |
| E. faecium 11 | 111 | 2783595 | 37.9 | 2719 | 2648 | 214 |
| E. faecium 12 | 182 | 2712126 | 38.3 | 2665 | 2597 | Unknown |
| E. faecium 13 | 28 | 2772865 | 37.7 | 2659 | 2591 | 955 |
| E. thailandicus 14 | 17 | 2603791 | 36.7 | 2495 | 2430 | – |
| E. villorum 15 | 42 | 2994157 | 34.9 | 2834 | 2765 | – |
| E. villorum 16 | 159 | 3056754 | 34.9 | 2907 | 2837 | – |
| E. faecalis 17 | 34 | 2913318 | 37.3 | 2788 | 2729 | 242 |
| E. gallinarum 18 | 41 | 3381991 | 40.5 | 3259 | 3197 | – |
| E. durans 19 | 43 | 2931269 | 37.9 | 2723 | 2657 | – |
| E. casseliflavus 20 | 85 | 3483586 | 42.6 | 3355 | 3295 | – |
| E. casseliflavus 21 | 50 | 3639801 | 42.2 | 3436 | 3375 | – |
a ST sequence type
DNA extraction and sequencing
Genomic DNA was isolated using phenol:chloroform extraction. Enterococcus spp. were inoculated into 5 mL brain heart infusion (BHI; BD, Franklin Lakes, New Jersey, USA) broth and grown for 24 h in a shaking incubator (250 rpm; Excella E24 Incubator Shaker, New Brunswick Scientific) at 37 °C. To increase cell yield, 150 μL aliquots were inoculated into duplicate tubes containing 6 mL BHI (BD) and grown for 24 h as described above. Cells were harvested by centrifugation at 10,000 × g for 5 min into a 2 mL microfuge tube and stored at −20 °C until genomic DNA was extracted. For extraction, the pellet was thawed on ice and resuspended in 1 mL of sterile 0.85% NaCl to remove residual growth media. The cells were repelleted by centrifugation (10,000 × g) for 1 min and the supernatant decanted. The washed cell pellet was resuspended in 665 μL of T10E25 (10 mM Tris-HCl pH 7.5; 25 mM EDTA) and 35 μL of lysozyme (50 mg/mL; Sigma-Aldrich, Co., St. Louis, Mo, USA) was added. The tubes were incubated at 55 °C for 60 min as a pre-lysis step. A 175 μL of 5 M NaCl, 35 μL of proteinase K (10 mg/mL; Sigma-Aldrich) and 44 μL of 20% SDS were added to the suspension and mixed by gentle inversion before being incubated at 65 °C for 1–2 h until cell lysis was complete. The lysed cells were extracted once with phenol, once with phenol:chloroform:isoamylalcohol (25:24:1) and twice with chloroform. Ammonium acetate (10 M) was added to the mixture so as to achieve a final concentration of 0.5 M, followed by one volume of isopropanol to precipitate DNA. To encourage precipitation, the tubes were chilled on ice for 10 min before centrifuging at 10,000 × g for 10 min. The supernatant was decanted and the DNA pellet washed with 70% ethanol and allowed to air dry before dissolving in 400 μL of TE (10 mM Tris-HCl; 1 mM EDTA). RNase A was added to achieve a final concentration of 30 μg/mL and the mixture was incubated for 20 min at 37 °C. Duplicate solutions for each sample were pooled before performing a second extraction, once with phenol:chloroform:isoamylalcohol and once with chloroform. Ammonium acetate (10 M) was added to the final aqueous solution to achieve a final concentration of 2 M followed by one volume of isopropanol and chilled on ice for 10 min to precipitate DNA. The DNA was pelleted by centrifugation, washed with 70% ethanol, air-dried, dissolved in 100 μL of sterile deionized water and stored at −80 °C until genomic library construction.
Genomic library construction was performed using the Illumina Nextera XT DNA sample preparation kit (Illumina, Inc., CA, USA) following the manufacturer’s instructions and sequenced on an Illumina MiSeq platform (Illumina). High-quality reads were de novo assembled using SPAdes genome assembler version 3.6.0 software [16] and annotated using Prokka version 1.10 [17]. Multi-locus sequence typing (MLST) was performed using the MLST database (version 1.8) [18].
Comparative analysis
Draft genome sequences of the 21 Enterococcus spp. were investigated for the presence of putative virulence genes and ARGs, mobile genetic elements (MGEs), bacteriophage, CRISPR-Cas and secondary metabolite biosynthetic gene clusters. Virulence genes were identified using VirulenceFinder (version 1.5) [19], and ARGs using a combination of ResFinder (version 2.1) [20] and the Comprehensive Antibiotic Resistance Database (CARDs) [21]. Results for ARGs were further verified using megaBLAST and hits were manually inspected. Genomes were investigated for integrative conjugative elements (ICEs) by homology searches using BLAST against 466 ICEs downloaded from the ICEberg database (version 1.0) [22]. To identify bacteriophage, the contigs of each draft genome were ordered based on alignment against a reference genome (see Additional file 1: Table S1) using progressive Mauve [23], and then analyzed for the presence of prophage using PHAST [24]. CRISPR-Cas were identified using the CRISPRdb [25] and secondary metabolite biosynthetic gene clusters using the Antibiotics and Secondary Metabolite Analysis Shell (antiSMASH) [26]. All alignments and BLAST searches were performed in Geneious version 9.0.4 (Biomatters, Ltd). Assignment of proteins into Clusters of Orthologous Groups (COGs) was performed using the Integrated Microbial Genomes (IMG) platform [27]. Blast atlases were generated by GView Java package software [28] using both alignment length and percent identity cut-off values at 80%. The phylogenetic analyses were conducted using a single nucleotide variant phylogenomics (SNVPhyl) pipeline [29]. Briefly the paired-end reads originating from Illumina sequencing of samples were aligned to the reference genome to generate read pileups (SMALT v.0.7.5; http://www.sanger.ac.uk/science/tools/smalt-0) followed by mapping quality filtering and coverage estimations. From the pileup, the variant calling, variant consolidation and single nucleotide variant (SNV) alignment generation of the final phylogeny was run through PhyML [30] using maximum likelihood. The resulting tree was visualized using FigTree software (http://tree.bio.ed.ac.uk/software/figtree/). When included in the analyses, complete genomes from NCBI database were run through Wombac Shred (https://github.com/tseemann/wombac/blob/master/bin/wombac-shred_fasta) or art illumina [31] to generate paired-end reads with 2 × 250 bp length and >30X coverage. The resulting reads along with the reads from experimental isolates and the reference genome were run through SNVPhyl pipeline to obtain phylogeny as described above.
Results and discussion
Sequencing statistics
A summary of the sequencing statistics for the 21 Enterococcus spp. genomes can be found in Table 1. The genomes ranged in size from 2.60 − 3.64 Mb with E. thailandicus exhibiting the smallest and E. casseliflavus the largest genome. There was considerable variation in the size of E. hirae genomes, suggesting large differences in the size of the chromosome between strains and/or the presence/absence of plasmids.
Phylogeny
Phylogenetic trees were constructed based on analysis of single-nucleotide variants (SNVs) of the core genes. A phylogenetic tree was constructed using all 21 sequenced Enterococcus spp. genomes as well as 27 complete enterococci genomes downloaded from the NCBI database (Fig. 1). The 27 compete genomes from NCBI included: E. hirae (2 strains; ATCC 9790, R17), E. faecium (13 strains; Aus0004, Aus0085, T110, 6E6, VRE001, E1, E745, E39, UW8175, NRRL B2354, ATCC 700221, EFE10021), E. faecalis (9 strains; LD33, L12, KB1, 62, D32, V583, DENG1, OG1RF, ATCC 29212), E. durans (1 strain; KLDS6_0933), E. gallinarum (1 strain; FDAARGOS163), and E. casseliflavus (1 strain; EC20). A probiotic strain, Enterococcus faecium T110 was used as an outgroup reference (Fig. 1). The assembled tree was consistent with the PFGE profile dendrogram observed from our previous study [10]. As expected, clustering was observed for genomes of the same species further verifying the identity of each species based on previous groES-EL spacer speciation [10].
Fig. 1.
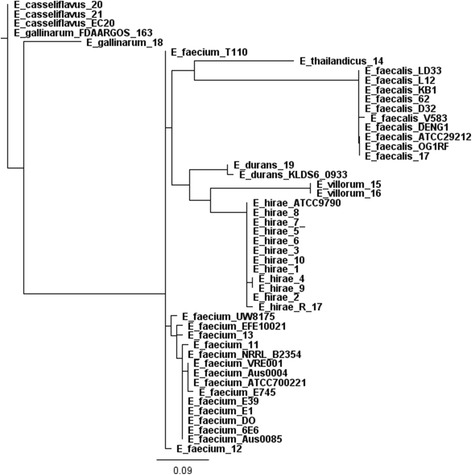
Phylogenetic tree constructed based on analysis of single-nucleotide polymorphisms (SNPs) of the core genes of 48 entercocci genomes, including the 21 isolates obtained from bovine feces in the present study. Entercoccus faecalis, Entercoccus faecium, Enterococcus hirae, Entercoccus durans, Entercoccus casseliflavus and Entercoccus gallinarum were compared using E. faecium strain T110 as a reference
Phylogenetic analyses were also conducted within species for E. hirae, E. faecalis and E. faecium. For E. hirae, as no genome from a clinical isolate was available and the origin of the type strain ATCC 9790 was unknown, E. hirae strain R17 (BioSample SAMN04892752) isolated from retail raw meat was used as an outgroup reference (Fig. 2a). Two distinct clades were identified with the majority of E. hirae constituting one clade and two genomes in the second (E. hirae 4 and E. hirae 9). The only E. faecalis isolate sequenced in this study, E. faecalis_17, clustered closely with a vancomycin resistant human clinical isolate, E. faecalis strain V583 (Fig. 2b). The other strains included in the analysis were of human-clinical, human-, swine- and mouse- commensal and dairy-related origin. Comparative phylogenetic studies with more E. faecalis isolates from bovine sources are required to understand the degree of relatedness between clinical and bovine isolates.
Fig. 2.
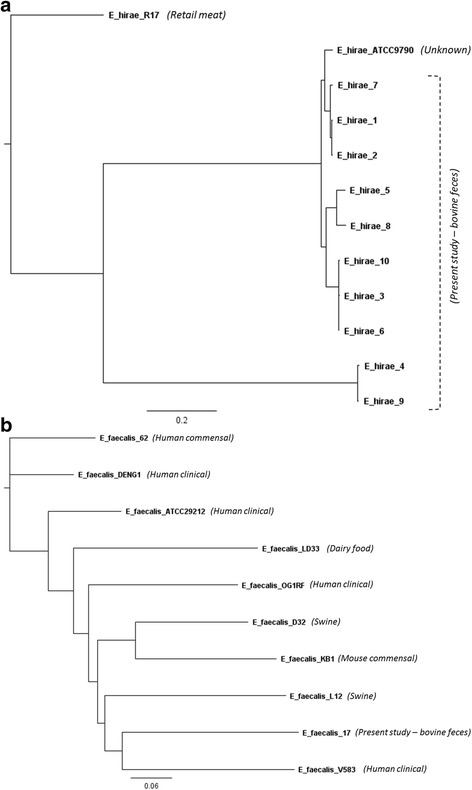
Phylogenetic trees of a Entercoccus hirae and b Entercoccus faecalis genome sequences from present study and complete genome sequences from the NCBI database based on analysis of single-nucleotide varients (SNVs) of the core genes. Origin of isolates are as indicated in the figures
All three E. faecium genomes sequenced in this study were more closely related to NCBI genomes from commensal, probiotic and dairy isolates as compared to clinical isolates (Fig. 3a). To explore this further, we focused our phylogenetic analyses on the presence or absence of accessory genes, which further enhanced the distinction of these two clusters (Fig. 3b). Three complete NCBI E. faecalis genomes from non-clinical sources including human, swine, a probiotic and dairy clustered together with the bovine fecal isolates obtained in the current study. All other isolates included in the relatedness tree analysis were from various human clinical sources and generated a separate cluster. In the process of evolution, bacteria have accessorized their genomes with DNA from other bacteria with the help of mobile genetic elements (MGE) including plasmids, transposons, genomic islands and bacteriophages. The MGE-based accessory genome offers a very useful resource to bacteria for improving their fitness and adaptation within various environments, potentially through the development of pathogenicity and virulence. It is speculated that majority of E. faecium accessory genes contributing to the distinct clustering of clinical and commensal isolates in present study may be plasmid- or chromosomal MGE-borne. Further studies are required to identify those accessory genes to understand their role in enabling enterococci to adapt to specific environments.
Fig. 3.
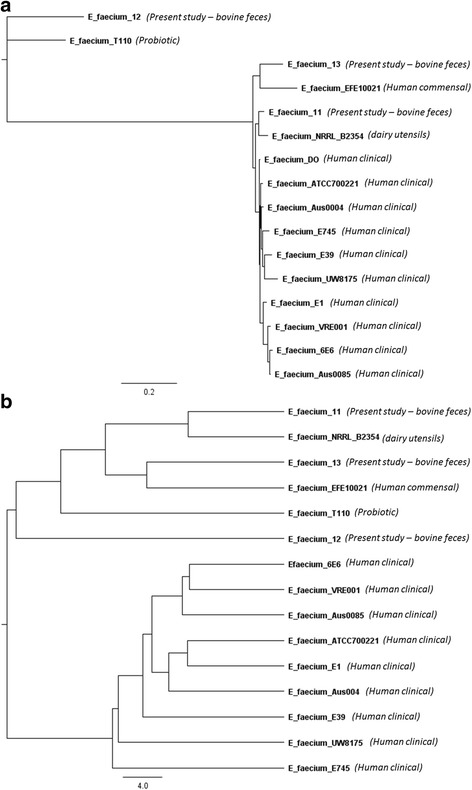
a Phylogenetic tree of Entercoccus faecium genome sequences from the present study and complete genome sequences from the NCBI database based on analysis of single-nucleotide varients (SNVs) of the core genes. b Relatedness tree of E. faecium genome sequences from present study and complete genome sequences from the NCBI database based on Pearson correlation similarity matrix analysis of accessory genes. Origin of isolates are as indicated in the figures
BLAST atlas
BLAST atlases were constructed using the genomes of isolated E. hirae and E. faecium and the reference genomes of E. hirae ATCC 9790 and E. faecium DO from the NCBI database (Fig. 4a & b). Of the E. hirae strains, E. hirae 7 exhibited the highest relatedness to the reference strain. E. hirae 7 and E. hirae 8 also shared phage-related genes with the reference strain (Fig. 4a). Several transposon-related loci were also shown to be shared with the reference genome. There were few variable regions identified among strains of E. hirae, illustrating their similarity in gene content. Likewise, the gene content among strains of E. faecium was also highly similar. However, several phage and transposon related loci from the reference strain appeared to be absent in commensal strains but present in clinical strains (Fig. 4b). This observation further supports their distinct segregation into independent clades.
Fig. 4.
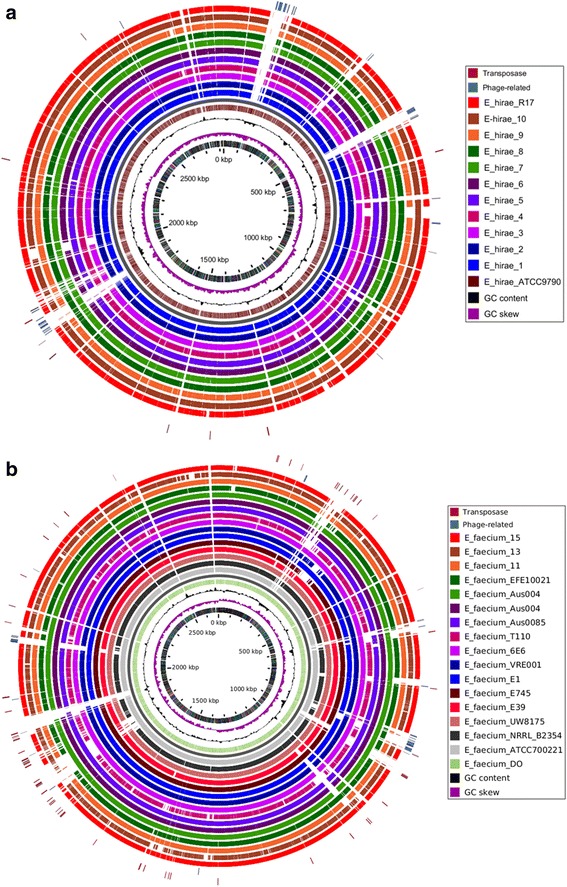
a Blast atlas of 10 Enterococcus hirae strains isolated from bovine feces and E. hirae strain R17 mapped against E. hirae ATCC9790. b Blast atlas of the genomes of 3 Entercoccus faecium isolates from bovine feces and 12 complete E. faecium genomes from the NCBI database mapped against reference sequence E. faecium DO. Blast atlases were generated by GView Java package software [28] using both alignment length and percent identity cut-off values of 80%. Based on the reference genomes, phage and transposon related regions/loci are indicated on the altas diagram
Clusters of Orthologous Groups (COGs)
Clusters of Orthologous Groups (COGs) are broad functional categories used to assign proteins related by function [32]. Functional categorization of proteins into different COGs (Additional file 2: Figure S1) revealed variation in the functional profile among Enterococcus spp., but the percentage of COGs assigned to cell cycle control, cell division, chromosome partitioning; extracellular structures; and intracellular trafficking, secretion and vesicular transport were similar among species. The percentage of COGs assigned to cell motility was greatest for E. gallinarum and E. casseliflavus, two species of Enterococcus that are known to be motile [12]. The percentage of COGs for cell motility was low for all other non-motile enterococci species [33]. There was little difference in the functional profile among strains of the same species with the exception of the mobilome: prophages, transposons category, in which inter-species variation was observed. Two E. hirae strains, E. hirae 4 and E. hirae 9, clustered together (Fig. 2a), two E. faecium strains (E. faecium 11 and E. faecium 12) and an E. villorum, E. faecalis and E. casseliflavus strain (E. villorum 16, E. faecalis 17 and E. casseliflavus 20, respectively) exhibited the greatest percentage of proteins assigned as phage and transposases.
Using the compare genomes function available in the IMG platform, we produced an abundance profile overview of the gene count for different COGs for all 21 Enterococcus spp. genomes (Additional file 1: Table S2). Van Schaik et al. [14] performed a COG-based functional comparison between E. faecium and E. faecalis in an effort to identify characteristics that distinguished the two species. In their analysis, they identified differences in sugar metabolism for the pentose sugar arabinose. They found COGs responsible for the metabolism (COG2160 and COG3957), uptake (COG4213 and COG4214) and degradation (COG3940) of arabinose to be present in E. faecium and absent in E. faecalis, attributing this to the inability of E. faecalis to metabolise arabinose [34]. Genes for these COGs, with the exception of COG4214 in E. faecium 12, were present in the E. faecium strains examined in this study and absent in our E. faecalis strain. Genes for these COGs were also present in E. gallinarum and E. casseliflavus strains, suggesting these species of Enterococcus also have the ability to metabolise arabinose (Additional file 1: Table S2). Ford et al. [35] previously documented that strains of E. gallinarum and E. casseliflavus that they examined were able to metabolise arabinose, but demonstrated poor growth compared to E. faecium. In the current study, E. hirae, E. villorum, E. durans and E. thailandicus all lacked genes for these COGs suggesting that they lacked the ability to metabolise arabinose, an outcome that has been biochemically confirmed by others [36–38]. Arabinose is a subunit of the plant polysaccharide hemicellulose and therefore would be in abundance in the GI tract of cattle [14]. Despite E. faecium being able to utilize arabinose as an energy source, this trait does not appear to provide a competitive advantage for this species to proliferate in the GI tract of cattle, considering E. hirae is the predominant species identified [10].
Van Schaik et al. [14] investigated other COGs involved in the metabolism of carbon sources from plants including COG4677, which is predicted to be involved in the metabolism of pectin, and COG3479, which is involved in the breakdown of coumaric acid and other components of lignocellulose. In our study, COG4677 was present in E. faecium, E. durans and E. casseliflavus and absent from E. hirae, E. thailandicus, E. villorum, E. faecalis and E. gallinarum, whilst COG3479 was present in E. hirae, E. faecium, E. villorum and E. durans and absent from E. faecalis, E. thailandicus, E. gallinarum and E. casseliflavus. These authors also highlighted a number of COGs present in E. faecalis that were absent in E. faecium including COGs for the utilization of ethanolamine as a carbon source. In the current study, E. faecalis possessed COGs for the utilization of ethanolamine, which were confirmed to be absent in E. faecium. Ethanolamine utilization has been demonstrated for E. faecalis [39], but not for other Enterococcus species. In the current study, these COGs were also identified in E. gallinarum suggesting this Enterococcus species may also utilize ethanolamine as an energy source, but to our knowledge this has not been biochemically confirmed. It is clear that different Enterococcus spp. have the ability to utilize various carbon sources allowing them to inhabit and survive in many diverse environments, including the GI tract of cattle. From this study, it was not apparent if E. hirae possessed specific traits for carbohydrate metabolism that may promote its abundance in the GI tract of cattle over other Enterococcus spp.
Van Schaik et al. [14] also investigated proteins involved in protection against oxidative stress. They identified the enzyme catalase (COG0753) was present in E. faecalis and absent in E. faecium. Examination of the different Enterococcus spp. in this study confirmed catalase to be specific for E. faecalis as it was absent from all other species. In the presence of heme, E. faecalis exhibits catalase activity [40]. Catalase production has been speculated to play a role in virulence in pathogenic bacteria including Staphylococcus aureus [41, 42]. E. faecalis can be exposed to oxidative stress as part of the host defence against invasion [40]. Catalase production may offer some protection against oxidation during invasion, contributing to the virulence of E. faecalis. Other mechanisms in E. faecium may play a role in the oxidative stress response, including the production of glutathione peroxidase (COG0386) [14]. With the exception of E. faecalis, this COG was present in all species of Enterococcus examined in this study, demonstrating the different strategies Enterococcus spp. use to combat oxidative stress.
Multi-locus sequence typing (MLST)
Multi-locus sequence typing (MLST) has been used to study the population structure and evolution of E. faecium and E. faecalis [43, 44]. This technique involves sequencing and analysis of housekeeping genes and assignment of a sequence type (ST) [44, 45]. In the current study E. faecium 11, E. faecium 12 and E. faecium 13 were classified as ST214, unknown and ST955, respectively, and E. faecalis 17 as ST242 (Table 1). The lack of an assignment of a ST for E. faecium 12 suggests there are STs that have yet to be defined within the MLST database. STs can be assigned to a clonal complex (CC) based on their similarity to a central alleic profile [46]. MLST analysis of the population structure of E. faecium has identified that the majority of strains associated with nosocomial infections belong to the Clonal Complex 17 (CC17) [43]. For E. faecalis it appears that two complexes, CC2 and CC9, represent hospital-derived strains [44, 47]. The STs assigned to E. faecium and E. faecalis identified in the current study have been described previously [47–50] and are not associated with complexes of hospital-derived strains. There is currently no typing scheme available for other Enterococcus spp.
Virulence genes
Virulence genes contribute to the pathogenicity of an organism. In this study, virulence genes were only detected in E. faecium and E. faecalis. All three E. faecium strains contained the efaA and acm genes, whilst E. faecalis contained a number of virulence genes including efaA, ace, ebp pili genes, gelE and fsrB. The acm and ace genes described in E. faecium and E. faecalis, respectively, are important for facilitating cell wall adhesion to host tissues [51, 52]. The efaA gene found in both E. faecalis and E. faecium also plays a role in adherence to host tissues and is a virulence factor involved in endocarditis [53, 54]. The ebp pili genes described in E. faecalis, comprising of ebpA, ebpB and ebpC, assist in adherence and biofilm formation [55]. The gelE gene in E. faecalis encodes for gelatinase, which hydrolyses gelatin, collagen, casein and haemoglobin [56]. Its expression is regulated by the two-component fsr system, with both gelE and fsr genes important in biofilm formation [57–59].
In addition to these virulence genes, a number of bacterial sex pheromone genes were also present in E. faecalis including cad, camE, cCF10 and cOB1. Certain conjugative plasmids found in E. faecalis respond to the secretion of bacterial sex pheromone genes from plasmid-free enterococci, inducing their transfer [60]. Sex pheromone response plasmids have rarely been described in other Enterococcus spp, but a few have been reported for E. faecium [61, 62]. The bacterial sex pheromones detected in the E. faecalis genome target the sex pheromone plasmids pAD1, pAM373, pCF10 and pOB1, respectively. Some of these plasmids encode features that can contribute to virulence such as pAD1 and pOB1, both encoding for a bacteriocin and hemolysin, and pCF10, encoding tetracycline resistance [63]. The pheromone cAD1 precursor lipoprotein cad gene was detected in all of the Enterococcus spp. isolates sequenced in this study, with 98–59% amino acid identities to E. faecalis strain FA2-2. Presence of the cAD1 precursor lipoprotein in these Enterococcus spp. increases their potential of receiving the highly conjugative pheromone-responding plasmid pAD1. The hemolysin/bacteriocin (cytolysin) encoded by this plasmid has been shown to contribute to virulence in animal models [64]. Therefore acquisition of this plasmid by these Enterococcus spp. could increase their virulence. Further analysis is required to determine if this sex pheromone precursor is able to induce transfer of pAD1 to Enterococcus spp. other than E. faecalis.
Virulence genes have mostly been characterized in E. faecalis and E. faecium, with little information available on the nature of these genes in other enterococcal species. A whole cytolysin operon has been reported in E. durans and cytolysin genes have been identified in dairy-associated E. hirae and E. gallinarum. Other virulence genes were also commonly detected in E. durans, such as the esp gene which is important for adhesion [65]. With the exception of E. faecalis and E. faecium, virulence genes were not detected in the sequence of the other Enterococcus isolates from the bovine GI tract. In addition to human clinical enterococci, virulence genes have been identified in enterococci from non-clinical environments [65–67] with E. faecalis having a greater prevalence of virulence genes than E. faecium [68, 69], an observation that aligns with our study.
Antibiotic resistance genes
Enterococci can exhibit resistance to a number of antibiotics, partly due to their innate resistance to many commonly used antibiotics such as penicillin, but also due to their ability to successfully acquire resistance through horizontal exchange of ARGs on MGEs [70]. In this study we screened the 21 Enterococcus genomes against the ResFinder and CARDs databases for resistance genes (Table 2). Genes conferring resistance to vancomycin were only found in the genomes of E. gallinarum and E. casseliflavus, where the vanC operon was present. The vanC operon is intrinsic to these species of Enterococcus and provides resistance to low concentrations of vancomycin [71, 72]. Of the isolates examined in this study, only E. casseliflavus_20 displayed phenotypic resistance to vancomycin (Additional file 1: Table S3). The intrinsic resistance of E. casseliflavus and E. gallinarum can provide protection to concentrations of vancomycin as high as 32 μg/mL [73]. Vancomycin was present at 30 μg in the disks used for susceptibility testing [10], a concentration that was sufficient to inhibit the growth of E. gallinarum_18 and E. casseliflavus_21 inspite of the vanC operon in these isolates. However, vanC in E. casseliflavus_20 provided adequate resistance to allow growth of this isolate in the presence of vancomycin. The lack of vancomycin resistance genes in Enterococcus isolated from bovine feces was not surprising as avoparcin, a glycopeptide related to vancomycin, has not been used in cattle in North America [74].
Table 2.
Antibiotic resistance gene profile of Enterococcus spp. isolated from bovine feces. Values represent % pairwise identity
| Resistance gene | aph(3′)-III | aac(6′)-Ii | aac(6′)-Iid | aac(6′)-Iih | ant(9) -Ia | adeC | erm(B) | msrC | lsa(A) a | lsa(E) | lnu(B) | tet(L) | tet(M) | tet(O) | sat4 | vanC operon b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain | ||||||||||||||||
| E. hirae 1 | 99.5 | 100 | 100 | 96.5 | ||||||||||||
| E. hirae 2 | 99.5 | 100 | ||||||||||||||
| E. hirae 3 | 100 | 100 | 93.0 | |||||||||||||
| E. hirae 4 | 98.9 | 100 | 100 | |||||||||||||
| E. hirae 5 | 100 | 99.5 | 91.4 | |||||||||||||
| E. hirae 6 | 100 | |||||||||||||||
| E. hirae 7 | 99.5 | |||||||||||||||
| E. hirae 8 | 100 | |||||||||||||||
| E. hirae 9 | 98.9 | 96.9 | ||||||||||||||
| E. hirae 10 | 100 | 92.8 | ||||||||||||||
| E. faecium 11 | 100 | 100 | 100 | 99.9 | 100 | 98.9 | 98.9 | 99.9 | 100 | 95.1 | 99.0 | |||||
| E. faecium 12 | 100 | 92.9 | 95.4 | |||||||||||||
| E. faecium 13 | 99.8 | 99.8 | 99.3 | |||||||||||||
| E. thailandicus 14 | ||||||||||||||||
| E. villorum 15 | ||||||||||||||||
| E. villorum 16 | 99.6 | 100 | ||||||||||||||
| E. faecalis 17 | 99.5 | |||||||||||||||
| E. gallinarum 18 | 98.6 | present | ||||||||||||||
| E. durans 19 | 100 | |||||||||||||||
| E. casseliflavus 20 | present | |||||||||||||||
| E. casseliflavus 21 | 98.6 | present |
aIntrinsic to E. faecalis
b vanC operon consists of vanC, vanR-C, vanS-C, vanXY-C and is intrinsic to E.gallinarum and E. casseliflavus; GenBank accession numbers for resistance genes: aph(3′)-III (M26832.1), aac(6′)-Ii (L12710.1), aac(6′)-Iid (AJ584701.2), aac(6′)-Iih (AJ584700.2), ant(9)-Ia (JQ861959.1), adeC (CP003583.1), erm(B) (U86375.1), msrC (AY004350.1), lsa(A) (AY225127.1), lsa(E) (JX560992.1), lnu(B) (AJ238249.1), tet(L) (M29725.1), tet(M) (EU182585.1), tet(O) (Y07780.1), tet(32) (AJ295238.3), sat4 (U01945.1), vanC operon E. gallinarum (AF162694.1), vanC operon E. casseliflavus (EU151753.1)
Resistance genes to macrolides were present in a number of Enterococcus genomes sequenced, a finding that coincides with the fact that cattle were administered tylosin phosphate in their diets [10]. Erm(B) confers resistance to macrolide-lincosamide-streptogramin B (MLSB) antibiotics and was found in isolates of E. hirae, E. faecium, E. villorum, E. gallinarum and E. casseliflavus. In contrast, msrC, a macrolide efflux pump, was only detected in E. faecium (Table 2). This is consistent with Portillo et al. [75] who described erm(B) as the predominant gene conferring resistance to erythromycin in Enterococcus spp. and msrC in E. faecium. The presence of these resistance genes corresponds with the phenotypic resistance observed in these isolates (Additional file 1: Table S3). Interestingly, E. hirae_6, E. durans_19 and E. casseliflavus_20 exhibited resistance to macrolides even though no resistance genes to macrolides matched those in either the ResFinder or CARDs databases.
We previously reported that the E. thailandicus isolate sequenced in this study exhibited intermediate resistance to erythromycin (Additional file 1: Table S3) [10, 76]. Although there were no obvious macrolide resistance genes present, there were a number of genes identified as having multidrug efflux functions which may have contributed to intermediate resistance to erythromycin [76]. There is also the possibility that this phenotype was as a result of an unknown gene that codes for erythromycin resistance.
Genes conferring resistance to high concentrations of aminoglycosides were not detected in any of the genomes. Susceptibility to high concentrations of aminoglycosides was confirmed by the lack of phenotypic resistance to gentamicin and streptomycin (Additional file 1: Table S3). Enterococci are intrinsically resistant to low concentrations of aminoglycosides which is conferred by the genes aac(6′)-Ii, aac(6′)-Iid and aac(6′)-Iih present in E. faecium, E. durans and E. hirae, respectively (Table 2) [77, 78].
Genes coding for tetracycline resistance were detected in a number of genomes, including E. hirae, E. faecium and E. villorum (Table 2). Tet(L) encodes for an efflux protein whilst tet(M) and tet(O) encode for ribosomal protection proteins [79]. Anderson et al. [80] found tet(O) was the most prevalent gene encoding for tetracycline resistance in enterococci isolated from cattle, a finding that agrees with ours. Anderson et al. [80] reported E. hirae as the predominant species isolated from cattle and tet(O) was only resistance determinant associated with E. hirae in the current study. Detection of tet(M) and tet(L) in other isolates is not unexpected as both genes are also frequently detected in enterococci from animals including poultry, pigs, dogs, cats, rabbits, badgers, wildcats and birds [81–83]. Disk susceptibility testing revealed isolates containing tet(M) were resistant to doxycycline whilst those containing tet(L) or tet(O) were susceptible (Additional file 1: Table S3). It is possible that isolates that are sensitive to doxycycline are susceptible to other members of the tetracycline family. In general, bacteria that are resistant to doxycycline are also resistant to tetracycline and oxytetracycline [84, 85].
Only a few of the selected genomes contained ARGs to two or more antibiotics. Of particular interest was E. faecium_11, which contained at least 11 ARGs as inferred from the analysis of genome sequences (Table 2), including those conferring aminoglycoside, MLSB, pleuromutilin, streptogramin A, tetracycline and streptothricin resistance.
Mobile genetic elements
Mobile genetic elements (MGEs) play an important role in horizontal gene transfer (HGT) of ARGs within and between bacteria from human and/or animal hosts [86–88]. MGEs include plasmids, transposable elements, prophages and various genomic islands such as integrative and conjugative elements (ICEs) [89]. A number of MGEs have been described in enterococci including transposons, plasmids and bacteriophage [90].
The well-known Tn3-like transposon, Tn917, which is widely distributed in enterococci was identified in several of the sequenced genomes. Four E. hirae strains (E. hirae 1, E. hirae 2, E. hirae 3 and E. hirae 4) and one E. faecium strain (E. faecium 11) had high sequence homology (>95%) to the Tn917 transposon, previously described in E. faecalis [91]. All of these strains exhibited erythromycin resistance (Additional file 1: Table S3) [10], conferred by the erm(B) resistance gene present in Tn917. Other distinguishing features of this transposon include a transposase (TnpA) and a resolvase (TnpR) involved in the replicative mode of transposition [92].
The erm(B) gene was present in a number of other genomes including E. hirae_5, E. villorum_16, E. gallinarum_18 and E. casseliflavus_21. However, it did not align with the Tn917 transposon. In E. hirae_5, the erm(B) gene was found on a contig associated with chromosomal genes. The tetracycline resistance gene tet(O) was also found in the vicinity of erm(B). Based on sequence information, erm(B) in the other three genomes appeared to be plasmid mediated. In E. villorum_16, the erm(B) and tet(L) genes were found on contigs associated with a plasmid sequence from an E. faecium strain UW8175 (GenBank accession no. CP011830.1). In E. gallinarum_18 and E. casseliflavus_21, the erm(B) gene was found on contigs associated with the plasmid sequence of pRE25 from an E. faecalis (GenBank accession no. X92945.2).
The tetracycline resistance genes tet(L) and tet(M) found in E. hirae_1 were located on a contig which shared 21,418 identical bp with the 25,963 bp transposon Tn6248 of E. faecium strain E506 (GenBank accession no. KP834592). The genes responsible for transposition (tnpA) and insertion and excision of Tn6248 (tndX) were absent, as was the chloramphenicol acetyltransferase gene (cat). This same contig also appeared to be associated with a plasmid sequence in E. hirae strain R17 (GenBank accession no. CP015517.1), suggesting this remnant transposon may be on a plasmid.
Integrative conjugative elements (ICEs) are self-transmissible elements that contain modules for their maintenance, dissemination and regulation [93]. In major Gram-positive human pathogens (e.g., Enterococcus spp., Staphylococcus spp. and Streptococcus spp.), tetracycline resistance is known to arise from the acquisition of the Tn916-family ICE carrying the tet(M) gene. The gene synteny in this family of ICE is well conserved, but there are differences in integrase (int) and excisionase (xis) gene sequences, insertion site specificity, and host range among family members [94–96]. The Tn916 ICE was originally identified as an 18-kb conjugative transposon in E. faecalis DS16 [97, 98]. Variants of some Tn916-tet(M) members, including Tn916, Tn5397, Tn6000 or Tn5801, are widely spread among several genera within the Firmicutes, suggesting widespread dissemination of these elements. Many Tn916-like ICEs have a broad host range and are responsible for dissemination of tetracycline resistance through tet(M) in Gram-positive bacteria associated with humans and animals [88, 98, 99]. Recently, almost identical Tn5801-like genomic islands have been identified in different Gram-positive bacterial species of pet (Staphylococcus pseudintermedius) and human (E. faecalis, S. aureus, Staphylococcus agalactiae) origin, suggesting a horizontal transfer of these elements [100]. In our study, two ICEs belonging to the Tn916-family were identified in E. faecium_11. These ICEs exhibited homology to Tn916 and Tn5801, each harboring a tet(M) variant, and appeared to be located within the chromosome. In Group B Streptococcus, the vast majority of Tn916 and Tn5801 are inserted into the core genome [101]. Once inserted in the genome, it is thought that Tn916 and Tn5801 are retained, as they impose a minimal impact on the biological fitness of the host bacteria [88, 101, 102].
A gene cluster aadE– sat4–aphA-3 encoding resistance to streptomycin, streptothricin and kanamycin, previously described in E. faecium [103], was also found in E. faecium_11 associated with plasmid related contigs. This gene cluster has also been described in Tn5405 within S. aureus [104] and Tn1545 from Streptococcus pneumoniae [105], suggesting that it is widespread among Gram-positive bacteria.
Bacteriophages
Bacteriophage mediated transduction of antibiotic resistance has been demonstrated in enterococci [106], and potential virulence determinants have been identified in phage associated with E. faecalis [107]. Phage found in enterococci usually belong to the Podoviridae, Siphoviridae or Myoviridae, but others including Inoviridae, Leviviridae, Guttaviridae and Fuselloviridae have also been reported [108, 109].
All Enterococcus genomes sequenced contained at least one putative phage, ranging in size from 8.0 to 70.3 kb (Additional file 1: Table S4). A total of 37 intact prophages were identified across the 21 sequenced genomes. E. hirae and E. faecium contained one to three intact prophages, whereas E. faecalis and E. gallinarum each contained two intact prophages and E. durans contained one intact prophage. E. villorum and E. casseliflavus contained up to four intact prophages whilst no intact prophages were detected in E. thailandicus. The intact prophages detected were from the Siphoviridae, Myoviridae or Podoviridae families, with prophage from the Siphoviridae family being most prevalent across all species examined (Additional file 1: Table S4). Prophages of the Phycodnaviridae family were identified in E. faecium and E. villorum. Its status was intact for only one of the E. faecium strains whilst it was questionable or incomplete in others (Additional file 1: Table S4). To our knowledge, phage from the Phycodnaviridae family have yet to be described in enterococci species. However, their presence in the rumen microbiome has been reported following metagenomic analysis [110].
CRISPR-Cas
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) genes are a type of adaptive immune response described in bacteria against invading genetic elements such as phage and plasmids [111]. A CRISPR locus includes a CRISPR array flanked by various cas genes, with the array comprised of short direct repeats alternating with short variable DNA sequences called ‘spacers’ [111]. Three types of CRISPR-Cas systems have been described, distinguished by the presence of different Cas genes namely cas3 for type I, cas9 for type II and cas10 for type III [112]. Recently, two additional types have been proposed to this classification system that includes type IV and type V [111]. CRISPR-Cas systems typically described in enterococci are of the type II variety. However, a recent report identified a type I system in Enterococcus cecorum [113, 114].
All E. hirae strains contained CRISPR arrays, except for E. hirae_8. CRISPR arrays were also detected in E. thailandicus, E. villorum and E. durans (Additional file 1: Table S5). The CRISPR arrays from these genomes were flanked by Cas genes, consisting of cas9, cas1, cas2 and csn2 with the exception of E. villorum which lacked the csn2 gene. CRISPR arrays flanked by these four Cas genes are classified as a type II-A system and are predicted to be functional as indicated by the presence of the core Cas genes cas1 and cas2 [112, 115]. Following the same nomenclature, the CRISPR-Cas system identified in E. villorum would also be classified as a type-II system, but its subtype is unclear.
Multiple CRISPR arrays can often be detected in bacterial genomes. However, not all CRISPR arrays may be accompanied by Cas genes. It is predicted that these arrays lie dormant or that Cas genes from other similar arrays may be sufficient for their activity [116]. Orphan CRISPR arrays (without Cas genes) [114] were identified by the CRISPRdb in a number of genomes, including two E. hirae strains and in E. thailandicus, E. faecalis and E. durans (Additional file 1: Table S4). No functional CRISPR arrays were detected for E. faecium, E. faecalis, E. gallinarum or E. casseliflavus.
Comparison of CRISPR arrays flanked by Cas genes revealed unique arrays between Enterococcus species, but some arrays were shared among strains of the same species (Fig. 5). Amongst the nine E. hirae strains, only four unique arrays were present. The arrays identified in E. villorum were identical for both strains. The largest array was identified in E. thailandicus. Arrays identified in the sequenced Enterococcus genomes contained between three and ten direct repeat (DR) sequences, alternating with spacer sequences (Fig. 5; Additional file 1: Table S6). A total of 26 unique spacer sequences associated with functional CRISPR arrays and an additional 38 unique spacers associated with orphan CRISPR arrays were identified (Additional file 1: Table S7).
Fig. 5.

Schematic of CRISPR-Cas systems identified in whole genome sequence analysis of 21 Enterococcus spp. genomes. a Functional CRISPR array spacer and direct repeat organization. Diamonds represent direct repeats interspaced with numbers representing spacers. Spacer numbers correlate with sequences displayed in Additional file 1: Table S6. b Orphan CRISPR array spacer and direct repeat organization. Diamonds represent direct repeats interspaced with numbers representing spacers. Spacer numbers correlate with sequences displayed in Additional file 1: Table S6. c Numbered direct repeats. Numbers correlate with sequences displayed in Additional file 2: Table S5
In enterococci, it is hypothesized that the absence of CRISPR-Cas systems is associated with increased antibiotic resistance in isolates of E. faecium and E. faecalis [117]. In this study, E. faecium 11 lacked CRISPR-Cas and harbored several antibiotic resistance genes, reflecting this association. Palmer and Gilmore [117] detected identities between CRISPR spacer sequences and sequences of known pheromone-responsive plasmids and phage, suggesting CRISPR-Cas systems provide defence against these invading genetic elements. The authors hypothesized that the absence of CRISPR-Cas systems resulted in a compromised genome defence, enabling the acquisition of ARGs on MGEs. Palmer and Gilmore [117] did not detect spacer sequences with identities to transposons and hypothesized CRISPR-Cas systems may not provide defence against transposons. Several E. hirae strains in the current study contained functional CRISPR-Cas systems and the erm(B) resistance gene on a Tn3-like transposon, supporting this theory.
Functional CRISPR arrays and intact prophage were identified in most of the genomes sequenced in this study, with the exception of E. thailandicus. It is not surprising that these genomes contained prophage, as bacteriophage have developed strategies to avoid CRISPR regulation through the development of anti-CRISPR systems to enable integration into the genome [118]. In the case of E. thailandicus, spacers identified in CRISPR arrays aligned to incomplete prophage sequences with 100% sequence similarity and may possibly explain the lack of intact prophage in this genome. Spacer 60 aligned with both regions 3 and 4 of E. thailandicus prophage whilst spacer 12 aligned with region 4. None of the remaining spacers identified in CRISPR arrays had any sequence similarity to identified prophage.
Secondary metabolites
Bacteriocins are ribosomally synthesized antimicrobial peptides produced by Gram-positive and Gram-negative bacteria that have antimicrobial activity against closely related bacteria [119]. In Gram-positive bacteria, they are classified into three major classes. Class I consists of the heat stable, modified peptides or lantibiotics, Class II describes the heat stable, unmodified non-lantibioitics and Class III consists of large proteins that are heat unstable [119, 120]. It is believed the production of bacteriocins by bacteria provides a competitive advantage to their survival in certain ecological niches [121].
Putative lantibiotics were identified in E. hirae, E. thailandicus and E. gallinarum whilst none were predicted in E. faecium, E. villorum, E. faecalis, E. durans or E. casseliflavus. Putative class II bacteriocins were identified in seven E. hirae strains (E. hirae_3, E. hirae_4, E. hirae_5, E. hirae_6, E. hirae_8, E. hirae_9, E. hirae_10), two E. faecium strains (E. faecium_11, E. faecium_13), E. thailandicus, E. villorum and E. durans. A putative bacteriocin identified in E. faecium_11 and E. faecium_13 had an amino acid identity of 99% to Enterocin A (Genbank accession no. AAF44686.1). Enterocin A was first described in an E. faecium strain isolated from fermented Spanish sausage [122]. Enterocin A inhibits a broad spectrum of Gram-positive bacteria including species of Clostridium, Propionibacterium, Listeria and Staphylococcus [123].
Until recently, terpenes were mainly considered secondary metabolites associated with plants and fungi, and were described in prokaryotes in only a few instances. These compounds serve a number of purposes including acting as antibiotics, hormones, flavor or odor constituents and pigments [124]. Since the advent of genomic sequencing, a number of presumptive terpene synthase genes have been discovered in bacteria [125]. Putative terpenes were identified in all E. hirae, E. villorum, E. gallinarum, E. durans and E. casseliflavus genomes sequenced in this study. None were predicted in E. faecium, E. thailandicus and E. faecalis genomes. The role of terpenes in enterococci remains unclear.
Conclusions
This study has provided valuable insight about genetic differences observed among Enterococcus spp. isolated from bovine feces. We hypothesized that enterococci originating from bovine feces would lack genes coding for virulence, but would contain MGEs that could promote the dissemination of ARGs. We confirmed the majority of Enterococcus spp. isolated from bovine feces lacked virulence traits. The virulence traits that were identified were primarily associated with E. faecium and E. faecalis. As E. faecium and E. faecalis are not the predominant species of the bovine GI tract, the risk of transmission to humans through contamination of food products is likely low. Of most concern perhaps is dissemination of ARGs on MGEs. We identified that both E. faecium and E. hirae contained the Tn917 transposon conferring MLSB resistance suggesting that transfer of ARGs may occur in the bovine GI tract between Enterococcus spp. We also identified two ICE of the Tn916 family that conferred tetracycline resistance in one isolate of E. faecium. As only a small number of isolates were examined in this study it is possible that other enterococci may be present in the bovine GI tract with ICE harbouring ARGs. As the cost of genomic sequencing continues to decline, further investigation of ICE using whole genome sequencing will help determine if there are linkages between enterococci isolates from bovine, the surrounding environment and human clinical sources.
Acknowledgements
We thank Shaun R. Cook for his technical assistance.
Funding
This study was conducted with funding from Beef Cattle Research Council (BCRC) Beef Cluster of Agriculture and Agri-Food Canada and the GRDI program of the Government of Canada and is gratefully acknowledged. The funders had no role in the design, collection of samples, analysis, interpretation or writing of the manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are include within the article and its additional files. The draft genome sequences of E. hirae_1, E. hirae_2, E. hirae_3, E. hirae_4, E. hirae_5, E. hirae_6, E. hirae_7, E. hirae_8, E. hirae_9, E. hirae_10, E. faecium_11, E. faecium_12, E. faecium_13, E. thailandicus_14, E. villorum_15, E. villorum_16, E. faecalis_17, E. gallinarum_18, E. durans_19, E. casseliflavus_20, E. cassilflavus_21 were submitted to GenBank (BioProjects IDs PRJNA342049 and PRJNA224116).
Authors’ contributions
AB, RZ and TM designed the experiments. AB and RZ performed the bioinformatics analysis. AB, NG and RZ conducted the experiments. AB drafted the manuscript. RZ, NG, KA, AC, MW and TM revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
The research study was reviewed and approved by the Lethbridge Research Centre Animal Care Committee, an evaluation body that is accredited by the Canadian Council of Animal Care. Consent to participate was not required as all isolates for the studies were obtained from the Lethbridgre Research and Development Centre culture collection.
Abbreviations
- GI
Gastointestinal
- HGT
Horizontal gene transfer
- ICE
Integrative congugative element
- IMG
Integrated microbial genomes
- MGEs
Mobile genetic elements
- MLSB
Macrolide-lincosamide-streptogramin B
- MLST
Multi-locus sequence typing
- PFGE
Pulsed-field gel electophoresis
- SNPs
Single-nucleotide polymorphisms
Additional files
Reference sequences for contig ordering using progressive Mauve. Table S2. Clusters of Orthologous Groups (COGs) gene abundance profile overview for all 21 Enterococcus spp. isolated from bovine feces. Table S3. Raw antibiogram data from disk susceptibility testing conducted previously [10]. Table S4. Putative prophage detected in Enterococcus spp. isolated from bovine feces. Table S5. Presence and absence of CRISPR arrays and intact prophage in Enterococcus spp. isolated from bovine feces. Table S6. Direct repeat sequences of CRISPR arrays found in Enterococcus spp. isolated from bovine feces. Table S7. Spacer sequences of CRISPR arrays found in Enterococcus spp. isolated from bovine feces. (XLSX 466 kb)
{kind=link}
Organisation of protein coding genes by Clusters of Orthologous Groups (COGs) category. (PNG 731 kb)
Contributor Information
Alicia G. Beukers, Email: abeu9727@uni.sydney.edu.au
Rahat Zaheer, Email: Rahat.Zaheer@AGR.GC.CA.
Noriko Goji, Email: Noriko.Goji@inspection.gc.ca.
Kingsley K. Amoako, Email: amoakok@inspection.gc.ca
Alexandre V. Chaves, Email: alex.chaves@sydney.edu.au
Tim A. McAllister, Email: tim.mcallister@agr.gc.ca
References
- 1.Franz CM, Huch M, Abriouel H, Holzapfel W, Galvez A. Enterococci as probiotics and their implications in food safety. Int J Food Microbiol. 2011;151(2):125–40. doi: 10.1016/j.ijfoodmicro.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Agudelo Higuita NI, Huycke MM. Enterococcal Disease, Epidemiology, and Implications for Treatment. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: from commensals to leading causes of drug resistant infection. Boston: Massachusetts Eye and Ear Infirmary; 2014. [Google Scholar]
- 3.Poh CH, Oh HM, Tan AL. Epidemiology and clinical outcome of enterococcal bacteraemia in an acute care hospital. J Infect. 2006;52(5):383–6. doi: 10.1016/j.jinf.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Sivert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Cont Hosp Ep. 2013;34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 5.Arias CA, Murray BE. Emergence and management of drug-resistant enterococcal infections. Expert Rev Anti Infect Ther. 2008;6(5):637–55. doi: 10.1586/14787210.6.5.637. [DOI] [PubMed] [Google Scholar]
- 6.Bager F, Madsen M, Christensen J, Aarestrup FM. Avoparcin used as a growth promoter is associated with the occurrence of vancomycin-resistant Enterococcus faecium on Danish poultry and pig farms. Prev Vet Med. 1997;31(1–2):95–112. doi: 10.1016/S0167-5877(96)01119-1. [DOI] [PubMed] [Google Scholar]
- 7.Klare I, Heier H, Claus H, Bohme G, Marin S, Seltmann G, et al. Enterococcus faecium strains with vanA-mediated high-level glycopeptide resistance isolated from animal foodstuffs and fecal samples of humans in the community. Microb Drug Resist. 1995;1(3):265–72. doi: 10.1089/mdr.1995.1.265. [DOI] [PubMed] [Google Scholar]
- 8.Schouten MA, Voss A. VRE and meat. Lancet. 1997;349(9060):1258. doi: 10.1016/S0140-6736(05)62461-0. [DOI] [PubMed] [Google Scholar]
- 9.European Commission . Ban on the antibiotic “Avoparcin” in animal feed. 1997. [Google Scholar]
- 10.Beukers AG, Zaheer R, Cook SR, Stanford K, Chaves AV, Ward MP, et al. Effect of in-feed administration and withdrawal of tylosin phosphate on antibiotic resistance in enterococci isolated from feedlot steers. Front Microbiol. 2015;6:483. doi: 10.3389/fmicb.2015.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coburn PS, Baghdayan AS, Dolan GT, Shankar N. Horizontal transfer of virulence genes encoded on the Enterococcus faecalis pathogenicity island. Mol Microbiol. 2007;63(2):530–44. doi: 10.1111/j.1365-2958.2006.05520.x. [DOI] [PubMed] [Google Scholar]
- 12.Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, et al. Comparative genomcis of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. MBio. 2012;3(1):e00318–11. doi: 10.1128/mBio.00318-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin X, Galloway-Pena JR, Sillanpaa J, Roh JH, Nallapareddy SR, Chowdhury S, et al. Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes. BMC Microbiol. 2012;12:135. doi: 10.1186/1471-2180-12-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Schaik W, Top J, Riley DR, Boekhorst J, Vrijenhoek JE, Schapendonk CM, et al. Pyrosequencing-based comparative genome analysis of the nosocomial pathogen Enterococcus faecium and identification of a large transferable pathogenicity island. BMC Genomics. 2010;11:239. doi: 10.1186/1471-2164-11-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmer KL, van Schaik W, Willems RJ, Gilmore MS. Enterococcal genomics. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: from commensals to leading causes of drug resistant infection. Boston: Massachusetts Eye and Ear Infirmary; 2014. [PubMed] [Google Scholar]
- 16.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 18.Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, et al. Multilocus sequence typing of total genome sequenced bacteria. J Clin Microbiol. 2012;50(4):1355–61. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. 2014;52(5):1501–10. doi: 10.1128/JCM.03617-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–4. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57(7):3348–57. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bi D, Xu Z, Harrison EM, Tai C, Wei Y, He X, et al. ICEberg: a web-based resource for integrative and conjugative elements found in bacteria. Nucleic Acids Res. 2012;40:D621–6. doi: 10.1093/nar/gkr846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–52. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–7. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite bio-synthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–46. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbioal genomes database and cpmarative analysis system. Nucl Acids Res. 2012;40(D1):D115–22. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petkau A, Stuart-Edwards M, Stothard P, Van Domselaar G. Interactive microbial genome visualization with GView. Bioinformatics. 2010;26(24):3125–6. doi: 10.1093/bioinformatics/btq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petkau A, Mabon P, Sieffert C, Knox N, Cabral J, Iskander M, Iskander M, Weedmark K, Zaheer R, Katz LS, Nadon C, Reimer A, Taboada E, Beiko RG, Hsiao W, Brinkman F, Graham M, The IRIDA Consortium, Van Domselaar D. 2016. bioRxiv. Cold Spring Harbor Laboratory Press. doi: https://doi.org/10.1101/092940.
- 30.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 31.Huang W, Li L, Myers JR, Marth GT. ART: a next-generation sequencing read simulator. Bioinformatics. 2012;28(4):593–4. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acid Res. 2001;29(1):22–8. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devriese LA, Pot B, Collins MD. Phenotypic identification of the genus Enterococcus and differentiation of phylogenetically distinct enterococcal species and species groups. J Appl Bacteriol. 1993;75(5):399–408. doi: 10.1111/j.1365-2672.1993.tb02794.x. [DOI] [PubMed] [Google Scholar]
- 34.Deibel RH, Lake DE, Niven CF., Jr Physiology of the enterococci as related to their taxonomy. J Bacteriol. 1963;86:1275–82. doi: 10.1128/jb.86.6.1275-1282.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ford M, Perry JD, Gould FK. Use of cephalexin-aztreonam-arabinose agar for selective isolation of Enterococcus faecium. J Clin Microbiol. 1994;32(12):2999–3001. doi: 10.1128/jcm.32.12.2999-3001.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Devriese LA, Vancanneyt M, Descheemaeker P, Baele M, Van Landuyt HW, Gordts B, et al. Differentiation and identification of Enterococcus durans, E. hirae and E. villorum. J Appl Microbiol. 2002;92(5):821–7. doi: 10.1046/j.1365-2672.2002.01586.x. [DOI] [PubMed] [Google Scholar]
- 37.Farrow JA, Collins MD. Enterococcus hirae, a new species that includes amino assay strain NCDO 1258 and strains causing growth depression in young chickens. Int J Syst Evol Microbiol. 1985;35(1):73–5. [Google Scholar]
- 38.Tanasupawat S, Sukontasing S, Lee J-S. Enterococcus thailandicus sp. nov., isolated from fermented sausage (‘mum’) in Thailand. Int J Syst Evol Microbiol. 2008;58:1630–4. doi: 10.1099/ijs.0.65535-0. [DOI] [PubMed] [Google Scholar]
- 39.Florencia Del Papa M, Perego M. Ethanolamine activates a sensor histidine kinase regulating its utilization in Enterococcus faecalis. J Bacteriol. 2008;190(21):7147–56. doi: 10.1128/JB.00952-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frankenberg L, Brugna M, Hederstedt L. Enterococcus faecalis heme-dependent catalase. J Bacteriol. 2002;184(22):6351–6. doi: 10.1128/JB.184.22.6351-6356.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clements MO, Foster SJ. Stress resistance in Staphylococcus aureus. Trends Microbiol. 1999;7(11):458–62. doi: 10.1016/S0966-842X(99)01607-8. [DOI] [PubMed] [Google Scholar]
- 42.Kanafani H, Martin SE. Catalase and superoxide dismutase activities in virulent and nonvirulent Staphylococcus aureus isolates. J Clin Microbiol. 1985;21(4):607–10. doi: 10.1128/jcm.21.4.607-610.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willems RJ, Top J, van Santen M, Robinson DA, Coque TM, Baquero F, et al. Global spread of vancomycin-resistant Enterococcus faecium from distinct nosocomial genetic complex. Emerg Infect Dis. 2005;11(6):821–8. doi: 10.3201/1106.041204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruiz-Garbajosa P, Bonten MJ, Robinson DA, Top J, Nallapareddy SR, Torres C, et al. Multilocus sequence typing scheme for Enterococcus faecalis reveals hospital-adapted genetic complexes in a background of high rates of recombination. J Clin Microbiol. 2006;44(6):2220–8. doi: 10.1128/JCM.02596-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Homan WL, Tribe D, Poznanski S, Li M, Hogg G, Spalburg E, et al. Multilocus sequence typing scheme for Enterococcus faecium. J Clin Microbiol. 2002;40(60):1963–71. doi: 10.1128/JCM.40.6.1963-1971.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.PubMLST. Clonal complex designation. 2016. http://pubmlst.org/neisseria/info/complexes.shtml. Accessed 5 Sep 2016.
- 47.Leavis HL, Bonten MJ, Willems RJ. Identification of high-risk enterococcal clonal complexes: global dispersion and antibiotic resistance. Curr Opin Microbiol. 2006;9(5):454–60. doi: 10.1016/j.mib.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 48.Boyd DA, Levesque S, Picard AC, Golding GR. Vancomycin-resistance Enterococcus faecium harbouring vanN in Canada: a case and complete sequence of pEfm12493 harbouring the vanN operon. J Antimicrob Chemother. 2015;70(7):2163–5. doi: 10.1093/jac/dkv057. [DOI] [PubMed] [Google Scholar]
- 49.Camargo IL, Gilmore MS, Darini AL. Multilocus sequence typing and analysis of putative virulence factors in vancomcyin-resistant and vancomcyin-sensistive Enterococcus faecium isolates from Brazil. Clin Microbiol Infect. 2006;12(11):1123–30. doi: 10.1111/j.1469-0691.2006.01496.x. [DOI] [PubMed] [Google Scholar]
- 50.Sun J, Song X, Kristiansen BE, Kjaereng A, Willems RJ, Eriksen HM, et al. Enterococci in marginal and apical periodontitis: occurrence, population structure and antimicrobial resistance. J Clin Microbiol. 2009;47(7):2218–25. doi: 10.1128/JCM.00388-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nallapareddy SR, Weinstock GM, Murray BE. Clinical isolates of Enterococcus faecium exhibit strain-specific collagen binding mediated by Acm, a new member of the MSCRAMM family. Mol Microbiol. 2003;47(6):1733–47. doi: 10.1046/j.1365-2958.2003.03417.x. [DOI] [PubMed] [Google Scholar]
- 52.Rich RL, Kreikemeyer B, Owens RT, LaBrenz S, Narayana SV, Weinstock GM, et al. Ace is a collagen-binding MSCRAMM from Enterococcus faecalis. J Biol Chem. 1999;274(38):26939–45. doi: 10.1074/jbc.274.38.26939. [DOI] [PubMed] [Google Scholar]
- 53.Lowe AM, Lambert PA, Smith AW. Cloning of an Enterococcus faecalis endocarditis antigen: homology with adhesins from some oral streptococci. Infect Immun. 1995;63(2):703–6. doi: 10.1128/iai.63.2.703-706.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh KV, Coque TM, Weinstock GM, Murray BE. In vivo testing of an Enterococcus faecalis efaA mutant and use of efaA homologs for species identification. FEMS Immunol Med Microbiol. 1998;21(4):323–31. doi: 10.1111/j.1574-695X.1998.tb01180.x. [DOI] [PubMed] [Google Scholar]
- 55.Nallapareddy SR, Singh KV, Sillanpaa J, Garsin DA, Hook M, Erlandsen SL, et al. Endocarditis and biofilm-associated pili of Enterococcus faecalis. J Clin Invest. 2006;116(10):2799–807. doi: 10.1172/JCI29021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su YA, Sulavik MC, He P, Makinen KK, Makinen P-L, Fiedler S, et al. Nucleotide sequence of the gelatinase gene (gelE) from Enterococcus faecalis subsp. Liquefaciens Infect Immun. 1991;59(1):415–20. doi: 10.1128/iai.59.1.415-420.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hancock LE, Perego M. Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J Bacteriol. 2004;186:7951–8. doi: 10.1128/JB.186.23.7951-7958.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakayama J, Chen S, Oyama N, Nishiguchi K, Azab EA, Tanaka E, et al. Revised model for Enterococcus faecalis fsr quorum-sensing system: the small open reading frame fsrD encodes the gelatinase biosynthesis-activating pheromone propeptided corresponding to staphylococcal AgrD. J Bacteriol. 2006;188(23):8321–6. doi: 10.1128/JB.00865-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin X, Singh KV, Weinstock GM, Murray BE. Effects of Enterococcus faecalis fsr genes on production of gelatinase and a serine protease and virulence. Infect Immun. 2000;68(5):2579–86. doi: 10.1128/IAI.68.5.2579-2586.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clewell DB. Bacterial sex pheromone – induced plasmid transfer. Cell. 1993;73:9–12. doi: 10.1016/0092-8674(93)90153-H. [DOI] [PubMed] [Google Scholar]
- 61.Handwerger S, Pucci MJ, Kolokathis A. Vancomycin resistance is encoded on a pheromone response plasmid in Enterococcus faecium 228. Antimicrob Agents Chemother. 1990;34(2):358–60. doi: 10.1128/AAC.34.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Magi G, Capretti R, Paoletti C, Pietrella M, Ferrante L, Biavasco F, et al. Presence of a vanA-carrying pheromone response plasmid (pBRG1) in a clinical isolate of Enterococcus faecium. Antimicrob Agents Chemother. 2003;47(5):1571–6. doi: 10.1128/AAC.47.5.1571-1576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wirth R. The sex pheromone system of Enterococcus faecalis. More than just a plasmid-collection mechanism? Eur J Biochem. 1994;222(2):235–46. doi: 10.1111/j.1432-1033.1994.tb18862.x. [DOI] [PubMed] [Google Scholar]
- 64.Clewell DB. Properties of Enterococcus faecalis plasmid pAD1, a member of a widely disseminated family of pheromone-responding, conjugative, virulence elements encoding cytolysin. Plasmid. 2007;58(3):205–27. doi: 10.1016/j.plasmid.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 65.Semedo T, Santos MA, Lopes MF, Figueiredo Marques JJ, Barreto Crespo MT, Tenreiro R. Virulence factors in food, clinical and reference enterococci: a common trait in the genus? System Appl Microbiol. 2003;26(1):13–22. doi: 10.1078/072320203322337263. [DOI] [PubMed] [Google Scholar]
- 66.Iweriebor BC, Obi LC, Okoh AI. Virulence and antimicrobial resistance factors in Enterococcus spp. Isolated from fecal samples from piggery farms in Eastern Cape, South Africa. BMC Microbiol. 2015;15:136. doi: 10.1186/s12866-015-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jimenez E, Ladero V, Chico I, Maldonado-Barragan A, Lopez M, Martin V, et al. Antibiotic resistance, virulence determinants and production of biogenic amines among enterococci from ovine, feline, canine, porcine and human milk. BMC Microbiol. 2013;13:288. doi: 10.1186/1471-2180-13-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eaton TJ, Gasson MJ. Molecular screening of Enterococcus virulence determinants and potential for genetic exchange between food and medical isolates. Appl Environ Microbiol. 2001;67(4):1628–35. doi: 10.1128/AEM.67.4.1628-1635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Franz CM, Muscholl-Silberhorn AB, Yousif NM, Vancanneyt M, Swings J, Holzapfel WH. Incidence of virulence factors and antibiotic resistance among enterococci isolated from food. Appl Environ Microbiol. 2001;67(9):4385–9. doi: 10.1128/AEM.67.9.4385-4389.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kristich CJ, Rice LB, Arias CA. Enterococcal infection – treatment and antibiotic resistance. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: from commensals to leading causes of drug resistant infection. Boston: Massachusetts Eye and Ear Infirmary; 2014. [PubMed] [Google Scholar]
- 71.Leclercq R, Dutka-Malen S, Duval J, Courvalin P. Vancomycin resistance gene vanC is specific to Enterococcus gallinarum. Antimicrob Agents Chemother. 1992;36(9):2005–8. doi: 10.1128/AAC.36.9.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Navarro F, Courvalin P. Analysis of genes encoding D-Alanine-D-Alanine ligase-related enzymes in Enterococcus casseliflavus and Enterococcus flavescens. Antimicrob Agents Chemother. 1994;38(8):1788–93. doi: 10.1128/AAC.38.8.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gold HS. Vancomycin-resistant enterococci: mechanisms and clinical observations. Clin Infect Dis. 2001;33(2):210–9. doi: 10.1086/321815. [DOI] [PubMed] [Google Scholar]
- 74.Health Canada . Uses of antimicrobials in food animals in Canada: impact on resistance and human health. 2002. [Google Scholar]
- 75.Portillo A, Ruiz-Larrea F, Zarazaga M, Alonso A, Martinez JL, Torres C. Macrolide resistance genes in Enterococcus spp. Antimicrob Agents Chemother. 2000;44(4):967–71. doi: 10.1128/AAC.44.4.967-971.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beukers AG, Zaheer R, Goji N, Cook SR, Amoako KK, Chaves AV, et al. Draft genome sequence of an Enterococcus thailandicus strain isolated from bovine feces. Genome Announc. 2016;4(4):e00576–16. doi: 10.1128/genomeA.00576-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Costa Y, Galimand M, Leclercq R, Duval J, Courvalin P. Characterization of the chromosomal aac(6′)-Ii gene specific for Enterococcus faecium. Antimicrob Agents Chemother. 1993;37(9):1896–903. doi: 10.1128/AAC.37.9.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Del Campo R, Galan JC, Tenorio C, Ruiz-Garbajosa P, Zarazaga M, Torres C, et al. New aac(6′)-I gene in Enterococcus hirae and Enterococcus durans: effect on β-lactam/aminoglycoside synergy. J Antimicrob Chemother. 2005;55(6):1053–5. doi: 10.1093/jac/dki138. [DOI] [PubMed] [Google Scholar]
- 79.Roberts MC. Update on acquired tetracycline resistance genes. FEMS Microbol Lett. 2005;245(2):195–203. doi: 10.1016/j.femsle.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 80.Anderson JF, Parrish TD, Akhtar M, Zureck L, Hirt H. Antibiotic resistance of enterococci in American bison (Bison bison) from a nature preserve compared to that of enterococci in pastured cattle. Appl Environ Microbiol. 2008;74(6):1726–30. doi: 10.1128/AEM.02164-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aaerestrup FM, Agerso Y, Gerner-Smidt P, Madsen M, Jensen LB. Comparision of antimicrobial resistance phenotypes and resistance genes in Enterocococcus faecalis and Enterococcus faecium from humans in the community, broilers, and pigs in Denmark. Diagn Micobiol Infect Dis. 2000;37(2):127–37. doi: 10.1016/S0732-8893(00)00130-9. [DOI] [PubMed] [Google Scholar]
- 82.Poeta P, Costa D, Saenz Y, Klibi N, Ruiz-Larrea F, Rodrigues J, et al. Characterization of antibiotic resistance genes and virulence factors in faecal enterococci of wild animals in Portugal. J Vet Med. 2005;52(9):396–402. doi: 10.1111/j.1439-0450.2005.00881.x. [DOI] [PubMed] [Google Scholar]
- 83.Poeta P, Costa D, Rodrigues J, Torres C. Antimicrobial resistance and the mechanisms implicated in faecal enterococci from healthy humans, poultry and pets in Portugal. Int J Antimicrob Agents. 2006;27(2):131–7. doi: 10.1016/j.ijantimicag.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 84.Holzel CS, Harms KS, Kuchenhoff H, Kunz A, Muller C, Meyer K, et al. Phenotypic and genotypic bacterial antimicrobial reisstance in liquid pig manure is variously associated with contents of tetracyclines and sulfonamides. J Appl Microbiol. 2010;108(5):1642–56. doi: 10.1111/j.1365-2672.2009.04570.x. [DOI] [PubMed] [Google Scholar]
- 85.Roberts MC. Resistance to tetracyclines, macrolide-lincosamide-streptogramin, trimethoprim, and sulfonamide drug classes. Mol Biotechnol. 2002;20(3):261–83. doi: 10.1385/MB:20:3:261. [DOI] [PubMed] [Google Scholar]
- 86.Burrus V, Pavlovic G, Decaris B, Guedon G. Conjugative transposons: the tip of the iceberg. Mol Microbiol. 2002;46(3):601–10. doi: 10.1046/j.1365-2958.2002.03191.x. [DOI] [PubMed] [Google Scholar]
- 87.Bennett PM. Plasmid encoded antibiotic resistance: acquisition and transfer of antibiotic resistance genes in bacteria. Br J Pharmacol. 2008;153:S347–57. doi: 10.1038/sj.bjp.0707607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roberts AP, Mullany P. Tn916-like genetic elements: a diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol Rev. 2011;35(5):856–71. doi: 10.1111/j.1574-6976.2011.00283.x. [DOI] [PubMed] [Google Scholar]
- 89.Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Microbiol. 2010;8(8):552–63. doi: 10.1038/nrmicro2382. [DOI] [PubMed] [Google Scholar]
- 90.Werner G, Coque TM, Franz CM, Grohmann E, Hegstad K, Jensen L, et al. Antibiotic resistant enterococci – tales of a drug resistance gene trafficker. Int J Med Microbiol. 2013;303(6–7):360–79. doi: 10.1016/j.ijmm.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 91.Shaw JH, Clewell DB. Complete nucleotide sequence of macrolide-lincosamide-streptogramin B-resistance transposon Tn917 in Streptococcus faecalis. J Bacteriol. 1985;164(2):782–96. doi: 10.1128/jb.164.2.782-796.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nicolas E, Lambin M, Dandoy D, Galloy C, Nguyen N, Oger CA, et al. The Tn3-family of replicative transposons. Microbiol. Spectrum 2015;3(4): doi: 10.1128/microbiolspec.MDNA3-0060-2014. [DOI] [PubMed]
- 93.Burrus V, Waldor MK. Shaping bacterial genomes with integrative and conjugative elements. Res Microbiol. 2004;155(5):376–86. doi: 10.1016/j.resmic.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 94.Ciric L, Jasni A, ElviradeVries L, Agerso Y, Mullany P, Roberts AP. The Tn916/Tn1545 family of conjugative transposons. In: Roberts AP, Mullany P, editors. Bacterial and integrative mobile genetic elements. Austin: Landes Bioscience; 2013. [Google Scholar]
- 95.Hegstad K, Mikalsen T, Coque TM, Werner G, Sundsfjord A. Mobile genetic elements and their contribution to the emergence of antimicrobial resistant Enterococcus faecalis and Enterococcus faecium. Clin Microbiol Infect. 2010;16(6):541–54. doi: 10.1111/j.1469-0691.2010.03226.x. [DOI] [PubMed] [Google Scholar]
- 96.Novais C, Freitas AR, Silveira E, Bacquero F, Peixe L, Roberts AP, et al. Different genetic supports for the tet(S) gene in enterococci. Antimicrob Agents Chemother. 2012;56(11):6014–8. doi: 10.1128/AAC.00758-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Flannagan SE, Zitzow LA, Su YA, Clewell DB. Nucleotide sequence of the 18-kb conjugative transposon Tn916 from Enterococcus faecalis. Plasmid. 1994;32(3):350–4. doi: 10.1006/plas.1994.1077. [DOI] [PubMed] [Google Scholar]
- 98.Franke AE, Clewell DB. Evidence for a chromosome-borne resistance transposon (Tn916) in Streptococcus faecalis that is capable of “conjugal” transfer in the absence of a conjugative plasmid. J Bacteriol. 1981;145(1):494–502. doi: 10.1128/jb.145.1.494-502.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rice LB. Tn916 family conjugative transposons and dissemination of antimicrobial resistance determinants. Antimicrob Agents Chemother. 1998;42(8):1871–7. doi: 10.1128/aac.42.8.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.de Vries LE, Hasman H, Jurado Rabadán S, Agersø Y. Sequence-based characterization of Tn5801-like genomic islands in tetracycline-resistant Staphylococcus pseudintermedius and other Gram-positive bacteria from humans and animals. Front Microbiol. 2016;7:576. doi: 10.3389/fmicb.2016.00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Da Cunha V, Davies MR, Douarre PE, Rosinski-Chupin I, Margarit I, Spinali S, et al. Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline. Nat Commun. 2014;5:4544. doi: 10.1038/ncomms5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Celli J, Trieu-Cuot P. Circularization of Tn916 is required for expression of the transposon-encoded transfer functions: characterization of long tetracycline-inducible transcripts reading through the attachment site. Mol Microbiol. 1998;28(1):103–17. doi: 10.1046/j.1365-2958.1998.00778.x. [DOI] [PubMed] [Google Scholar]
- 103.Werner G, Hildebrandt B, Witte W. Aminoglycoside-streptothricin resistance gene cluster aadE-sat4-aphA-3 disseminated among multiresistant isolates of Enterococcus faecium. Antimicrob Agents Chemother. 2001;45(11):3267–9. doi: 10.1128/AAC.45.11.3267-3269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Derbise A, Aubert S, El Solh N. Mapping the regions carrying the three contiguous antibiotic resistance genes aadE, sat4, and aphA-3 in the genomes of staphylococci. Antimicrob Agents Chemother. 1997;41(5):1024–32. doi: 10.1128/aac.41.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palmieri C, Mingoia M, Massidda O, Giovanetti E, Varaldo PE. Streptococcus pneumoniae transposon Tn1545/Tn6003 changes to Tn6002 due to spontaneous excision in circular form of the erm(B)- and aphA3- containing macrolide-aminoglycoside-streptothricin (MAS) element. Antimicrob Agents Chemother. 2012;56(11):5994–7. doi: 10.1128/AAC.01487-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mazaheri Nezhad Fard R, Barton MD, Heuzenroeder MW. Bacteriophage-mediated transduction of antibioitic resistance in enterococci. Lett Appl Microbiol. 2011;52(6):559–64. doi: 10.1111/j.1472-765X.2011.03043.x. [DOI] [PubMed] [Google Scholar]
- 107.Yasmin A, Kenny JG, Shankar J, Darby AC, Hall N, Edwards C, et al. Comparative genomics and transduction potential of Enterococcus faecalis temperate bacteriophages. J Bacteriol. 2010;192(4):1122–30. doi: 10.1128/JB.01293-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Duerkop BA, Palmer KL, Hornsburgh MJ. Enterococcal bacteriophages and genome defense. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: from commensals to leading causes of drug resistant infection. Boston: Massachusetts Eye and Ear Infirmary; 2014. [PubMed] [Google Scholar]
- 109.Mazaheri Nezhad Fard R, Barton MD, Heuzenroeder MW. Novel bacteriophage in Enterococcus spp. Curr Microbiol. 2010;60(6):400–6. doi: 10.1007/s00284-009-9555-z. [DOI] [PubMed] [Google Scholar]
- 110.Berg Miller ME, Yeoman CJ, Chia N, Tringe SG, Angly FE, Edwards RA, et al. Phage-bacteria relationships and CRISPR elements revealed by a metagenomic survey of the rumen microbiome. Environ Microbiol. 2012;14(1):207–27. doi: 10.1111/j.1462-2920.2011.02593.x. [DOI] [PubMed] [Google Scholar]
- 111.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, et al. An updated evolutionary classifcation of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13(11):722–36. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9(6):467–77. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Borst LB, Suyemoto MM, Scholl EH, Fuller FJ, Barnes HJ. Comparative genomic analysis identifies divergent genomic features of pathogenic Enterococcus cecorum including a type IC CRISPR-Cas system, a capsule locus, an epa-like locus, and putative host tissue binding proteins. PLoS One. 2015;10(4):e0121294. doi: 10.1371/journal.pone.0121294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Katyal I, Chaban B, Ng B, Hill JE. CRISPRs of Enterococcus faecalis and E. hirae isolates from pig feces have species-specific repeats but share some common spacer sequences. Microb. Ecol. 2013;66(1):182–8. doi: 10.1007/s00248-013-0217-0. [DOI] [PubMed] [Google Scholar]
- 115.Chylinski K, Makarova KS, Charpentier E, Koonin EV. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42(10):6091–105. doi: 10.1093/nar/gku241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–97. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 117.Palmer KL, Gilmore MS. Multidrug-resistant enterococci lack CRISPR-cas. mBio. 2010;1(4):e00227–10. doi: 10.1128/mBio.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sorek R, Kunin V, Hugenholtz P. CRISPR – a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6(3):181–6. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- 119.Yang SC, Lin CH, Sung ST, Fang JY. Antibacterial activities of bacteriocins: application in foods and pharmaceuticals. Front Microbiol. 2014;5:241. doi: 10.3389/fmicb.2014.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nes IF, Diep DB, Ike Y. Enterococcal bacteriocins and antimicrobial proteins that contribute to niche control. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: from commensals to leading causes of drug resistant infection. Boston: Massachusetts Eye and Ear Infirmary; 2014. [PubMed] [Google Scholar]
- 121.Eijsink VG, Axelsson L, Diep DB, Havarstein LS, Holo H, Nes IF. Production of class II bacteriocins by lactic acid bacteria; an example of biological warfare and communication. Antonie Van Leeuwenhoek. 2002;81(1–4):639–54. doi: 10.1023/A:1020582211262. [DOI] [PubMed] [Google Scholar]
- 122.Aymerich T, Holo H, Havarstein LS, Hugas M, Garriga M, Nes IF. Biochemical and genetic characterization of Enterocin A from Enterococcus faecium, a new antilisterial bacteriocin in the pediocin family of bacteriocins. Appl Environ Microbiol. 1996;62(5):1676–82. doi: 10.1128/aem.62.5.1676-1682.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Casaus P, Nilsen T, Cintas LM, Nes IF, Hernandez PE, Holo H. Enterocin B, a new bacteriocin from Enterococcus faecium T136 which can act synergistically with enterocin A. Microbiology. 1997;143:2287–94. doi: 10.1099/00221287-143-7-2287. [DOI] [PubMed] [Google Scholar]
- 124.Yamada Y, Cane DE, Ikeda H. Diversity and analysis of bacteria terpene syntases. Methods Enzymol. 2012;515:123–62. doi: 10.1016/B978-0-12-394290-6.00007-0. [DOI] [PubMed] [Google Scholar]
- 125.Yamada Y, Kuzuyama T, Komatsu M, Shin-Ya K, Omura S, Cane DE, et al. Terpene synthases are widely distributed in bacteria. Proc Natl Acad Sci U S A. 2015;112(3):857–62. doi: 10.1073/pnas.1422108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are include within the article and its additional files. The draft genome sequences of E. hirae_1, E. hirae_2, E. hirae_3, E. hirae_4, E. hirae_5, E. hirae_6, E. hirae_7, E. hirae_8, E. hirae_9, E. hirae_10, E. faecium_11, E. faecium_12, E. faecium_13, E. thailandicus_14, E. villorum_15, E. villorum_16, E. faecalis_17, E. gallinarum_18, E. durans_19, E. casseliflavus_20, E. cassilflavus_21 were submitted to GenBank (BioProjects IDs PRJNA342049 and PRJNA224116).