

Abstract
Background and Purpose
Stroke and Alzheimer's disease (AD) are related pathologies in which the cerebrovascular system is involved. Plasma levels of semicarbazide‐sensitive amine oxidase/vascular adhesion protein 1 (SSAO/VAP‐1, also known as Primary Amine Oxidase ‐PrAO) are increased in both stroke and AD patients and contribute to the vascular damage. During inflammation, its enzymatic activity mediates leukocyte recruitment to the injured tissue, inducing damage in the blood‐brain barrier (BBB) and neuronal tissue. We hypothesized that by altering cerebrovascular function, SSAO/VAP‐1 might play a role in the stroke–AD transition. Therefore, we evaluated the protective effect of the novel multitarget‐directed ligand DPH‐4, initially designed for AD therapy, on the BBB.
Experimental Approach
A human microvascular brain endothelial cell line expressing human SSAO/VAP‐1 was generated, as the expression of SSAO/VAP‐1 is lost in cultured cells. To simulate ischaemic damage, these cells were subjected to oxygen and glucose deprivation (OGD) and re‐oxygenation conditions. The protective role of DPH‐4 was then evaluated in the presence of methylamine, an SSAO substrate, and/or β‐amyloid (Aβ).
Key Results
Under our conditions, DPH‐4 protected brain endothelial cells from OGD and re‐oxygenation‐induced damage, and also decreased SSAO‐dependent leukocyte adhesion. DPH‐4 was also effective at preventing the damage induced by OGD and re‐oxygenation in the presence of Aβ as a model of AD pathology.
Conclusions and Implications
From these results, we concluded that the multitarget compound DPH‐4 might be of therapeutic benefit to delay the onset and/or progression of the neurological pathologies associated with stroke and AD, which appear to be linked.
Abbreviations
- Aβ
β amyloid peptide
- AD
Alzheimer's disease
- BuChe
butyrylcholinesterase
- G418
geneticine
- hCMEC/D3
human cerebral microvascular endothelial cells/D3
- MA
methylamine
- MTDL
multitarget‐directed ligand
- OGD
oxygen–glucose deprivation
- SC
semicarbazide
- SMC
smooth muscle cells
- WT
wild type
Tables of Links
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
There is increasing evidence suggesting that the neurovasculature plays an important role in the onset and progression of neurological disorders like Alzheimer's disease (AD) (Zlokovic, 2008; Grammas, 2011; Marchesi, 2014). In this regard, the concept of ‘neurovascular unit’, integrated by neurons, astrocytes and vascular cells, constitutes a functional unit able to maintain the homeostasis of the brain microenvironment (Iadecola, 2010). Stroke is a vascular disorder and the second leading cause of death worldwide and is prevalent in elderly people. Inflammation and oxidative stress accumulate during human ageing and exacerbate the vascular damage following a stroke incident (DiNapoli et al., 2008). Also, the immune system may contribute to infarct progression (Iadecola and Anrather, 2011). In fact, the observation that a high percentage of patients that have suffered stroke subsequently develop AD suggests that there is a strong link between these two pathologies. In this regard, hypoxia and ischaemic injury induce the up‐regulation of Beta‐secretase 1 (BACE‐1) that then increases the formation of β‐amyloid (Aβ) (Guglielmotto et al., 2009). In addition, endothelial cells activated by hypoxia during stroke produce free radical species, which induce the expression of adhesion molecules such as vascular adhesion protein 1 (VAP‐1) that mediates the recruitment of leukocytes. These leukocytes then infiltrate through the blood‐brain barrier (BBB) to the injured tissue, mediate the release of cytokines, which further damage the tissues.
VAP‐1, a homodimeric glycoprotein, is an enzyme that binds leukocytes through its semicarbazide ‐sensitive amine oxidase (SSAO; EC 1.4.3.21; also known as PrAO) activity (Smith et al., 1998; Jalkanen and Salmi, 2008). As an enzyme, VAP‐1 metabolizes primary amines producing aldehydes, hydrogen peroxide and ammonia, which are able to induce cellular damage when overproduced (Yu and Deng, 1998). SSAO/VAP‐1, either present at the cell membrane or in a soluble form, released into blood plasma, is altered in several human pathologies (Kurkijärvi et al., 1998; Boomsma et al., 2003) including AD (Ferrer et al., 2002; Hernández et al., 2005) and cerebral ischaemia (Airas et al., 2008). The mediators that induce these alterations in SSAO/VAP‐1 levels are still unknown, but it is believed that increased SSAO/VAP‐1 levels may contribute to the physiopathology of these diseases (Conklin et al., 1998; Solé et al., 2008), therefore, representing a potential therapeutic target. In addition, increased human plasma SSAO activity is a strong predictor of parenchymal haemorrhage in ischaemic stroke patients following treatment with tissue plasminogen activator (Hernández‐Guillamón et al., 2010). Furthermore, plasma SSAO activity, also elevated in haemorrhagic stroke patients, predicts neurological outcome (Hernández‐Guillamón et al., 2012). Altogether, these data suggest that SSAO/VAP‐1 by altering the function of cerebrovascular tissue, may contribute to the association between stroke and the development of AD.
AD is a complex disease for which therapy has been long focused on one of its multiple pathological signs, the depletion of basal forebrain cholinergic neurons with the subsequent decrease in cholinergic transmission (Perry et al., 1977; Geula and Mesulam, 1999). At present, no drug has been able to successfully prevent or cure the neurodegenerative process of AD. The Food and Drug Administration (FDA, US)‐approved drugs for the treatment of AD are based on the cholinergic hypothesis of AD and, therefore, are aimed at increasing cholinergic transmission to recover cognitive function. To date, only four drugs (rivastigmine, galantamine, donepezil and memantine) have been approved by the FDA for the treatment of AD (Birks and Harvey, 2006). Among them, donepezil has shown some temporary efficacy but fails to reverse the cognitive decline. In the search for more effective therapies, the ‘one drug, multiple target’ strategy, also called multitarget‐directed ligand (MTDL) approach, might be more appropriate (Buccafusco and Terry, 2000; Youdim and Buccafusco, 2005; Bolea et al., 2013; León et al. 2013). This strategy suggests the use of compounds with multiple activities for different biological targets (Cavalli et al., 2008). In this context, we recently designed a novel series of derivatives based on the hybridization of selected moieties from donepezil, propargylamine and 8‐hydroxyquinoline (DPH); these were synthesised and pharmacologically evaluated for the potential prevention and treatment of AD (Wang et al., 2014). They behaved as dual inhibitors of cholinesterase (AChE and BuChE) and MAO (MAO A and MAO B) activities, both altered in AD, and indicated potential for the prevention and treatment of this neurological disorder. Among them, the hybrid DPH‐4 showed strong biometal chelating properties against Cu2+ and Fe2+, good absorption, distribution, metabolism, elimination and toxicity (ADMET) properties and brain penetration capacity, as well as significantly attenuating scopolamine‐induced learning deficits in healthy adult mice (Wang et al., 2014). Additionally, compound DPH‐4 (Figure 1) inhibited bovine SSAO activity in the low micromolar range (IC50 = 2.8 ± 0.7 μM).
Figure 1.
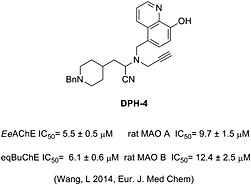
Structure of the multifunctional compound DPH‐4. DPH‐4 inhibits AChE (Ee, electric eel), BuChE (eq, equine) and MAO (A and B) enzymes, showing biometal‐chelating properties, as described in the corresponding publication.
Taking into account that stroke and AD are related pathologies and that one can trigger the progression of the other, the aim of this work was to analyse whether the beneficial effect of DPH‐4, previously observed in an experimental model of AD (Wang et al., 2014), could also exhibit a protective effect on a new in vitro experimental model of cerebral ischemia. This model uses human cerebral microvascular endothelial cells expressing the human SSAO/VAP‐1 protein [hCMEC/D3 h SSAO/VAP‐1] as a model of the BBB. Obtaining SSAO/VAP‐1‐expressing cells was an essential preliminary step as the expression of this protein is lost in cultured cells.
Methods
Cell culture and transfection
The human cerebral microvascular endothelial cell line hCMEC/D3 was obtained from co‐authors from the Institut COCHIN (Paris, France) as previously described (Weksler et al., 2005) as a model of the BBB. hCMEC/D3 cells were cultured as recommended, on 150 µg · mL−1 collagen type I (Rat Tail; Corning New York, NY, USA)‐coated plates in EBM‐2 (Lonza Basel, Switzerland) medium supplemented with 5% FBS (Life Technologies (Thermo) Waltham, MA, USA), 1.4 μM hydrocortisone (Sigma St Louis, MO, USA), 5 µg · mL−1 ascorbic acid (Sigma), 1% chemically defined lipid concentrate (Life Technologies), 10 mM HEPES (Life Technologies) and 1 ng · mL−1 human basic fibroblast growth factor (Sigma). human SSAO/VAP‐1‐expressing HUVEC and rat smooth muscle cells (SMC) [A7r5 hSSAO/VAP‐1] were previously developed in our group, and cultured as described previously (Solé et al., 2007; Solé and Unzeta, 2011). THP‐1 monocytic cells were obtained from the American Type Culture Collection (ATCC Manassas, VA, USA) and grown in RPMI 1640 medium (Life Technologies) supplemented with 10% FBS. All cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Geneticine (G418, 100 µg · mL−1; Invitrogen) was added to the culture medium of HUVEC and SMC cells to ensure the maintenance of hSSAO/VAP‐1 DNA.
In order to obtain the hCMEC/D3 cell line stably expressing the hSSAO/VAP‐1, wild type (WT), hCMEC/D3 cells were transfected with a PcDNA3.1(+) vector containing the hSSAO/VAP‐1 cDNA (Solé et al., 2007) using the Fugene® HD transfection reagent (Roche Indianapolis, IN, USA) according to the manufacturer's conditions. After transfection, cells were selected by the addition of G418 antibiotic (800 µg · mL−1) for 1–2 months. Then, cells were diluted to allow the formation of monoclonal colonies in the presence of 200 µg · mL−1 G418, an antibiotic concentration that was used thereafter for cell maintenance. Cell colonies were amplified and checked for SSAO/VAP‐1 expression and activity before being frozen.
Cell lysates and concentrated culture medium
Cells were collected and homogenized in 50 mM Tris‐HCl (pH 7.5), containing 1% Triton X‐100, 10 mM EDTA and a protease inhibitor cocktail (Sigma) (1:100) and were sonicated for 10 s. To obtain concentrated culture medium samples, culture media were collected after cell treatments and centrifuged at 4400 g for 10 min to eliminate dead cells and debris. Then media samples were lyophilized by evaporation in a Refrigerated CentriVap Concentrator (Labconco Kansas City, MO, USA) and reconstituted in a smaller, known volume of distilled water to obtain 10‐fold concentrated culture medium.
Sub‐cellular fractions
Membrane‐enriched preparations were obtained by homogenization of cells in 10 mM HEPES, 1.5 mM MgCl2 and 10 mM KCl buffer at pH 7.9, containing protease inhibitor cocktail. After centrifugation at 2000 g for 15 min at 4 °C, the resulting supernatant was ultracentrifuged at 100 000 g (Sorvall Discovery M120 SE Thermo, Waltham, MA, USA) for 30 min at 4 °C to separate the soluble cytosolic fraction from the pellet containing the membrane‐enriched fraction.
Lipid raft‐enriched fractions were obtained by scraping the cells in PBS, recovering them by centrifugation for 5 min at 800 g and then reconstituting the pellet in 450 μL of 50 mM Tris‐HCl, 150 mM NaCl, 1 mM EDTA and 1% Brij 98 buffer at pH 7.2, containing protease inhibitor cocktail. After 15 min incubation at 37 °C under continuous agitation, samples were centrifuged for 10 min at 2000 g to discard nuclei. The supernatants were mixed with 450 μL of 90% sucrose in Tris‐HCl buffer to obtain 45% sucrose fractions, which were deposited at the bottom of ultracentrifuge tubes. Two additional fractions of 35% (2 mL) and 5% (0.8 mL) sucrose were added to the former to generate a sucrose gradient, and then the samples were centrifuged for 19 h at 120 000 g. Ten 370 μL fractions were recovered and analysed by Western blot to identify the lipid raft and soluble membrane‐enriched fractions.
OGD model and cell treatment
Combined oxygen and glucose deprivation (OGD) and re‐oxygenation have been used as an experimental approach to ischaemic stroke. For HUVEC hSSAO/VAP‐1 cells, the ischaemic condition was carried out by subjecting the cells to OGD for 24 h followed by 7 h of re‐oxygenation, as previously described (Sun et al., 2014). For WT and h SSAO/VAP‐1 hCMEC/D3 cells, the OGD treatment was performed in glucose‐free DMEM (Life Technologies) after cells had been washed with glucose‐free PBS, and then the cells were transferred to a temperature‐controlled (37 ± 1 °C) Invivo2 hypoxia workstation (RUSKINN Sanford, ME, USA) containing a gas mixture composed of 5% CO2, 95% N2 and 0.5% O2. Control cells were maintained in DMEM (5 mM glucose) in the incubator under normoxic conditions (5% CO2/95% air). In experiments including re‐oxygenation, cells that had undergone OGD were returned to normoxic conditions after glucose‐free DMEM had been replaced by serum‐free DMEM (5 mM glucose) and adding the same treatments present during OGD. In experiments analysing release of soluble SSAO/VAP‐1, DMEM was not replaced for the re‐oxygenation period, but glucose was added to the OGD samples.
For cell viability and adhesion assays, HUVEC hSSAO/VAP‐1 were seeded at 53 200 cells · mL−1, hCMECs were seeded at 50 000 cells · mL−1, and both were grown for 48 h before the addition of treatments for another 24 h. For immunoblot analysis, cells were seeded at 60 000 cells · mL−1 and grown to 80–90% confluence before treatments. Methylamine (MA), semicarbazide (SC), Aβ1–40D (Aβ1–40 peptide containing the Dutch mutation, Anaspec Fremont, CA, USA) and/or DPH‐4 (Wang et al., 2014) were added into DMEM before the start of OGD. In the re‐oxygenation process, the compounds were maintained at the same concentrations as during OGD. For the preparation of Aβ1–40D, it was pretreated with 1,1,1,3,3,3‐hexa‐fluoro‐2‐propanol (Sigma‐Aldrich St Louis, MO, USA) for more than 4 h but less than 6 h, then aliquoted, evaporated at room temperature and stored at −80 °C until ready for use, then dissolved in sterile PBS containing 0.1% ammonium hydroxide.
Cell viability assay
3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) reduction assay was employed to evaluate the cell viability. Briefly, at the end of the treatments, cells were incubated with 0.5 mg · mL−1 MTT for 3 h in HUVECs and for 1.5 h in hCMECs at 37 °C. The medium was then replaced by DMSO to dissolve the blue formazan precipitate, and colour was spectophotometrically quantified at 560 and 620 nm in a microplate reader (Labsystems Multiskan RC BioTek, Winooski, VT, USA) (Plumb et al., 1989).
Antibodies and Western blot analysis
The antibodies used were rabbit anti‐VAP‐1 (Abcam Cambridge, UK, 1:1000), rabbit anti‐bovine SSAO (1:1000) (Lizcano et al., 1998), rabbit anti‐VCAM‐1 (Epitomics (Abcam, Cambridge, UK), 1:1000), rabbit anti‐ICAM‐1 (GeneTex Irvine, CA, USA, 1:1000), rabbit anti‐P‐selectin (BioVision Milpitas, CA, USA, 1:1000), rabbit anti‐E‐selectin (Santa Cruz Biotechnology Dallas, TX, USA, 1:500), rabbit anti‐IGF1R (insulin‐like growth factor 1 receptor) (Santa Cruz Biotechnology, 1:1000), mouse anti‐flotillin (BD Biosciences Franklin Lakes, NJ, USA, 1:1000), mouse anti‐GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase) (Ambion (Thermo, Waltham, MA, USA), 1:40 000), mouse anti‐transferrin receptor (TfR1; ZYMED (Thermo, Waltham, MA, USA), 1:1000), HRP (horseradish peroxidase)‐conjugated anti‐rabbit IgG (BD Biosciences, 1:2000) and HRP anti‐mouse IgG (Dako Glostrup, Denmark, 1:2000). Equal amounts of protein (20 µg per lane) determined by the Bradford method, or equal volumes of media (45 μL per lane), were separated by SDS‐PAGE and transferred onto nitrocellulose membranes. Membranes were blocked for 1 h with TBS/0.1%‐Tween buffer containing 5% (w v‐1) non‐fatty milk and incubated overnight at 4 °C with the corresponding primary antibodies. After incubation with the secondary antibodies, blots were developed using ECL® Chemiluminescent detection reagents and HPLC Films (GE Healthcare Barcelona, Spain). The imagej software Schneider et al., 2012 was used to quantify the Western blot signals.
Determination of SSAO enzymatic activity
For determining SSAO activity, cells were collected and homogenized in 100 mM Tris‐HCl, pH 9, and containing protease inhibitors cocktail. Enzymatic activity was determined radiochemically by using a modification of the Otsuka and Kobayashi method (Otsuka and Kobayashi, 1964). Briefly, [14C]‐benzylamine hydrochloride (100 μM and 2 mCi · mmol−1, American Radiolabeled Chemicals St Louis, MO, USA) was used as substrate, and 1 μM deprenyl was added to avoid MAO B interference. A 30‐min inhibitory pretreatment of samples was performed at 37 °C with 1 μM deprenyl or deprenyl plus DPH‐4. Reactions were performed at 37 °C for 120 min in 100 mM Tris‐HCl buffer, pH 9.0, adding 25 μL of substrate to the 200 μL of reaction. Eighty to a hundred micrograms of HUVEC hSSAO/VAP‐1 or hCMEC/D3 hSSAO/VAP‐1 cell lysates were used in each reaction. The reaction was stopped by adding 100 μL of 2 M citric acid. The [14C]‐aldehyde products were extracted into 4 mL of toluene/ethylacetate (1:1, v v‐1) solution containing 0.6% (w v‐1) of diphenyloxazole. The amount of [14C]‐aldehyde was quantified using a Tri‐Carb 2810TR liquid scintillation counter (Perkin Elmer Waltham, MA, USA) and the quanta smart 3.0 software (Perkin Elmer).
Adhesion assays
THP‐1 monocytes were labelled with 1 μM calcein‐AM, and at the end of treatments, endothelial cells were incubated with the calcein‐AM‐labelled THP‐1 cells (2.5 × 105 per well in 24‐well plates) for 30 min at 37 °C. Then, unbound monocytes were removed by turning over the plates onto absorbent paper, carefully adding FBS‐free RPMI 1640 medium to the plates with an auto‐pipette, and repeating the washing at least three times. The fluorescence intensity was measured using a fluorescence microplate reader (λex/λem: 495/530 nm). Results are presented as the percentage of fluorescence intensity, referring values to those obtained for the untreated cells.
Analysis of data
Results are given as mean ± SEM of independent experiments. Statistical analysis was performed by one‐way ANOVA and further Newman–Keuls multiple comparison test. P < 0.05 was considered to be statistically significant. Statistical analyses and graphic representations were obtained with the graph‐pad prism 6.0 software GraphPad Software, Inc. La Jolla, CA, USA.
Results
Generation and characterization of the human SSAO/VAP‐1‐expressing hCMEC/D3 cell line
In order to obtain a brain endothelial cell model to study the role of SSAO/VAP‐1 in cerebrovascular tissue in vitro, the immortalized human brain endothelial cell line hCMEC/D3 (Weksler et al., 2005) was stably transfected with hSSAO/VAP‐1 cDNA. This step was necessary because SSAO/VAP‐1 expression is lost in cultured cells (El Hadri et al., 2002). After selection and amplification of G418‐resistant cells, different clones were analysed for SSAO activity and expression (Figure 2A,B). Clones 1 and 2 showed the highest SSAO enzymatic activity using benzylamine as specific substrate, as well as the highest protein expression of SSAO/VAP‐1 by Western blot analysis. The activity levels observed were significantly higher than those obtained with previously generated HUVECs (H) or A7r5 smooth muscle (SMC) SSAO/VAP‐1‐transfected cell lines (Solé et al., 2007; Solé and Unzeta, 2011).
Figure 2.
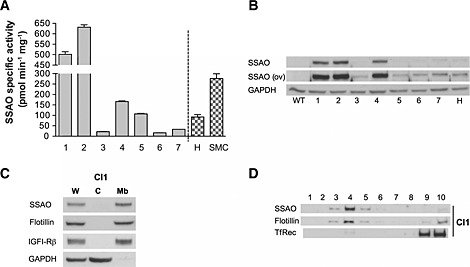
Characterization of the hCMEC/D3 cell line stably transfected with human SSAO/VAP‐1. (A) SSAO‐specific activity (expressed as pmol · min−1 · mg−1 of protein) of the antibiotic‐selected and amplified positive clones (1–7). HUVEC endothelial (H) or A7r5 smooth muscle (SMC) SSAO/VAP‐1‐transfected cell lines are shown for comparison. (B) SSAO/VAP‐1 protein visualized by Western blot corresponding to the same clones as those in (A). Overexposure (ov) of the SSAO Western blot is shown to highlight the weaker bands. GAPDH was used as loading control. WT, wild type (non‐transfected cells). (C) The transfected SSAO protein is located in the membrane fraction and absent from the cytosol in clone 1 cells, shown as example. Flotillin and IGF1Rβ were used as membrane‐positive markers; GAPDH was used as a cytosolic marker. W, whole fraction; C, cytosolic fraction; Mb, membrane fraction. (D) The transfected SSAO/VAP‐1 is located in the lipid raft fractions of the cell membrane; clone 1 is shown as an example. Flotillin was used as a raft‐positive protein, and transferrin receptor (TfR) was used as marker of the soluble membrane fraction.
The correct sub‐cellular localization of the expressed SSAO/VAP‐1 protein was checked by two distinct cell fractionation procedures. In one of these, the membrane and cytosolic fractions were separated, and their subsequent analysis by Western blot showed that SSAO/VAP‐1 expression was associated with the membrane fraction, while it was absent in the cytosol (Figure 2C). Furthermore, SSAO/VAP‐1 was located in the lipid rafts regions as revealed by its presence in flotillin‐containing fractions, a characteristic raft‐positive protein (Figure 2D). The presence of other amine oxidases was also assessed in both WT‐expressing and SSAO/VAP‐1‐expressing cells showing an absence of MAO‐B and moderate MAO‐A activity with slightly increased levels in SSAO/VAP‐1‐expressing cells compared with WT (data not shown).
Effect of OGD and re‐oxygenation on the cell viability of WT‐ and hSSAO/VAP‐1‐expressing hCMEC/D3 cells, and influence of SSAO activity
Both WT and hSSAO/VAP‐1 hCMEC/D3 cells underwent OGD for different time periods (1 to 24 h). This treatment induced a decrease in cell viability reaching 50% of cell death after 24 h of OGD exposure. No differences between either cell type were observed at any of the time‐points analysed (Figure 3A).
Figure 3.
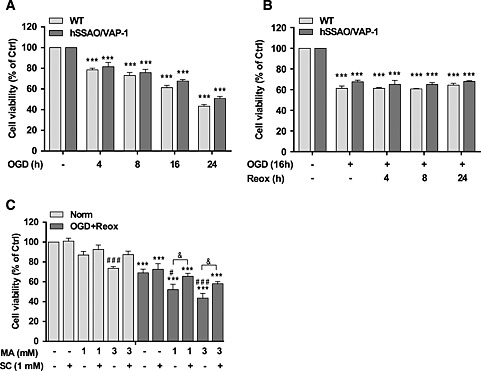
Effect of OGD, OGD with re‐oxygenation and MA on cell viability of WT and human SSAO/VAP‐1‐transfected hCMEC/D3 cells [hCMEC/D3 hSSAO/VAP‐1]. Different durations of OGD (A) or 16 h OGD with different durations of re‐oxygenation (B) induced the death of WT and hSSAO/VAP‐1‐transfected hCMEC/D3 cells. MTT reduction assay was used to assess cell viability under the different assay conditions. Cells without OGD were maintained under normoxic conditions and were considered to be control cells (Ctrl). The metabolism of MA subjected to 16 h OGD plus 24 h of re‐oxygenation (OGD + Reox) (C) induced a reduction in cell viability in hSSAO/VAP‐1‐expressing hCMEC/D3 cells, which was partially prevented by the SSAO activity inhibitor SC. MA (1 or 3 mM) and SC (1 mM) were added before OGD and maintained during re‐oxygenation. Control cells are untreated cells in normoxia (Norm). Data are expressed as mean ± SEM of at least three independent experiments. *** P < 0.001 versus control of the corresponding cell type. # P < 0.05 and ### P < 0.001 versus untreated hSSAO/VAP‐1‐expressing hCMEC/D3 cells in the corresponding condition (Norm or OGD + Reox); & P < 0.05 between the indicated treatments. Statistical analyses were performed by a one‐way ANOVA and the addition of Newman–Keuls multiple comparison test.
A 16 h OGD treatment inducing 40% cell death was selected to further study the effect of different re‐oxygenation times (4 to 24 h) after OGD. Results shown in Figure 3B revealed that the addition of re‐oxygenation did not increase cell death following OGD treatment. In order to assess whether the SSAO catalytic activity versus MA as specific substrate could contribute to the vascular damage under these conditions, a 16 h OGD with 24 h re‐oxygenation treatment was selected. SSAO/VAP‐1‐expressing cells were incubated in the presence of MA (1 and 3 mM) and/or semicarbazide (1 mM) under the OGD with re‐oxygenation conditions selected. Figure 3C shows that the presence of MA enhanced the loss of cell viability induced by OGD with reoxygenation in a dose‐dependent manner, which was partially recovered by SC, a specific SSAO inhibitor. These results confirmed that the SSAO catalytic activity enhanced the cell death induced by OGD with re‐oxygenation conditions.
OGD with re‐oxygenation induces the release of soluble SSAO/VAP‐1 into the culture media by hCMEC/D3 SSAO/VAP‐1 cells
The soluble form of SSAO/VAP‐1 is derived from the membrane‐bound protein by a shedding process that may be activated under some pathological conditions (Abella et al., 2004; Sun et al., 2014). In order to assess whether stroke can induce SSAO/VAP‐1 release from human brain endothelial cells, concentrated culture media of SSAO/VAP‐1‐expressing cells subjected to different periods of OGD and OGD with re‐oxygenation were analysed using TNF‐α as positive control (Figure 4A).
Figure 4.
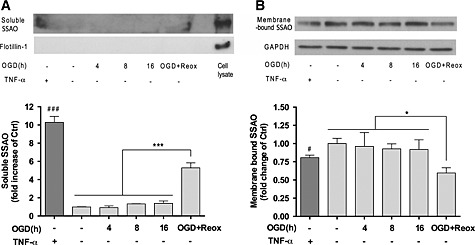
OGD with re‐oxygenation induces the release of soluble SSAO to the culture media by hCMEC/D3 hSSAO/VAP‐1 cells. (A) Levels of soluble hSSAO/VAP‐1 in 10‐fold concentrated culture media, in response to TNF‐α treatment (24 h in normoxia, 100 ng · mL−1), different OGD times and 16 h OGD with 24 h re‐oxygenation (OGD + Reox). TNF‐α was used as a positive control of SSAO/VAP‐1 release. Flotillin‐1 was used as control of cell debris absence in the media. (B) Presence of membrane‐bound SSAO/VAP‐1 in hCMEC/D3 hSSAO/VAP‐1 cell lysates under the same experimental conditions as in (A). The presence of membrane‐bound SSAO was normalized to the GAPDH levels. Untreated cells or media under normoxia conditions were considered control samples (Ctrl). Data in graphs represent the quantification of Western blots and are expressed as mean ± SEM of data obtained from three independent experiments. * P < 0.05 and *** P < 0.001 as indicated; # P < 0.05 and ### P < 0.001 versus control cells, by a one‐way ANOVA and the addition of Newman–Keuls multiple comparison test.
The Western blot results showed soluble SSAO/VAP‐1 release into the medium occurred after 16 h OGD and 24 h re‐oxygenation in contrast to OGD alone. This result correlated with the loss of signal observed in the membrane‐bound form, obtained by analysing the cell lysates under the same experimental conditions (Figure 4B).
DPH‐4 attenuates the cell death induced by the SSAO‐mediated methylamine metabolism in both normoxia and OGD with re‐oxygenation conditions, in a dose‐dependent manner
In order to determine a possible protective effect of DPH‐4 in the stroke conditions, the previously established experimental model with hSSAO/VAP‐1‐expressing hCMEC/D3 cells was compared with a peripheral endothelial model using hSSAO/VAP‐1‐expressing HUVEC cells, in the same conditions previously described (Sun et al., 2014). Both endothelial cell lines were pre‐incubated with DPH‐4 (1 μM) before the addition of MA (3 mM) and subjected to OGD and re‐oxygenation. A significant loss of cell viability was observed in the presence of MA (Figure 5A,B) in normoxia as well as an enhancement of cell toxicity induced by OGD and re‐oxygenation.
Figure 5.
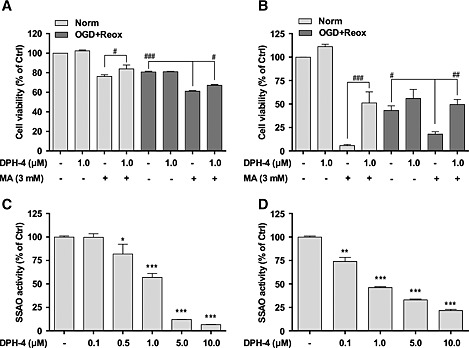
DPH‐4 attenuates the cell death induced by the metabolism of MA in normoxia or in OGD with re‐oxygenation conditions in different endothelial cell types. MTT reduction assay was used to determine the cell viability of hCMEC/D3 hSSAO/VAP‐1 cells subjected to 16 h OGD with 24 h re‐oxygenation (A), and HUVEC hSSAO/VAP‐1 cells subjected to 24 h OGD with 7 h re‐oxygenation (B). MA (3 mM) and DPH‐4 (1 μM) were added before OGD and maintained during re‐oxygenation. Untreated endothelial cells under normoxia were used as control (Ctrl) cells. The inhibition of SSAO activity mediated by DPH‐4 was determined by investigating SSAO activity remaining in hCMEC/D3 hSSAO/VAP‐1 cell lysates after being incubated with DPH‐4 (0.1–10 μM) for 30 min (C), or by evaluating SSAO activity present in HUVEC hSSAO/VAP‐1 cell lysates from DPH‐4‐treated (0.1–10 μM) cells for 24 h (D). SSAO activity was analysed by a radiometric method, using [14C]‐benzylamine as a substrate. Untreated cell lysate was considered the control (Ctrl) sample. Data in graphs are expressed as mean ± SEM and represent data obtained from three independent experiments. * P < 0.05, ** P < 0.01 and *** P < 0.001 versus non‐treated samples; # P < 0.05, ## P < 0.01 and ### P < 0.001 as indicated, by a one‐way ANOVA and the addition of Newman–Keuls multiple comparison test.
The effect of MA was higher in HUVECs (Figure 5B) than in hCMEC/D3 cells (Figure 5A), revealing a greater resistance of the latter, even though they showed higher SSAO/VAP‐1 expression and activity levels (Figure 2). Interestingly, the loss of cell viability was partially restored in both cell types in the presence of DPH‐4, although the protective effect was surprisingly more significant in HUVECs than in hCMEC/D3 (Figure 5B), reaching 50% recovery in normoxia and almost 100% under OGD with re‐oxygenation conditions (Figure 5A), hCMEC/D3 showed less cell toxicity with MA. These results confirmed the protective effect of DPH‐4 on human brain endothelial cells expressing SSAO/VAP‐1 in this experimental stroke model. The protective behaviour of DPH‐4 might be explained by the inhibition of SSAO activity, because it was clearly observed in the presence of MA. Thus, different concentrations of DPH‐4 (0.1–10 μM) were used to determine the inhibition of SSAO activity using [14C]‐benzlylamine as substrate in both hSSAO/VAP‐1‐expressing hCMEC/D3 (Figure 5C) and HUVEC (Figure 5D) cells. Results revealed that DPH‐4 inhibited SSAO activity with a rough IC50 value of 1 μM in both endothelial cell types from different vascular origin.
In order to determine whether this effect was concentration‐dependent, we performed the same analysis using the hCMEC/D3 and different concentrations of DPH‐4 (Figure 6). These results showed that the 5 μM concentration of DPH‐4 was the most effective in protecting hCMEC/D3 cells from OGD and re‐oxygenation when MA was present. No effect was observed in the absence of MA.
Figure 6.
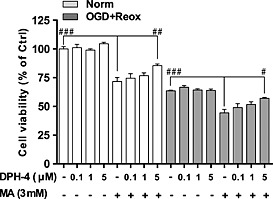
Dose‐response effect of DPH‐4 on hCMEC/D3 cell viability under OGD with re‐oxygenation. MTT reduction assay was used to determine the cell viability of hSSAO/VAP‐1‐expressing hCMEC/D3 cells subjected to normoxia (Norm) or 16 h OGD with 24 h re‐oxygenation (OGD + Reox). MA (3 mM) and DPH‐4 (0.1, 1 and 5 μM) were added before OGD and maintained during re‐oxygenation. Data in graph are expressed as mean ± SEM and represent data obtained from three independent experiments. # P < 0.05, ## P < 0.01 and ### P < 0.001 as indicated, by one‐way ANOVA and the addition of Newman–Keuls multiple comparison test.
DPH‐4 shows an anti‐inflammatory effect, preventing MA‐induced leukocyte binding to hSSAO/VAP‐1‐expressing hCMEC/D3 cells subjected to OGD
BBB dysfunction and the occurrence of haemorrhages after stroke have been associated with leukocyte adhesion to brain endothelium and subsequent transmigration into the injured tissue. Because SSAO/VAP‐1 mediates leukocyte extravasation, the anti‐inflammatory behaviour of DPH‐4 was assessed in hSSAO/VAP‐1‐expressing hCMEC/D3 cells under normoxic and OGD conditions, as well as in the presence of MA, by quantifying the binding of calcein‐AM‐labelled THP‐1 leukocytes to the endothelial cells (Figure 7). The presence of MA induced an increase in leukocyte adhesion to the endothelium in hSSAO/VAP‐1‐expressing cells after short OGD (5 h, Figure 7A), long OGD (16 h, Figure 7b) and OGD with re‐oxygenation (16 + 24 h, Figure 7C), while no effect was observed in WT cells after MA treatment, indicating that this inflammatory effect was induced by the catalytic action of SSAO with MA as a substrate. This effect was prevented when cells were incubated in presence of DPH‐4 in all the conditions assayed.
Figure 7.
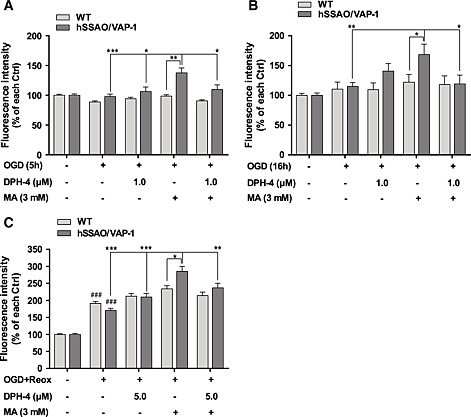
DPH‐4 prevents increased leukocyte adhesion to endothelial cells under different OGD conditions in the presence of the SSAO substrate. Leukocyte–endothelium adhesion assay was performed to analyse the anti‐inflammatory effect of DPH‐4 against the SSAO‐mediated pro‐inflammatory action. WT and hSSAO/VAP‐1 ‐expressing hCMEC/D3 cells treated with MA (3 mM) and/or DPH‐4 (1 or 5 μM) that were subjected to 5 h OGD (A), 16 h OGD (B) or 16 h OGD with 24 h‐re‐oxygenation (OGD + Reox, C), and the binding of calcein‐AM‐labelled THP‐1 leukocytes on these cells was quantified. Untreated cells under normoxic conditions were considered as control (Ctrl) for each type of cell. Data are expressed as mean ± SEM of three independent experiments. * P < 0.05, ** P < 0.01 and *** P < 0.001 versus hCMEC/D3 hSSAO/VAP‐1 treated with MA under OGD conditions; ### P < 0.001 versus control of the corresponding cell type, by one‐way ANOVA and the addition of Newman–Keuls multiple comparison test.
In order to analyse whether other adhesion proteins are involved in this adhesion process after OGD + re‐oxygenation, we also measured SSAO/VAP‐1, VCAM‐1, ICAM‐1, P‐selectin and E‐selectin levels in SSAO/VAP‐1‐expressing hCMEC/D3 cells (Figure 8). ICAM‐1 and E‐selectin showed very low expression levels with no changes among the performed treatments (data not shown). SSAO/VAP‐1 and VCAM‐1 displayed similar behaviour, being decreased after the OGD + re‐oxygenation treatment and with no changes among the pharmacological treatments under these conditions. P‐selectin showed non‐significant changes after OGD + re‐oxygenation or other treatments.
Figure 8.
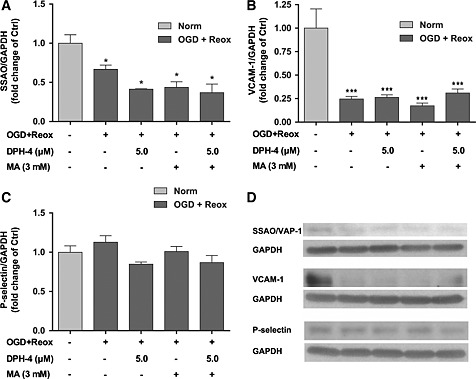
Effect of DPH‐4 on the response of other adhesion molecules to the OGD with re‐oxygenation stimulus and their role in the leukocyte adhesion to hCMEC/D3 endothelial cells. The presence of membrane‐bound SSAO/VAP‐1 (A), VCAM‐1 (B) and P‐selectin (C) in hCMEC/D3 hSSAO/VAP‐1 cell lysates subjected to 16 h OGD with 24 h re‐oxygenation (OGD + Reox) in the presence of DPH‐4 (5 μM) and MA (3 mM). (D) Representative Western blots. The presence of each protein was normalized to the GAPDH levels. Untreated cells were considered to be control samples (Ctrl). Data in graphs represent the quantification of Western blots and are expressed as mean ± SEM of data obtained from at least two independent experiments. * P < 0.05, *** P < 0.001 versus Ctrl, by a one‐way ANOVA test and the addition of Newman–Keuls multiple comparison test.
DPH‐4 protects against cell death induced by the co‐treatment of hCMEC/D3 hSSAO/VAP‐1 cells subjected to OGD with re‐oxygenation with SSAO substrate MA and Aβ1–40D
In order to determine whether the presence of Aβ and SSAO/VAP‐1 promoted vascular damage under ischaemic conditions, as we previously described for normoxia (Solé et al., 2015), cells subjected to normoxia and OGD with re‐oxygenation were incubated in the presence of Aβ1–40D, and cell viability was determined. Both the co‐treatment of MA with Aβ1–40D (Figure 9A) and the 24 h pretreatment with Aβ1–40D (Figure 9B) induced an increased loss of cell viability compared with that induced by either treatment under OGD with re‐oxygenation. The addition of DPH‐4 in the presence of both treatments significantly increased cell viability; similar results were obtained when semicarbazide was used (data not shown).
Figure 9.
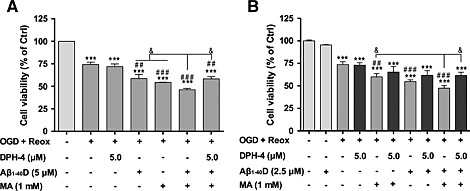
DPH‐4 protects against death caused by increased SSAO activity and Aβ1–40D of hCMEC/D3 hSSAO/VAP‐1 cells subjected to OGD with re‐oxygenation. MTT reduction assay was used to evaluate the cell viability of hCMEC/D3 hSSAO/VAP‐1 cells subjected to 16 h OGD with 24 h re‐oxygenation (OGD + Reox) in the presence of Aβ1–40 containing the Dutch mutation (Aβ1–40D, 2.5 or 5 μM), MA (1 mM) and/or DPH‐4 (5 μM). (A) Aβ1–40D (5 μM), MA and DPH‐4 were added at the same time before OGD. (B) A 24 h pretreatment was performed with Aβ1–40D (2.5 μM), and cells were then subjected to 8 h OGD with 24 h re‐oxygenation. All treatments were maintained during re‐oxygenation. Untreated cells under normoxia were considered as control (Ctrl). Data are expressed as mean ± SEM of three independent experiments. *** P < 0.001 versus Ctrl; ## P < 0.05 and ### P < 0.001 versus untreated cells under OGD + Reox; & P < 0.05 as indicated, by one‐way ANOVA and the addition of Newman–Keuls multiple comparison test.
Discussion and conclusions
Cerebral hypoperfusion, atherosclerosis, oxidative stress, vascular Aβ deposition or a failure of its clearance are insults that can disturb the transport of nutrients across the BBB. These alterations can lead to an acute and big failure of cerebrovascular function, due to brain ischaemia or haemorrhage, contributing to cognitive decline and dementia. Moreover, a thorough meta‐analysis recently reported that stroke significantly and independently increases the risk for AD and in turn, the risk of intracerebral haemorrhage (Zhou et al., 2015). Furthermore, both hypoxia and ischaemic injury induce the up‐regulation of BACE‐1 and increase the generation of Aβ (Guglielmotto et al., 2009), confirming a link between AD and stroke.
Although the pathogenesis of AD is not yet fully understood, there is a clear consensus that it is a multifactorial disease caused by several agents, including BBB dysfunction. At present, it is widely accepted that a more effective therapy for the multifactorial nature of AD would result from the development of molecules that have the ability to interact with several or all the systems altered in this disorder. Among them, the hybrid DPH‐4 has emerged as an interesting compound able to dually inhibit cholinesterase and MAO activities, and with metal chelating properties and hence, is of great therapeutic interest for AD treatment (Wang et al., 2014).
Because of the levels of SSAO/VAP‐1 are altered in AD (Ferrer et al., 2002) and stroke (Hernández‐Guillamón et al., 2010), and because of its high expression in cerebrovascular tissue, we hypothesized that this protein could be a potential link and therapeutic target for both pathologies. However, the expression of some proteins is lost when cells are cultured, as is the case for SSAO/VAP‐1; therefore, we report for the first time the preparation of a human brain endothelial‐immortalized cell line derived from the hCMEC/D3 cell line (Weksler et al., 2005), which stably expresses the hSSAO/VAP‐1 gene. The hCMEC/D3 hSSAO/VAP‐1 cell line was characterized and compared with previously developed endothelial and SMC lines expressing SSAO/VAP‐1 (Solé, et al., 2007; Solé and Unzeta, 2011). Then, we established a new experimental model of stroke consisting of subjecting these brain endothelial cells to OGD and re‐oxygenation conditions. The hCMEC/D3 cell line is the first stable well‐differentiated and well‐characterized human brain endothelial cell line (Weksler et al., 2005) in terms of expression of endothelial markers and up‐regulation of adhesion molecules in response to inflammatory cytokines, as well as BBB characteristics, so it was chosen as an accepted BBB cellular model (Weksler et al., 2013), instead of peripheral endothelial HUVEC cells, because genes expressed by cerebral endothelial cells are important and distinct in several processes such as vasculogenesis and angiogenesis (VEGF) and immunoregulation (decorin, IL‐6) or have growth‐supporting properties (Brain‐derived neurotrophic factor (BDNF), transforming growth factor‐β) (Kallmann et al., 2002). Although this cell line is a good approximation to a BBB model, it has some limitations, and co‐cultures including neurons and glia would be a better physiological model.
Under OGD with re‐oxygenation conditions, cell viability was observed to be significantly decreased in the presence of MA, the main physiological substrate for SSAO/VAP‐1, and restored when cells were pre‐incubated in the presence of semicarbazide, a specific SSAO/VAP‐1 inhibitor. These results confirmed that this enzyme plays an important role in enhancing endothelial cell death under ischaemia. Moreover, under these hypoxic conditions, membrane‐bound SSAO/VAP‐1 is released because of an increase in its shedding, probably resulting from the activation of metalloproteinase 2 (Sun et al., 2014). This soluble form of the enzyme may also contribute to the vascular cell damage through its catalytic activity, as previously described by some of us (Hernández et al., 2006). At the concentrations used, DPH‐4 inhibited about 50% of the SSAO activity. DPH‐4 pretreatment mediated a dose‐dependent protective effect on hSSAO/VAP‐1‐expressing hCMEC/D3 cells in the presence of MA in both normoxic and OGD with re‐oxygenation conditions. This protection was found to be even more significant when the same experiment was carried out with hSSAO/VAP‐1‐expressing HUVEC cells. This different behaviour might be because cerebral endothelial cells are somewhat protected from cell death by the SV40T antigen that was used in their immortalization, so are more hardy under difficult conditions. However, the possibility that the cells have a different sensitivity to the toxicities generated by SSAO activity cannot be ruled out.
The beneficial effect of DPH‐4 was also noticeable in terms of inflammation, because it significantly reduced the leukocyte adhesion to the endothelia subjected to different OGD conditions in the presence of MA, as a result of its inhibitory effect on SSAO activity. In this regard, the participation of other adhesion molecules cannot be ruled out, but it would be more likely to happen after the OGD + re‐oxygenation stimulus, where both the WT and the SSAO/VAP‐1‐expressing cells increase the leukocyte adhesion, rather than in response to MA treatment, where leukocyte adhesion increases only in SSAO‐expressing cells. Moreover, although the Western blot results mainly showed a reduction in the membrane‐bound form of these adhesion molecules, especially SSAO/VAP‐1 and VCAM‐1, it is worth to point out that both proteins can be shed to generate a soluble form, as we demonstrated for SSAO/VAP‐1. It is known that soluble SSAO/VAP‐1 contributes to the adhesion process through the generation of H2O2 and probably other unknown mechanisms. Therefore, it is reasonable to think that VCAM‐1 could show a similar behaviour. This anti‐inflammatory effect may also contribute to the protective effect observed in this experimental model of stroke.
In addition, when Aβ1–40D treatment was introduced into this experimental model of ischaemia, that simulates a pre‐existing AD pathology, DPH‐4 showed a protective effect on the synergistic damaging effect induced by MA and Aβ1–40D. These results allow us to conclude that Aβ1–40D together with the catalytic activity of SSAO/VAP‐1 induces more vascular damage under OGD with re‐oxygenation conditions and that the protective effect of DPH‐4 is higher than that observed when each toxic molecule is applied separately.
To sum up, herein, we report for the first time that DPH‐4, a new MTDL hybrid, designed by juxtaposition of selected pharmacophoric groups present in donepezil, propargylamine and 8‐hydroxyquinoline, protects hCMEC/D3 hSSAO/VAP‐1 cells under hypoxic conditions through its inhibitory and anti‐inflammatory effect on SSAO/VAP‐1. In the context of the close relationship between AD and stroke, and the involvement of SSAO/VAP‐1 in both disorders, DPH‐4 could be considered as a promising multivalent drug with potential therapeutic interest for use in both neurological pathologies.
Author contributions
P. S. and G. E. performed the research study. M. U. and M. S. designed the research study and contributed to data analysis. T. I. provided compound DPH‐4. J. M‐C. designed compound DPH‐4. B. B. W., I. A. R. and P. O. C. provided the hCMEC/D3 cellular model. All authors contributed to the drafting and revising of the manuscript and accepted the final manuscript
Conflict of interests
The authors declare that they have no conflict of interest.
Acknowledgement
This study was supported by the MINECO (Spanish Ministry of Economy and Competitiveness): projects SAF2006‐08764‐C02‐01, SAF2009‐07271 and SAF2012‐33304.
Sun, P. , Esteban, G. , Inokuchi, T. , Marco‐Contelles, J. , Weksler, B. B. , Romero, I. A. , Couraud, P. O. , Unzeta, M. , and Solé, M. (2015) Protective effect of the multitarget compound DPH‐4 on human SSAO/VAP‐1‐expressing hCMEC/D3 cells under oxygen–glucose deprivation conditions: an in vitro experimental model of cerebral ischaemia. British Journal of Pharmacology, 172: 5390–5402. doi: 10.1111/bph.13328.
References
- Abella A, García‐Vicente S, Viguerie N, Ros‐Baró A, Camps M, Palacín M et al. (2004). Adipocytes release a soluble form of VAP‐1/SSAO by a metalloprotease‐dependent process and in a regulated manner. Diabetologia 47: 429–438. [DOI] [PubMed] [Google Scholar]
- Airas L, Lindsberg PJ, Karjalainen‐Lindsberg ML, Mononen I, Kotisaari K, Smith DJ et al. (2008). Vascular adhesion protein‐1 in human ischaemic stroke. Neuropathol Appl Neurobiol 34: 394–402. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birks J, Harvey RJ (2006). Donepezil for dementia due to Alzheimer's disease. Cochrane Database Sys Rev 1 CD001190: 1–165. [DOI] [PubMed] [Google Scholar]
- Bolea I, Gella A, Unzeta M (2013). Propargylamine‐derived multitarget‐directed ligands: fighting Alzheimer's disease with monoamine oxidase inhibitors. J Neural Transm 120: 893–902. [DOI] [PubMed] [Google Scholar]
- Boomsma F, Bhaggoe UM, van der Houwen AM, van den Meiracker AH (2003). Plasma semicarbazide‐sensitive amine oxidase in human (patho)physiology. Biochim Biophys Acta 1647: 48–54. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Terry AV Jr (2000). Multiple central nervous system targets for eliciting beneficial effects on memory and cognition. J Pharmacol Exp Ther 295: 438–446. [PubMed] [Google Scholar]
- Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M et al. (2008). Multi‐target‐directed ligands to combat neurodegenerative diseases. J Med Chem 51: 347–372. [DOI] [PubMed] [Google Scholar]
- Conklin DJ, Langford SD, Boor PJ (1998). Contribution of serum and cellular semicarbazide‐sensitive amine oxidase to amine metabolism and cardiovascular toxicity. Toxicol Sci 46: 386–392. [DOI] [PubMed] [Google Scholar]
- DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL (2008). Early disruptions of the blood‐brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging 29: 753–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hadri K, Moldes M, Mercier N, Andreani M, Pairault J, Feve B (2002). Semicarbazide sensitive amino oxidase in vascular smooth muscle cells: differentiation‐dependent expression and role in glucose uptake. Arterioscler Thromb Vasc Biol 22: 89–94. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Lizcano JM, Hernández M, Unzeta M (2002). Overexpression of semicarbazide sensitive amine oxidase in the cerebral blood vessels in patients with Alzheimer's disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Neurosci Lett 321: 21–24. [DOI] [PubMed] [Google Scholar]
- Geula C, Mesulam MM (1999). Cholinergic systems in Alzheimer's disease In: Terry SD et al. (eds.). Alzheimer Disease, 2nd edn. Lippincot, Williams, & Wilkins: Philadelphia, pp. 69–292. [Google Scholar]
- Grammas P (2011). Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer's disease. J Neuroinflammation 8: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmotto M, Aragno M, Autelli R, Giliberto L, Novo E, Colombatto S et al. (2009). The up‐regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1 alpha. J Neurochem 108: 1045–1056. [DOI] [PubMed] [Google Scholar]
- Hernández M, Esteban M, Szabo P, Boada M, Unzeta M (2005). Human plasma semicarbazide sensitive amine oxidase (SSAO), beta‐amyloid protein and aging. Neurosci Lett 384: 183–187. [DOI] [PubMed] [Google Scholar]
- Hernández M, Solé M, Boada M, Unzeta M (2006). Soluble semicarbazide sensitive amine oxidase (SSAO) catalysis induces apoptosis in vascular smooth muscle cells. Biochim Biophys Acta 1763: 164–173. [DOI] [PubMed] [Google Scholar]
- Hernández‐Guillamón M, García‐Bonilla L, Solé M, Sosti V, Parés M, Campos M et al. (2010). Plasma VAP‐1/SSAO activity predicts intracranial hemorrhages and adverse neurological outcome after tissue plasminogen activator treatment in stroke. Stroke 41: 1528–1535. [DOI] [PubMed] [Google Scholar]
- Hernández‐Guillamón M, Solé M, Delgado P, García‐Bonilla L, Giralt D, Boada C et al. (2012). VAP‐1/SSAO plasma activity and brain expression in human hemorrhagic stroke. Cerebrovasc Dis 33: 55–63. [DOI] [PubMed] [Google Scholar]
- Iadecola C (2010). The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 120: 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anrather J (2011). The immunology of stroke: from mechanisms to translation. Nat Medicine 17: 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalkanen S, Salmi M (2008). VAP‐1 and CD73, endothelial cell surface enzymes in leukocyte extravasation. Arterioscler Thromb Vasc Biol 28: 18–26. [DOI] [PubMed] [Google Scholar]
- Kallmann BA, Wagner S, Hummel V, Buttmann M, Bayas A, Tonn JC et al. (2002). Characteristic gene expression profile of primary human cerebral endothelial cells. FASEB J 16: 589–591. [DOI] [PubMed] [Google Scholar]
- Kurkijärvi R, Adams DH, Leino R, Möttönen T, Jalkanen S, Salmi M (1998). Circulating form of human vascular adhesion protein‐1 (VAP‐1): increased serum levels in inflammatory liver diseases. J Immunol 161: 1549–1557. [PubMed] [Google Scholar]
- León R, García AG, Marco‐Contelles J (2013). Recent advances in the multitarget‐directed ligands approach for the treatment of Alzheimer's disease. Med Res Rev 33: 139–189. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Tipton KF, Unzeta M (1998). Purification and characterization of membrane‐bound semicarbazide‐sensitive amine oxidase (SSAO) from bovine lung. Biochem J 331: 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi VT (2014). Alzheimer's disease and CADASIL are heritable, adult‐onset dementias that both involve damaged small blood vessels. Cell Mol Life Sci 71: 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, Kobayashi Y (1964). Radioisotopic assay for monoamine oxidase determinations in human plasma. Biochem Pharmacol 13: 995–1006. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry EK, Perry RH, Blessed G, Tomlinson BE (1977). Necropsy evidence of central cholinergic deficits in senile dementia. Lancet 1: 189. [DOI] [PubMed] [Google Scholar]
- Plumb JA, Milroy R, Kaye SB (1989). Effects of the pH dependence of 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐tetrazolium bromide‐formazan absorption on chemosensitivity determined by a novel tetrazolium‐based assay. Cancer Res 49: 4435–4440. [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Salmi M, Bono P, Hellman J, Leu T, Jalkanen S (1998). Cloning of vascular adhesion protein 1 reveals a novel multifunctional adhesion molecule. J Exp Med 188: 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé M, Hernández M, Boada M, Unzeta M (2007). Characterization of A7r5 cell line transfected in a stable form by hSSAO/VAP‐1 gene (A7r5 hSSAO/VAP‐1 cell line). J Neural Transm 114: 763–767. [DOI] [PubMed] [Google Scholar]
- Solé M, Hernández‐Guillamón M, Boada M, Unzeta M (2008). p53 phosphorylation is involved in vascular cell death induced by the catalytic activity of membrane‐bound SSAO/VAP‐1. Biochim Biophys Acta 1783: 1085–1094. [DOI] [PubMed] [Google Scholar]
- Solé M, Unzeta M (2011). Vascular cell lines expressing SSAO/VAP‐1: a new experimental tool to study its involvement in vascular diseases. Biol Cell 103: 543–557. [DOI] [PubMed] [Google Scholar]
- Solé M, Miñano AJ, Unzeta M (2015). A cross‐talk between Aβ and endothelial SSAO/VAP‐1 accelerates vascular damage and Aβ aggregation related to CAA‐AD. Neurobiol Aging 36: 762–775. [DOI] [PubMed] [Google Scholar]
- Sun P, Solé M, Unzeta M (2014). Involvement of SSAO/VAP‐1 in oxygen‐glucose deprivation‐mediated damage using the endothelial hSSAO/VAP‐1‐expressing cells as experimental model of cerebral ischemia. Cerebrovasc Dis 37: 171–180. [DOI] [PubMed] [Google Scholar]
- Wang L, Esteban G, Ojima M, Bautista‐Aguilera OM, Inokuchi T, Moraleda I et al. (2014). Donepezil + propargylamine + 8‐hydroxyquinoline hybrids as new multifunctional metal‐chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer's disease. Eur J Med Chem 80: 543–561. [DOI] [PubMed] [Google Scholar]
- Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M et al. (2005). Blood‐brain barrier‐specific properties of a human adult brain endothelial cell line. FASEB J 19: 1872–1874. [DOI] [PubMed] [Google Scholar]
- Weksler BB, Romero IA, Couraud PO (2013). The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 10: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MB, Buccafusco JJ (2005). CNS targets for multi‐functional drugs in the treatment of Alzheimer's and Parkinson's diseases. J Neural Transm 112: 519–537. [DOI] [PubMed] [Google Scholar]
- Yu PH, Deng YL (1998). Endogenous formaldehyde as a potential factor of vulnerability of atherosclerosis: involvement of semicarbazide‐sensitive amine oxidase‐mediated methylamine turnover. Atherosclerosis 140: 357–363. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV (2008). The blood‐brain barrier in health and chronic neurodegenerative disorders. Neuron 57: 178–201. [DOI] [PubMed] [Google Scholar]
- Zhou J, Yu JT, Wang HF, Meng SF, Tan CC, Wang J et al. (2015). Association between stroke and Alzheimer's disease: systematic review and meta‐analysis. J Alzheimers Dis 43: 479–489. [DOI] [PubMed] [Google Scholar]