

Abstract
Background and Purpose
Abnormal Ca2+ metabolism has been involved in the pathogenesis of vascular dysfunction associated with oxidative stress. Here, we have investigated the actions of H2O2 on store‐operated Ca2+ (SOC) entry in coronary arteries and assessed whether it is impaired in arteries from a rat model of metabolic syndrome.
Experimental Approach
Simultaneous measurements of intracellular Ca2+ concentration and contractile responses were made in coronary arteries from Wistar and obese Zucker rats, mounted in microvascular myographs, and the effects of H2O2 were assessed.
Key Results
H2O2 raised intracellular Ca2+ concentrations, accompanied by simultaneous vasoconstriction that was markedly reduced in a Ca2+‐free medium. Upon Ca2+ re‐addition, a nifedipine‐resistant sustained Ca2+ entry, not coupled to contraction, was obtained in endothelium‐denuded coronary arteries. The effect of H2O2 on this voltage‐independent Ca2+ influx was concentration‐dependent, and high micromolar H2O2 concentrations were inhibitory and reduced SOC entry evoked by inhibition of the sarcoplasmic reticulum ATPase (SERCA). H2O2‐induced increases in Fura signals were mimicked by Ba2+ and reduced by heparin, Gd3+ ions and by Pyr6, a selective inhibitor of the Orai1‐mediated Ca2+ entry,. In coronary arteries from obese Zucker rats, intracellular Ca2+ mobilization and SOC entry activated by acute exposure to H2O2 were augmented and associated with local oxidative stress.
Conclusion and Implications
H2O2 exerted dual concentration‐dependent stimulatory/inhibitory effects on store‐operated, IP3 receptor‐mediated and Orai1‐mediated Ca2+ entry, not coupled to vasoconstriction in coronary vascular smooth muscle. SOC entry activated by H2O2 was enhanced and associated with vascular oxidative stress in coronary arteries in metabolic syndrome.
Abbreviations
- [Ca2+]i
intracellular Ca2+ concentration
- CPA
cyclopiazonic acid
- CRAC
Ca2+ release Ca2+‐activated
- EDH
endothelium‐derived hyperpolarization
- ER
endoplasmic reticulum
- IP3
inositol trisphosphate
- KPSS
125 mM K+ physiological saline solution
- LZR
lean Zucker rat
- OZR
obese Zucker rat
- PSS
physiological saline solution
- Pyr6
N‐(4‐(3,5‐bis(trifluoromethyl)‐1H‐pyrazol‐1‐yl)phenyl)‐3‐fluorisonicotinamide
- SERCA
sarcoendoplasmic reticulum Ca2+‐ATPase
- SOC
store‐operated Ca2+
- SR
sarcoplasmic reticulum
- STIM1
stromal interaction molecule 1
Tables of Links
| TARGETS |
|---|
| Ligand‐gated ion channels a |
| IP3receptor |
| Ion channels b |
| L‐type Ca 2+ channels (Ca V 1.x) |
| TRPC1 |
| TRPM2 |
| LIGANDS |
|---|
| CPA, cyclopiazonic acid |
| IP3 |
| Nifedipine |
| H2O2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a bAlexander et al., 2013a, 2013b).
Introduction
Reactive oxygen species (ROS) derived from various sources in the vascular wall are involved in the regulation of redox‐sensitive physiological processes such as vasoconstriction, proliferation and migration. A role of ROS as intracellular signalling molecules in the regulation of the intracellular Ca2+ concentration ([Ca2+]i) of different cell types is now well established (Trebak et al., 2010; Bogeski et al., 2011). For instance, H2O2 and other ROS can activate Ca2+ mobilization from intracellular stores and stimulate Ca2+ entry through ion channels in the plasma membrane to increase the overall [Ca2+]i. Conversely, the activity of ROS‐generating enzymes such as NADPH oxidase and NOS can be regulated by stimuli inducing changes in [Ca2+]i (Touyz, 2005; Trebak et al., 2010).
ROS have been reported to regulate Ca2+ influx through both voltage‐dependent and non‐voltage‐dependent plasmalemmal Ca2+ channels (Trebak et al., 2010). Voltage‐gated l‐type Ca2+ channels play a key role in increasing the global [Ca2+]i during vascular smooth muscle (VSM) contraction, and both superoxide anion (O2 −) and H2O2 exogenously applied or endogenously produced in response to vasoconstrictors and growth factors have been shown to activate L‐type Ca2+ entry in VSM (Chaplin and Amberg, 2012; Santiago et al., 2013), as well as in cardiac myocytes (Hool, 2000; Hool and Arthur, 2002; Viola et al., 2007). Ca2+ entry through non‐voltage‐dependent channels is produced following receptor activation and/or falling of Ca2+ levels of the intracellular stores in the endoplasmic reticulum (ER), the latter known as capacitative Ca2+ entry that takes place through store‐operated Ca2+ (SOC) channels (Parekh and Putney, 2005). Modulation of Ca2+ entry through non‐voltage–gated channels by ROS has been characterized mainly in mammalian non‐excitable cells including endothelial cells (Hu et al., 1998; Az‐ma et al., 1999; Redondo et al., 2004a,2004b) and to a lesser extent in excitable VSM cells, where information remains scarce (Lin et al., 2007; Pourmahram et al., 2008).
Accumulated evidence suggests that ROS are critically involved in the physiological regulation of coronary blood flow. H2O2 plays a role along with NO and adenosine in coronary autoregulation (Yada et al., 2003) and is released by shear stress in coronary arterioles (Miura et al., 2003) and involved in the pacing‐induced metabolic coronary vasodilatation to couple coronary blood flow to myocardial O2 consumption (Saitoh et al., 2006; Yada et al., 2007). H2O2 is an endogenous mediator of the endothelium‐derived hyperpolarization (EDH) of coronary microvessels (Matoba et al., 2003; Shimokawa, 2010), through activation of Ca2+‐activated K+ channels (Barlow and White, 1998) and of the Na+/K+ pump (Wong et al., 2014). We have recently demonstrated that H2O2 can also induce endothelium‐dependent vasoconstriction tightly coupled to increased Ca2+ influx through voltage‐dependent l‐type Ca2+ channels in coronary arteries (Santiago et al., 2013), a mechanism shared by cardiac myocytes where endogenous peroxide activates l‐type Ca2+ currents and has been proposed to sensitize these Ca2+ channels and increase the responses to β‐adrenoceptor stimulation (Hool, 2000; Hool and Arthur, 2002). However, H2O2 also induced a nifedipine‐resistant [Ca2+]i rise in coronary VSM (Santiago et al., 2013) consistent with the fact that H2O2 might mobilize intracellular Ca2+ and subsequent Ca2+ entry through non‐selective cation channels, as reported in pulmonary arterial myocytes (Lin et al., 2007; Pourmahram et al., 2008).
In the present study, we sought to determine whether H2O2 mobilized Ca2+ from intracellular stores and activated store depletion‐dependent Ca2+ entry in coronary arteries. Furthermore, the effect of H2O2 on SOC entry in coronary arteries was also assessed under pathological conditions of high vascular oxidative stress such as metabolic syndrome (Furukawa et al., 2004), a constellation of metabolic and cardiovascular abnormalities including obesity, insulin resistance, dyslipidemia and hypertension, which represent a risk condition for ischaemic heart disease (Arbel et al., 2015).
Methods
Animals and tissue preparation
All animal care and experimental protocols conformed to the European Union Guidelines for the Care and the Use of Laboratory Animals (European Union Directive 2010/63/EU) and were approved by the IACUC at Complutense University (Madrid, Spain). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of N animals were used in the experiments described here.
Male Wistar rats 12–14 weeks old (n = 55) and Zucker rats 17–18 weeks old (n = 20, provided by Charles River Laboratories France, 69592 L'Arbresle Cedex France) were used. Animals were anesthetized with sodium pentobarbital (40 mg·kg−1, i.p.) and killed by cervical dislocation and exsanguination. The heart was quickly removed and transferred into cold (4°C) physiological saline solution (PSS) of the following composition (mM): 119 NaCl, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 1.5 CaCl2, 24.9 NaHCO3, 0.027 EDTA, and 11 glucose, pH = 7.4. Second‐order branches of the left anterior descending coronary artery were dissected and mounted in microvascular myographs (Danish Myotechnology, Denmark) for isometric tension recording. The arteries were equilibrated for 30 min in PSS at 37°C while continuously gassed with a mixture of 95% O2–5% CO2. The internal circumference L100 corresponding to a transmural pressure of 100 mmHg for a relaxed vessel in situ was calculated, and the arteries were set to L1 equal to 0.9 times L100 (L1 = 0.9 × L100) (Mulvany and Halpern, 1977).
The obese Zucker rat (OZR) was used as a well‐established model of insulin resistance and the metabolic syndrome, caused by a dysfunctional gene of the leptin receptor. Male OZR (fa/fa, n = 13)) and their control counterparts, lean Zucker rats (LZR) (fa/‐, n = 7)), were purchased from Charles River Laboratories (Barcelona, Spain) at 8–10 weeks of age, housed at the Pharmacy School animal care facility and maintained on standard chow and water ad libitum, until they were used for study, at 16–18 weeks of age (Villalba et al., 2009).
Simultaneous measurements of [Ca2+]i and tension
Simultaneous measurements of [Ca2+]i and tension were performed in intact arterial segments using Fura‐2‐acetoxymethylester (Fura‐2‐AM) fluorescence as described previously (Villalba et al., 2008; Santiago et al., 2013). Coronary arteries were incubated in the dark at 37°C in PSS containing the indicator 4 μM Fura‐2‐AM and 0.05% Cremophor EL for a 2 h period. They were washed three times in PSS to remove remaining Fura‐2‐AM, and the solution was changed to PSS with fresh Fura‐2‐AM after 1 h. After Fura‐2‐AM loading, arteries were washed for 45 min in PSS. Experiments were performed in PSS (37°C) continuously gassed with a mixture of 95% O2–5% CO2 to maintain pH at 7.4. The myograph was mounted on an inverted microscope (Zeiss Axiovert S100 TV) equipped for dual excitation wavelength microfluorimetry (Deltascan, Photon Technology International).The coronary artery was alternately illuminated at two different wavelengths, 340 and 380 nm, and the intensity of emitted fluorescence was collected through a 510 nm filter using a photomultiplier and monitored together with the tension. At the end of each experiment, Ca2+‐insensitive signals determined by quenching of Fura‐2‐AM with Mn2+ were subtracted from the measured emission levels at 340 (F340) and 380 nm (F380). The ratio (R) F340/F380 corrected for autofluorescence was taken as a measure of [Ca2+]i.
Experimental procedures for the functional experiments
At the beginning of each experiment, the viability of each artery was tested by stimulating it twice with 124 mM K+ PSS (KPSS), similar to PSS except that NaCl was replaced by KCl on an equimolar basis. In some experiments, the endothelial cell layer was mechanically removed by guiding a human hair through the vessel lumen and gently moving it back and forward as described previously. Absence of functional endothelium was assessed by the lack of relaxation to ACh (10 μM). The role of intracellular Ca2+ in the H2O2‐induced vasoconstriction was assessed by stimulating the arteries with a single dose of H2O2 (100 μM) in the absence or presence of extracellular Ca2+. In order to evaluate whether store depletion by H2O2 induces ‘capacitative Ca2+ entry’, the bath solution was exchanged for Ca2+‐free PSS for 5 min and then for nominally Ca2+‐free PSS with nifedipine (1 μM), a selective blocker of l‐type voltage‐operated Ca2+ channels, for a period of 10 min. H2O2 (30, 100 μM and 300 μM) or cyclopiazonic acid (CPA; 20 μM), a selective inhibitor of the sarcoendoplasmic reticulum Ca2+‐ATPase (SERCA) was then added for a period of 8 min, after which 1.5 mM Ca2+ or Ba2+ was re‐introduced into the bath solution and kept for 20 min.
The effects of H2O2 on the SOC entry in coronary VSM were characterized in endothelium‐denuded coronary arteries. The inositol trisphosphate receptor (IP3R) antagonist heparin (200 µg·mL−1) (Saleem et al., 2014), Gd3+ ions (50 μM), as a non‐selective cation channel blocker and Pyr6 (3 μM; N‐(4‐(3,5‐bis(trifluoromethyl)‐1H‐pyrazol‐1‐yl)phenyl)‐3‐fluorisonicotinamide), a new selective blocker of the stromal interaction molecule 1 (STIM1) and Orai1‐coupled Ca2+ release Ca2+‐activated (CRAC)‐mediated SOC entry (Schleifer et al., 2012) were tested on the non‐l‐type Ca2+ entry induced by store depletion with either H2O2 or CPA. In order to assess thiol oxidation‐dependency of H2O2 actions on Ca2+ entry, the effect of H2O2 on the SOC entry in coronary arteries was assessed in arteries treated with the reducing agent DTT (100 μM).
Measurement of O2 − production by chemiluminescence
Levels of production O2 − by coronary arteries and myocardial tissue from control (LZR) and metabolic syndrome (OZR) rats under basal conditions were detected by lucigenin‐enhanced chemiluminescence as previously described in heart tissue (Santiago et al., 2013). Briefly, segments of coronary arteries and samples of myocardial tissue were dissected and equilibrated in PSS for 30 min at room temperature and then incubated in the presence or absence of tempol (30 μM) at 37°C. Samples were then transferred to microtiter plate wells containing 5 μM lucigenin in air‐equilibrated Krebs solution buffered with 10 mM HEPES‐NaOH, in the absence and presence of tempol, and H2O2 (100 μM) was then applied. Chemiluminescence was measured in a luminometer (BMG Fluostar Optima). Baseline values were subtracted from the observed values under the different experimental conditions, and O2 − production was normalized to the weight of the tissue samples.
Data analysis
Mechanical responses of the arteries were measured as force and expressed as active wall tension, which is the increase in force, ΔF, divided by twice the segment length (Mulvany and Halpern, 1977). Changes in [Ca2+]i were measured as the increase in the Fura‐2‐AM ratio taking as baseline the average value of the baseline in PSS and baseline in Ca2+‐free PSS (ΔF340/F380). The results are expressed as either absolute values of tension (as N m−1) or in units of ratio of fluorescence (F340/F380), or as a percentage of the responses induced by KPSS. All the results are reported as means ± SEM; n represents the number of animals. One arterial segment per animal was used in the calcium experiments, and one to two arterial or myocardial samples per animal were used in the chemiluminescence experiments for O2 − measurement. Statistical differences between means were analyzed by unpaired Student's t‐test for comparison between two groups when assessing the effect of various treatments on the changes in [Ca2+]i and tension activated by H2O2 or CPA. Two‐factor repeated measures one‐way ANOVA followed by a Bonferroni post hoc test was applied to assess statistically significant differences in changes in either [Ca2+]i or tension activated by H2O2 or CPA (factor 1) over time (factor 2). The time at which Ca2+ re‐addition was made was taken as time 0. P <0.05 was considered significant. All calculations were made using a standard software package (Prism 5.0; GraphPad Software).
Materials
The sources of the compounds used were as follows: barium chloride, gadolinium chloride, H2O2, indomethacin and lucigenin were obtained from Sigma Aldrich (Madrid, Spain), Fura‐2‐AM and ionomycin from Invitrogen (Life Technologies SA, Madrid, Spain), CPA, heparin and nifedipine from Tocris (Bristol, UK) and Pyr6 from Calbiochem (Merck‐Millipore, Madrid, Spain).
Results
H2O2 stimulates Ca2+ entry and mobilizes Ca2+ from intracellular stores in coronary arteries
To assess whether H2O2 might induce Ca2+ mobilization from intracellular stores thus activating SOC entry, coronary arteries were stimulated with a single submaximal vasoconstrictor concentration of H2O2 (100 μM) (Santiago et al., 2013), in the absence and presence of extracelullar Ca2+. In PSS containing 1.5 mM Ca2+, H2O2 induced an increase in [Ca2+]i (∆F340/F380 = 0.33 ± 0.06, n = 8) that was accompanied by a simultaneous sustained vasoconstriction on baseline tension (0.67 ± 0.11 Nm−1, n = 8) (Figure 1). Both the [Ca2+]i rise and the increase in tension induced by H2O2 were markedly reduced after removal of Ca2+ from the extracellular medium, although a small fraction of the [Ca2+]i rise (∆F340/F380 = 0.05 ± 0.01, n = 8) and contraction (0.11 ± 0.02 Nm−1, n = 8) still persisted in Ca2+‐free medium (Figure 1). These data suggest that H2O2 induces Ca2+ mobilization from intracellular stores.
Figure 1.
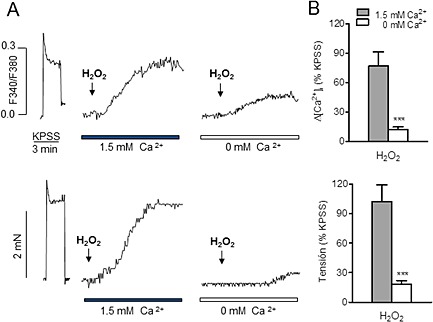
Rise in [Ca2+]i and vasoconstriction induced by H2O2 are in part to due to mobilization of Ca2+ from intracellular stores in coronary arteries. (A) Simultaneous recordings showing changes in [Ca2+]i (top) and tension (bottom) induced by KPSS and H2O2 in the presence and absence of extracellular Ca2+ in coronary arteries with intact endothelium. (B) Summarized data showing the changes in [Ca2+]i (top) and tension (bottom) in response to H2O2 (100 μM) in the presence and absence of extracellular Ca2+. Results are expressed as a percentage of the KPSS‐induced contraction and are means ± SEM of eight animals. ***P < 0.001; significantly different from control; Student's t‐test.
H2O2 stimulates SOC entry not coupled to contraction in coronary arteries
In Ca2+‐free PSS, addition of H2O2 produced a small Ca2+ mobilization and further re‐addition of Ca2+ (1.5 mM) to the medium resulted in a marked and sustained increase in [Ca2+]i after 20 min (∆F340/F380 = 0.48 ± 0.16, n = 5) similar in magnitude to the increase in [Ca2+]i produced by stimulation with KPSS in the same arteries (∆F340/F380 = 0.52 ± 0.16, n = 5) (Figure 2A, top). However, vasoconstriction induced by H2O2 under these conditions (0.29 ± 0.07 Nm−1) was about one‐third of the KPSS‐elicited contraction (0.88 ± 0.22, n = 5) (Figure 2A, bottom). The changes in [Ca2+]i induced by H2O2 were not affected by the presence of the voltage‐dependent l‐type Ca2+ channel blocker nifedipine (Figure 2B, top), suggesting that Ca2+ entry occurs through voltage‐independent Ca2+ channels activated by emptying of intracellular Ca2+ stores induced by H2O2. Nifedipine blunted the small contraction induced by H2O2 (Figure 2B, bottom).
Figure 2.
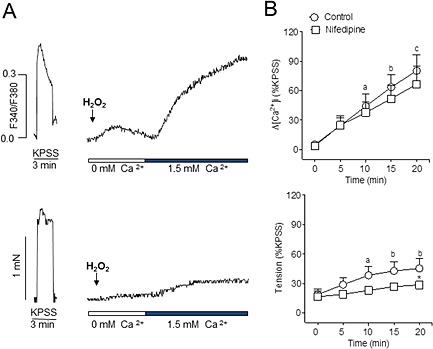
H2O2 induces l‐type voltage channel‐independent Ca2+ entry in coronary arteries. (A) Simultaneous recordings of [Ca2+]i (A, top) and tension (A, bottom) showing the marked increase in [Ca2+]i and moderate vasoconstriction produced by H2O2 (100 μM) in a Ca2+‐free medium after the re‐addition of 1.5 mM Ca2+ in endothelium‐intact coronary arteries. (B) Summarized data showing the changes in [Ca2+]i (top) and tension (bottom) induced by H2O2 (100 μM) in response to the re‐addition of Ca2+ to the extracellular medium, in the absence and presence of the l‐type channel blocker nifedipine (1 μM). Results are expressed as a percentage of the KPSS‐induced contraction and are means ± SEM of five animals. a P < 0.05, b P < 0.01, c P < 0.001; significantly different from [Ca2+]i or tension at time 0 (Ca2+ re‐addition); one‐way ANOVA. *P < 0.05; significantly different from control (absence of nifedipine); Student's t‐test.
Characterization of SOC entry activated by H2O2 in coronary VSM
Because vascular endothelium is involved in the Ca2+ mobilization induced by H2O2 in the coronary arterial wall (Santiago et al., 2013), experiments were performed in coronary arteries in which endothelium was mechanically removed in order to assess the SOC entry activated by H2O2 in coronary VSM. In endothelium‐denuded arteries kept in a Ca2+‐free medium under conditions of l‐type channel blockade, H2O2 (100 μM) mobilized intracellular Ca2+, and readmission of Ca2+ to the medium evoked a large and sustained increase in [Ca2+]i (Figure 3A) similar to that obtained in endothelium‐intact arteries and not significantly different from the one induced by depolarization with KPSS in the same arteries. This [Ca2+]i increase was not accompanied by significant increases in tension (Figure 3B). These data suggest that H2O2 activates SOC entry in VSM not coupled to contraction. Re‐introduction of Ca2+ in the absence of peroxide evoked a minor [Ca2+]i increase in endothelium‐denuded coronary arteries (Figure 3A).
Figure 3.
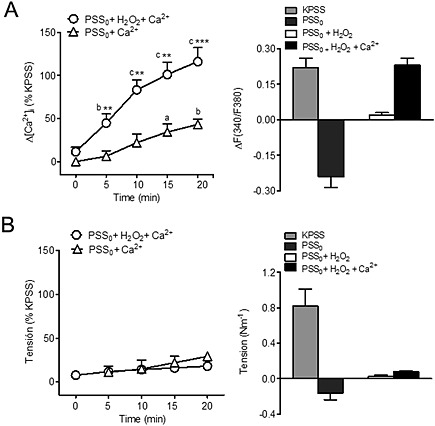
H2O2 promotes store‐operated Ca2+ entry not coupled to contraction in coronary vascular smooth muscle. Average changes in (A) [Ca2+]i and (B) contraction evoked by Ca2+ re‐addition to endothelium‐denuded coronary arteries kept in Ca2+‐free medium (PSS0) and activated by H2O2 under conditions of l‐type channel blockade. In the presence of nifedipine (1 μM) and 100 μM H2O2, re‐admission of 1.5 mM Ca2+ to the extracellular medium caused a large rise in [Ca2+]i (A, left) similar to the one induced by KPSS (A, right), which was not accompanied by significant changes in tension (B). In control experiments, Ca2+ re‐addition was conducted in arteries kept in Ca2+‐free PSS in the absence of activating H2O2. Results are expressed as a percentage of the KPSS‐induced responses (A, B right) or as absolute values of F340/380 (A, left) and of tension (as N m−1: B, left) and are means ± SEM of four to nine animals. a P < 0.05, b P < 0.01, c P < 0.001; significantly different from [Ca2+]i or tension at time 0 (Ca2+ re‐addition); one‐way ANOVA.*P < 0.05, **P < 0.01, ***P < 0.001; significantly different from control (absence of H2O2); Student's t‐test.
SOC entry activated by H2O2 in coronary VSM was compared with the Ca2+ entry stimulated by passive sarcoplasmic reticulum (SR) Ca2+ depletion by SERCA inhibition with CPA. In endothelium‐denuded coronary arteries kept in Ca2+‐free PSS, CPA (20 μM) induced a moderate increase in [Ca2+]i significantly larger than that evoked by 100 μM H2O2 (Figure 4A–C). Re‐addition of Ca2+ to the medium resulted in a marked rise in [Ca2+]i in the presence of CPA (Figure 4B) that was about 1.5‐fold larger than the one induced by H2O2 (Figure 4C and D).
Figure 4.
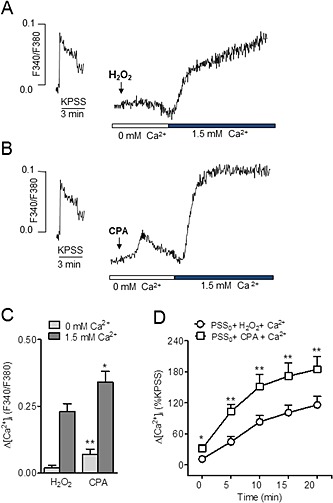
Store‐operated Ca2+ entry induced by SERCA inhibition and by H2O2 in coronary vascular smooth muscle. (A, B) Representative traces showing changes in [Ca2+]i induced by H2O2 (100 μM) (A) and by the SERCA nhibitor CPA (20 μM) (B) in response to the re‐addition of Ca2+ to the extracellular Ca2+‐free medium in an endothelium‐denuded coronary artery. (C) Comparative average effects of CPA and 100 μM H2O2, on [Ca2+]i in the absence of extracellular Ca2+ and after Ca2+ re‐addition. (D) Average time‐dependent changes in [Ca2+]i in response to the re‐addition of Ca2+ to the extracellular medium in endothelium‐denuded arteries activated with CPA or H2O2. Results are expressed as absolute values of F340/380 (C) or as a percentage of the KPSS‐induced ΔF340/380 (D) and are means ± SEM of four to nine animals. *P < 0.05, **P < 0.01; significantly different from control (100 μM H2O2); Student's t‐test.
To assess whether the action of H2O2 on SOC entry was concentration dependent, the effect of three different concentrations of H2O2 on the Ca2+ entry upon Ca2+ re‐addition to the extracellular medium was compared. While 100 μM H2O2 increased [Ca2+]i about 116% of the KPPS‐induced response, 30 μM H2O2 induced about half this response, while 300 μM H2O2 had an inhibitory effect compared with the effect at 100 μM (Figure 5A–D). Moreover, treatment of endothelium‐denuded coronary arteries with 300 μM H2O2 for 30 min reduced by half SOC entry induced by SERCA inhibition with CPA (Figure 5E). At 1 mM H2O2, SOC entry was nearly abolished (not shown). Subsequent experiments were performed at 100 μM H2O2.
Figure 5.
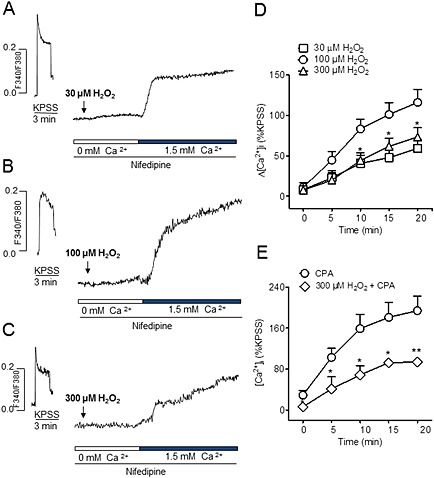
H2O2 has dual concentration‐dependent effects on non‐l‐type Ca2+ entry in coronary vascular smooth muscle. (A, B, C) Representative traces showing the increase in [Ca2+]i produced by the re‐addition of Ca2+ (1.5 mM) to the extracellular medium after stimulation with 30 (A), 100 (B) or 300 μM H2O2 (C) in endothelium‐denuded coronary arteries. (D) Mean values of the changes in [Ca2+]i in response to the re‐addition of Ca2+ (1.5 mM) into the extracellular medium after stimulation 30, 100 and 300 μM H2O2. (E) Mean values of the changes in [Ca2+]i in response to the re‐addition of Ca2+ (1.5 mM) into the extracellular medium in the presence of CPA (20 μM) in control conditions and after 30 min treatment with 300 μM H2O2. Results are expressed as a percentage of the KPSS‐induced ΔF340/380 and are means ± SEM of three to nine animals. *P < 0.05, **P < 0.01, significantly different from control (D, 100 µm H2O2) (E, 20 μM CPA); Student's t‐test.
Activation of IP3Rs and Orai1 is involved in the H2O2‐induced Ca2+ entry of coronary VSM
To investigate whether stimulation of IP3Rs and the subsequent intracellular Ca2+ release underlie H2O2‐induced Ca2+ entry, coronary arteries were treated with the IP3R antagonist heparin (200 µg·mL−1). Acute exposure to heparin reduced the nifedipine‐resistant Ca2+ influx activated by H2O2 (Figure 6A) and caused a large inhibition of SOC entry obtained by SERCA inhibition with CPA (Figure 6B). Heparin also reduced the rises in intracellular Ca2+ induced by CPA but not by H2O2 in the absence of extracellular Ca2+ (Figure 6C).
Figure 6.
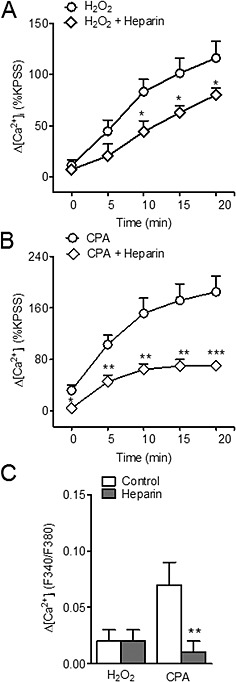
Non‐l‐type Ca2+ entry induced by H2O2 is inhibited by the IP3R antagonist heparin. (A, B) Average values of the changes in [Ca2+]i induced by H2O2 (100 μM) (A) and CPA (20 μM) (B) in response to the re‐addition of Ca2+ to the extracellular medium in the absence and after 10 min exposure to heparin (200 µg•mL−1) in coronary arteries. (C) Comparative average effects of heparin on the CPA‐induced and H2O2‐induced rises [Ca2+]i in the absence of extracellular Ca2+. Results are expressed as a percentage of the KPSS‐induced ΔF340/380 and are means ± SEM of four animals. *P < 0.05, **P < 0.01, ***P < 0.001, significant effects of heparin; Student's t‐test.
The contribution of non‐selective cation channels to the Ca2+ entry stimulated by H2O2 in coronary VSM was evaluated by using Ba2+ fluorimetry instead of Ca2+ to investigate the influx of divalent cations activated by peroxide treatment without the intervention of Ca2+ extrusion and of Ca2+ storage mechanisms (Villalba et al., 2008). In endothelium‐denuded coronary arteries kept in Ca2+‐free PSS under conditions of l‐type channel blockade and stimulated with H2O2 (100 μM), addition of Ba2+ (1.5 mM) to the Ca2+‐free solution increased the Fura‐2‐AM ratio to a similar extent to the rise elicited by Ca2+ re‐addition (Figure 7A, B and D). Moreover, treatment of coronary arteries with Gd3+ (50 μM), a non‐selective blocker of cation channels, significantly reduced the increase in [Ca2+]i activated by H2O2 upon extracellular Ca2+ re‐addition (Figure 7C and E). These results suggest the involvement of non‐selective cation channels in the capacitative Ca2+ influx activated by H2O2 in coronary VSM.
Figure 7.
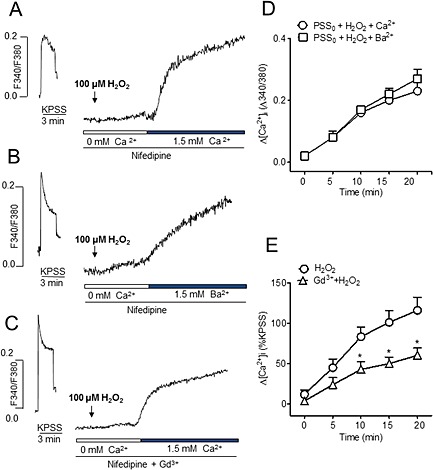
Non‐l‐type Ca2+ entry induced by H2O2 is mediated by non‐selective cation channels. (A, B, C) Representative traces showing (A) the increase in [Ca2+]i induced by 100 μM H2O2 in a Ca2+‐free medium upon Ca2+ re‐addition, (B) the increase in Fura‐2‐AM fluorescence in arteries stimulated with 100 μM H2O2 in a Ca2+‐free medium after re‐addition of 1.5 mM Ba2+ and (C) the increase in [Ca2+]i induced by 100 μM H2O2 in a Ca2+‐free medium upon Ca2+ re‐addition after treatment with 50 μM Gd3+ in endothelium‐denuded coronary arteries. (D) Average comparative effects on the increase in F340/380 upon Ca2+ and Ba2+ re‐addition in coronary arteries activated with 100 μM H2O2 in a Ca2+‐free medium. (E) Mean values of changes in [Ca2+]i in response to H2O2 in the presence and absence of the non‐selective cation channel inhibitor Gd3+ (50 μM). Results are expressed as a percentage of the KPSS‐induced ΔF340/380 and are means ± SEM of three to four animals. *P < 0.05, **P < 0.01, significant effects of Gd3+; Student's t‐test).
The role of Orai1 channels in the nifedipine‐resistant Ca2+ entry induced by H2O2 was further assessed by treatment of coronary arteries with the new selective inhibitor of the Orai1‐mediated SOC entry Pyr6. In the presence of Pyr6 (3 μM), Ca2+ influx activated by the re‐addition of Ca2+ in coronary arteries activated with either H2O2 (Figure 8A, C) or CPA (Figure 8 B,D) was abolished and reduced to levels similar to those obtained by Ca2+ re‐addition in the absence of either H2O2 or CPA in control experiments (Figure 3A).
Figure 8.
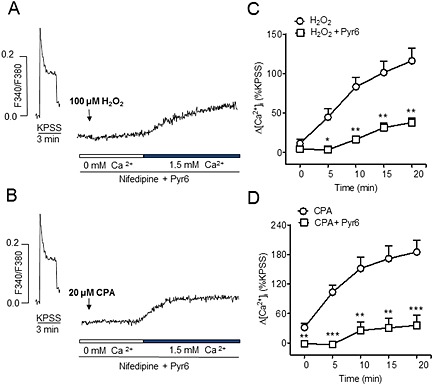
Non‐l‐type Ca2+ entry induced by H2O2 is inhibited by Pyr6, a selective blocker of Orai1‐mediated Ca2+ entry. (A, B) Representative traces showing the increase in [Ca2+]i induced by H2O2 (A) and by CPA (B) in the presence of Pyr6 (3 μM) in response to re‐addition of Ca2+ to the extracellular medium. (C, D) Mean values of the changes in [Ca2+]i induced by H2O2 (100 μM) (C) and CPA (20 μM) (D) in response to the re‐addition of Ca2+ to the extracellular medium in the absence and presence of Pyr6 (3 μM) in coronary arteries. Results are expressed as a percentage of the KPSS‐induced ΔF340/380 and are means ± SEM of four animals. *P < 0.05, **P < 0.01, ***P < 0.001, significant effects of Pyr6; Student's t‐test).
Thiol oxidation‐dependent effects of H2O2 on SOC entry in coronary VSM
Treatment of endothelium‐denuded coronary arteries with the thiol‐specific reducing agent DTT (100 μM) induced a marked inhibition of the voltage‐independent Ca2+ entry stimulated by H2O2 (Figure 9A) and to a lesser but not significant extent that induced by SERCA inhibition with CPA (Figure 9B), which suggests that the action of H2O2 on SOC entry in VSM is due to oxidation of thiol groups.
Figure 9.
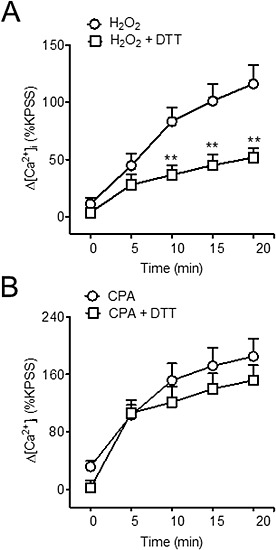
H2O2‐induced SOC entry is due to thiol oxidation. Average changes in [Ca2+]i induced by H2O2 (100 μM) (A) and CPA (B) in response to the re‐addition of Ca2+ to the extracellular medium in the absence and after treatment with the thiol specific reducing agent DTT (100 μM) in endothelium‐denuded coronary arteries. Results are expressed as a percentage of the KPSS‐induced ΔF340/380 and are means ± SEM of four to eight animals. *P < 0.05, **P < 0.01, significant effects of DTT; Student's t‐test.
Effect of H2O2 on SOC entry in coronary VSM in metabolic syndrome
Figure 10 shows that basal O2 − production was markedly enhanced in coronary arteries (Figure 10A) but not myocardium (Figure 10B) from insulin resistant OZR compared with their lean counterparts and was quenched by the O2 − scavenger tempol. These data suggest higher levels of vascular oxidative stress in animals with metabolic syndrome.
Figure 10.
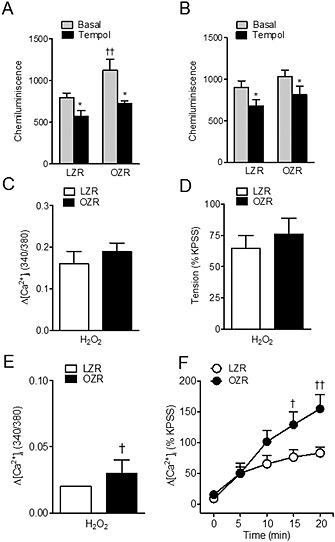
ROS generation and effects of H2O2 on [Ca2+]i and vasoconstriction in coronary arteries from LZR and OZR. (A, B) Basal O2 − production is increased in coronary arteries (A) but not myocardium (B) from OZR. Effect of the free radical scavenger tempol (30 μM) on basal superoxide production in coronary arteries (A) and myocardium (B) from LZR and OZR. (C, D) H2O2 induces rises in [Ca2+]i and vasoconstriction of similar magnitude in LZR and OZR. Changes induced by H2O2 (100 μM) in resting [Ca2+]i (C) and basal tension (D) in the presence of Ca2+ in the extracellular medium in endothelium‐denuded coronary arteries from LZR and OZR. (E) Intracellular Ca2+ mobilization induced by H2O2 in a Ca2+‐free medium was larger in coronary arteries from OZR. (F) Increases in [Ca2+]i induced by H2O2 (100 μM) in response to the re‐addition of Ca2+ (1,5 mM) to the extracellular medium in the presence of nifedipine (1 μM) was enhanced in coronary arteries from OZR compared with LZR. (A, B) Results are expressed as cpm (mg tissue)‐1 and bars represent mean ± SEM of 4–13 animals. (C–F) Results are expressed either as absolute values of ratio F340/380 (C, E) or as a percentage of the KPSS‐induced responses (D, F) and are means ± SEM of three to four animals. *P < 0.05, significantly different from basal; †P < 0.05, ††P < 0.01, significantly different from LZR; one‐way ANOVA.
In order to determine whether the increase in [Ca2+]i and vasoconstriction elicited by H2O2 in coronary VSM was affected by the conditions of metabolic syndrome, endothelium‐denuded coronary arteries from LZR and OZR were stimulated with a single concentration of H2O2 (100 μM). H2O2 induced a sustained increase in [Ca2+]i (Figure 10C) that was accompanied by a simultaneous and sustained vasoconstriction (Figure 10D) of similar magnitude in coronary arteries from LZR and OZR. Ca2+ responses to H2O2 were largely inhibited upon removal of extracellular Ca2+, and mobilization of intracellular Ca2+ by H2O2 was higher in arteries from OZR compared with LZR (Figure 10E). Furthermore, SOC entry activated by H2O2 in coronary VSM from OZR was impaired. Thus, in a Ca2+‐free medium in the presence of nifedipine, Ca2+ entry stimulated by H2O2 upon Ca2+ re‐addition after 20 min was significantly larger in coronary VSM from OZR than in that from LZR (Figure 10F). However, increases in [Ca2+]i induced by depolarization with KPSS were unchanged in OZR (∆F340/F380 0.20 ± 0.02, n = 5) compared with LZR (∆F340/F380 = 0.21 ± 0.05, n = 6).
Discussion
Interactions between ROS and the molecular machinery involved in non‐l‐type Ca2+ entry in VSM are relatively little explored. The present study provides evidence for a redox regulation of SOC entry in coronary VSM, as well as oxidative stress‐associated changes in this store depletion‐dependent Ca2+ influx under conditions of vascular dysfunction in the metabolic syndrome.
ROS regulation of voltage‐independent Ca2+ entry has previously been reported mainly in non‐excitable cells including blood and endothelial cells, where SOC entry is a major route for Ca2+ entry (Hu et al., 1998, 2000; Az‐ma et al., 1999; Redondo et al., 2004a,2004b; Florea and Blatter, 2008; Sun et al., 2011). In the present study, we show that H2O2 mobilizes Ca2+ from intracellular stores and activates a significant non‐l‐type Ca2+ entry in intact coronary arteries. Because H2O2‐induced [Ca2+]i rises in coronary arteries are partially dependent on the endothelium (Santiago et al., 2013), SOC entry was further characterized in endothelium‐denuded coronary arteries. Our data confirmed a nifedipine‐resistant Ca2+ entry activated by H2O2 in coronary VSM of similar magnitude to the one obtained by maximum high‐K+ depolarization leading to Ca2+ entry through l‐type channels and about two‐thirds the SOC entry obtained by inhibition of SERCA and passive depletion of the SR with CPA.
Although H2O2 is known to produce vasoconstriction via activation of Ca2+ entry through l‐type Ca2+ channels in coronary arteries (Santiago et al., 2013), the current results reveal an uncoupling between the nifedipine‐resistant Ca2+ entry and contraction activated by H2O2 in coronary VSM. H2O2 is a well‐recognized endogenous mediator of the EDH response in coronary arteries (Matoba et al., 2003; Miura et al., 2003; Yada et al., 2003; Shimokawa, 2010), and therefore, the possibility that H2O2 activates vasodilator mechanisms involving KCa channels (Barlow and White, 1998) or the Na+/K+ pump (Wong et al., 2014) cannot be ruled out. However, uncoupling of the nifedipine‐resistant Ca2+ entry stimulated by H2O2 and contraction in coronary arteries, as shown in the present study, is consistent with the uncoupling between the capacitative Ca2+ entry induced by SERCA inhibition with CPA and vasoconstriction in the same arteries. This dissociation between [Ca2+]i and contraction is in agreement with that earlier observed in other small arteries and arterioles and suggests that Ca2+ entering through SOC channels is confined to a subcellular compartment that has limited access to the contractile apparatus (Flemming et al., 2002; Snetkov et al., 2003; Villalba et al., 2007). In fact, SOC entry, CRAC currents and channel proteins involved in SOC entry have been reported to be up‐regulated in synthetic/proliferative VSM cells compared with quiescent/contractile vascular myocytes and associated to gene expression, proliferation and migration (Berra‐Romani et al., 2008; Potier et al., 2009; Li et al, 2011).
Studies on the role of ROS in SOC entry in non‐excitable cells have often provided controversial data, and both enhanced and attenuated ROS‐induced capacitative Ca2+ entry have been reported, these reciprocal effects being ascribed to the concentration, the time of exposure and the type of oxidant species used in acute experimental protocols. In human platelets (Redondo et al., 2004a), endothelial cells (Elliott et al., 1989; Hu et al., 1998; Florea and Blatter, 2008) and pulmonary artery myocytes (Lin et al., 2007), concentrations of H2O2 in the micromolar range up to 500 μM, dose‐dependently stimulated intracellular Ca2+ release and store depletion‐dependent Ca2+ entry, while higher millimolar concentrations had an inhibitory effect on SOC entry that was also time‐dependent (Florea and Blatter, 2008). In agreement with these observations, the present results indicate that redox regulation of SOC entry in coronary VSM is also dependent on the local levels of ROS and, whereas a maximum nifedipine‐resistant Ca2+ entry was obtained upon exposure to 100 μM H2O2 for 30 min, this Ca2+ response and also SOC entry elicited by SERCA inhibition with CPA were decreased at higher concentrations and practically abolished at millimolar concentrations of H2O2 in coronary arteries, which suggests an inhibition of store‐dependent Ca2+ entry under conditions of elevated vascular oxidative stress. Concentrations of H2O2 up to 100 μM have been reported in inflammatory microenvironments surrounding macrophages (Dröge, 2003). The inhibitory effects of high levels of H2O2 are currently explained on the basis of irreversible oxidations of thiol groups of proteins at higher oxidant conditions, considered as oxidative damage (Nunes and Demaurex, 2014). Consistent with this view, concentrations of H2O2 up to 100 μM have been shown to activate the Na+/K+ ATPase and the EDH‐type relaxant responses in porcine coronary arteries, while exposure to millimolar concentrations of H2O2 inhibited the pump and caused non‐specific relaxations by impairing VSM contraction (Ellis et al., 2003; Wong et al., 2014). The present findings further suggest that high oxidant stress might induce coronary relaxation by reducing Ca2+ entry in VSM.
ROS can influence SOC entry via either direct effects on the molecular machinery of SOC including Orai, the STIM1 and transient receptor potential canonic (TRPC) channels or indirectly by influencing the content of the intracellular SR Ca2+ stores (Trebak et al., 2010; Nunes and Demaurex, 2014). H2O2 has been reported to induce intracellular Ca2+ release via IP3R activation in blood cells (Parekh and Penner, 1995; Redondo et al., 2004b), and NADPH‐derived endothelial H2O2 has been shown to increase the sensitivity of intracellular Ca2+ stores to IP3 in human endothelial cells (Hu et al., 2000; Zheng and Shen, 2005). In the present study, the inhibitory effect of the IP3R antagonist heparin suggests that H2O2‐induced SOC Ca2+ entry in coronary VSM is partially mediated by store depletion through activation of IP3R. These findings are consistent with data obtained in DT40 B‐lymphocytes where both pharmacological inhibition of IP3R and molecular knock‐out of all three IP3Rs strongly reduced H2O2‐induced CRAC currents (Grupe et al., 2010). Heparin also significantly reduced intracellular Ca2+ mobilization and SOC entry activated by CPA in coronary arteries, which suggests that passive depletion of the SR by SERCA inhibition is blunted upon blockade of the IP3R.
The molecular identity of the non‐voltage‐gated Ca2+ channels mediating CRAC currents and SOC entry has been clarified during the last decade, and the ER Ca2+ sensor STIM1 (Roos et al., 2005), the plasma membrane Ca2+ selective pore‐forming subunit Orai1 (Vig et al., 2006) and also the channel proteins of the TRPC subfamily, including TRPC1 (Ong et al., 2007; Jardin et al., 2008), are known to interact to allow SOC entry. In the present study, the nifedipine‐resistant Ca2+ entry activated by H2O2 in coronary VSM exhibited classical pharmacological features of SOC entry in other cells, namely, inhibition by low concentrations of lanthanides and reproduction by Ba2+ fluorescence, indicative of Ba2+ influx through non‐selective cation channels (Parekh, 2006). Furthermore, the selective inhibitor of Orai1‐mediated Ca2+ entry, Pyr6 (Schleifer et al., 2012), abolished Ca2+ entry obtained by SERCA inhibition and also the non‐l‐type‐dependent Ca2+ entry activated in the presence of H2O2, thus indicating that H2O2 activates SOC entry in a Orai1‐dependent manner in coronary VSM. These data are consistent with recent observations in several native cell lines demonstrating that at micromolar concentrations, H2O2 activates Orai1‐mediated Ca2+ influx in a STIM1‐dependent manner, also dependent on activation of IP3R activity rather than modification of Orai1/STIM1 itself (Grupe et al., 2010). On the other hand, the present data showing that H2O2 mobilizes Ca2+ from intracellular stores and activates a non‐l‐type Ca2+ entry in coronary VSM agreed in part with that earlier reported in vascular myocytes from rat intralobar pulmonary arteries, where H2O2 releases Ca2+ from IP3‐sensitive and ryanodine‐sensitive intracellular stores and further activates a nifedipine‐resistant Ca2+ entry and a Ni2+‐sensitive cation current (Lin et al., 2007). However, this voltage‐independent Ca2+ entry was not inhibited by either lanthanides or TRPC blockers, and hence, TRPM2 channels were proposed to be involved in Ca2+ influx activated by H2O2 in pulmonary arterial myocytes.
It is well established that H2O2 regulates intracellular signalling pathways through reactions with the highly reactive cysteine residues in target proteins, these oxidative modifications including the reversible formation of disulfide bridges or oxidation products such as sulphenic acid, or the irreversible production of sulphinic or sulphonic acids (Trebak et al., 2010; Bogeski et al., 2011). The data provided here suggest that thiol oxidation produced store depletion‐dependent Ca2+ entry in coronary VSM, because the nifedipine‐resistant [Ca2+]i rise induced by H2O2, but not by SERCA inhibition with CPA, was abolished by the oxidant thiol reductant DTT. These findings are in line with functional studies demonstrating redox‐dependent regulation of coronary blood flow (Saitoh et al., 2006, 2007) and H2O2‐mediated EDH‐dependent relaxation (Wong et al., 2014) through oxidation of thiol groups in the redox sensitive p38 MAPK and the Na+/K+ ATPase respectively.
The fact that H2O2 mobilizes Ca2+ from intracellular stores through IP3R activation along with the inhibitory effect of DTT on H2O2‐induced SOC entry in coronary arteries further suggests that H2O2 might regulate IP3R activity through thiol oxidation to induce store depletion, as earlier shown in blood (Parekh and Penner, 1995; Redondo et al., 2004a) and endothelial cells where Ca2+‐induced Ca2+ release via the IP3R is enhanced by oxidant‐induced glutathionylation (Lock et al., 2012). However, involvement of other Ca2+ transport protein oxidation in the activation of SOC entry induced by H2O2 in coronary VSM, such as ryanodine receptors (Du et al., 2005), SERCA (Lancel et al., 2010), the Na+–Ca2+ exchanger (Kuster et al., 2010), Orai1 (Bogeski et al., 2010) and/or STIM1 Hawkins et al., 2010), cannot be ruled out.
Oxidative stress underlies the pathogenesis of vascular disease in diabetes, obesity and other insulin resistant states (Furukawa et al., 2004; Muñoz et al., 2015). On the other hand, accumulated evidence suggests a critical involvement of abnormal VSM [Ca2+]i homeostasis in the augmented vasoconstriction, remodelling and vascular dysfunction in insulin‐resistant states (Touyz, 2005; Villalba et al., 2008; Linde et al., 2012; Contreras et al., 2013). The present findings confirm high levels of oxidative stress in coronary arteries from OZR, an experimental model of the metabolic syndrome and insulin resistance, consistent with the high levels of ROS reported in the myocardium of these rats (Serpillon et al., 2009), despite the compensatory increase in antioxidant defences including the H2O2‐generating enzymes superoxide dismutases (SOD) (Conti et al., 2004). In our study, we found that mobilization of Ca2+ from intracellular stores and SOC entry obtained upon acute exposure to H2O2 were enhanced in arteries from obese compared with lean rats, as shown by the higher [Ca2+]i rise in a Ca2+‐free medium and by the larger nifedipine‐resistant Ca2+ entry induced by H2O2, and in contrast to the preserved l‐type‐mediated Ca2+ entry elicited by KPSS depolarization. These observations suggest that SR Ca2+ release and SOC entry activated by exogenous H2O2 are sensitized under conditions of increased vascular levels of ROS and would be consistent with the increased sensitivity of the intracellular Ca2+ stores to IP3R activation by H2O2 reported in human endothelial cells (Hu et al., 2000). Likewise, enhanced intracellular Ca2+ mobilization and Ca2+ entry have been found along with increased IP3R and SERCA2 expression in mesenteric artery myocytes from genetic hypertensive rats (Linde et al., 2012), where high levels of vascular oxidative stress have been reported (Touyz, 2005). Augmented Ca2+ signalling was associated with increased vasoconstriction in hypertensive rats, in contrast to the augmented H2O2‐induced SR Ca2+ release and entry not coupled to contraction in coronary arteries from obese rats. The latter would be consistent with the increased SOC entry and CRAC currents found in proliferative VSM myocytes (Berra‐Romani et al., 2008; Potier et al., 2009; Li et al, 2011).
In conclusion, we provide evidence that Orai1‐mediated SOC entry in coronary VSM is redox sensitive and can be activated by micromolar concentrations of H2O2 in part through the release of Ca2+ from IP3‐sensitive intracellular stores, whereas higher H2O2 concentrations are inhibitory and blunt SOC entry. Under conditions of vascular oxidative stress in the metabolic syndrome, SR Ca2+ release and store‐dependent Ca2+ entry responses to acute exposure to H2O2 are sensitized, which suggests abnormal Ca2+ handling, not related to augmented coronary vasoconstriction.
Author contributions
E. S. and D. P. conceived and designed the experiments. E. S., B. C. and M. M. performed the experiments. E. S., B. C., M. M. and D. P. analysed the data. B. C., A. G. S., L. R. and D. P. contributed reagents/materials/analysis tools. E. S. and D. P. wrote the manuscript. E. S., B. C., A. G. S., L. R. and D. P. revised the article critically for important intellectual content.
Conflict of interest
None declared.
Acknowledgements
This work was supported by grant SAF 2012‐31631 from MINECO, Spain. We thank Francisco Puente and Manuel Perales for their expert technical assistance.
Santiago, E. , Climent, B. , Muñoz, M. , García‐Sacristán, A. , Rivera, L. , and Prieto, D. (2015) Hydrogen peroxide activates store‐operated Ca2+ entry in coronary arteries. British Journal of Pharmacology, 172: 5318–5332. doi: 10.1111/bph.13322.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Ligand‐Gated Ion Channels. Br J Pharmacol 170: 1582–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbel Y, Havakuk O, Halkin A, Revivo M, Berliner S, Herz I et al. (2015). Relation of metabolic syndrome with long‐term mortality in acute and stable coronary disease. Am J Cardiol 115: 283–287. [DOI] [PubMed] [Google Scholar]
- Az‐ma T, Saeki N, Yuge O (1999). Cytosolic Ca2+ movements of endothelial cells exposed to reactive oxygen intermediates: role of hydroxyl radical‐mediated redox. Br J Pharmacol 126: 1462–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow R, White R (1998). Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity. Am J Physiol 275: 1283–1289. [DOI] [PubMed] [Google Scholar]
- Berra‐Romani R, Mazzocco‐Spezzia A, Pulina MV, Golovina VA (2008). Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am J Physiol Cell Physiol 295: C779–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogeski I, Kappl R, Kummerow C, Gulaboski R, Hoth M, Niemeyer B (2011). Redox regulation of calcium ion channels: chemical and physiological aspects. Cell Calcium 50: 407–23. [DOI] [PubMed] [Google Scholar]
- Bogeski I, Kummerow C, Al‐Ansary D, Schwarz E, Koehler R, Kozai D et al. (2010). Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal 3: ra24. doi: 10.1126/scisignal.2000672 [DOI] [PubMed] [Google Scholar]
- Chaplin N, Amberg G (2012). Hydrogen peroxide mediates oxidant‐dependent stimulation of arterial smooth muscle L‐type calcium channels. Am J Physiol Cell Physiol 302: C1382–C1393. [DOI] [PubMed] [Google Scholar]
- Conti M, Renaud IM, Poirier B, Michel O, Belair M‐F, Mandet C et al. (2004). High levels of myocardial antioxidant defense in aging nondiabetic normotensive Zucker obese rats. Am J Physiol Regul Integr Comp Physiol 286: R793–R800. [DOI] [PubMed] [Google Scholar]
- Contreras C, Sánchez A, Martínez P, Climent B, Benedito S, García‐Sacristán A et al. (2013). Impaired endothelin calcium signaling coupled to endothelin type B receptors in penile arteries from insulin‐resistant obese Zucker rats. J Sex Med 10: 2141–53. [DOI] [PubMed] [Google Scholar]
- Dröge W (2003). Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95. [DOI] [PubMed] [Google Scholar]
- Du W, Frazier M, Mcmahon T, Eu J (2005). Redox activation of intracellular calcium release channels (ryanodine receptors ) in the sustained phase of hypoxia‐induced pulmonary vasoconstriction. Chest 128: 556S–558S. [DOI] [PubMed] [Google Scholar]
- Elliott S, Eskin S, Schilling W (1989). Effect of t‐butyl‐hydroperoxide on bradykinin‐stimulated changes in cytosolic calcium in vascular endothelial cells. J Biol Chem 264: 3806–10. [PubMed] [Google Scholar]
- Ellis A, Pannirselvam M, Anderson TJ, Triggle CR (2003). Catalase has negligible inhibitory effects on endothelium‐dependent relaxations in mouse isolated aorta and small mesenteric artery. Br J Pharmacol 140: 1193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming R, Cheong A, Dedman A, Beech D (2002). Discrete store‐operated calcium influx into an intracellular compartment in rabbit arteriolar smooth muscle. J Physiol 543: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florea SM, Blatter L (2008). The effect of oxidative stress on Ca2+ release and capacitative Ca2+ entry in vascular endothelial cells. Cell Calcium 43: 405–415. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y et al. (2004). Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupe M, Myers G, Penner R, Fleig A (2010). Activation of store‐operated I(CRAC) by hydrogen peroxide. Cell Calcium 48: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD et al. (2010). S‐glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol 190: 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool L (2000). Hypoxia increases the sensitivity of the L‐type Ca(2+) current to beta‐adrenergic receptor stimulation via a C2 region‐containing protein kinase C isoform. Circ Res 87: 1164–71. [DOI] [PubMed] [Google Scholar]
- Hool L, Arthur P (2002). Decreasing cellular hydrogen peroxide with catalase mimics the effects of hypoxia on the sensitivity of the L‐type Ca2+ channel to beta‐adrenergic receptor stimulation in cardiac myocytes. Circ Res 91: 601–9. [DOI] [PubMed] [Google Scholar]
- Hu Q, Corda S, Zweier JL, Capogrossi MC, Ziegelstein RC (1998). Hydrogen peroxide induces intracellular calcium oscillations in human aortic endothelial cells. Circulation 97: 268–275. [DOI] [PubMed] [Google Scholar]
- Hu Q, Zheng G, Zweier J, Deshpande S, Irani K, Ziegelstein R (2000). NADPH oxidase activation increases the sensitivity of intracellular Ca2+ stores to inositol 1,4,5‐trisphosphate in human endothelial cells. J Biol Chem 275: 15749–57. [DOI] [PubMed] [Google Scholar]
- Jardin I, Lopez J, Salido G, Rosado J (2008). Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1‐forming Ca2+ channels. J Biol Chem 283: 25296–304. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuster GM, Lancel S, Zhang J, Communal C, Trucillo MP, Lim CC et al. (2010). Redox‐mediated reciprocal regulation of SERCA and Na+–Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic Biol Med 48: 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancel S, Qin F, Lennon S, Zhang J, Tong X, Mazzini M et al. (2010). Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq‐overexpressing mice. Circ Res 107: 228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McKeown L, Ojelabi O, Stacey M, Foster R, O'Regan D et al. (2011). Nanomolar potency and selectivity of a Ca2+ release‐activated Ca2+ channel inhibitor against store‐operated Ca2+ entry and migration of vascular smooth muscle cells. Br J Pharmacol 164: 382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Yang X, Cao Y, Sham J (2007). Hydrogen peroxide‐induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 292: L1598–608. [DOI] [PubMed] [Google Scholar]
- Lock JT, Sinkins WG, Schilling WP (2012). Protein S‐glutathionylation enhances Ca2+‐induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J Physiol 590: 3431–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde CI, Karashima E, Raina H, Zulian A, Wier WG, Hamlyn JM et al. (2012). Increased arterial smooth muscle Ca2+ signaling, vasoconstriction, and myogenic reactivity in Milan hypertensive rats. Am J Physiol Heart Circ Physiol 302: H611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Morikawa K, Kubota H, Kunihiro I, Urakami‐Harasawa L et al. (2003). Electron spin resonance detection of hydrogen peroxide as an endothelium‐derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler Thromb Vasc Biol 23: 1224–30. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W (1977). Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res 41: 19–26. [DOI] [PubMed] [Google Scholar]
- Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD (2003). Role for hydrogen peroxide in flow‐induced dilation of human coronary arterioles. Circ Res 92: e31–40. [DOI] [PubMed] [Google Scholar]
- Muñoz M, Sánchez A, Martínez P, Benedito S, López‐Oliva ME, García‐Sacristán A et al. (2015). COX‐2 is involved in vascular oxidative stress and endothelial dysfunction of renal interlobar arteries from obese Zucker rats. Free Radic Biol Med 84: 77–90. [DOI] [PubMed] [Google Scholar]
- Nunes P, Demaurex N (2014). Redox regulation of store‐operated Ca2+ entry. Antioxid Redox Signal 21: 915–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong H, Cheng K, Liu X, Bandyopadhyay B, Paria B, Soboloff J et al. (2007). Dynamic assembly of TRPC1–STIM1–Orai1 ternary complex is involved in store‐operated calcium influx. Evidence for similarities in store‐operated and calcium release‐activated calcium channel components. J Biol Chem 282: 9105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A (2006). On the activation mechanism of store‐operated calcium channels. Pflugers Arch 453: 303–11. [DOI] [PubMed] [Google Scholar]
- Parekh A, Penner R (1995). Activation of store‐operated calcium influx at resting InsP3 levels by sensitization of the InsP3 receptor in rat basophilic leukaemia cells. J Physiol 489: 377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A, Putney JJ (2005). Store‐operated calcium channels. Physiol Rev 85: 757–810. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier M, Gonzalez J, Motiani R, Abdullaev I, Bisaillon J, Singer H et al. (2009). Evidence for STIM1‐ and Orai1‐dependent store‐operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J 23: 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourmahram G, Snetkov V, Shaifta Y, Drndarski S, Knock G, Aaronson P et al. (2008). Constriction of pulmonary artery by peroxide: role of Ca2+ release and PKC. Free Radic Biol Med 45: 1468–1476. [DOI] [PubMed] [Google Scholar]
- Redondo P, Salido G, Pariente J, Rosado J (2004a). Dual effect of hydrogen peroxide on store‐mediated calcium entry in human platelets. Biochem Pharmacol 67: 1065–1076. [DOI] [PubMed] [Google Scholar]
- Redondo PC, Salido GM, Rosado JA, Pariente JA (2004b). Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem Pharmacol 67: 491–502. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio P, Yeromin A, Ohlsen K, Lioudyno M, Zhang S et al. (2005). STIM1, an essential and conserved component of store‐operated Ca2+ channel function. J Cell Biol 169: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh S, Kiyooka T, Rocic P, Rogers P, Zhang C, Swafford A et al. (2007). Redox‐dependent coronary metabolic dilation. Am J Physiol Heart Circ Physiol 293: H3720–H3725. [DOI] [PubMed] [Google Scholar]
- Saitoh S, Zhang C, Tune J, Potter B, Kiyooka T, Rogers P et al. (2006). Hydrogen peroxide: a feed‐forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol 26: 2614–2621. [DOI] [PubMed] [Google Scholar]
- Saleem H, Tovey SC, Molinski TF, Taylor CW (2014). Interactions of antagonists with subtypes of inositol 1,4,5‐trisphosphate (IP3) receptor. Br J Pharmacol 171: 3298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago E, Contreras C, García‐Sacristán A, Sánchez A, Rivera L, Climent B et al. (2013). Signaling pathways involved in the H2O2‐induced vasoconstriction of rat coronary arteries. Free Radic Biol Med 60: 136–146. [DOI] [PubMed] [Google Scholar]
- Schleifer H, Doleschal B, Lichtenegger M, Oppenrieder R, Derler I, Frischauf I et al. (2012). Novel pyrazole compounds for pharmacological discrimination between receptor‐operated and store‐operated Ca(2+) entry pathways. Br J Pharmacol 167: 1712–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpillon S, Floyd B, Gupte R, George S, Kozicky M, Neito V et al. (2009). Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose‐6‐phosphate dehydrogenase‐derived NADPH. Am J Physiol Heart Circ Physiol 297: H153–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H (2010). Hydrogen peroxide as an endothelium‐derived hyperpolarizing factor. Pflugers Arch 459: 915–22. [DOI] [PubMed] [Google Scholar]
- Snetkov V, Aaronson P, Ward J, Knock G, Robertson T (2003). Capacitative calcium entry as a pulmonary specific vasoconstrictor mechanism in small muscular arteries of the rat. Br J Pharmacol 140: 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Yau HY, Lau OC, Huang Y, Yao X (2011). Effect of hydrogen peroxide and superoxide anions on cytosolic Ca2+: comparison of endothelial cells from large‐sized and small‐sized arteries. PLoS One 6: e25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz R (2005). Reactive oxygen species as mediators of calcium signaling by angiotensin II: implications in vascular physiology and pathophysiology. Antioxid Redox Signal 7: 1302–14. [DOI] [PubMed] [Google Scholar]
- Trebak M, Ginnan R, Singer H, Jourd D (2010). Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid Redox Signal 12: 657–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa D, Rabah D, Koblan– Huberson M et al. (2006). CRACM1 is a plasma membrane protein essential for store‐operated Ca2+ entry. Science 312: 1220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba N, Martínez P, Bríones A, Sánchez A, Salaíces M, García‐Sacristán A et al. (2009). Differential structural and functional changes in penile and coronary arteries from obese Zucker rats. Am J Physiol Heart Circ Physiol 297: H696–707. [DOI] [PubMed] [Google Scholar]
- Villalba N, Stankevicius E, Garcia‐Sacristán A, Simonsen U, Prieto D (2007). Contribution of both Ca2+ entry and Ca2+ sensitization to the alpha1‐adrenergic vasoconstriction of rat penile small arteries. Am J Physiol Heart Circ Physiol 292: H1157–69. [DOI] [PubMed] [Google Scholar]
- Villalba N, Stankevicius E, Simonsen U, Prieto D (2008). Rho kinase is involved in Ca2+ entry of rat penile small arteries. Am J Physiol Heart Circ Physiol 294: 1923–1932. [DOI] [PubMed] [Google Scholar]
- Viola H, Arthur P, Hool L (2007). Transient exposure to hydrogen peroxide causes an increase in mitochondria‐ derived superoxide as a result of sustained alteration in L‐type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ Res 100: 1036–44. [DOI] [PubMed] [Google Scholar]
- Wong PS, Garle MJ, Alexander SPH, Randall MD, Roberts RE (2014). A role for the sodium pump in H2O2‐induced vasorelaxation in porcine isolated coronary arteries. Pharmacol Res 90: 25–35. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Shen X (2005). H2O2 directly activates inositol 1,4,5‐trisphosphate receptors in endothelial cells. Redox Rep 10: 29–36. [DOI] [PubMed] [Google Scholar]
- Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M et al. (2003). Hydrogen peroxide, an endogenous endothelium‐derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation 107: 1040–1045. [DOI] [PubMed] [Google Scholar]
- Yada T, Shimokawa H, Hiramatsu O, Shinozaki Y, Mori H, Goto M et al. (2007). Important role of endogenous hydrogen peroxide in pacing‐induced metabolic coronary vasodilation in dogs in vivo. J Am Coll Cardiol 50: 1272–1278. [DOI] [PubMed] [Google Scholar]